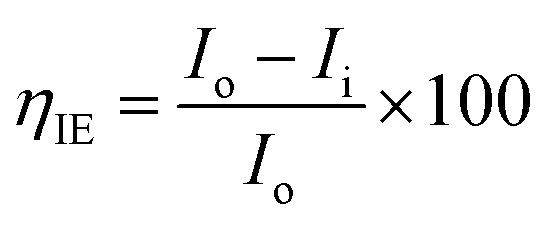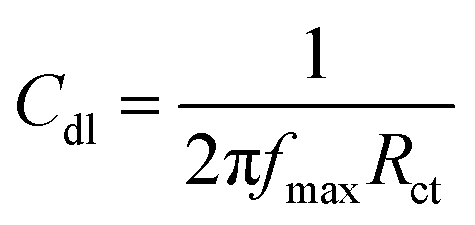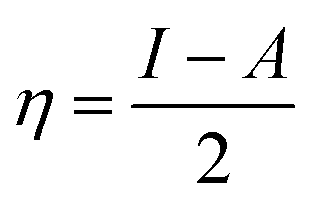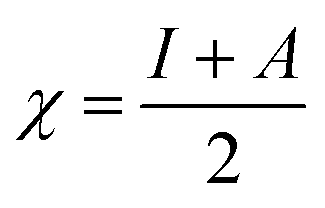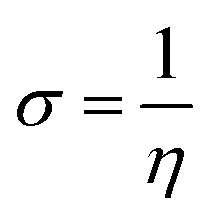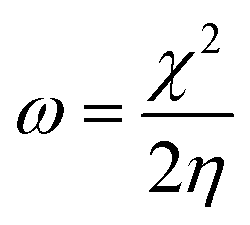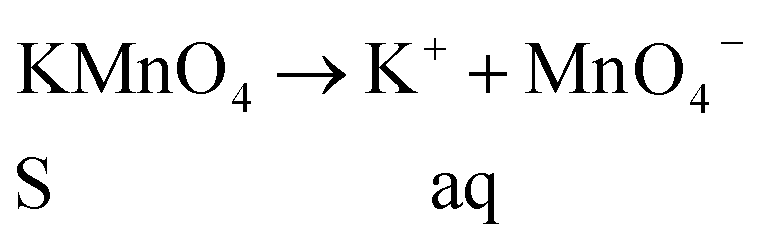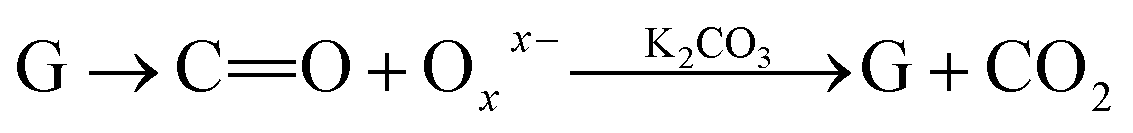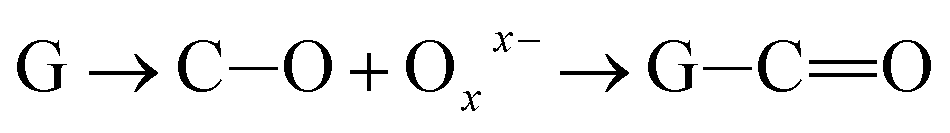DOI:
10.1039/C8RA05118F
(Paper)
RSC Adv., 2018,
8, 34275-34286
Praseodymium-decorated graphene oxide as a corrosion inhibitor in acidic media for the magnesium AZ31 alloy
Received
14th June 2018
, Accepted 14th September 2018
First published on 9th October 2018
Abstract
In the present work, Pr-decorated graphene oxide was synthesized and tested as a corrosion barrier layer in acidic media for the magnesium AZ31 alloy. The morphology, composition and structure of Pr-decorated graphene oxide sheets were characterized via HRTEM, FESEM, Raman, XRD, DLS, UV and FTIR studies. The corrosion inhibition efficiency on the alloy surface was monitored via microstructural and electrochemical methods. The results indicate that Pr-decorated graphene oxide provides improved protection for the Mg AZ31 alloy compared to conventional epoxy coatings. The proposed mechanism arises from a combination of the barrier activities of the composite, GO + Pr, and the epoxy coating on the Mg alloy in acidic media.
1. Introduction
Magnesium alloys are alternative materials for use in biomaterials, hydrogen storage, automobiles, and the airbus manufacturing industry, due to their prominent properties, such as light weight and high metallurgic strength.1 Magnesium alloy native oxide films are not stable in strongly acidic and salty environments, and accelerated corrosion occurs due to the poor stability of the native oxide layer in acidic media. As a result of this, 14 and 15 block and Zn elements are added to make new Mg alloys to improve the corrosion resistance of magnesium alloys in salty media.2 In addition, Cr nanoparticles have been used as corrosion inhibitors; however, these are carcinogenic agents, and several countries have banned Cr based coating materials.3 Other solutions for Mg alloy corrosion inhibition improvement involve using inhibitors, such as ammonium ions, phytic acid, amino acids, organic silicates, inorganic Zn, anionic surfactants, and 2-hydroxy-4-methoxy-acetophenone. Further, Mg alloy microstructures are protected using tetraphenylporphyrin, sodium fluoride, and inorganic and organic inhibitors.4–12 Hence, hydroxyapatite and Ca+ cations are added to improve corrosion resistance.13,14 Furthermore, the effects of NaCl electrolytes in the corrosion medium on Mg alloy dissolution in the Mg+ state, and the associated microstructures have been analysed via SVET methods.15–19 In addition, d, s and f block elements are used to enhance corrosion resistance in NaCl environments, such as zirconium, arsenic, phosphates, germanium, lutetium, neodymium, cerium, La3+, Dy and mischmetal.20–29 Further, cathodic hydrogen evolution is controlled using inhibitor molecules.30–32 Coatings have been used on magnesium surfaces as self-healing agents to increase long term protection.33–35 Hence, rare earth chlorides, carboxylate complexes, anionic surfactants, and cerium complexes are used as green corrosion inhibitors. And large-surface areas, high electron densities and rare earth complexes can also enhance the corrosion inhibition efficiency.36–47 In recent years, functionalized graphene oxide materials have been reported, such as poly(urea-formaldehyde), tin oxide, plasma based, and epoxy + graphene composite coatings, graphene ink, covalently functionalized N-graphene oxide, and ionic liquids, which are excellent corrosion barrier layers on alloy surfaces because they are impermeable to electrolytes and gas molecues.48–58 Furthermore, GO functional groups can also enhance electron donation and accepting from the alloy surface.59,60 The predominant properties of functional graphene oxide materials have a significant impact on overcoming corrosion failure. We have created decorated praseodymium based GO sheet materials as a low cost corrosion inhibition barrier layer. They could enhance the life span of the coating on the alloy surface, and overcome conventional epoxy coating failure. Our research aim is the facile synthesis of praseodymium-decorated graphene oxide, to study the possible corrosion inhibition effects on Mg alloys in acidic media, as metallurgy, automobile industry pickling, and other works take place in strong acidic media. Further, Mg alloy corrosion mitigation has not yet been achieved due to traditional epoxy coating failure. We first investigated a GO + Pr composite coating on Mg alloys as a corrosion inhibition barrier in acidic media. The novelty of the GO decorated material is the intercalation of small sized Pr3+ between the graphene oxide layers to enhance corrosion resistance; the coating lifespan increased in acidic media compared to epoxy coatings.
2. Experimental section
2.1. GO synthesis
Graphite powder, H2SO4, H3PO4, H2O2, NaNO3, KMnO4, and Pr(NO3)3 were purchased from Alfa Aesar Company and used without further purification. GO was synthesized via a modified Hummers' method.61 In brief, 1 g of graphite powder was placed into a 250 mL round bottom flask (RB), and 40 mL of H2SO4 and 60 mL of H3PO4 were slowly added. The mixture was kept in an ice bath and 3 g of KMnO4 and 0.1 g of NaNO3 were slowly added to the mixture, which was then kept under reflux at 60 °C for 12 h. After the mixture was cooled to room temperature, it was poured into 1000 mL of ice water, and then 10 mL of 30% H2O2 was added to terminate the reaction. The resultant GO was washed with plenty of deionized (DI) water until neutrality was achieved.
2.2. GO-praseodymium decoration
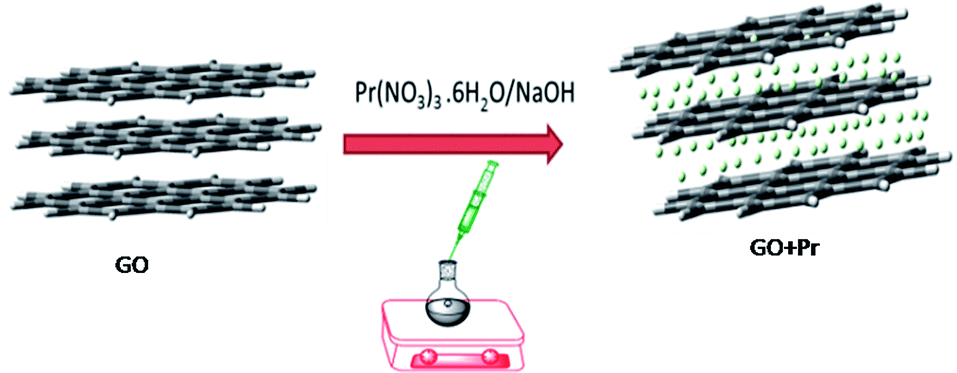
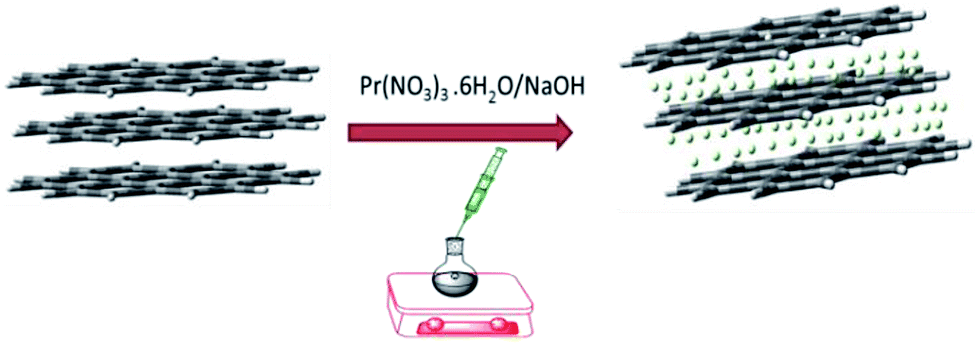
For decoration, 200 mg of GO powder was placed into a round bottom flask containing 100 mL of DI water. The mixture was sonicated to finely disperse the GO using an SB-300 DTY multi-ultrasonic cleaner operating at 40 W and a frequency of 40 Hz. After that, 100 mg of Pr(NO3)3 was added into the GO solution, followed by 10 mL of 1 M NaOH solution as an activator. The mixture was continually stirred for 24 h, as shown in Scheme 1.
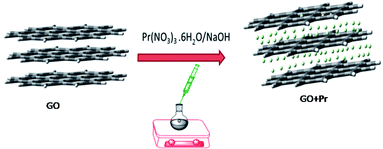 |
| | Scheme 1 | |
The resulting product was washed and centrifuged at 5000 rpm 5 times to remove remaining alkali. The GO + Pr material was dried at 80 °C in a vacuum oven for 24 h and kept in a desiccator before being used as an anti-corrosion coating material (Fig. 1).
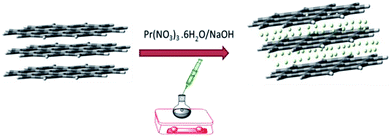 |
| | Fig. 1 Schematic illustration of the GO decoration. | |
2.3. Material characterization
Graphite, GO and the GO-Pr material were characterized via powder X-ray diffraction with a D8 instrument from Bruker, using Cu Kα radiation (λ = 1.5406 Å). The optical absorption was studied by finely dispersing 0.1 mg of graphene oxide flake in DI water to make a stable, transparent dispersion, and studying this using a milli-Q A10 TOC spectrometer in the 200–600 nm range. The molecular structure, i.e. the functional groups, was studied using an FTIR Spectrum 65 instrument from PerkinElmer. Thermal behaviour was tested using SII TG/DTA7300 apparatus. The microstructures of the materials were studied via confocal Raman spectroscopy, using a 532 nm laser as the excitation source, with a WITec Raman microscope. The morphologies and compositions of the materials were studied using a Carl Zeiss FESEM Marline compact microscope equipped with an EDX detector. The size distributions and zeta potentials of aqueous solutions were studied using a Microtrac Zetatrac U2771. Thin film analysis was conducted using Horiba UVi Cell2 apparatus over a range from 1.5000–6.0000 eV at increments of 0.0500 eV. The GO sheet morphology and GO + Pr composite material crystallinity were observed using an FEI Techani F Twin 500 transmission electron microscope.
