
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and biological evaluation of novel nitric oxide-donating podophyllotoxin derivatives as potential antiproliferative agents against multi-drug resistant leukemia cells†
Lei Zhang *a,
Ying Rongb,
Jie Zhengc,
Chengli Yanga,
Yongzheng Chena,
Jing Wang*a and
Gang Wei*d
*a,
Ying Rongb,
Jie Zhengc,
Chengli Yanga,
Yongzheng Chena,
Jing Wang*a and
Gang Wei*d
aGeneric Drug Research Center of Guizhou Province, Green Pharmaceuticals Engineering Research Center of Guizhou Province, School of Pharmacy, Zunyi Medical University, Zunyi 563003, PR China. E-mail: lzhang@zmc.edu.cn; wangjing@zmc.edu.cn
bSecond Department of Pediatrics, Affiliated Hospital of Zunyi Medical University, Zunyi 563003, PR China
cDepartment of Nephrology & Rheumatology, Affiliated Hospital of Zunyi Medical University, Zunyi 563003, PR China
dCSIRO Manufacturing, PO Box 218, Lindfield, NSW 2070, Australia. E-mail: gang.wei@csiro.au
First published on 5th October 2018
Abstract
Multidrug resistance remains a major obstacle for the effective treatment of carcinoma. To find new drugs for the chemotherapy of drug-resistant leukemia, in this study, two novel nitric oxide-donating podophyllotoxin derivatives were synthesized and preliminarily evaluated in vitro. Biological evaluation indicated that the more active molecule, S1, enhanced the intracellular NO level and significantly inhibited the proliferation of drug-resistant K562/VCR and K562/ADR cells with IC50 values of 0.008 ± 0.001 and 0.007 ± 0.001 μM, respectively, which were similar to that of sensitive K562 cells. Furthermore, it was observed that S1 blocked the G2 phase of the K562/ADR cell cycle by disruption of the microtubule organization and inhibition of CDK1 and CDK2 expression. Meanwhile, S1 induced apoptosis of K562/ADR cells via mitochondrial depolarization and activation of caspase-3. In addition, S1 suppressed the P-gp expression, induced autophagy by regulation of Beclin1 and LC3-II, and inhibited the mTOR and STAT3 signaling in K562/ADR cells. Overall, S1 may be a promising candidate against drug-resistant leukemia.
Introduction
Multidrug resistance (MDR) is a leading clinical problem resulting in the failure of carcinoma chemotherapy. MDR of cancer is attributed to several factors, such as evasion of apoptosis, DNA repair, overexpression of enzymes with elimination property, and increase of drug efflux.1 Recent studies have showed that overexpression of ATP-binding cassette (ABC) transporters is the major reason, which could pump chemotherapy agents (e.g. etoposide, doxorubicin and vincristine) out of cytoplasm, resulting in low levels of intracellular drugs.2 There are three major ABC transporters in cancer MDR, including P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), and multidrug resistance-associated protein 1 (MRP1).3 Therefore, one of the strategies to reverse MDR is the inhibition of efflux pumps expression and activity, however, there is no ABC transporter inhibitor in clinical trials.4 Accordingly, novel strategies are a pressing need to find new agents with the property of potent cytotoxicity against MDR cancer cells.5,6Podophyllotoxin (PPT, 1, Fig. 1), a naturally occurring cyclolignan isolated mainly from Podophyllum hexandrum and Podophyllum peltatum, shows significant antineoplastic and antiviral activities, attracting much attention from investigators.7,8 This unique product could disrupt microtubule formation via inhibiting tubulin polymerization.9 However, toxic side effects had limited its clinical application.10 Therefore, extensive modifications have been widely carried out for many years. Until now, many semi-synthetic derivatives of podophyllotoxin with potent antitumor activity have been prepared, such as etoposide (2, Fig. 1) and teniposide (3, Fig. 1), which have been widely used as antineoplastic agents for clinical treatment by inhibiting another target, DNA-topoisomerase II.11,12 Although the two drugs have been used for the therapy of a variety of malignancies, lower solubility, drug-resistance and severe side effects prompt urgently to seek for new podophyllotoxin derivatives.13 The structure–activity relationship (SAR) and molecular modeling analysis demonstrate that C-4 in C ring of podophyllotoxin is the primary modified site, which is tolerant to considerable structural diversification, including many bulky groups.14–16 Recent studies have showed that podophyllotoxin and its derivatives of C-4 modifications have significant antitumor activity against several drug-resistant cancer cell lines, suggesting that they might have the potential to reverse MDR.17–21 For example, Liu and coworkers reported the synthesis and antiproliferative activity of dichloroplatinum(II) complexes of podophyllotoxin. The most active complex, 4 (Fig. 1), displayed excellent cytotoxicity against resistant K562/ADR cells in vitro with an IC50 value of 0.091 μM.22 Our group discovered that the conjugate of podophyllotoxin and aspirin (5, Fig. 1) significantly reversed the resistance of Bel-7402/5-FU cells via several pathways. Treatment of Bel-7402/5-FU cells with 5 markedly induced apoptosis, disrupted microtubule network, and inhibited MRP1 expression.23 More recently, Wang et al. synthesized the hybrid of podophyllotoxin and indirubin, 6 (Fig. 1), which displayed significant cytotoxicity against resistant K562/VCR cells. It was observed that 6 induced apoptosis and cell cycle arrest, caused the accumulation of ROS, regulated AKT and JNK signaling, and suppressed the expression levels of P-gp and MRP1 proteins.24
 | ||
| Fig. 1 The structures of podophyllotoxin and its derivatives. | ||
Nitric oxide (NO), an important signaling and/or effector molecule, plays a critical role in many physiological and pathophysiological processes.25 Numerous investigations have showed that high concentrations of NO inhibit cancer progression via inducing apoptosis, sensitizing tumors to chemotherapy, and reversing resistance to chemotherapy in vitro and in vivo.26,27 NO displays antitumor effect by inhibition of key transcription factors, DNA repair enzymes and drug efflux pumps.28,29 Actually, the direct use of NO has been applied in clinical practice for many years. However, owing to the inconvenient handling and high reactivity, NO donors have gained increasing attention in recent decades. NO donors are chemical compounds which generate NO in vitro or in vivo, including organic nitrates, diazeniumdiolates, furoxans and S-nitrosothiols. And the application of NO donors in combination with anticancer drugs has been widely carried out in many preclinical and clinical studies.30,31 However, because NO has various biological processes, so the design of target molecules by hybridization of NO donor(s) with anticancer drug has been well investigated.32,33 For instance, Stewart et al. reported that compound 7 (Fig. 2), a NO-NSAID called NO-sulindac, showed antiproliferative and proapoptotic effects against PC-3 cells, and directly inhibited the hypoxia response of PC-3 cells by decreasing HIF-1α translation and Akt signalling pathway.34 Fang and coworkers synthesized nitrate/oleanolic acid hybrids with amino acid/dipeptide moiety linkers. Among them, compound 8 (Fig. 2) possessed significant cytotoxicity against A549 cells with an IC50 value of 7.5 ± 1.0 μM, which was probably due to its NO-releasing property.35 More importantly, NO-donating agents usually do not induce an acquired resistance in tumor cells. For example, Rothweiler group demonstrated that NO-modified saquinavir derivative (9, Fig. 2) had strong antitumor effect on drug-resistant cancer cells overexpressing ABC transporters and sensitized resistant cancer cells to chemotherapy with less toxic effects.36 In addition, compound 10 (Fig. 2), designed by conjugation of doxorubicin with NO donor nitrooxy moiety and synthesized by Riganti and coworkers, increased DOX accumulation in resistant HT29/DOX cancer cells, inducing high cytotoxicity. Preliminary studies suggested that 10 could inhibit the activity of drug efflux.37 Subsequently, the group demonstrated that 10 exhibited a faster uptake and localized in the mitochondria, where it damaged the mitochondria-associated ABC transporters, resulting in the activation of caspase-9 and -3 in HT29/DOX cells. The paper suggested that 10 may be a novel anthracycline with distinct cellular targets, and it had greater efficacy against MDR tumors.38 In addition, the research group of Zhang and Huang synthesized a lot of nitric oxide-releasing bifendate and 2-cyano-3,12-dioxooleana-9-dien-28-oic acid (CDDO) derivatives. Biological evaluations indicated that several compounds significantly inhibited the proliferation of drug-resistant MCF-7/ADR, HCT-8/5-FU or K562/ADR cells in vitro and in vivo.39–41 These findings suggested that NO-donating agents may be promising agents for the treatment of drug-resistant tumors.