2.4. Coating methods
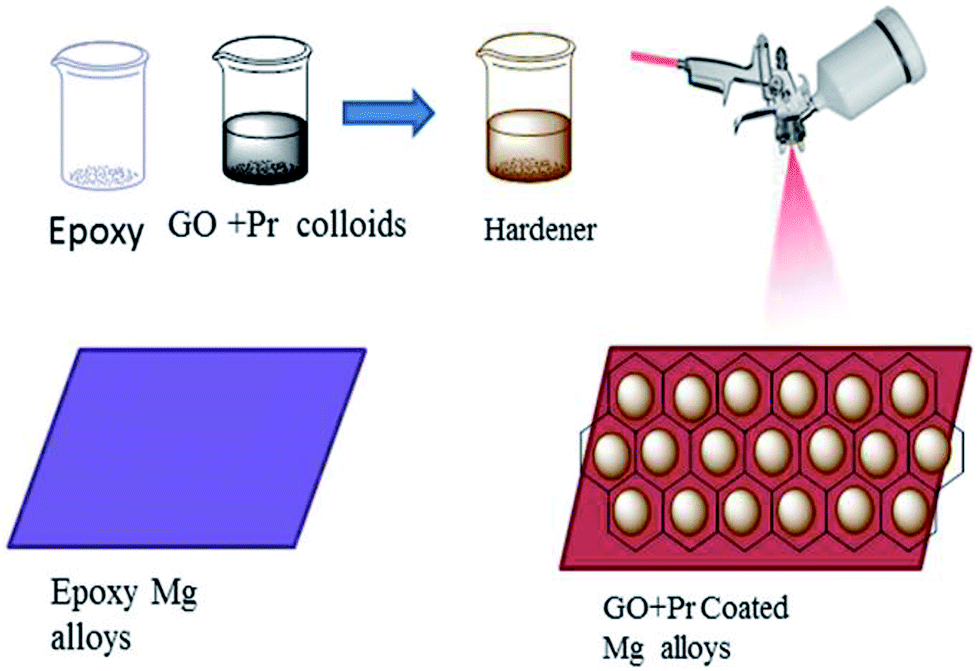
As shown in Fig. 2, before coating, the Mg alloys were polished with different grades of silicon paper, from 600 to 1200 microns. The epoxy powder was purchased from Mathura chemical Pv. Ltd. Hence the coating material, the (GO + Pr) composite, and a hardener, polyamine, were mixed in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (10 mg:5 mg) weight ratio. The mixture was finely dispersed using an SB-300 DTY multi-ultrasonic cleaner operating at 40 W and a frequency of 40 Hz; the colloids were taken as the coating material. The epoxy coating material was prepared via the same method. The coating was carried out using a brush, and the brush size was 0.2 μm. The coated Mg alloy was dried before being used in corrosion inhibition studies at a temperature of 80 °C in a hot air oven.
:1 (10 mg:5 mg) weight ratio. The mixture was finely dispersed using an SB-300 DTY multi-ultrasonic cleaner operating at 40 W and a frequency of 40 Hz; the colloids were taken as the coating material. The epoxy coating material was prepared via the same method. The coating was carried out using a brush, and the brush size was 0.2 μm. The coated Mg alloy was dried before being used in corrosion inhibition studies at a temperature of 80 °C in a hot air oven.
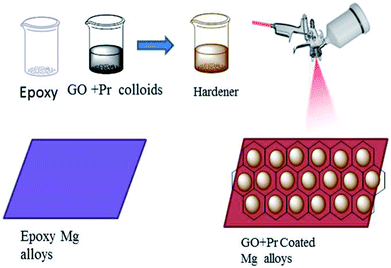 |
| | Fig. 2 A schematic illustration of the methods for GO + Pr composite and epoxy coating. | |
2.5. Corrosion inhibition
The magnesium AZ31 alloy samples were prepared according to the literature ref. 62. The test Mg alloys were 1 cm × 1 cm squares of Mg AZ31 alloy, with a 1 mm thickness, polished with a series of different grade silicon carbide paper, from 600 to 1200 microns. The corrosion medium was 1 M H2SO4 solution, and the GO-Pr and epoxy-coated Mg alloys were immersed for 3 days in the corrosion medium. The corrosion inhibition efficiency was analysed using CHI 920D electrochemical workstation software; the coated Mg alloy was set as the working electrode, platinum foil was the counter electrode and Ag/AgCl (KCl Sat) was reference electrode. The electrochemical reaction was carried out with respect to an open circuit potential of ± 250 mV at a 0.1 mV scan rate. The corrosion inhibition efficiency (ηIE), double layer capacitance (Cdl), quantum chemical calculation of electronegativity (χ), global hardness (η), softness (σ), and electrophilicity (ω) were calculated via the following equations:| |
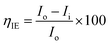 | (1) |
| |
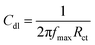 | (2) |
| |
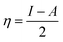 | (3) |
| |
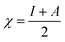 | (4) |
| |
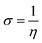 | (5) |
| |
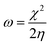 | (6) |
where fmax is the maximum frequency, Rct is charge transfer resistance, Io is the current density of the epoxy coating and Ii is the current density of the GO + Pr coating.
3. Results and discussion
3.1. Morphology and GO and GO + Pr composition
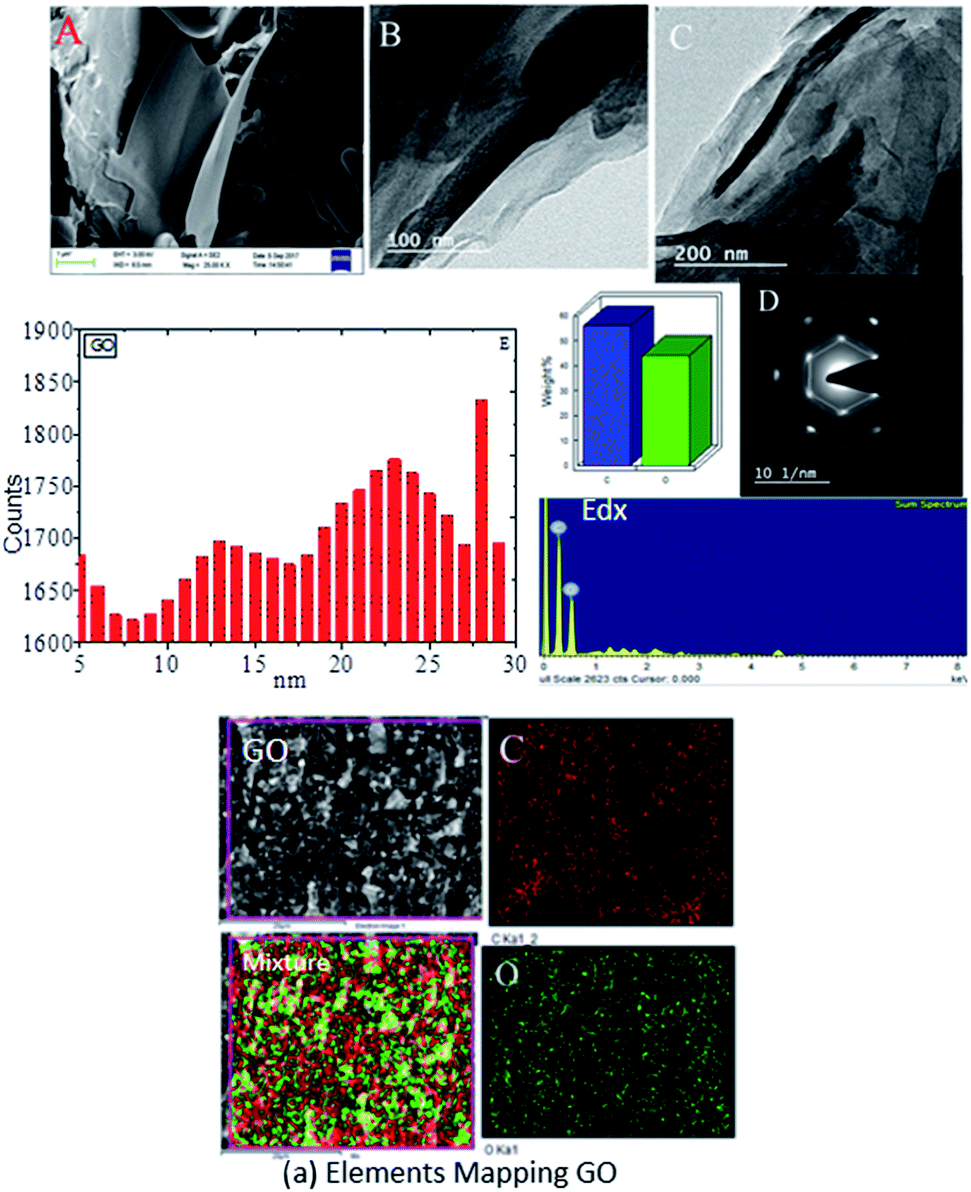
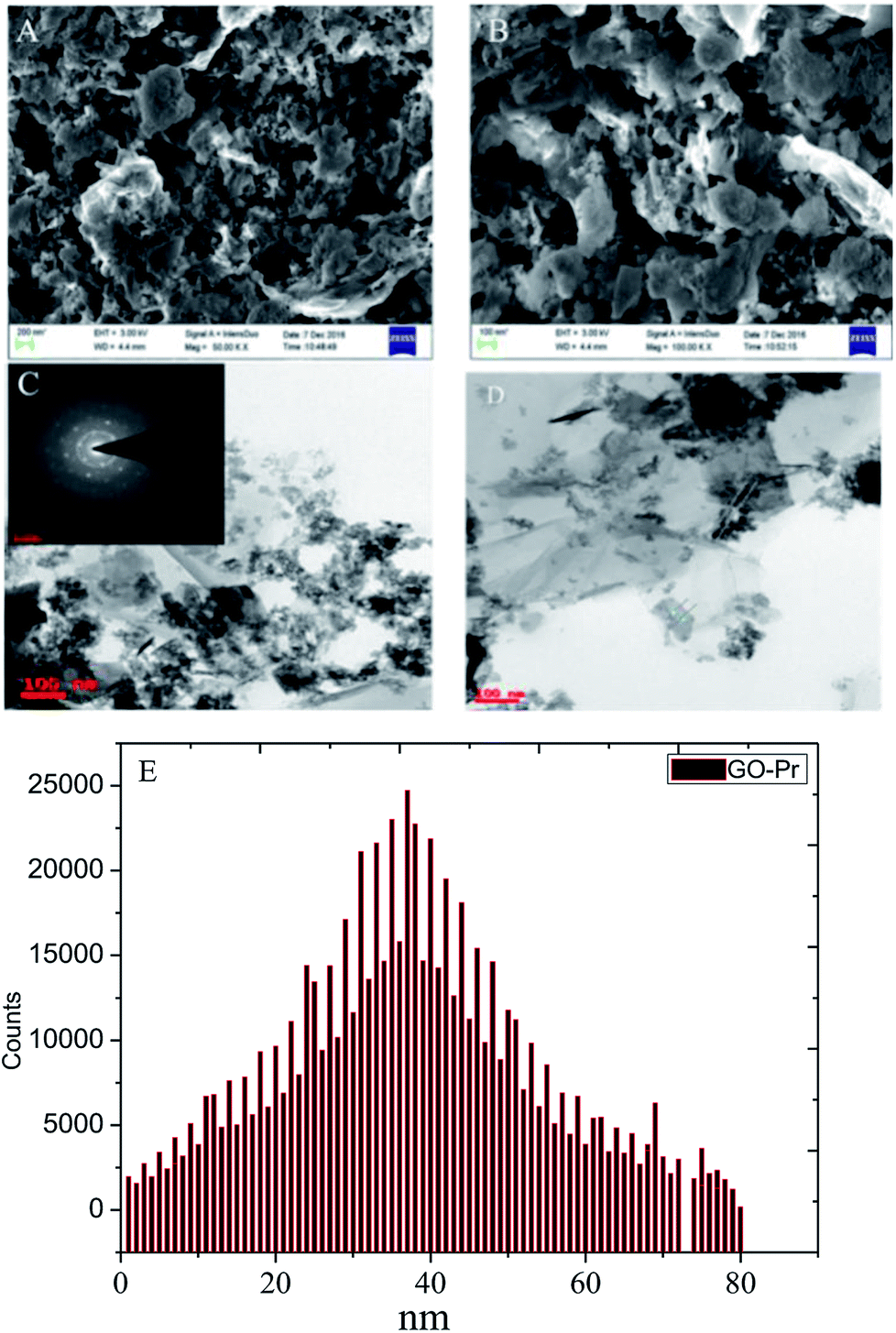
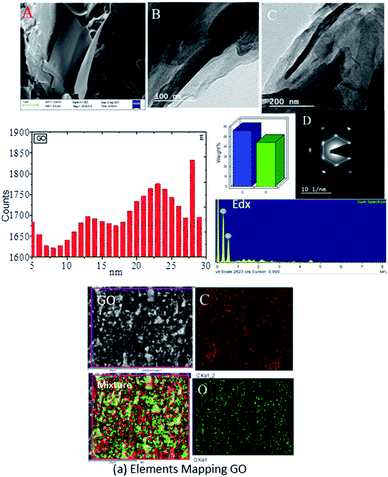
Graphene oxide (GO) microstructures are presented in Fig. 3. In Fig. 3A, an FESEM image of GO63–65 shows few layer graphene oxide sheets and continuing sheet form, due to oxidation. Fig. 3B and C provide HRTEM images of GO, showing few interlayers and an average sheet size of 100 nm to 200 nm. Further confirmation, as shown in Fig. 3D, through selected area electron diffraction (SAED) studies of GO indicates polycrystalline sheets and elements analysis by EDX. And the GO sheet size is analysed via Gatan microscope software; the GO nanosheet sizes are presented in Fig. 3E and a elemental mapping of GO. Further, praseodymium decorated graphene oxide (GO-Pr) FESEM images are presented in Fig. 4A and B. The Pr decorated GO sheets appear bright due to Pr nanoparticles being incorporated in the graphene oxide sheets, and in addition, a HRTEM micrograph and selected area electron diffraction (SAED) pattern of polycrystalline GO-Pr are presented in Fig. 4C. A GO nanosheet with corresponding Pr nanoparticles spread all over the GO sheets is presented in Fig. 4D. This suggests that well defined Pr nanoparticles are incorporated into the graphene oxide sheet. It also attributes polycrystallinity to the graphene oxide sheets.
 |
| | Fig. 3 FESEM images of GO (A) and GO (B and C) of HRTEM micrograph (D) SAED pattern and EDX of GO and (E) of GO nano sheet size. (a) Elemental mapping of GO. | |
 |
| | Fig. 4 FESEM images of GO + Pr (A and B), HRTEM images of (Pr + GO) (C and D), and (E) GO + Pr particle size data. | |
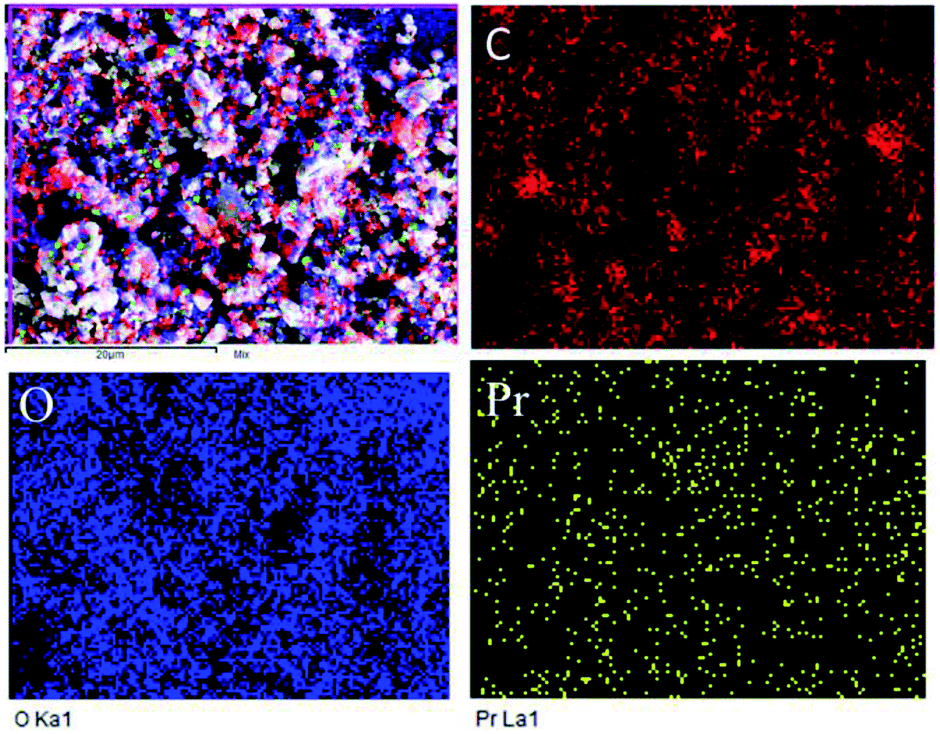
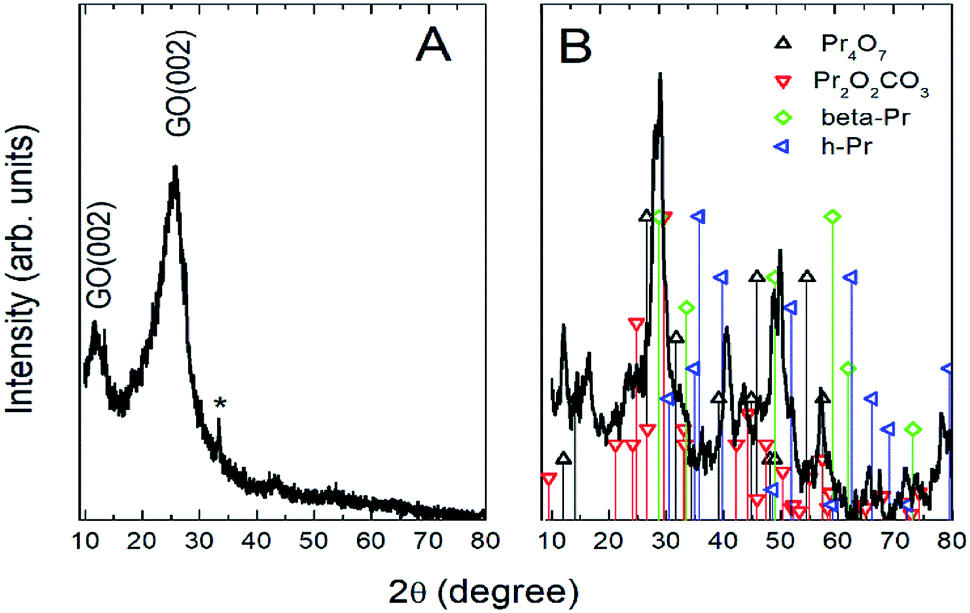
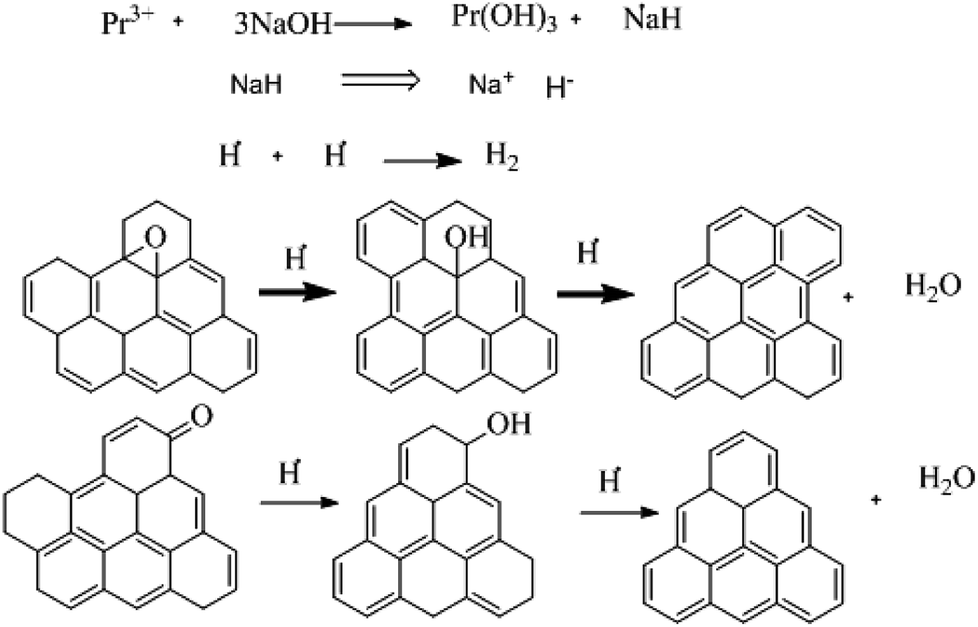
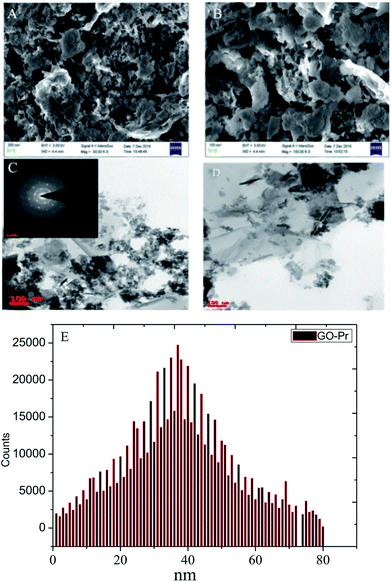
Further, the Pr particles are analyzed using Gatan digital microscope software, and the sizes are shown in Fig. 4E. Element mapping of GO + Pr is presented in Fig. 5. It suggests that praseodymium nanoparticles interact with the graphene oxide sheets, and the C, O, and Pr elements are uniformly distributed on the graphene oxide surface. The microscopy results suggest that GO and the composite GO + Pr material have similar microstructures. The X-ray diffraction spectrum of GO powder is presented in Fig. 6A; the characteristic graphene oxide (002) reflection at 2θ = 11.6° and the graphite related (002) reflection at 2θ = 25.7° show that GO is partially reduced during the synthesis.66 However, GO with well exfoliated sheets (GO (002) = 0.76 nm) is attained with this preparation, as observed in the HRTEM images. A GO-Pr diffraction spectrum is presented in Fig. 6B, where several diffraction peaks assigned to elemental Pr (beta and hexagonal phases), Pr4O7 and Pr-carbonate phases are observed, due to the effects of 1 M NaOH strong basic conditions, and a possible mechanism is shown in Fig. 7 for how different phase Pr particles formed,67 which is confirmed by the SAED pattern. There are other peaks that could not be indexed, which probably correspond to Pr-aromatic phases from Pr reactions within GO basal sheets.68–70 The presence of the different observed phases suggests that the decoration process could involve partial reduction by nascent oxygen moieties, with possible reactions proceeding as follows:
 |
| | Fig. 5 Composite elemental mapping of GO + Pr. | |
 |
| | Fig. 6 XRD of GO and (GO + Pr) decorated GO sheet. | |
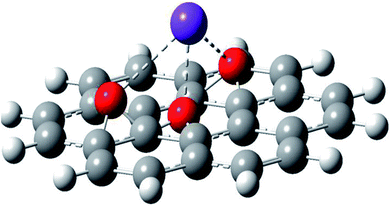 |
| | Fig. 7 The proposed reduction mechanism for GO. | |
KMnO4 dissociation leads to K+ cations and MnO4− anions: the cations co-ordinate with GO carbonyl groups to form carbonates. The carbonate nascent oxygen eliminates carbonyl groups as carbon dioxide, leaving defects on the GO surface, indicating reduced graphene oxide.
3.2. Molecular and microstructural characterization
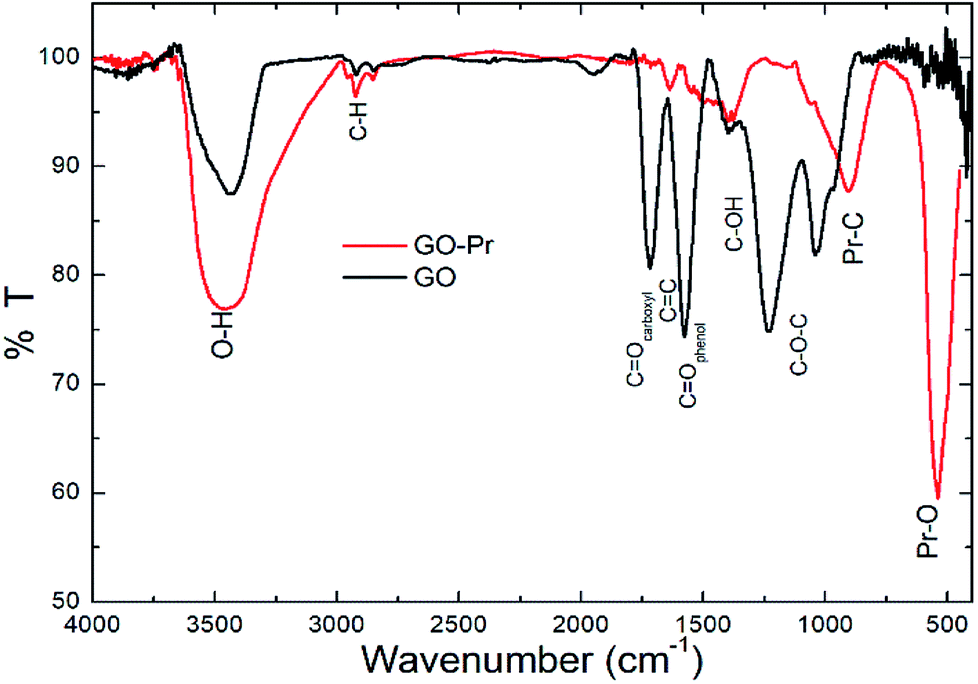
Infrared spectra of GO and GO-Pr samples are shown in Fig. 8. The wide band at 3500 cm−1 is due to O–H stretching of adsorbed water, and the small peaks at ca. 2800 cm−1 are attributed to alkyl stretching. The GO spectrum displays characteristic bands at 1740 cm−1 (carboxyl groups), 1630 cm−1 (aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), and 1570 cm−1 (CO in phenols), as well as hydroxyl and epoxy bands below 1500 cm−1.71–74 On the other hand, the GO-Pr spectrum displays GO, CC and hydroxyl bands related to the GO matrix, and new bands at 1300–1500 cm−1 and 1041 cm−1, attributed to carbonates, are observed, as well as another peak at around 550 cm−1 related to Pr–O bonding.75 The FTIR results are consistent with the phases observed from XRD studies.
C), and 1570 cm−1 (CO in phenols), as well as hydroxyl and epoxy bands below 1500 cm−1.71–74 On the other hand, the GO-Pr spectrum displays GO, CC and hydroxyl bands related to the GO matrix, and new bands at 1300–1500 cm−1 and 1041 cm−1, attributed to carbonates, are observed, as well as another peak at around 550 cm−1 related to Pr–O bonding.75 The FTIR results are consistent with the phases observed from XRD studies.