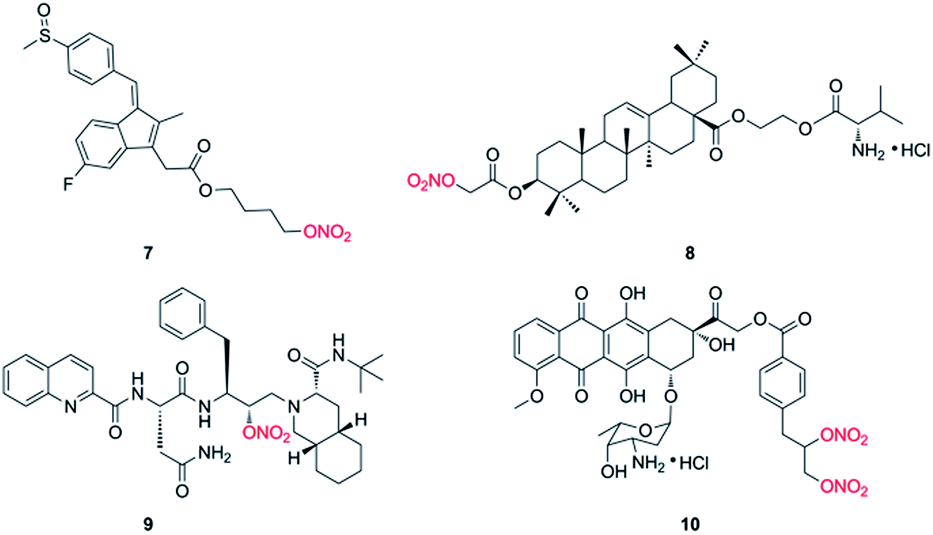 | ||
| Fig. 2 The structures of nitric oxide-donating agents. | ||
From above investigations, we hypothesized that hybridization of podophyllotoxin and NO donor moiety could generate new NO-donating podophyllotoxin derivatives, which might exert synergistic effect against MDR tumor cells. Therefore, to test the hypothesis, in present study, we synthesized novel NO-releasing podophyllotoxin derivatives by coupling podophyllotoxin with organic nitrate group (–ONO2) through aliphatic spacer using the molecular hybridization strategy (Fig. 3). Additionally, we further evaluated the inhibitory activities against drug-resistant K562 cells, and multiple molecular mechanisms involved in multidrug resistance reverting property of designed NO-donating podophyllotoxin derivatives.
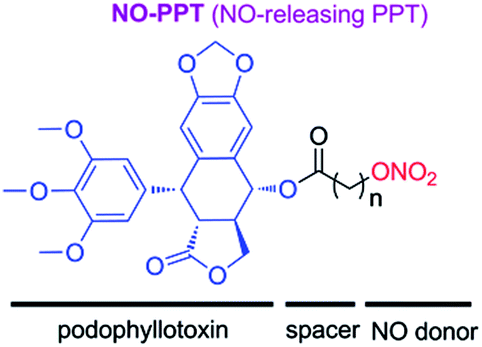 | ||
| Fig. 3 Design of nitric oxide-donating podophyllotoxin derivatives. | ||
Results and discussion
Synthesis of compounds S1-S2 is depicted in Scheme 1. As the starting material, podophyllotoxin was coupled with chloro acyl chloride in the presence of triethylamine (Et3N) to yield the corresponding intermediates, which were directly treated with the silver nitrate (AgNO3) to produce the target molecules S1-S2. The target compounds were purified by flash chromatography, and their structures were confirmed by 1H NMR, 13C NMR and high-resolution mass spectra (HRMS).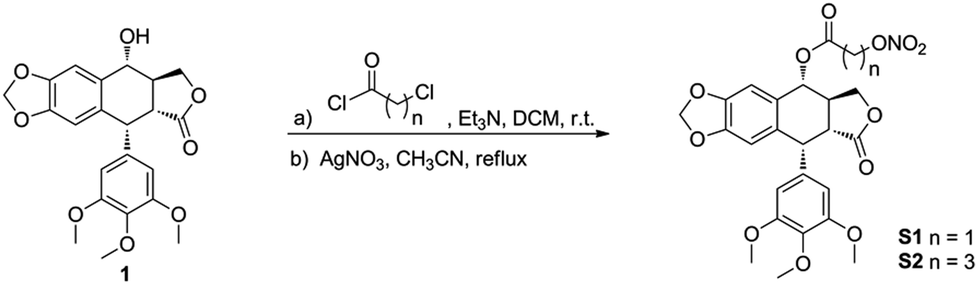 | ||
| Scheme 1 Synthesis of oxide-donating podophyllotoxin derivatives. | ||
The antiproliferative activities of the target compounds against sensitive human leukemic cells (K562) and drug-resistant leukemic cells (K562/VCR and K562/ADR) were evaluated by CCK-8 assay in vitro using podophyllotoxin (1), vincristine (VCR) and adriamycin (ADR) as positive controls. The IC50 values of test compounds were provided in Table 1. The two target molecules exhibited potent antiproliferative activity against above three leukemic cell lines with nanomolar IC50 values. Especially, the more potent compound S1, possessing two carbon chain, showed similar inhibitory activity against resistant K562/VCR and K562/ADR cells (IC50 = 0.008 ± 0.001 and 0.007 ± 0.001 μM, respectively) to sensitive K562 cells (IC50 = 0.007 ± 0.001 μM). However, it was notable that compound S2 showed less inhibitory activity against above cancer cells than compounds S1 and 1. Furthermore, as shown in Table 1, the antitumor activity of S1 against above three sensitive and resistant leukemic cell lines were higher than that of lead compound 1. That may be related to the distance between the ester bond and –ONO2 group in the compound. The two groups both owned the electron-withdrawing effect, so the hybrid with less length linker was more instability, which might release NO easily. Moreover, the longer group at C-4 might block the insertion of compound S2 into the colchicine binding site of the tubulin. Notably, S1 displayed about 1000 fold more cytotoxic against K562/VCR and K562/ADR cells than two positive clinical drugs, VCR and ADR.
| Compd. | IC50 (μM)a | ||
|---|---|---|---|
| K562 | K562/VCR (RFb) | K562/ADR (RFc) | |
| a CCK-8 method, drug exposure was for 72 h. Data were expressed as mean IC50 ± SD (μM).b RF = IC50(K562/VCR)/IC50(K562).c RF = IC50(K562/ADR)/IC50(K562).d Literature values.24 | |||
| S1 | 0.007 ± 0.001 | 0.008 ± 0.001 (1.14) | 0.007 ± 0.001 (1.00) |
| S2 | 0.018 ± 0.004 | 0.074 ± 0.008 (4.11) | 0.083 ± 0.007 (4.61) |
| 1 | 0.012 ± 0.001 | 0.032 ± 0.002 (2.66) | 0.025 ± 0.004 (2.08) |
| VCR | 0.178 ± 0.006d | 4.737 ± 0.647 (26.61)d | 6.860 ± 0.895 (38.53) |
| ADR | 0.037 ± 0.002 | 8.527 ± 1.505 (230.45) | 7.403 ± 0.285 (200.08) |
From Table 1, it could be found that VCR and ADR showed much weaker inhibitory activity against resistant K562/VCR and K562/ADR cell lines with resistant factor (RF) values of 26.61, 38.53 and 230.45, 200.08, respectively. In agreement with our previous reports,42–44 podophyllotoxin had significant antitumor activity against resistant K562/VCR and K562/ADR cells with RF values of 2.66 and 2.08, respectively. More importantly, it was noticed that S1 had similar antiproliferative activities against both sensitive human leukemic cells (K562) and drug-resistant leukemic cells (K562/VCR and K562/ADR) with RF values of 1.14 and 1.00, respectively, suggesting that S1 might be a promising drug candidate for overcoming the MDR in resistant leukemia.
To investigate whether the inhibition of cell growth by molecule S1 was caused by a cell-cycle effect, the cell cycle distribution was measured after staining the DNA with propidium iodide (PI) by flow cytometry. As shown in Fig. 4, treatment of K562/ADR cells with S1 at concentrations of 0.01 and 0.02 μM resulted in accumulation of 30.95% and 35.47% of cells at the G2 phase, respectively, as compared with 1.89% in the control group. These results indicated that S1 significantly induced K562/ADR cell cycle arrest at G2 phase.
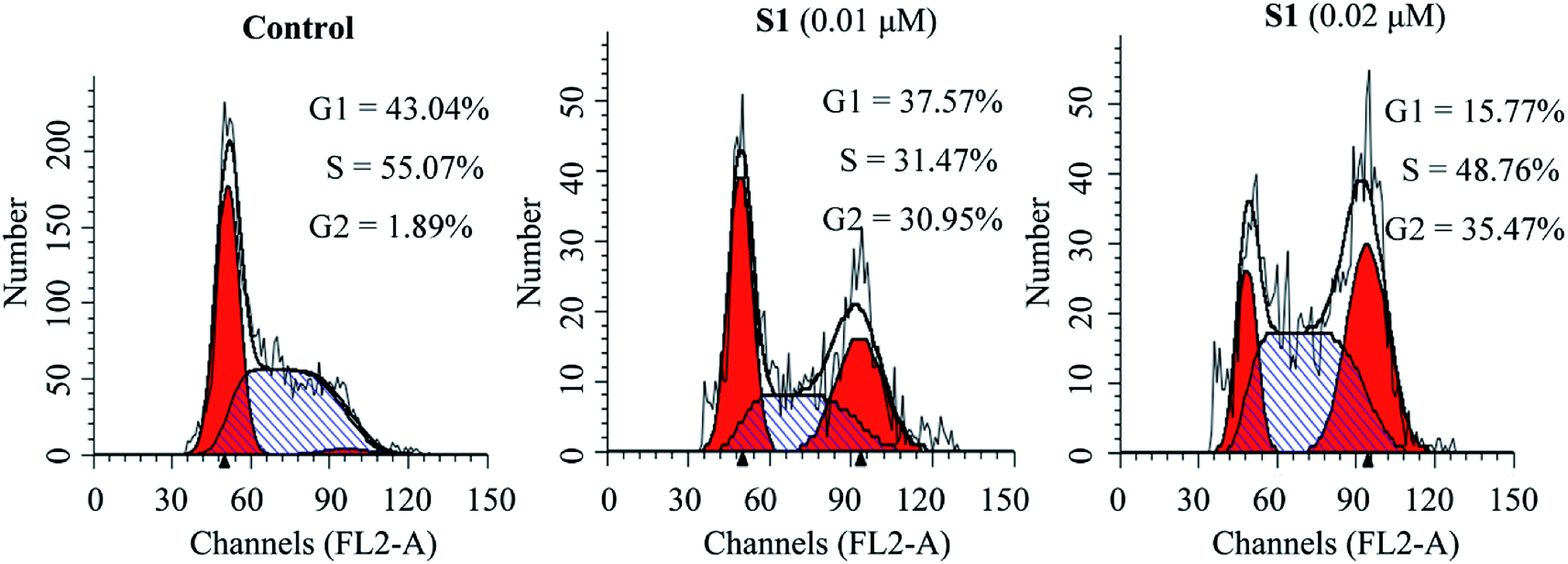 | ||
| Fig. 4 Effects of S1 on cell cycle of K562/ADR cells. K562/ADR cells were incubated with vehicle, 0.01 μM S1 and 0.02 μM S1 for 48 h, respectively, and stained with PI, followed by flow cytometry analysis. | ||
To explore whether S1 could be able to induce cancer cell apoptosis, K562/ADR cells were incubated with vehicle, 0.01 μM S1 or 0.02 μM S1 for 48 h, and stained with Annexin V-APC/7-AAD, followed by flow cytometric analysis. It was observed from Fig. 5 that treatment with S1 at concentrations of 0.01 and 0.02 μM resulted in 12.72% and 25.65% apoptotic cells, respectively, as compared with 5.83% in the control group, indicating that S1 induced apoptotic cell death in K562/ADR cells.
 | ||
| Fig. 5 Effects of S1 on apoptosis of K562/ADR cells. K562/ADR cells were incubated with vehicle, 0.01 μM S1 and 0.02 μM S1 for 48 h, respectively, and stained with Annexin V-APC/7-AAD, followed by flow cytometric analysis. | ||
Previous report showed that the loss of mitochondrial membrane potential (ΔΨm) played a significant role in the process of apoptosis.45 Here, we examined the effect of S1 on ΔΨm using the lipophilic mitochondrial probe JC-1. As seen in Fig. 6, after treatment with S1 at concentrations of 0.01 and 0.02 μM for 48 h, the number of K562/ADR cells with loss ΔΨm increased to 17.84% and 27.74%, respectively, as compared with 7.23% in the control group. These results suggested that S1 caused mitochondrial depolarization of K562/ADR cells in the process of apoptosis.