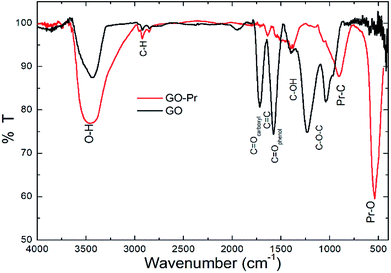 |
| | Fig. 8 FTIR spectra of GO and GO-Pr samples. | |
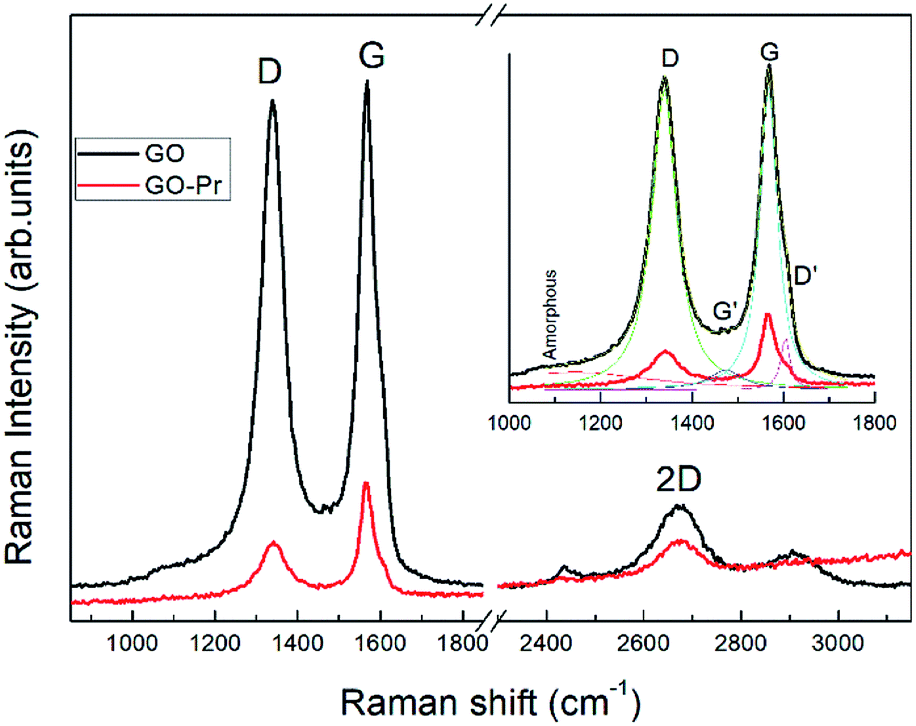
Raman spectra of GO and Pr + GO samples are presented in Fig. 10. D, G and 2D bands attributed to sp3 and carbon-vacancy defects in the graphitic structure, sp2 carbon symmetric vibrations, and high order vibrations of the carbon skeleton, respectively, are observed in both spectra. Evident reduction due to hydride anions is shown in the proposed mechanism in Fig. 9,76 related to the reduction degree of subjacent GO sheets upon Pr nanoparticle incorporation. The inset in Fig. 10 presents deconvolution of the spectra with regards to the D and G peaks, as well as secondary G′ and D′ bands related to structural defects in the crystalline regions. The AD/AG ratios were calculated to determine the relative crystalline ordering of the GO sample; AD/AG is 1.42 for GO and 1.03 for GO-Pr respectively, which accounts for the higher degree of crystallinity in the graphitic structure of the GO-Pr sample.77
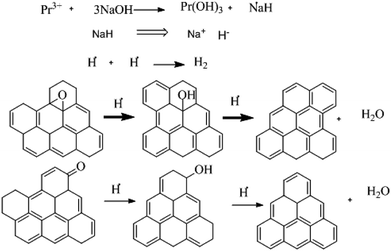 |
| | Fig. 9 The proposed mechanism for GO reduction. | |
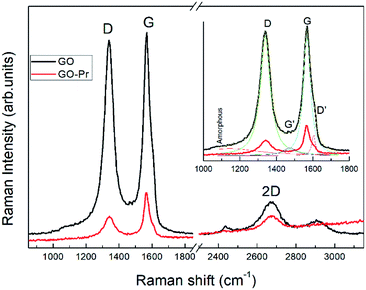 |
| | Fig. 10 Raman spectra of GO and GO-Pr samples showing spectral deconvolution to identify peaks. | |
3.3. Thermal, optical and hydrodynamic characterization
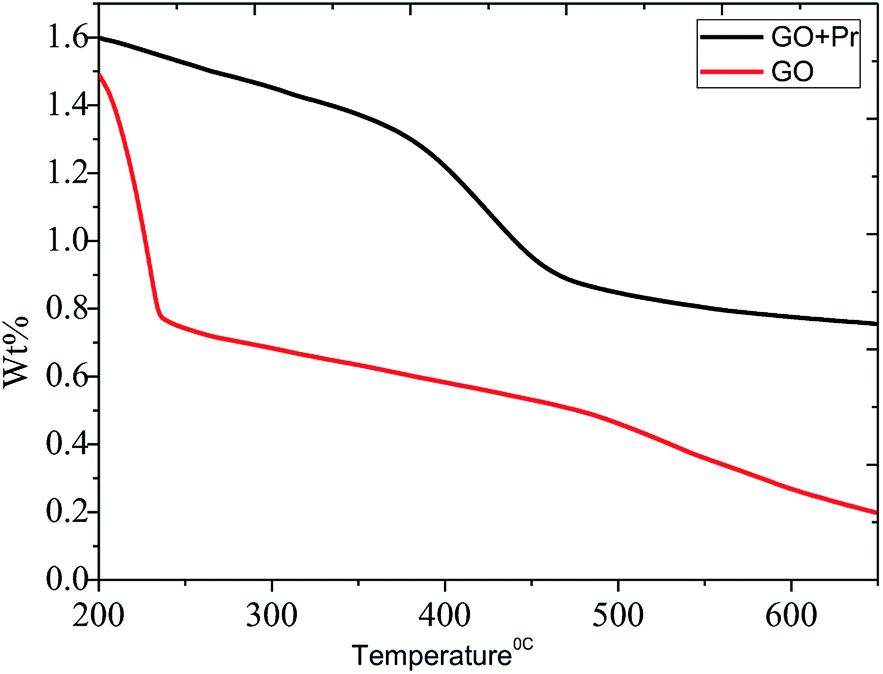
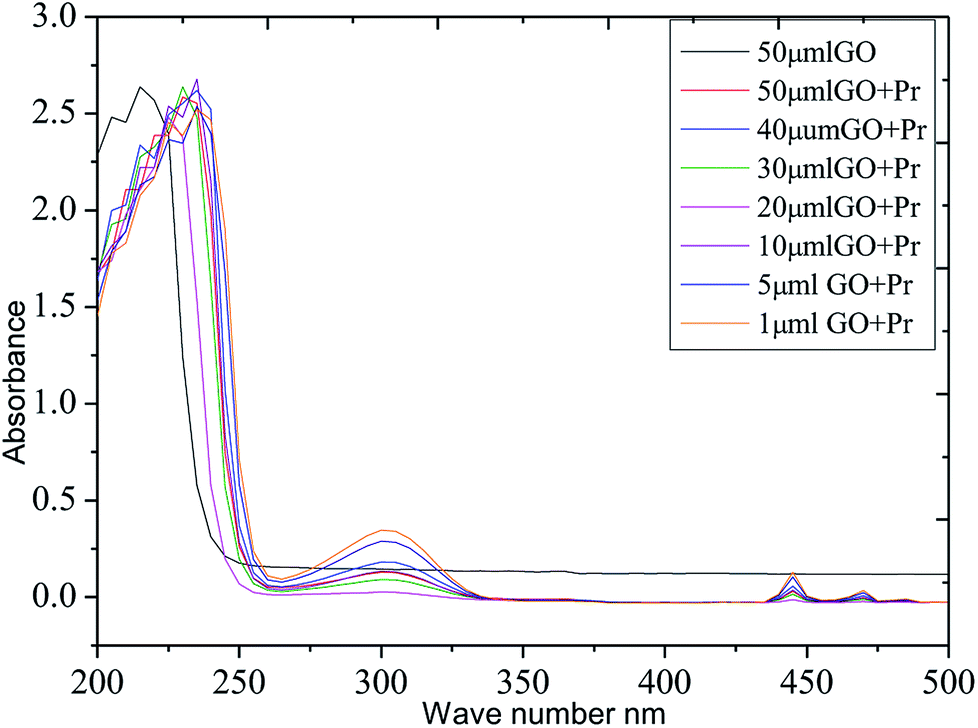
The thermal studies were conducted under an inert atmosphere to confirm the praseodymium oxide stable decomposition temperature. GO and GO-Pr thermal decomposition data are shown in Fig. 11, measured as relative weight loss percentages. The initial weight loss at 200 °C (ref. 78) is due to water moisture in the GO sheets. This weight loss in GO is shown to be 50%, and it constantly increases up to 500 °C. This is attributed to carboxylate and carbonyl group decomposition. The further stability of GO is due to the presence of strong sp2 carbon conjugated bonds, up to 600 °C.79 On the other hand, Pr + GO sheet weight loss was initiated at 350 °C, with gradual weight loss up to 450 °C; it is suggested that thermal decomposition of Pr oxide species suggests that Pr2(CO3)3·xH2O decomposition is the most probable feature observed in Fig. 11. For GO, the maximum decomposition rate occurs at 425 °C, while that of GO-Pr shifts to 444 °C; as mentioned before, GO-Pr is thermally more stable because of the refractory nature of Pr oxides, which are formed at a temperature of 480 °C.80 Optical absorption spectra of GO and GO-Pr solutions are shown in Fig. 12. High absorbance and the typical π to π* transition is observed at 230 nm in the GO suspension, as well as an abrupt decrease in absorbance at around 250 nm, due to partial GO reduction.81 Correspondingly, the GO-Pr suspension absorption increases as the concentration increases. GO + Pr absorbance peaks from 300 nm to 450 nm, and at 470 nm are closely related to those reported for Pr6O11.82,83
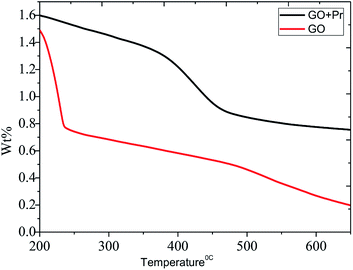 |
| | Fig. 11 TGA analyses of GO and GO + Pr. | |
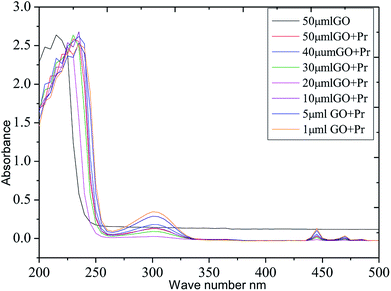 |
| | Fig. 12 UV-vis absorbance spectra of GO and GO + Pr suspensions. | |
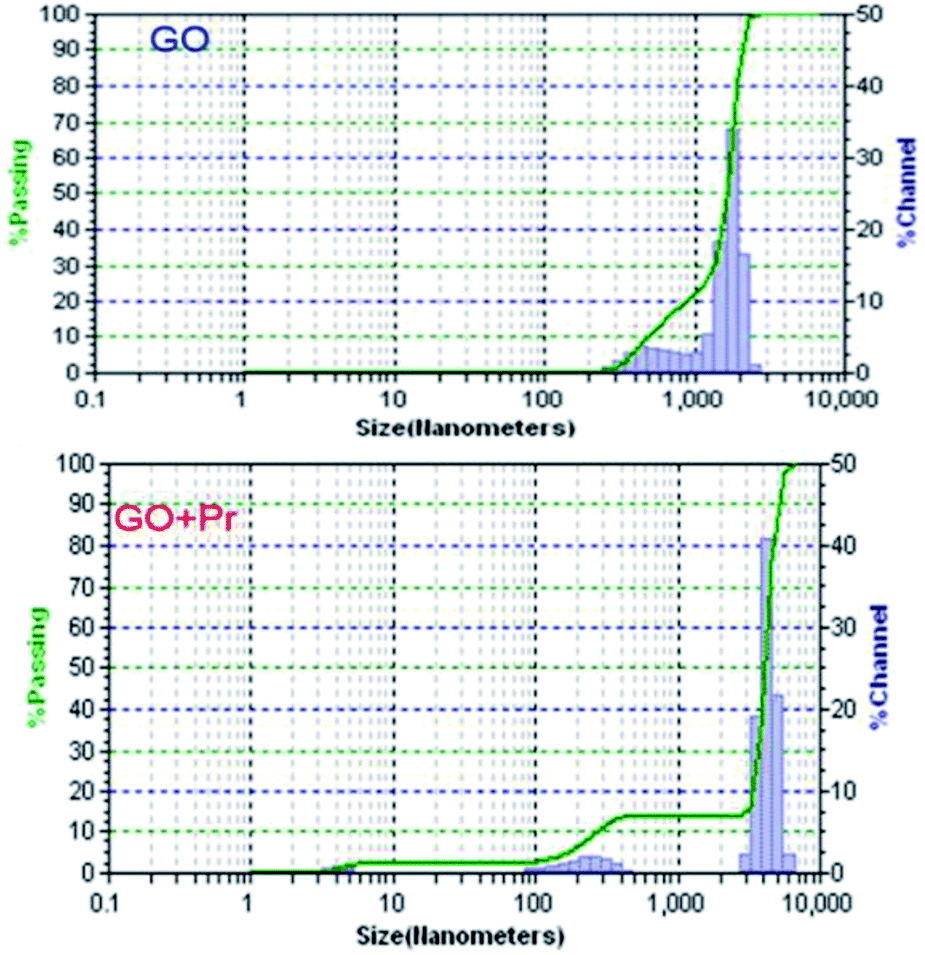
Hydrodynamic light scattering techniques (DLS) have been used to study the hydrodynamic particle sizes of GO + Pr and GO samples, as shown in Fig. 13. The size distribution of the GO sample has a bimodal shape, with one maximum centred at 500 nm with a maximum frequency of 10%, and the other one centred at 100 nm with a maximum of 70%. This behaviour suggests agglomeration between the GO sheets, mostly due to hydrogen bonding within adjacent sheets.84 Correspondingly, the GO-Pr sample has three maxima in its size distribution at 40 nm, 100 nm and 200 nm. The smallest sizes would correspond to Pr3+ adhered nanoparticles, as imaged in the HRTEM micrographs, while the 100 nm and 200 nm maxima would correspond to Pr-decorated sheets and agglomerated sheets, respectively.85
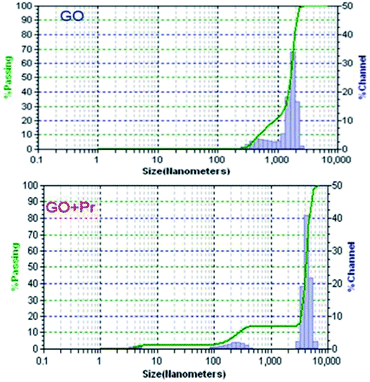 |
| | Fig. 13 Zeta potential data for GO and GO + Pr. | |
The colloid mobility increases with Pr incorporation, as well as the surface charge, because Pr GO sheet decoration produced smaller agglomerates on one side and more exfoliated sheets on the other, as described before. On the other hand, the Z potential and conductivity are reduced upon decoration, due to disruption of the aromaticity within the graphene oxide basal sheet. The positive charge is related to GO reduction (Table 1).