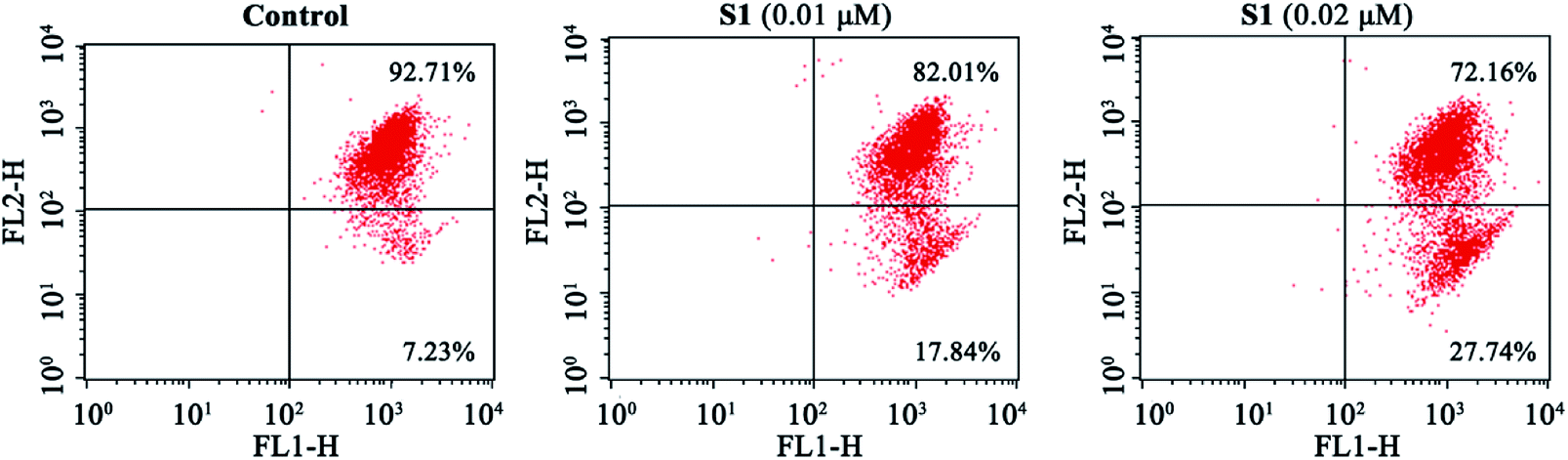 | ||
| Fig. 6 Effects of S1 on the mitochondrial membrane potential of K562/ADR cells. K562/ADR cells were incubated with vehicle, 0.01 μM S1 and 0.02 μM S1 for 48 h, respectively, and stained with fluorescence probe JC-1 dye, followed by flow cytometric analysis. | ||
To further obtain the effect of S1 on the microtubule, the cellular morphology of microtubule was further investigated by immunofluorescence staining. K562/ADR cells were incubated with vehicle, 0.01 μM S1 and 0.02 μM S1 for 24 h, respectively. As shown in Fig. 7, cells showed well-organized microtubule network in the control group, however, cells treated with 0.01 μM or 0.02 μM S1 displayed disrupted microtubule organization and reduced microtubule density, indicating that S1 could disrupt the microtubule network in K562/ADR cells.
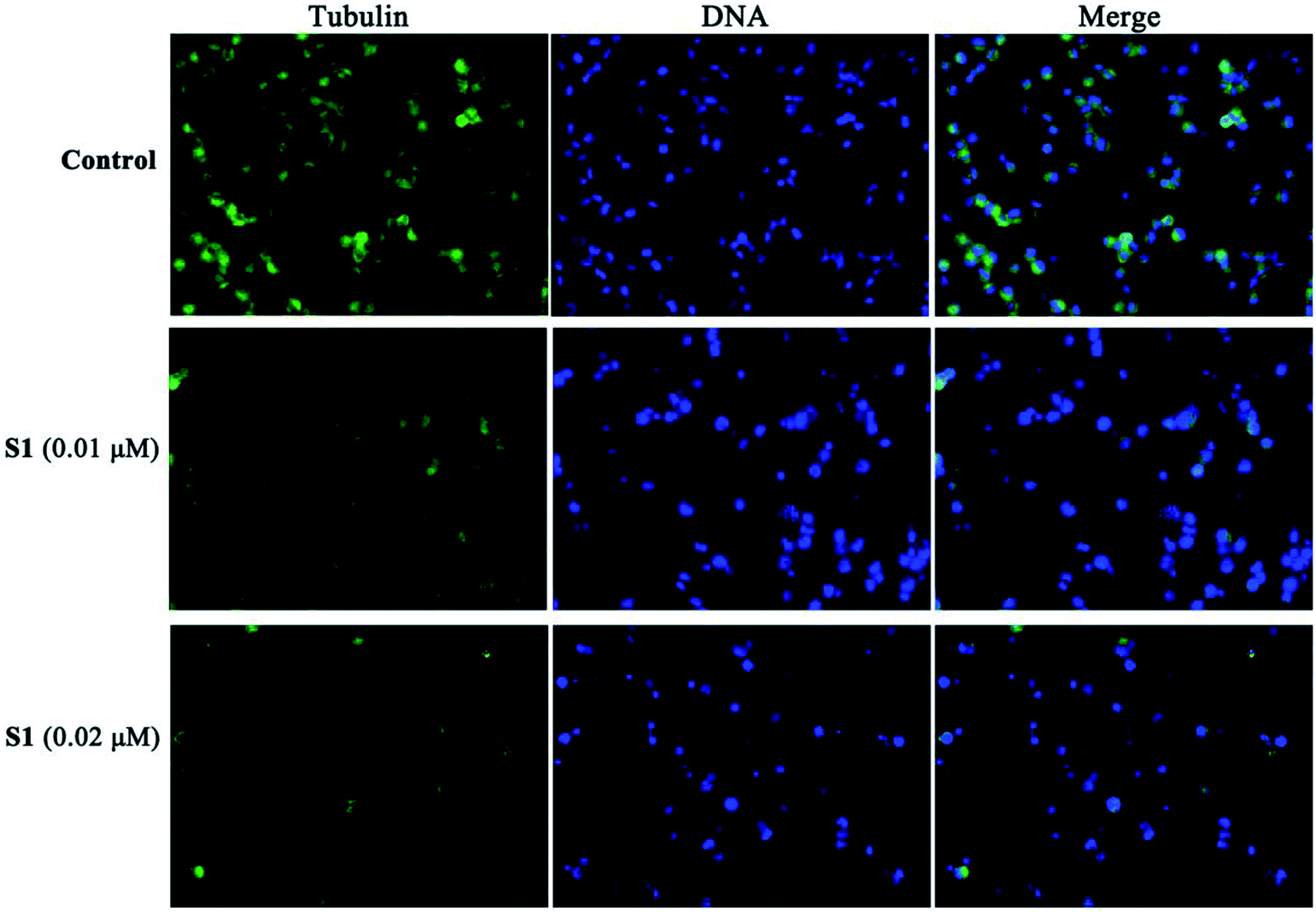 | ||
| Fig. 7 Effects of S1 on the organization of the cellular microtubule network in K562/ADR cells by immunofluorescence. K562/ADR cells were incubated with vehicle, 0.01 μM S1 and 0.02 μM S1 for 24 h, respectively. The cell nucleuses were stained with DAPI (blue), and microtubules were visualized with anti-a-tubulin conjugated with FITC (green). Magnification 400×. | ||
The intracellular NO levels were investigated using DAF-FM DA as a fluorescent probe. K562/ADR cells were incubated with vehicle, 0.01 μM S1, 0.02 μM S1 or 0.02 μM S2 for 24 h and stained with DAF-FM DA, followed by flow cytometric analysis. As described in Fig. 8, NO level increased to 40.85%, 41.29% and 29.31% after treatment with 0.01 μM S1, 0.02 μM S1 and 0.02 μM S2, respectively, as compared with 7.57% in control group. These results suggested that compound S1 could enhance the intracellular NO level in K562/ADR cells, additionally, compound S2 induced less effect on the intracellular NO level compared with compound S1.
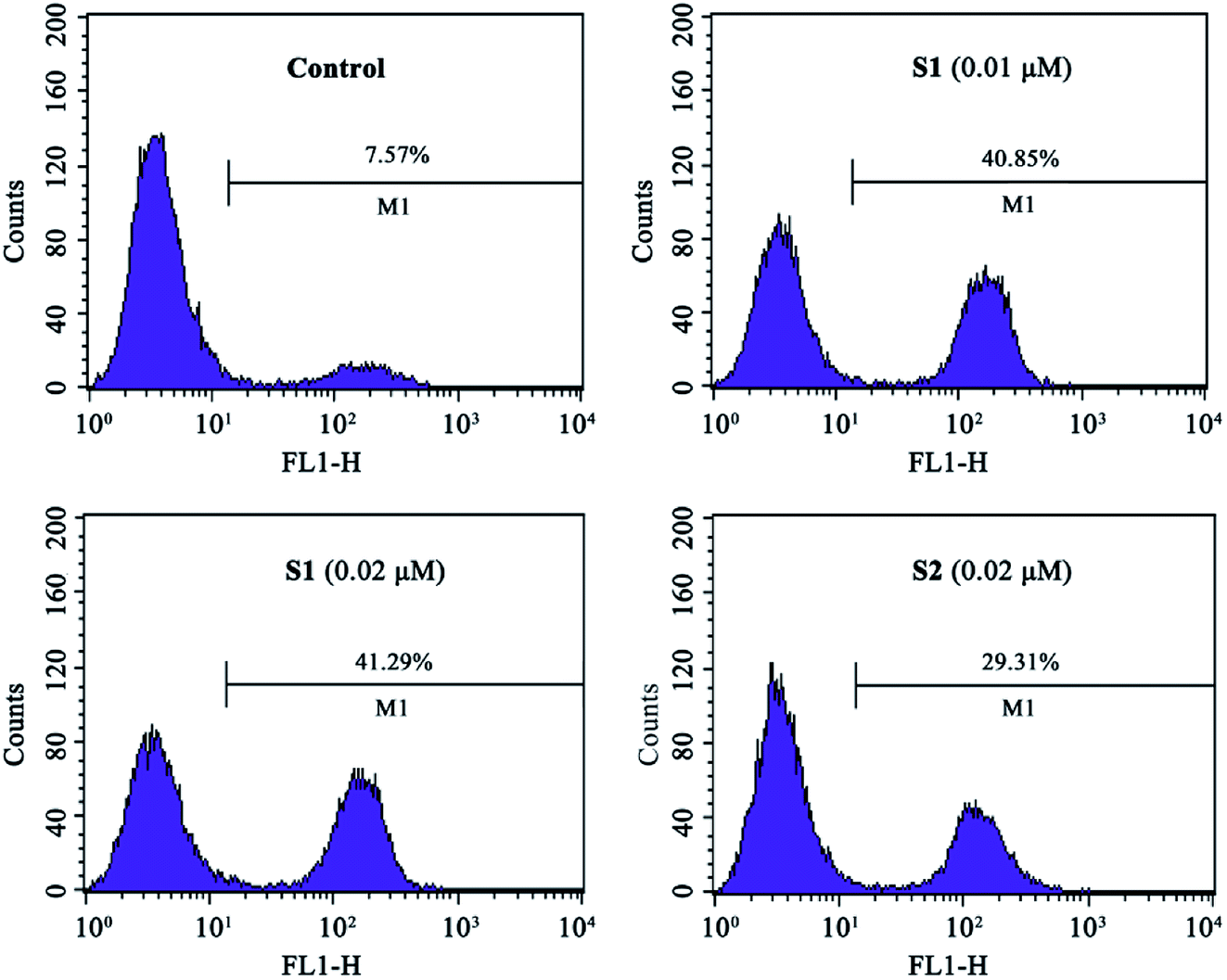 | ||
| Fig. 8 The generation of NO in K562/ADR cells. K562/ADR cells were incubated with vehicle, 0.01 μM S1, 0.02 μM S1 and 0.02 μM S2 for 24 h, respectively. The amount of NO was assayed by DAF-FM DA staining and detected using flow cytometry. | ||
Flow cytometry had shown that S1 could trigger cell cycle arrest and induce apoptosis in K562/ADR cells. In order to explore the molecular mechanisms involved in cell cycle and apoptosis, the expression levels of CDK1, CDK2 and cleaved caspase-3 were detected by western blotting. As shown in Fig. 9, S1 inhibited CDK1 and CDK2 expression, and promoted caspase-3 activation as compared with the control. The results indicated that S1 induced cell cycle arrest and apoptosis of K562/ADR cells through suppressing the levels of CDK1 and CDK2, and triggering cleavage of caspase-3.
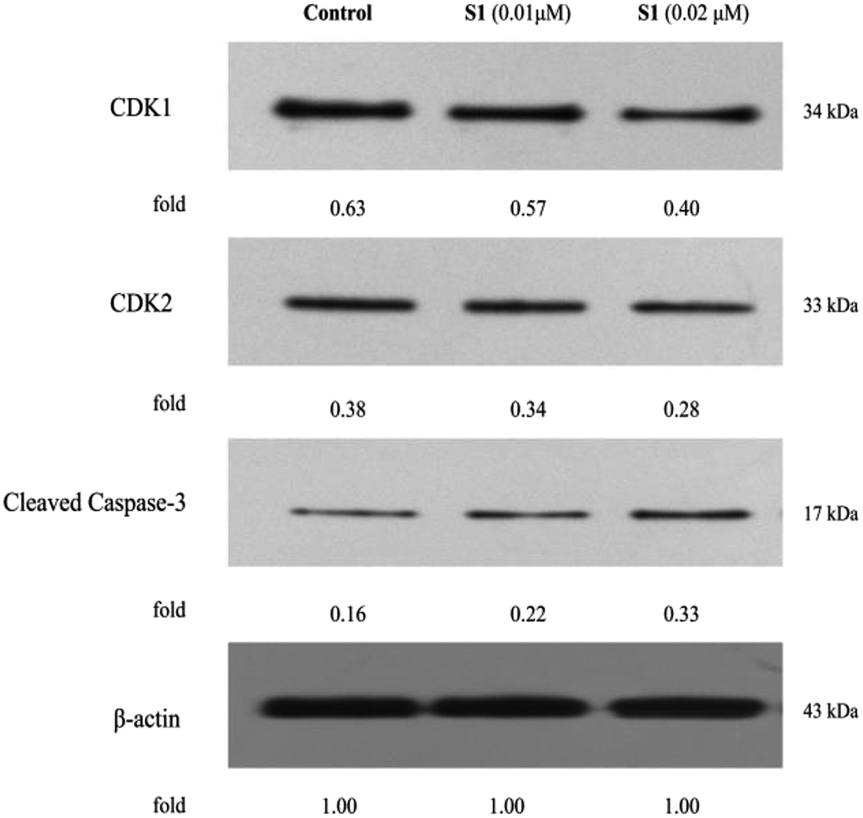 | ||
| Fig. 9 Effects of S1 on the levels of cell cycle- and apoptosis-related proteins of K562/ADR cells. K562/ADR cells were incubated with vehicle, 0.01 μM S1 and 0.02 μM S1 for 48 h, respectively, and the total cell lysates were prepared and analyzed by western blotting. | ||
To further validate the underlying mechanisms of S1, the impact of S1 on the expression levels of MDR-related proteins in K562/ADR cells was determined by western blotting (Fig. 10). It was found that S1 could significantly reduce the level of P-gp, however, S1 displayed no significant effect on the MRP1 expression. Our findings indicated that the anti-MDR property of S1 in resistant K562/ADR cells was attributed to suppressing the level of P-gp instead of MRP1.