Table 1 A summary of the Z-potential data
| S. no. |
Mobility |
Z |
Charge |
Polarity |
σ |
| GO |
1.92 |
24.55 |
0.272 |
+ |
37 |
| GO-Pr |
11.10 |
14.90 |
0.414 |
+ |
26 |
3.4. Ellipsometry studies
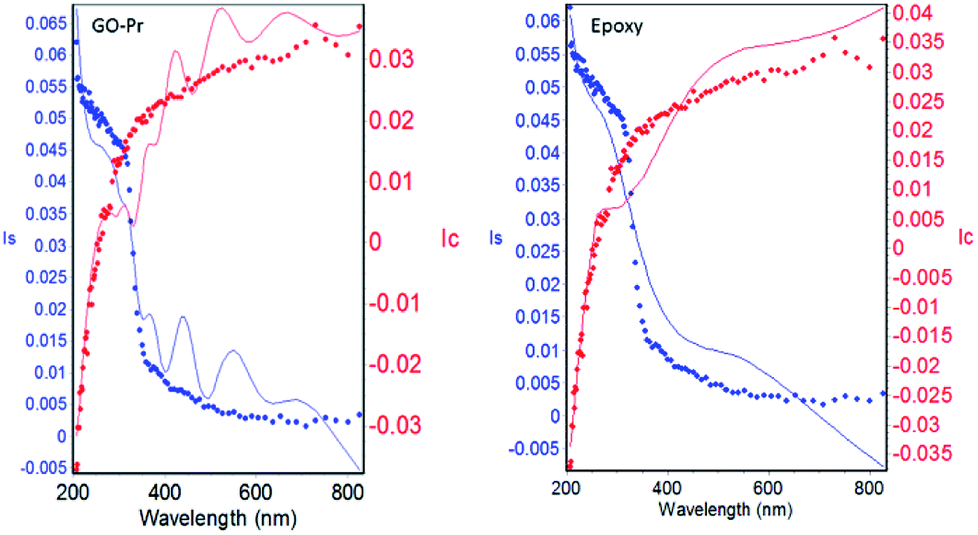
The epoxy and GO + Pr coating thicknesses are shown in Fig. 14. The solid lines are from experimental data and the dotted lines are from model fitting.86 A χ2 value of less than 5 is attributed to a close fit between model and experimental results. Our fitting results are less than 5, suggesting that the epoxy coating and GO + Pr coating are uniform, and the coating thickness χ2 values are shown in Table 2. The epoxy coating and GO + Pr coating are uniformly coated on the Mg alloy surfaces.87
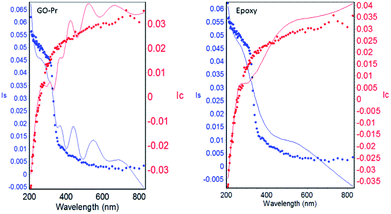 |
| | Fig. 14 GO + Pr coating and epoxy coating model and experimental data. | |
Table 2 Coating layer thickness values
| Sample |
χ2 fitting model |
Å coating thickness |
| Epoxy |
0.293564 |
1026.7331 |
| GO |
0.254214 |
733.9902 |
3.5. Electrochemical characterization
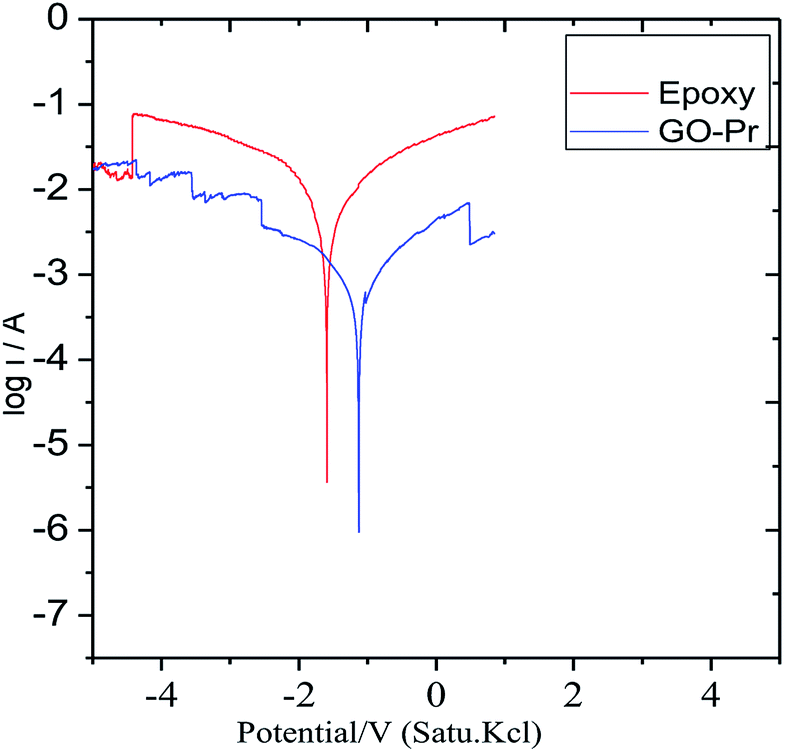
Epoxy and GO + Pr coated magnesium (Mg) alloys were continuously immersed for 3 days in 1 M H2SO4 solution. Afterwards, potentiodynamic polarization test were carried out, as shown in Fig. 15. The epoxy coated Mg alloy corrosion current increased and the corrosion potential values decreased. This indicates that the epoxy coating showed poor physisorption and chemisorption on Mg alloys in the acidic medium, and the Mg alloy initiated cathodic hydrogen evolution88–90 and anodic alloy dissolution. In the case of the GO + Pr composite coated Mg alloy, the corrosion current decreased and the corrosion potential increased due to strong physisorption and chemisorption. The potentiodynamic values are shown in Table 3. Further, GO + Pr coated Mg alloy hydrogen evolution and anodic alloy dissolution were controlled by the GO + Pr composite coating in the acidic medium.
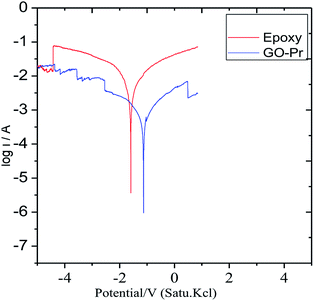 |
| | Fig. 15 The potentiodynamic polarization of the coated Mg alloys after 3 days of immersion in 1 M H2SO4. | |
Table 3 Magnesium alloy potentiodynamic and impedance values after 3 days of immersion in 1 M H2SO4
| Sample |
−Ecorr mV |
−Icorr mA |
ηIE % |
Rct Ω |
Cdl μF |
| Epoxy |
1.13 |
3.540 |
— |
489 |
6.58 |
| E + GO-Pr |
1.60 |
1.978 |
72.3 |
890 |
2.52 |
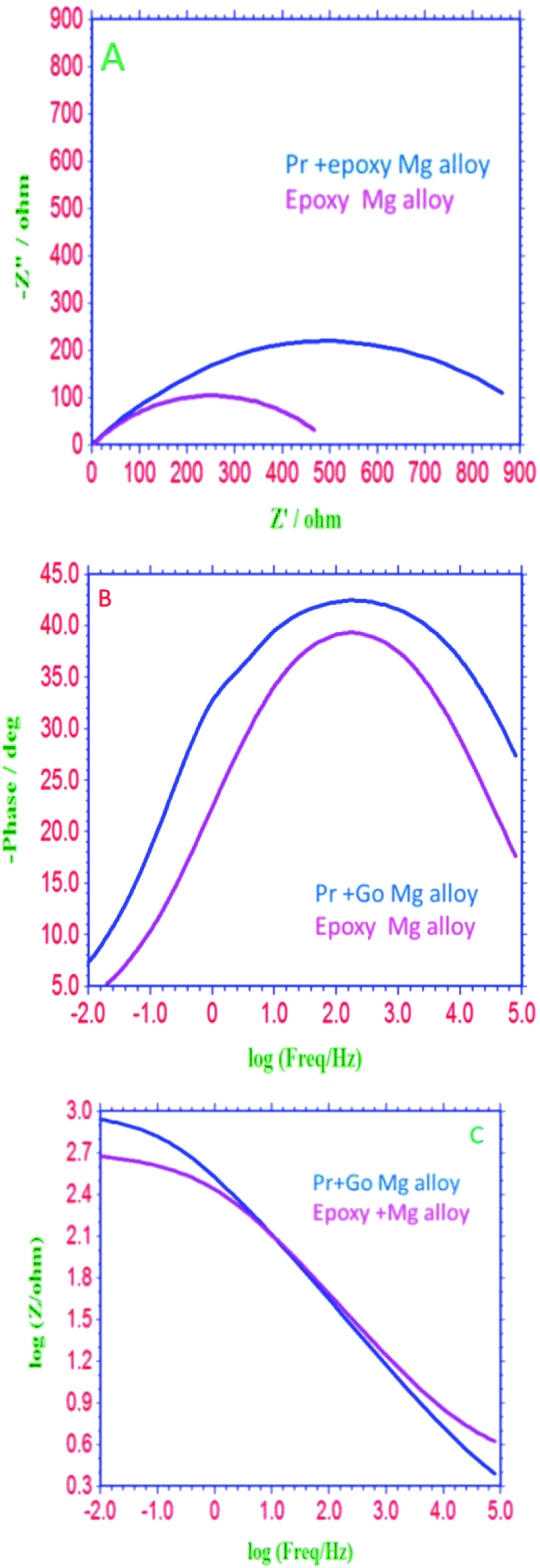
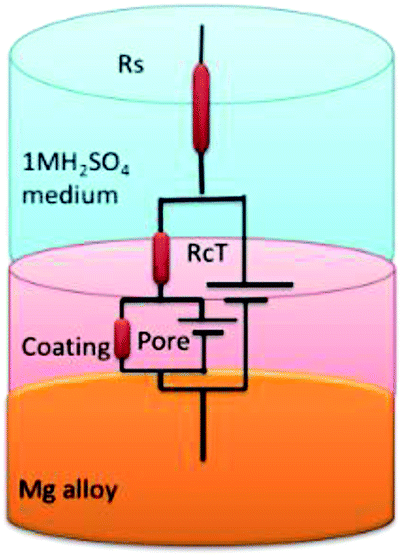
EIS spectra of epoxy and GO + Pr coated Mg alloys are presented in Fig. 16A; the Nyquist spectra indicates that the epoxy coated Mg alloy semicircle is suppressed, and this is attributed to pitting corrosion on the surface following attack by Cl ions. Hence, as shown in Fig. 16B and C, the epoxy coating Bode and frequency plots are also suppressed, due to localised corrosion initiated on the alloy surface due to epoxy coating failure in the acidic medium. On the other hand, charge transfer is decreased, and the double layer capacitance value is increased due to the poor physisorption of the epoxy coating on the alloy surface. This is attributed to the epoxy coating not being suitable as a long term coating material in acidic media. In the case of the GO + Pr coated Mg alloy, the Nyquist semicircle increased and the Bode and frequency plots are also increased due to the strong physisorption of the GO + Pr composite on the Mg alloy. Further, the charge transfer resistance Rct value is increased and the double layer capacitance value is decreased due to GO + Pr chemisorption on the alloy surface and the uniform surface.91–93 The charge transfer resistance and double layer capacitance values are shown in Table 3. Furthermore, the GO + Pr composite material shows excellent anticorrosion resistance in acidic environments due the Pr nanoparticle diffusing to the basal plane of the GO internal layer; this enhances passivation on the Mg alloy surface and controls H2 evolution. In the presented research, in decorated GO + Pr, the Pr nanoparticles provide synergetic effects due to their sub-nanometre size, and graphene oxide should act as diffusion barrier layer. Decorated GO-Pr, with sizes of around 200 nm, would provide mixed barrier inhibition, while free nonbonding electrons in Pr-GO sheet functional groups interacting with the alloy surface would act directly as inhibitors of hydrogen evolution Fig. 17 shows the equivalent circuit for fitting the electrochemical impedance values. The fitting values are presented in Table 3.