 | ||
| Fig. 10 Effects of S1 on the levels of MDR-related proteins of K562/ADR cells. K562/ADR cells were incubated with vehicle, 0.01 μM S1 and 0.02 μM S1 for 48 h, respectively, and the total cell lysates were prepared and analyzed by western blotting. | ||
Autophagy is another cell death-inducing pathway, which regulates cell homeostasis and survival during starvation via degradation and recycling of cellular components, such as cellular proteins, cytoplasm and organelles.46 Nevertheless, previous research showed that the pro-death or pro-survival property of autophagy was depending upon the cell type and context.47 Our group reported that hybrid of podophyllotoxin and indirubin, 6, could induce K562/VCR cells autophagy.24 However, no paper had demonstrated that podophyllotoxin or its derivative could induce autophagy in resistant K562/ADR cell line. In this study, we were eager to determine whether molecule S1 influenced autophagy in K562/ADR cells. Cells were incubated with vehicle, 0.01 μM S1 and 0.02 μM S1 for 48 h, respectively, and the total cell lysates were prepared and analyzed by western blotting. As shown in Fig. 11, we observed significant increases in the expression levels of Beclin1 and the cleavage of LC3 (LC3-II) in S1-treated K562/ADR cells, as compared to the control group, suggesting that the autophagic cell death pathway was also involved in the effect of S1 against resistant K562/ADR cells. The data showed that the nitric oxide-donating podophyllotoxin derivative, S1, could induce resistant K562/ADR cells autophagy.
 | ||
| Fig. 11 Effects of S1 on cellular levels of Beclin1 and LC3 in K562/ADR cells. K562/ADR cells were incubated with vehicle, 0.01 μM S1 and 0.02 μM S1 for 48 h, respectively, and the total cell lysates were prepared and analyzed by western blotting. | ||
The regulations of cell physiological and pathological processes were complicated and accurate, which were associated with various factors, including cellular signaling. Recently, some studies have shown that mTOR and STAT3 signaling are associated with the development of tumor cell apoptosis, autophagy and multiple drug resistance.48–50 To gain more insights into the mechanisms underlying the effect of S1, here, we determinated the regulatory effects of S1 on the mTOR and STAT3 signaling in K562/ADR cells. K562/ADR cells were incubated with vehicle, 0.01 μM S1 and 0.02 μM S1 for 48 h, respectively, and the total cell lysates were prepared and analyzed by western blotting. As shown in Fig. 12, treatment with S1 dramatically inhibited the mTOR and STAT3 phosphorylation in K562/ADR cells. These results showed that S1 could inhibit mTOR and STAT3 signaling pathways in resistant K562/ADR cells.
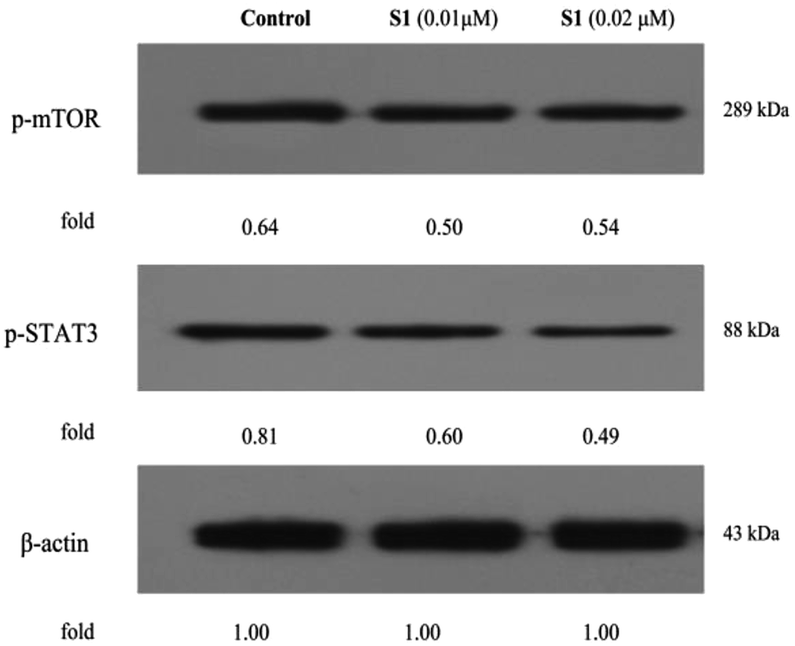 | ||
| Fig. 12 Effects of S1 on the mTOR and STAT3 signaling in K562/ADR cells. K562/ADR cells were incubated with vehicle, 0.01 μM S1 and 0.02 μM S1 for 48 h, respectively, and the total cell lysates were prepared and analyzed by western blotting. | ||
Experimental
General
Melting points were measured on a SGWX-4 melting point apparatus. 1H NMR and 13C NMR spectra were determined with an Agilent 400 NMR spectrometer, using TMS as an internal standard. High resolution mass spectrometry (HR-MS) was recorded on Agilent 6520 Accurate-Mass Q-TOF. All solvents were analytical grade.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcOEt = 10:1 v/v) to provide the pure compound.
:AcOEt = 10:1 v/v) to provide the pure compound.
4α-(2-Nitrooxy)-acetate-4-desoxypodophyllotoxin (S1). Thin yellow solid, yield 37%; mp: 102–104 °C; 1H NMR (400 MHz, CDCl3) δ 6.71 (s, 1H), 6.56 (s, 1H), 6.36 (s, 2H), 6.05–6.00 (m, 3H), 5.02 (s, 2H), 4.62 (s, 1H), 4.38 (t, J = 8.0 Hz, 1H), 4.19 (t, J = 9.6 Hz, 1H), 3.82 (s, 3H), 3.76 (s, 6H), 2.97–2.84 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 173.17, 166.46, 152.71, 148.55, 147.81, 134.40, 132.65, 126.74, 109.88, 107.94, 106.61, 101.77, 75.68, 70.85, 67.02, 60.76, 56.12, 45.45, 43.65, 38.39, 29.69; HRMS-ESI (m/z): calcd for C24H27N2O12 [M + NH4]+ 535.1559, found 535.1556.
4α-(4-Nitrooxy)-butyrate-4-desoxypodophyllotoxin (S2). Grey solid, yield 29%; mp: 72–73 °C; 1H NMR (400 MHz, CDCl3) δ 6.75 (s, 1H), 6.55 (s, 1H), 6.38 (s, 2H), 6.00 (d, J = 4.0 Hz, 2H), 5.92 (d, J = 8.8 Hz, 1H), 4.61 (s, 1H), 4.56 (t, J = 6.0 Hz, 2H), 4.38 (t, J = 7.2 Hz, 1H), 4.20 (t, J = 9.6 Hz, 1H), 3.81 (s, 3H), 3.76 (s, 6H), 2.95–2.79 (m, 2H), 2.66–2.51 (m, 2H), 2.14 (t, J = 6.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 173.53, 172.77, 152.64, 148.21, 147.63, 134.71, 132.39, 127.96, 109.77, 108.06, 106.87, 101.64, 74.12, 71.75, 71.25, 60.76, 56.16, 45.57, 43.69, 38.62, 30.41, 29.69, 22.31; HRMS-ESI (m/z): calcd for C26H27NNaO12 [M + Na]+ 568.1425, found 568.1430.
CCK-8 assay in vitro. The K562, K562/VCR and K562/ADR cell lines used in this study were purchased from KeyGen Biotech (Nanjing, China). The cells were plated in 96-well plates at a density of 2500–3000 cells per well and were incubated at 37 °C in a 5% CO2 incubator for 24 h. Cells were exposed to various concentrations of test compounds for 72 h, and 0.1% DMSO for control. Then, 10 μL of CCK-8 was added to each well and the plate was incubated for additional 2 h at 37 °C. The absorbance was read at 450 nm on a microplate reader. The IC50 values were calculated by GraphPad Prism version 5.0.
Cell cycle analysis. K562/ADR cells were cultured in 6-well plates overnight and treated with various concentrations of test compounds or 0.1% DMSO for 48 h. Cells were collected, washed with PBS, fixed with 70% ethanol for 2 h at 4 °C. Subsequently, cells were treated with 100 μL RNase A at 37 °C for 30 min, and then stained with 400 μL PI at 4 °C for 30 min. The cell cycles were analyzed by flow cytometry analysis.
Apoptosis analysis. K562/ADR cells were plated in 6-well plates overnight and incubated with various concentrations of test compounds or 0.1% DMSO for 48 h. Subsequently, cells were collected, washed twice with PBS and then resuspended in 500 μL of binding buffer. Then, 5 μL Annexin V-V-FITC and 5 μL PI were added to these cells, and the cells were incubated at room temperature in the dark for 15 min. Apoptotic cells were analyzed by a flow cytometer.
Mitochondrial membrane potential analysis. K562/ADR cells were cultured in 6 well plates and treated with various concentrations of test compounds or 0.1% DMSO for 48 h. The cells were collected and then resuspended in solution of incubation buffer containing JC-1 and further incubated at room temperature for 20 min. The cells were analyzed via flow cytometry.
Measurement of intracellular NO generation. K562/ADR cells were treated with various concentrations of test compounds or 0.1% DMSO at 37 °C for 24 h. Subsequently, cells were collected, washed with PBS and resuspended in solution of incubation buffer containing DAF-FM DA at 37 °C for 20 min. The cells were washed with PBS and intracellular NO was analyzed by flow cytometer.
Immunofluorescent analysis. K562/ADR cells were incubated with various concentrations of test compounds or 0.1% DMSO for 24 h. The cells were fixed with 4% paraformaldehyde for 15 min and washed thrice with PBS. Then, the cells were blocked by goat serum at room temperature for 20 min and incubated overnight with primary antibody (α-tubulin) at 4 °C, washed three times with PBS, and then incubated with Alexa Fluor 488 labeled secondary antibody at room temperature for 1 h. Subsequently, the nuclei of cells were stained with DAPI at room temperature for 5 min. The samples were visualized using a fluorescence microscopy.
Western blotting. After incubation with various concentrations of test compounds or 0.1% DMSO for 48 h, K562/ADR cells were collected, washed with PBS, and lysed with RIPA buffer. Then the total cellular protein were collected by centrifugation and determined by Bradford assay. The protein was separated on a sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel (SDS-PAGE) and transferred onto nitrocellulose membranes. The membranes were blocked with blocking buffer at room temperature for 2 h, and then incubated with primary antibodies (CDK1, CDK2, Cleaved caspase-3, MRP1, P-gp, Beclin1, LC3, p-MTOR, p-STAT3 or β-actin) overnight at 4 °C. After washing twice with PBS, the cells were treated with secondary antibodies at room temperature for 2 h. Protein bands were visualized using enhanced chemiluminescence detection reagents.
Conclusions
In summary, two novel nitric oxide-donating podophyllotoxin derivatives were synthesized and biologically evaluated. We found that hybrid S1 displayed strong antiproliferative activity against both sensitive K562, and drug-resistant K562/VCR and K562/ADR cells, associated with the improvement of intracellular NO level. Treatment with S1 promoted G2 cell cycle arrest and apoptosis. Moreover, S1 efficiently disrupted the microtubule network, depolarizated the mitochondrial membrane potential, and activated the caspase-3 in K562/ADR cells. Additionally, S1 induced autophagy, inhibited P-gp expression, and suppressed mTOR and STAT3 signaling. Together, these data indicated that NO-donating compound S1 may target several pathways to overcome the drug-resistance and display significant antiproliferative efficacy against leukemic K562/ADR cells, suggesting that it may be a promising antileukemic candidate for the treatment of drug-resistant leukemia. Subsequently, according to the preliminary results, we will further investigate the stability of the ester linkage in the cultivation medium and liver microsomes of rat, and elucidate the efficiency of the hybrid in vivo.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Ministry of Education “Chunhui Project” Foundation of China (No. Z2015008), National Natural Science Foundation of China (No. 81860622), Department of Science and Technology of Guizhou Province (No. [2017]1219), Joint Fund of the Department of Science and Technology of Zunyi City and Zunyi Medical University ([2018]27) and National First-Rate Construction Discipline of Guizhou Province (Pharmacy) (YLXKJS-YX-04).Notes and references
- G. Szakács, J. K. Paterson, J. A. Ludwig, C. Booth-Genthe and M. M. Gottesman, Nat. Rev. Drug Discovery, 2006, 5, 219–234 CrossRef PubMed.
- B. Sarkadi, L. Homolya, G. Szakács and A. Váradi, Physiol. Rev., 2006, 86, 1179–1236 CrossRef CAS PubMed.
- P. D. W. Eckford and F. J. Sharom, Chem. Rev., 2009, 109, 2989–3011 CrossRef CAS PubMed.
- M. Yu, A. Ocana and I. F. Tannock, Cancer Metastasis Rev., 2013, 32, 211–227 CrossRef CAS PubMed.
- C. P. Wu, A. M. Calcagno and S. V. Ambudkar, Curr. Mol. Pharmacol., 2008, 1, 93–105 CrossRef CAS PubMed.
- Q. Zhou, Y. Li, J. Jin, L. Lang, Z. Zhu, W. Fang and X. Chen, Biol. Pharm. Bull., 2012, 35, 2170–2179 CrossRef CAS.
- M. Gordaliza, P. A. García, J. M. M. Del Corral, M. A. Castro and M. A. Gómez-Zurita, Toxicon, 2004, 44, 441–459 CrossRef CAS PubMed.
- X. Zhang, K. P. Rakesh, C. S. Shantharam, H. M. Manukumar, A. M. Asiri, H. M. Marwani and H. L. Qin, Bioorg. Med. Chem., 2018, 26, 340–355 CrossRef CAS PubMed.
- L. Li, S. Jiang, X. Li, Y. Liu, J. Su and J. Chen, Eur. J. Med. Chem., 2018, 151, 482–494 CrossRef CAS PubMed.
- Y. You, Curr. Pharm. Des., 2005, 11, 1695–1717 CrossRef CAS PubMed.
- A. Kamal, S. M. Ali Hussaini, A. Rahim and S. Riyaz, Expert Opin. Ther. Pat., 2015, 25, 1025–1034 CrossRef CAS PubMed.
- C. Bailly, Chem. Rev., 2012, 112, 3611–3640 CrossRef CAS PubMed.
- X. Yu, Z. Che and H. Xu, Chem.–Eur. J., 2017, 23, 4467–4526 CrossRef CAS PubMed.
- W. X. Sun, Y. J. Ji, Y. Wan, H. W. Han, H. Y. Lin, G. H. Lu, J. L. Qi, X. M. Wang and Y. H. Yang, Bioorg. Med. Chem. Lett., 2017, 27, 4066–4074 CrossRef CAS PubMed.
- A. Abad, J. L. López-Pérez, E. del Olmo, L. F. García-Fernández, A. Francesch, C. Trigili, I. Barasoain, J. M. Andreu, J. F. Díaz and A. S. Feliciano, J. Med. Chem., 2012, 55, 6724–6737 CrossRef CAS PubMed.
- Y. Q. Liu, J. Tian, K. Qian, X. B. Zhao, S. L. Morris-Natschke, L. Yang, X. Nan, X. Tian and K. H. Lee, Med. Res. Rev., 2015, 35, 1–62 CrossRef PubMed.