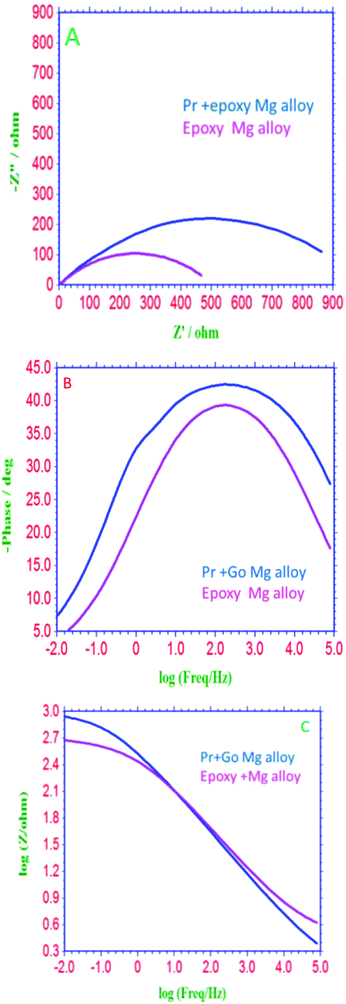 |
| | Fig. 16 (A) Nyquist, (B) Bode and (C) frequency plots of Mg alloys coated with epoxy and GO + Pr, after 3 days of immersion in 1 M H2SO4 medium. | |
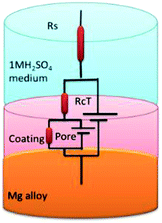 |
| | Fig. 17 The equivalent circuit. | |
3.6. Surface protection studies using the magnesium alloy AZ31
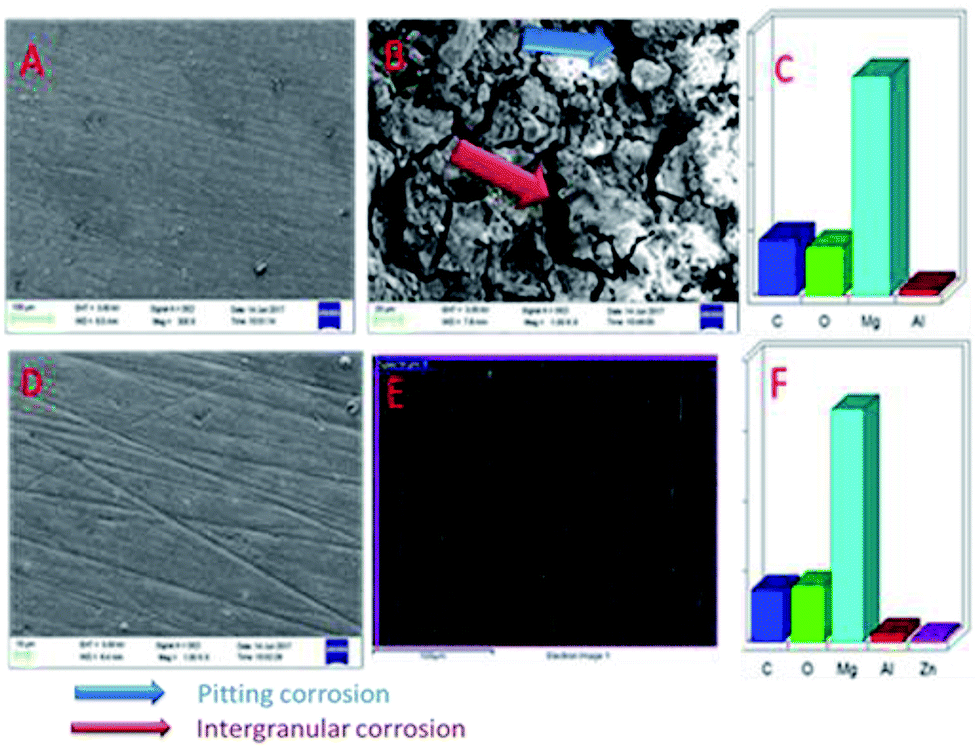
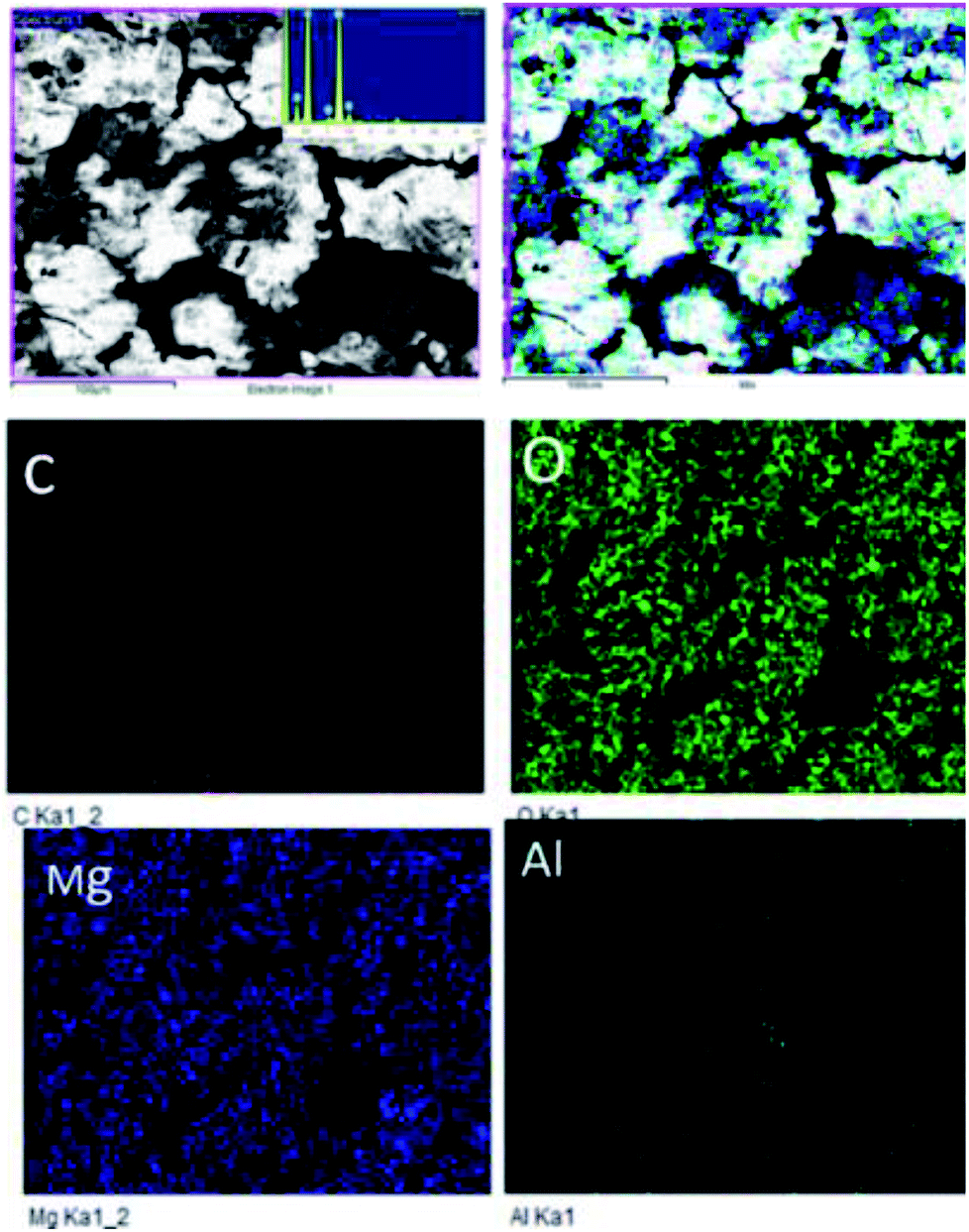
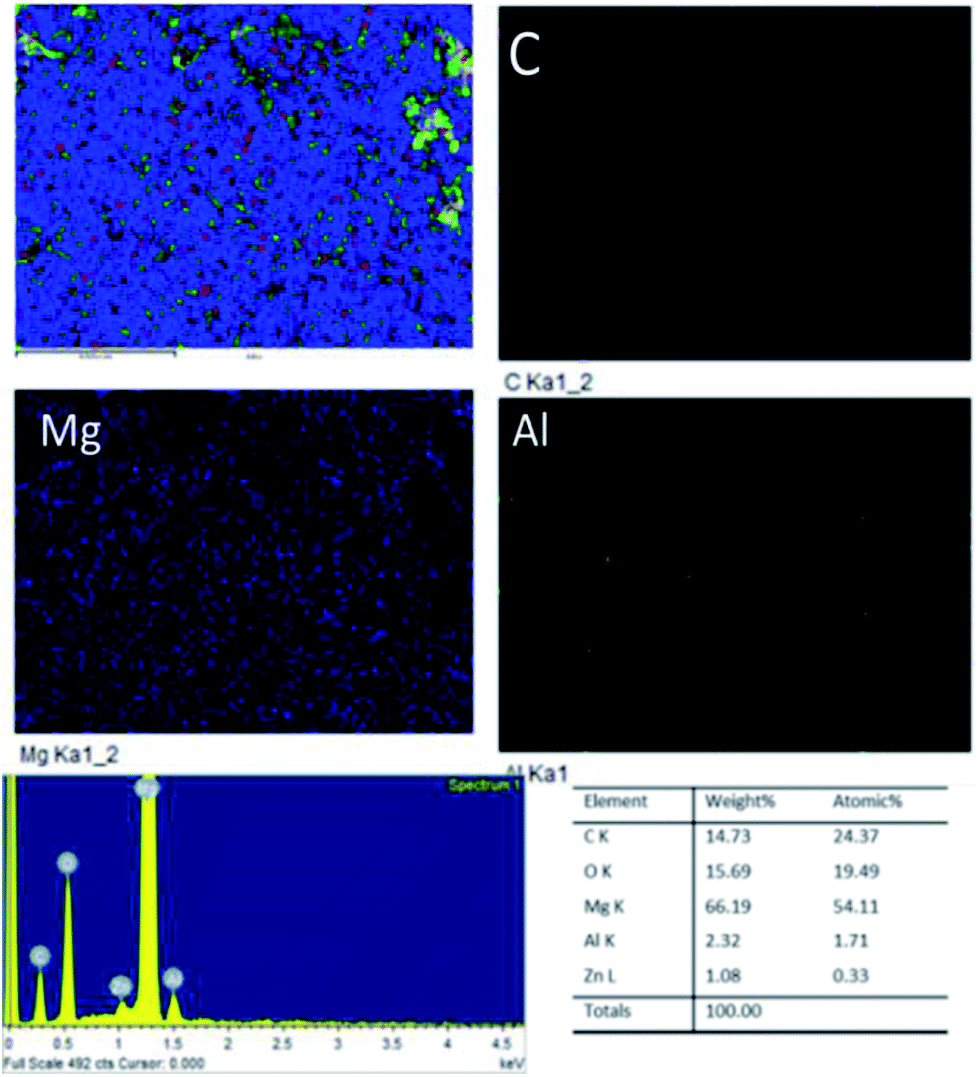
Fig. 18A shows an FESEM micrograph before the coating of the Mg alloy. Fig. 18B shows the epoxy coated Mg alloy after 3 days of immersion in 1 M H2SO4 medium, and Fig. 18C shows EDX data from the epoxy coated Mg alloy. These results indicate that the epoxy coating is not stable in an acidic environment. Further, at the epoxy coated Mg alloy grain boundary, localised corrosion is initiated on the alloy surface due to hydrogen evolution.94,95 In the case of the GO + Pr composite coated Mg alloy, results after 3 days of immersion in 1 M H2SO4 are presented in Fig. 18D–F. Here, the Mg alloy surface is homogenous and, at the grain boundary, pitting corrosion does not appear on the alloy surface. This is attributed to the Pr + GO coating showing strong physisorption on the alloy surface.96,97 According to the EDX results relating to the composition of the epoxy coated Mg alloy, the Zn element dissolved in the aggressive medium. In the case of the Pr + GO coated Mg alloy, the composition of elements is not changed due to the strong physisorption of the Pr + GO material. Furthermore, FESEM element mapping results suggesting that, from the element composition, Zn dissolution is high in the epoxy coated Mg alloy, compared with the Pr + GO Mg alloy, are presented in Fig. 19. Further, the Pr + GO composite coated Mg alloy element composition, showing uniform distribution, is presented in Fig. 20. From the microstructural results, it is suggested that Pr + GO shows strong physisorption and chemisorption compared with the epoxy coating. The GO + Pr composite coating is significantly corrosion resistant in acidic environments.
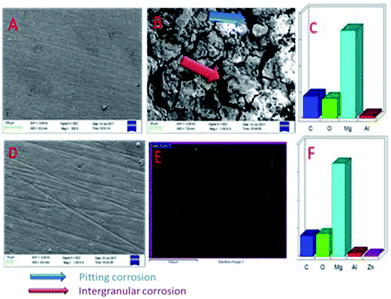 |
| | Fig. 18 SEM micrographs of the epoxy coated AZ31 alloy (A) before immersion and (B) after immersion in the acidic medium, and (C) EDX data. (D), (E), and (F) shows corresponding data for the GO + Pr coated alloy. | |
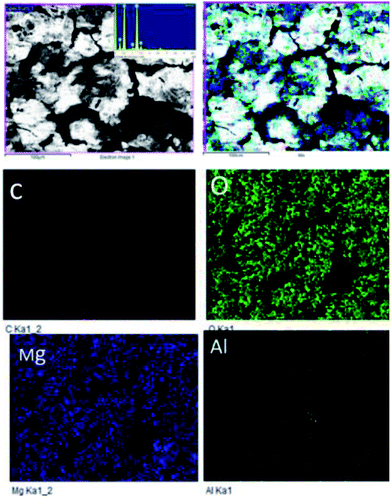 |
| | Fig. 19 Elemental mapping of the epoxy coated Mg alloy. | |
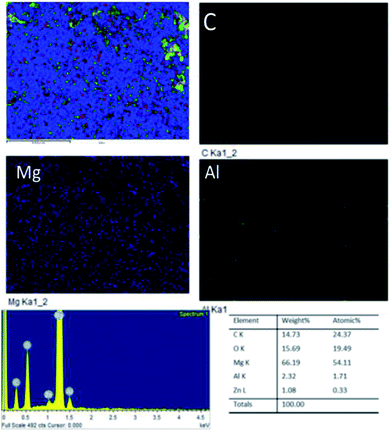 |
| | Fig. 20 Elemental mapping of the GO + Pr coated Mg alloy. | |
The Mg alloy microstructural results suggest that the Pr + GO coating exhibits excellent corrosion resistance compared with the epoxy coating on the Mg alloy in the acidic medium.
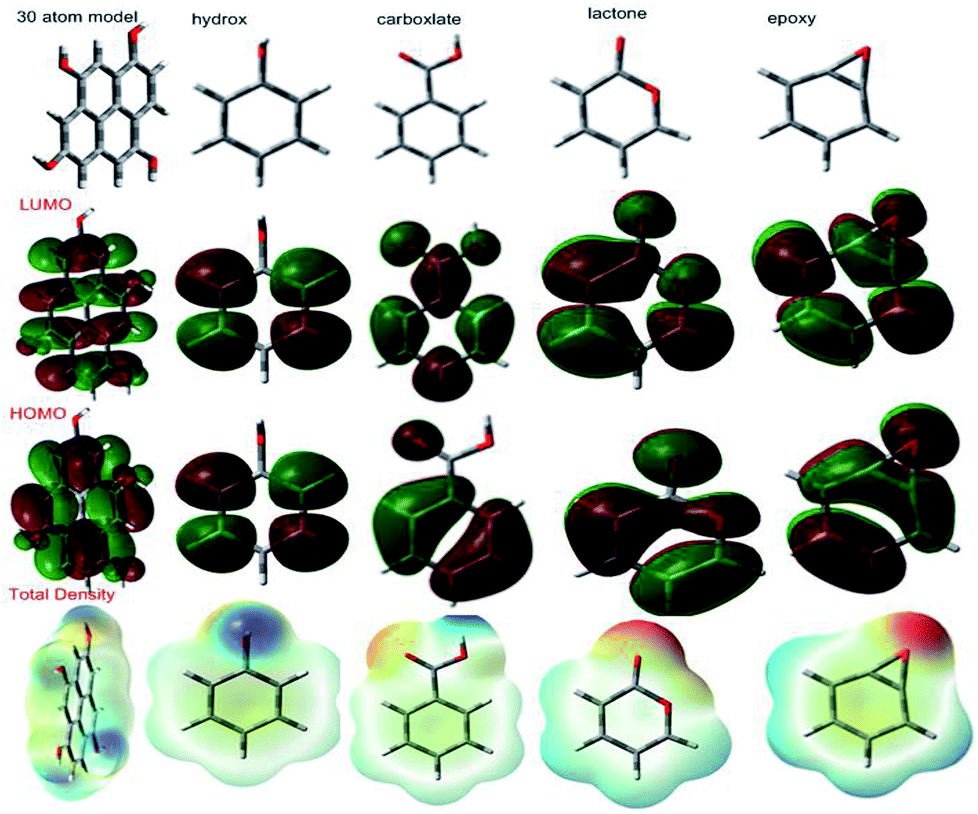
3.6.1 Quantum chemical graphene oxide model and functional groups. According to a literature reported model,98 we have investigated 30 atom graphene oxide models and their functional groups for possible electron donation and accepting involving the alloy and the surface, as shown in Fig. 21. The highest occupied molecular orbital (HOMO) values are assigned for electron donation to the vacant d-orbital of the alloy. The lowest unoccupied molecular orbital (LUMO) accepts electrons from the alloy surface. The model quantum chemical values are favourable for electron accepting and donating capabilities. The GO model global softness value increased; this indicates that graphene oxide could be adsorbed on the alloy surface. The global hardness value also decreased; this indicates that with graphene oxide functional groups, lone pair electrons tend to give partially filled vacant d orbitals in Mg alloys. Another parameter, dipole movement, is a significant factor for inhibitor molecules to increase the reactivity. The GO functional group dipole movements are gradually increased; this leads to increased adsorption on Mg alloy surfaces. Further, the total density values also support enhanced adsorption on the alloy surface. Hence, the GO sheet negative charge could repel electrolytes and water molecules from the alloy surface. The total density value gradually increases; this leads to protecting the surface from corrosive ion attacks. The quantum chemical 30 atom GO model and functional group results are in good agreement with the experimental results (Table 4).
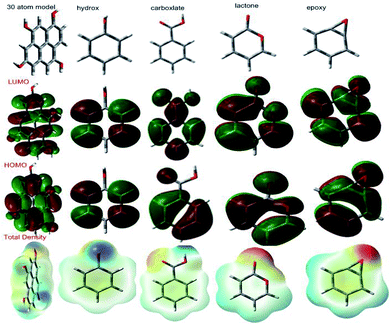 |
| | Fig. 21 GO functional group HOMO and LUMO levels. | |
Table 4 Quantum chemical GO functional group values
| S. No |
−HOMO eV |
−LUMO eV |
η |
χ |
ω |
σ |
D.M (μ) |
T.D |
| Model |
6.9644 |
2.1320 |
2.4162 |
7.4240 |
3.7119 |
0.4138 |
2.3010 |
906.1210 |
| Hydroxy |
6.5454 |
0.0054 |
3.2700 |
0.0176 |
0.0084 |
0.3058 |
1.7610 |
305.7674 |
| Epoxy |
6.0801 |
0.3044 |
2.8878 |
0.9253 |
0.4626 |
0.3462 |
2.4099 |
304.4123 |
| Ketone |
6.1260 |
0.2163 |
5.9009 |
0.6625 |
0.3306 |
0.1694 |
2.8162 |
308.1669 |
| Lactone |
6.5130 |
1.6775 |
2.4177 |
5.4627 |
2.7313 |
0.4136 |
4.4600 |
341.4320 |
| Carboxylate |
7.1685 |
1.2536 |
2.9574 |
4.4932 |
2.2465 |
0.3381 |
1.6361 |
418.5017 |
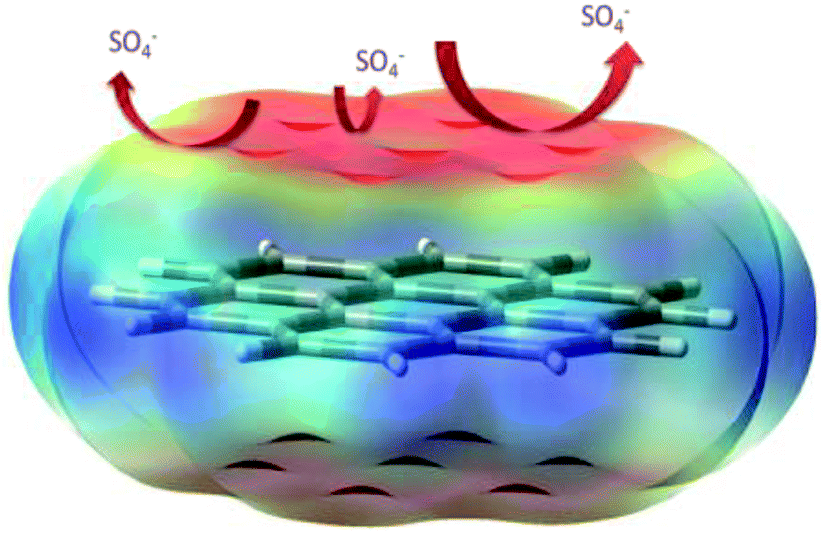
3.6.2 The possible mechanism. The 30 atom model of the graphene oxide sheet total density and the sulphate ion repulsion model are presented in Fig. 22. Graphene oxide has been studied as an excellent impermeable barrier layer on iron and SS alloys;98–101 in the graphene oxide edge functional groups, such as carbonyl, hydroxyl and carboxylate, free electrons increase the chemisorption on the alloy surface. This attributed negative charge on the graphene oxide could repel electrolyte ions. Further, the quantum chemical calculation values, such as global softness and hardness, HOMO and LUMO levels, and electrophility, are favourable for electron donation and accepting from the vacant d orbitals of the alloy. These results indicate that Pr + GO sheets are strongly adsorbed on the alloy surface. For that reason, the Pr–graphene oxide composite coating acts as an impermeable barrier layer. In the case of epoxy coatings, electrolytes can easily move through micropores towards the coating, initiating localised corrosion on the alloy surface.
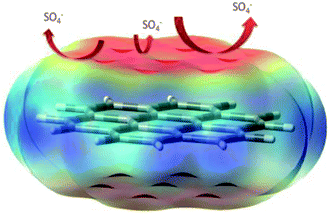 |
| | Fig. 22 A proposed scheme for the repulsion of corrosive ions. | |
4. Conclusions
Praseodymium decorated graphene oxide was prepared and tested for corrosion inhibition with Mg alloys. The praseodymium decorated graphene oxide coated AZ31 Mg alloy was immersed for 3 days in 1 M H2SO4. The results indicates that GO is partially reduced upon Pr-decoration and Pr carbonates and oxides are present, as well as some metallic Pr due to the disproportionation of Pr3+. Strongly adhered nanoparticles were observed via HRTEM and measured using DLS, where events related to decorated and agglomerated sheets were also recorded. From TGA analysis, it was found that decorated graphene oxide is thermally more stable than pristine GO. The Pr + GO composite coating was a significant barrier in acidic media compared to the epoxy coating, which was confirmed via electrochemical and microscopy methods. The GO + Pr composite coating corrosion potential is increased compared with the epoxy coating. And GO 30 atom model DFT study results suggest that electron accepting and donation occurs from the alloy surface. The experimental values and theoretical data suggest that the GO + Pr coating shows higher corrosion resistance in acidic media.
Conflicts of interest
The authors herein declare no conflicts of interest.
Acknowledgements
NP acknowledges the Central University of Gujarat Central Instrument facility, the UGC Network Resource Centre Hyderabad/University of Hyderabad, and a Non-Net fellowship, as well as Prof M. Sathiyendiran for support regarding the completion of this work. The authors thank Dr A. Srinivasan at the Materials Science and Technology Division CSIR-National Institute for Interdisciplinary Science and Technology (CSIRNIIST) in Thiruvananthapuram for providing the AZ31 Mg alloy; this work was partially financed by SIP-IPN Project 20161804.
References
- P. D. Ducharme and J. Mauzeroll, Anal. Chem., 2015, 87, 7499–7509 CrossRef PubMed.
- R. L. Liu, J. R. Scully, G. Williams and N. Birbilis, Electrochim. Acta, 2018, 260, 184–195 CrossRef CAS.
-
(a) S. Thomas, N. Birbilis, M. S. Venkatraman and I. S. Cole, Corros. Sci., 2013, 69, 11–22 CrossRef CAS;
(b) V. Chakarova, Tz. Boiadjieva-Scherzer, D. Kovacheva, H. Kronberger and M. Monev, Corros. Sci., 2018, 140, 73–78 CrossRef CAS;
(c) S. Dea and J. L. Lutkenhaus, Green Chem., 2018, 20, 506 RSC.
- A. Frignani, V. Grassi, F. Zanotto and F. Zucchi, Corros. Sci., 2012, 63, 29–39 CrossRef CAS.
- D. Buggio, M. Trueba and S. P. Trasatti, Corros. Sci., 2016, 104, 173–186 CrossRef CAS.
- X. Guo, K. Du, Q. Guo, Y. Wang, R. Wang and F. Wang, Corros. Sci., 2013, 76, 129–141 CrossRef CAS.
- N. H. Helal and W. A. Badawy, Electrochim. Acta, 2011, 56, 6581–6587 CrossRef CAS.
- J. Hu, D. Huang, G. L. Song and X. Guo, Corros. Sci., 2011, 53, 4093–4101 CrossRef CAS.
- J. Hu, D. Huang, G. Zhang, G. L. Song and X. Guo, Corros. Sci., 2012, 63, 367–378 CrossRef CAS.
- J. Hu, D. Zeng, Z. Zhang, T. Shi, G. L. Song and X. Guo, Corros. Sci., 2013, 74, 35–43 CrossRef CAS.
- D. Huang, J. Hu, G. L. Song and X. Guo, Electrochim. Acta, 2011, 56, 10166–10178 CrossRef CAS.
- J. Xu, Q. Yang, M. S. Javed, Y. Gong, K. Aslama and C. Chen, RSC Adv., 2017, 7, 5880 RSC.
- X.-B. Chen, N. Birbilis and T. B. Abbott, Corros. Sci., 2012, 55, 226–232 CrossRef CAS.
- X.-B. Chen, N. Birbilis and T. B. Abbott, Corros. Sci., 2011, 53, 2263–2268 CrossRef CAS.
- G. Williams, H. N. McMurray and R. Grace, Electrochim. Acta, 2010, 55, 7824–7833 CrossRef CAS.
- N. T. Kirkland, G. Williams and N. Birbilis, Corros. Sci., 2013, 77, 407–409 CrossRef CAS.
- N. T. Kirkland, G. Williams and N. Birbilis, Corros. Sci., 2012, 65, 5–9 CrossRef CAS.
- G. Williams, H. L. Dafydd and R. Grace, Electrochim. Acta, 2013, 109, 489–501 CrossRef CAS.
- G. Williams, N. Birbilis and H. Neil McMurray, Faraday Discuss., 2015, 180, 313–330 RSC.
- D. S. Gandel, M. A. Easton, M. A. Gibson, T. Abbott and N. Birbilis, Corros. Sci., 2014, 81, 27–35 CrossRef CAS.
- D. Eaves, G. Williams and H. N. McMurray, Electrochim. Acta, 2012, 79, 1–7 CrossRef CAS.
- G. Williams, H. L. Dafydd, H. N. McMurray and N. Birbilis, Electrochim. Acta, 2016, 219, 401–411 CrossRef CAS.
- R. L. Liu, Z. R. Zeng, J. R. Scully, G. Williams and N. Birbilis, Corros. Sci., 2018, 140, 18–29 CrossRef CAS.
- Y. Wang, R. K. Gupta, N. L. Sukiman, R. Zhang, H. J. Davies and N. Birbilis, Corros. Sci., 2013, 73, 181–187 CrossRef CAS.
- A. Samaniego, K. Gusieva, I. Llorente, S. Feliu and N. Birbilis, Corros. Sci., 2014, 89, 101–110 CrossRef CAS.
- Y. Qiao, W. Li, G. Wan and X. Zhang, J. Rare Earths, 2015, 33, 647–654 CrossRef CAS.
- S. Li, W. Zheng, B. Tang, D. Zeng and X. Guo, J. Rare Earths, 2007, 25, 227–232 CrossRef.
- Z. Weichao, L. Shuangshou, T. Bin, Z. Daben and G. Xutao, J. Rare Earths, 2006, 24, 346–351 CrossRef.
- G. bi, J. Jian, F. Zhang, D. Fang, Y. LI, Y. ma and Y. Hao, J. Rare Earths, 2016, 34, 931–937 CrossRef CAS.
- G. S. Frankel, A. Samaniego and N. Birbilis, Corros. Sci., 2013, 70, 104–111 CrossRef CAS.