- H. Chen, S. Zuo, X. Wang, X. Tang, M. Zhao, Y. Lu, L. Chen, J. Liu, Y. Liu, D. Liu, S. Zhang and T. Li, Eur. J. Med. Chem., 2011, 46, 4709–4714 CrossRef CAS PubMed.
- J. Li, H. M. Hua, Y. B. Tang, S. Zhang, E. Ohkoshi, K. H. Lee and Z. Y. Xiao, Bioorg. Med. Chem. Lett., 2012, 22, 4293–4295 CrossRef CAS PubMed.
- L. Kou, M. J. Wang, L. T. Wang, X. B. Zhao, X. Nan, L. Yang, Y. Q. Liu, S. L. Morris-Natschke and K. H. Lee, Eur. J. Med. Chem., 2014, 75, 282–288 CrossRef CAS PubMed.
- L. Zhang, F. Chen, Z. Zhang, Y. Chen and J. Wang, Bioorg. Med. Chem. Lett., 2016, 26, 38–42 CrossRef CAS PubMed.
- L. Zhang, Z. Zhang, J. Wang, Y. Chen, F. Chen, Y. Lin and X. Zhu, RSC Adv., 2016, 6, 2895–2903 RSC.
- X. Liu, L. L. Zhang, X. H. Xu, L. Hui, J. B. Zhang and S. W. Chen, Bioorg. Med. Chem. Lett., 2013, 23, 3780–3784 CrossRef CAS PubMed.
- L. Zhang, L. Liu, C. Zheng, Y. Wang, X. Nie, D. Shi, Y. Chen, G. Wei and J. Wang, Eur. J. Med. Chem., 2017, 131, 81–91 CrossRef CAS PubMed.
- J. Wang, L. Long, Y. Chen, Y. Xu and L. Zhang, Bioorg. Med. Chem. Lett., 2018, 28, 1817–1824 CrossRef CAS PubMed.
- D. Fukumura, S. Kashiwagi and R. K. Jain, Nat. Rev. Cancer, 2006, 6, 521–534 CrossRef CAS PubMed.
- A. J. Burke, F. J. Sullivan, F. J. Giles and S. A. Glynn, Carcinogenesis, 2013, 34, 503–512 CrossRef CAS PubMed.
- R. Fruttero, M. Crosetti, K. Chegaev, S. Guglielmo, A. Gasco, F. Berardi, M. Niso, R. Perrone, M. A. Panaro and N. A. Colabufo, J. Med. Chem., 2010, 53, 5467–5475 CrossRef CAS PubMed.
- C. Riganti, E. Miraglia, D. Viarisio, C. Costamagna, G. Pescarmona, D. Ghigo and A. Bosia, Cancer Res., 2005, 65, 516–525 CAS.
- R. Sullivan and C. H. Graham, Curr. Pharm. Des., 2008, 14, 1113–1123 CrossRef CAS PubMed.
- P. G. Wang, M. Xian, X. Tang, X. Wu, Z. Wen, T. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS PubMed.
- M. R. Miller and I. L. Megson, Br. J. Pharmacol., 2007, 151, 305–321 CrossRef CAS PubMed.
- T. B. Cai, X. Tang, J. Nagorski, P. G. Brauschweiger and P. G. Wang, Bioorg. Med. Chem., 2003, 11, 4971–4975 CrossRef CAS PubMed.
- X. Li, X. Wang, C. Xu, J. Huang, C. Wang, X. Wang, L. He and Y. Ling, MedChemComm, 2015, 6, 1130–1136 RSC.
- G. D. Stewart, J. Nanda, D. J. G. Brown, A. C. P. Riddick, J. A. Ross and F. K. Habib, Int. J. Cancer, 2009, 124, 223–232 CrossRef CAS PubMed.
- L. Fang, M. Wang, S. Gou, X. Liu, H. Zhang and F. Cao, J. Med. Chem., 2014, 57, 1116–1120 CrossRef CAS PubMed.
- F. Rothweiler, M. Michaelis, P. Brauer, J. Otte, K. Weber, B. Fehse, H. W. Doerr, M. Wiese, J. Kreuter, Y. Al-Abed, F. Nicoletti and J. Cinatl, Neoplasia, 2010, 12, 1023–1030 CrossRef CAS PubMed.
- K. Chegaev, C. Riganti, L. Lazzarato, B. Rolando, S. Guglielmo, I. Campia, R. Fruttero, A. Bosia and A. Gasco, ACS Med. Chem. Lett., 2011, 2, 494–497 CrossRef CAS PubMed.
- C. Riganti, B. Rolando, J. Kopecka, I. Campia, K. Chegaev, L. Lazzarato, A. Federico, R. Fruttero and D. Ghigo, Mol. Pharmaceutics, 2013, 10, 161–174 CrossRef CAS PubMed.
- X. Tang, X. Gu, H. Ai, G. Wang, H. Peng, Y. Lai and Y. Zhang, Bioorg. Med. Chem. Lett., 2012, 22, 801–805 CrossRef PubMed.
- Y. Ai, F. Kang, Z. Huang, X. Xue, Y. Lai, S. Peng, J. Tian and Y. Zhang, J. Med. Chem., 2015, 58, 2452–2464 CrossRef CAS PubMed.
- X. Gu, Z. Huang, Z. Ren, X. Tang, R. Xue, X. Luo, S. Peng, H. Peng, B. Lu, J. Tian and Y. Zhang, J. Med. Chem., 2017, 60, 928–940 CrossRef CAS PubMed.
- L. Zhang, F. Chen, J. Wang, Y. Chen, Z. Zhang, Y. Lin and X. Zhu, RSC Adv., 2015, 5, 97816–97823 RSC.
- L. Zhang, Z. Zhang, F. Chen, Y. Chen, Y. Lin and J. Wang, Eur. J. Med. Chem., 2016, 123, 226–235 CrossRef CAS PubMed.
- L. Zhang, Z. Zhang, F. Chen, Y. Chen, Y. Lin and J. Wang, Bioorg. Med. Chem. Lett., 2016, 26, 4466–4471 CrossRef CAS PubMed.
- S. Y. Jeong and D. W. Seol, BMB Rep., 2008, 41, 11–22 CrossRef CAS PubMed.
- N. Mizushima and M. Komatsu, Cell, 2011, 147, 728–741 CrossRef CAS PubMed.
- F. Nazio, F. Strappazzon, M. Antonioli, P. Bielli, V. Cianfanelli, M. Bordi, C. Gretzmeier, J. Dengjel, M. Piacentini, G. Fimia and F. Cecconi, Nat. Cell Biol., 2013, 15, 406–416 CrossRef CAS PubMed.
- M. Granato, C. Rizzello, M. S. G. Montani, C. Cuomo, M. Vitillo, R. Santarelli, R. Gonnella, G. D'Orazi, A. Faggioni and M. Cirone, J. Nutr. Biochem., 2017, 41, 124–136 CrossRef CAS PubMed.
- H. Miyata, T. Ashizawa, A. Iizuka, R. Kondou, C. Nonomura, T. Sugino, K. Urakami, A. Asai, N. Hayashi, K. Mitsuya, Y. Nakasu, K. Yamaguchi and Y. Akiyama, Cancer Genomics Proteomics, 2017, 14, 83–91 CrossRef CAS PubMed.
- M. Zou, C. Hu, Q. You, A. Zhang, X. Wang and Q. Guo, Mol. Carcinog., 2015, 54, 1363–1375 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06360e |
| This journal is © The Royal Society of Chemistry 2018 |