- S. H. Salleh, S. Thomas, J. A. Yuwono, K. Venkatesan and N. Birbilis, Electrochim. Acta, 2015, 161, 144–152 CrossRef CAS.
- S. Fajardo, C. F. Glover, G. Williams and G. S. Frankel, Electrochim. Acta, 2016, 212, 510–521 CrossRef CAS.
- Y.-J. Wu, X.-B. Chen, G. Williams, J. R. Scully, T. Gengenbach and N. Birbilis, RSC Adv., 2016, 6, 43408–43417 RSC.
- J. Y. Chen, X. Chen, J.-L. Li, B. Tang, N. Birbilis and X. Wang, J. Mater. Chem. A, 2014, 2, 5738–5743 RSC.
- N. T. Kirkland, T. Schiller, N. Medhekar and N. Birbilis, Corros. Sci., 2012, 56, 1–4 CrossRef CAS.
- Y. Zhu, J. Zhuang, Y. Yu and X. Zeng, J. Rare Earths, 2013, 31, 734–740 CrossRef CAS.
- E. Zhang, Y. Chen and Y. Tang, J. Rare Earths, 2012, 30, 269–273 CrossRef CAS.
- G. Kong, L. Liu, J. Lu, C. Che and Z. Zhong, J. Rare Earths, 2010, 28, 461–465 CrossRef CAS.
- G. Kong, J. Lu and H. Wu, J. Rare Earths, 2009, 27, 164–168 CrossRef.
- X. Xing, Z. Han, H. Wang and P. Lu, J. Rare Earths, 2015, 33, 1122–1128 CrossRef CAS.
- A. K. Mishra and R. Balasubramaniam, Corros. Sci., 2007, 49, 1027–1044 CrossRef CAS.
- A. L. Rudda, C. B. Breslin and F. Mansfield, Corros. Sci., 2000, 42, 275–288 CrossRef.
- X. Li, S. Deng, H. Fu and G. Mu, Corros. Sci., 2008, 50, 3599–3609 CrossRef CAS.
- X. Li, S. Deng, H. Fu and G. Mu, Corros. Sci., 2009, 51, 2639–2651 CrossRef CAS.
- N. D. Nama, A. Somers, M. Matheshc, M. Seter, B. Hinton, M. Forsyth and M. Y. J. Tana, Corros. Sci., 2014, 45, 128–138 CrossRef.
- M. Machkova, E. A. Matter, S. Kozhukharov and V. Kozhukharov, Corros. Sci., 2013, 69, 396–405 CrossRef CAS.
- A. E. Somers, B. R. W. Hinton, C. B. Dickason, G. B. Deacon, P. C. Junk and M. Forsyth, Corros. Sci., 2018, 139, 430–437 CrossRef CAS.
- H. Zheng, M. Guo, Y. Shao, Y. Wang, B. Liu and G. Meng, Corros. Sci., 2018, 139, 1–12 CrossRef CAS.
- L. Yang, Y. Wan, Z. Qin, Q. Xu and Y. Min, Corros. Sci., 2018, 130, 85–94 CrossRef CAS.
- I. Wlasny, P. Dabrowski, M. Rogala, I. Pasternak, W. Strupinski, J. M. Baranowski and Z. Klusek, Corros. Sci., 2015, 92, 69–75 CrossRef CAS.
- C. F. Glover, C. A. J. Richards, G. Williams and H. N. McMurray, Corros. Sci., 2018, 136, 304–310 CrossRef CAS.
- C. F. Glover, C. Richards, J. Baker, G. Williams and H. N. McMurray, Corros. Sci., 2017, 114, 169–172 CrossRef CAS.
- J. Zhao, X. Xie and C. Zhang, Corros. Sci., 2017, 114, 146–155 CrossRef CAS.
- S. Pourhashem, M. R. Vaezi, A. Rashidi and M. R. Bagherzadeh, Corros. Sci., 2017, 115, 78–92 CrossRef CAS.
- E. G. Yazdi, Z. S. Ghahfarokhia and M. Bagherzadeh, New J. Chem., 2017, 41, 12470–12480 RSC.
- S. Mayavan, T. Siva and S. Sathiyanarayanan, RSC Adv., 2013, 3, 24868–24871 RSC.
- A. Madhan Kumar, R. Suresh Babu, I. B. Obot and Z. M. Gasem, RSC Adv., 2015, 5, 19264–19272 RSC.
- C. Liu, S. Qiu, P. Du, H. Zhao and L. Wang, Nanoscale, 2018, 10, 8115–8124 RSC.
- R. K. Gupta, M. Malviya, C. Varma and M. A. Quraishi, Mater. Chem. Phys., 2017, 198, 360–373 CrossRef CAS.
- M. J. Nine, M. A. Cole, D. N. H. Tran and D. Losic, J. Mater. Chem. A, 2015, 3, 12580–12602 RSC.
- D. Ponnamma, K. K. Sadasivuni, M. Strankowski, P. Moldenaers, S. Thomas and Y. Grohens, RSC Adv., 2013, 3, 16068–16079 RSC.
- J. Jayaraj, S. Amruth Raj, A. Srinivasan, S. Ananthakumar, U. T. S. Pillai, N. G. K. Dhaipule and U. KamachiMudali, Corros. Sci., 2016, 113, 104–115 CrossRef CAS.
- W. Sun, L. Wang, T. Wu, Y. Pan and G. Liu, J. Mater. Chem. A, 2015, 3, 16843–16848 RSC.
- R. K. Gupta, M. Malviya, C. Verma, N. K. Gupta and M. A. Quraishi, RSC Adv., 2017, 7, 39063 RSC.
- A. Kumar, R. Anant, K. l Kumar, S. S. Chauhan, S. Kumar and R. Kumara, RSC Adv., 2016, 6, 113405 RSC.
-
(a) J. C. Soares, K. P. F. Siqueira, P. C. de Sousa Filho, R. L. Moreira and A. Dias, Dalton Trans., 2017, 46, 825–835 RSC;
(b) Q. Xu, M. Zeng, Z. Feng, D. Yin, Y. Huang, Y. Chen, C. Yan, R. Li and G. U. Yi, RSC Adv., 2016, 6, 31484–31496 RSC.
-
(a) N. Ahmad, M. M. Ahmad and P. N. Kotru, RSC Adv., 2015, 5, 93808–93817 RSC;
(b) Y. Borchert, P. Sonstro, M. Wilhelm, H. Borchert and M. Baumer, J. Phys. Chem. C, 2008, 112, 3054–3063 CrossRef CAS.
- H. Al Kutubi, L. Rassaei, W. Olthuis, G. W. Nelson, J. S. Foord, P. Holdway, M. Carta, R. Malpass-Evans, N. B. McKeown, S. C. Tsang, R. Castaing, T. R. Forder, M. D. Jones, D. He and F. Marken, RSC Adv., 2015, 5, 73323–73326 RSC.
- Y. Guan, T. Tsuboi, Y. Huang and W. Huang, Dalton Trans., 2014, 43, 3698–3703 RSC.
-
(a) M. A. Gusowski, H. C. Swart, L. S. Karlsson and M. T. Gusowska, Nanoscale, 2012, 4, 541–546 RSC;
(b) G. A. M. Hussein, J. Anal. Appl. Pyrolysis, 1994, 29, 89–102 CrossRef CAS.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291–312 RSC.
- M. Balaguer, C. Solís, S. Roitsch and J. M. Serra, Dalton Trans., 2014, 43, 4305–4312 RSC.
-
(a) J. M. C. Moreno, V. G. Pol, S. H. Suh and M. Popa, Inorg. Chem., 2010, 49, 10067–10073 CrossRef PubMed;
(b) A. Dodd, J. Colloid Interface Sci., 2013, 392, 137–140 CrossRef CAS PubMed.
- S. Z. Ajabshir and M. S. Niasari, New J. Chem., 2015, 39, 3948–3955 RSC.
- Y. A. El-Nadi, J. Rare Earths, 2010, 28, 215 CrossRef CAS.
-
(a) M. A. Krishnan, K. S. Aneja, A. Shaikh, S. Bohm, K. Sarkar, H. L. Mallika Bohm and V. S. Raja, RSC Adv., 2018, 8, 499 RSC;
(b) D. He, Z. Peng, W. Gong, Y. Luo, P. Zhao and L. Kong, RSC Adv., 2015, 5, 11966 RSC;
(c) L. R. Radovic, A. Suarez, F. Burgos and J. O. Sofo, Carbon, 2011, 49, 4226–4238 CrossRef CAS.
-
(a) V. H. Pham, H. D. Pham, T. T. Dang, S. H. Hur, E. J. Kim, B. S. Kong, S. Kima and J. S. Chung, J. Mater. Chem., 2012, 22, 10530–10536 RSC;
(b) C. H. Lee, J. Yu, Y. Wang, A. Y. L. Tang, C. W. Kan and J. H. Xin, RSC Adv., 2018, 8, 16593 RSC.
-
(a) S. Z. Ajabshir, M. S. Niasari and M. Hamadanian, RSC Adv., 2015, 5, 33792–33800 RSC;
(b) X. Zhou, L. J. Broadbelt and R. Vinu, Adv. Chem. Eng., 2016, 49, 95–198 CrossRef.
- L. Guangxiang, Z. Hong and R. Xiaoming, J. Rare Earths, 2011, 29, 1100 CrossRef.
-
(a) V. H. Pham, H. D. Pham, T. T. Dang, S. H. Hur, E. J. Kim, B. S. Kong, S. Kima and J. S. Chung, J. Mater. Chem., 2012, 22, 10530–10536 RSC;
(b) B. M. A. Zied, Y. A. Mohamed and A. M. Asiri, J. Rare Earths, 2013, 31, 701 CrossRef.
- A. Sarkar, C. Loho, L. Velasco, T. Thomas, S. S. Bhattacharya, H. Hahn and R. Djenadic, Dalton Trans., 2017, 46, 12167–12176 RSC.
-
(a) Y. Haibin, Z. Jianhui, Y. Ruijin and S. Qiang, J. Rare Earths, 2009, 27, 308 CrossRef;
(b) B. S. Balachandran, K. Thirumalai and M. Swaminathan, RSC Adv., 2014, 4, 27642–27653 RSC.
-
(a) R. F. Gonzalez, B. J. anLopez and E. C. P. Escribano, J. Mater. Chem., 2011, 21, 497–504 RSC;
(b) Y. Borchert, P. Sonstrom, M. Wilhelm, H. Borchert and M. Baumer, J. Phys. Chem. C, 2008, 112, 3054–3063 CrossRef CAS.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342–3347 CrossRef CAS.
-
(a) C. Shih, S. Lin, R. Sharma, M. S. Strano and D. Blankschtein, Langmuir, 2012, 28, 235–241 CrossRef CAS PubMed;
(b) N. Hara, Y. Kobayashi, D. Kagaya and N. Akao, Corros. Sci., 2007, 49, 166–175 CrossRef CAS.
- N. Palaniappan, L. R. Chowhan, S. Jothi, I. G. Bosco and I. S. Cole, Surf. Interfaces, 2016, 000, 1–10 Search PubMed.
- P. W. Chu, E. L. Mire and E. A. Marquis, Corros. Sci., 2017, 128, 253–264 CrossRef CAS.
- J. Liao and M. Hotta, Corros. Sci., 2016, 112, 276–288 CrossRef CAS.
- F. Singer, M. Schlesak, C. Mebert, S. Hohn and S. Virtanen, ACS Appl. Mater. Interfaces, 2015, 26758–26766 CrossRef CAS PubMed.
- Q. Liu, D. Chen and Z. Kang, ACS Appl. Mater. Interfaces, 2015, 7, 1859–1867 CrossRef CAS PubMed.
- L. B. Tong, J. B. Zhang, C. Xu, X. Wang, S. Y. Song, Z. H. Jiang, S. Kamado, L. R. Cheng and H. J. Zhang, Carbon, 2016, 109, 340–351 CrossRef CAS.
- P. Porızka, I. Rocnakova, J. Klus, D. Prochazka, L. Sladkova, P. Sperka, Z. Spotz, L. Celko, K. Novotny and J. Kaiser, J. Anal. At. Spectrom., 2015, 30, 2099–2106 RSC.
- P. W. Chu, E. L. Mire and E. A. Marquis, Corros. Sci., 2017, 128, 253–264 CrossRef CAS.
- H. Qi, Y. Qian, J. Xua, J. Zuo and M. L. Lia, Corros. Sci., 2018, 138, 170–177 CrossRef CAS.
- Z. Qiu, R. Wang, J. Wu, Y. Zhang, Y. Qu and X. Wu, RSC Adv., 2015, 5, 44149 RSC.
- W. Shang, C. He, Y. Wen, Y. Wang and Z. Zhang, RSC Adv., 2016, 6, 113967–113980 RSC.
- C. Zhang, D. M. Dabbs, L. M. Liu, I. A. Aksay, R. Car and A. Selloni, J. Phys. Chem. C, 2015, 119, 18167–18176 CrossRef CAS.
- Y. H. Yu, Y. Y. Lin, C. H. Lin, C. C. Chan and Y. C. Huang, Polym. Chem., 2014, 5, 535–550 RSC.
- R. Kumar Gupta, M. Malviya, C. Verma, N. K. Gupta and M. A. Quraishi, RSC Adv., 2017, 7, 39063–39074 RSC.
- A. Madhan Kumar, R. Suresh Babu, I. B. Obot and Z. M. Gasema, RSC Adv., 2015, 5, 19264–19272 RSC.
- N. Palaniappan, L. Raju Chowhan, S. Jothi, I. G. Bosco and I. S. Cole, Surf. Interfaces, 2016, 000, 1–10 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Ivan. S. Coleb,
F. Caballero-Brionesc,
Balasubaramanian K.d and
C. Lale
*a,
Ivan. S. Coleb,
F. Caballero-Brionesc,
Balasubaramanian K.d and
C. Lale