
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical bond parameters, bond energy and the local crystal sites of Eu3+ in Ca5(BO3)3F:1% Eu3+ phosphor
Yuhan Zhu,
Yu Pan,
Wenjun Wang,
Haibing Xu,
Liqun Zhou,
Xiaoguang Liu* and
Ling Li *
*
Hubei Collaborative Innovation Center for Advanced Organochemical Materials, Ministry-of-Education Key Laboratory for the Synthesis and Applications of Organic Functional Molecules, Hubei University, Wuhan, Hubei, China. E-mail: liling402431@hotmail.com; liuxiaoguang402@hotmail.com
First published on 18th September 2018
Abstract
The local crystal sites occupied by Eu3+ in Ca5(BO3)3F:1% Eu3+ phosphor were investigated experimentally and theoretically. Ca5(BO3)3F:1% Eu3+ was synthesized by high-temperature solid-state method in air. The crystal structure and optical properties of the phosphor were studied by X-ray powder diffraction and photoluminescence, respectively. Two different O2− → Eu3+ CT broad bands with the peaks at 266 and 283 nm in Ca5(BO3)3F:1% Eu3+ were detected, indicating the Eu3+ sites occupied Ca2 and Ca1, respectively. The different sharp f–f emission spectra under the excitation of 283 and 266 nm proved that there are two different local lattice environments around Eu3+ existing in Ca5(BO3)3F:1% Eu3+. Environmental factor he, the standard deviation of environmental factor (EFSD) 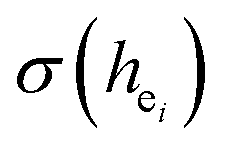 and the bond energy were used to illustrate and explain the site occupancy mechanism of Eu3+ into the host lattice. By comparing the intensity ratios of 5D0 → 7F2 transition to the 5D0 → 7F1 transition, I(5D0/7F2)/I(5D0/7F1) of Eu3+ at Ca2 (7.381) was found to be 2.5 times stronger than that of Eu3+ at Ca1 site (2.933).
and the bond energy were used to illustrate and explain the site occupancy mechanism of Eu3+ into the host lattice. By comparing the intensity ratios of 5D0 → 7F2 transition to the 5D0 → 7F1 transition, I(5D0/7F2)/I(5D0/7F1) of Eu3+ at Ca2 (7.381) was found to be 2.5 times stronger than that of Eu3+ at Ca1 site (2.933). 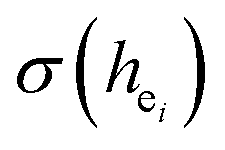 was calculated to analyze the I(5D0/7F2)/I(5D0/7F1) value. On the basis of the bond valence model, a bond-energy method was used to study the occupancy of the Eu ion, which indicated that the preferential sites of Eu ion occupancy in the Ca5(BO3)3F are the Ca2 and Ca1 sites. All three theoretical calculation results are consistent with each other.
was calculated to analyze the I(5D0/7F2)/I(5D0/7F1) value. On the basis of the bond valence model, a bond-energy method was used to study the occupancy of the Eu ion, which indicated that the preferential sites of Eu ion occupancy in the Ca5(BO3)3F are the Ca2 and Ca1 sites. All three theoretical calculation results are consistent with each other.
1. Introduction
White LEDs have the advantages of high brightness, low energy consumption, long life, small size, no radiation, no pollution, etc. It is considered the new generation of green light sources. The two commonly used methods for implementing white LEDs are: blu-ray chip and YAG:Ce3+ yellow phosphor combination, and near-ultraviolet LED chip combined with the trichromatic phosphor. However, the former lacks the red portion, resulting in a lower color rendering index (less than 80) and a higher color temperature (greater than 40), while the latter is less commercially available and lacks suitable red light materials.1–6 Therefore, finding a suitable red luminescent material and improving the color rendering index has become an important issue in the development of trichromatic white LEDs. In the rare earth family, Eu3+ ions are widely used in various fields of luminescent materials because they have good luminescence properties and can emit red fluorescence with good monochromaticity.7–9 The luminescence of Eu3+ derived from the 4f6 transition consists of sharp peaks of the Eu3+-doped inorganic compound in the red region.10–12Rare earth ion-doped borate luminescent materials have high UV transparency, nonlinear characteristics, good stability and optical properties, and they are expected to become a promising fluorescent material, attracting more and more research worldwide.13–15 Among the nonlinear optical crystal materials that have been found, ultraviolet and deep ultraviolet nonlinear optical crystals that have excellent performance are almost all borate compounds. Ca5(BO3)3F was first reported by Lei et al. in 1989. The crystal was also a new type of nonlinear optical crystal. The powder doubling effect was measured to be 2–3 KDP, and the transmission range was 190–3600 nm.16,17 There are many published papers focusing on energy transfer and color-tunability of rare earth (e.g., Bi3+, Ce3+, Tb3+)-doped Ca5(BO3)3F.18,19 There are three kinds of octahedrons surrounding the Ca ions that could be substituted by Eu3+. They have different covalence, average bond lengths, central ion coordination numbers, and charges of ligands in chemical bonds. These result in Eu3+ ions with different photoluminescence properties. For the above reasons, Ca5(BO3)3F compound with Ca2+ and B3+ cationic sites can be selected as a host lattice, and Eu3+ ions are used as a good activator. However, studies on the effects of Eu3+ on the CT properties and the local crystal sites occupied by Eu3+ in Ca5(BO3)3F:1% Eu3+ phosphor are rarely reported.
The environmental factor (he) was used to investigate the sites of Eu3+ based on the relationship between the CT bands of Eu3+ and the crystal structure of the host lattice. he can be calculated using the complex crystal chemical bond theory. The smaller the he, the larger the charge transfer energy from O2− to Eu3+. Through the literature, we know that the PL intensity ratio between the 5D0 → 7F2 and 5D0 → 7F1 transition of Eu3+ increases as the crystal distortion increases. The degree of distortion was calculated by the environmental factor standard deviation (EFSD) 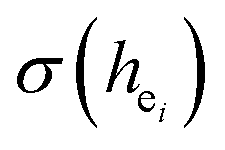 . This can provide information on the Eu ion sites. At the same time, the bond energy theory can be used to discuss the site occupancy of dopants into the host. In previous papers, this method has been proven; for example, the site preferential occupancy for Eu in Sr2V2O7, Sr9Gd(VO4)7 and Sr2V2O7/Sr9Gd(VO4)7, CaAl2Si2O8 phosphors,20 as well as Bi2+ in β-Ca2P2O7 crystals,21 have been solved though the bond energy method. Furthermore, this method was also used in study of bond energy and preferential occupancy of Eu3+ doped in the Ca10M(PO4)7 (M = Li, Na, K) systems.22
. This can provide information on the Eu ion sites. At the same time, the bond energy theory can be used to discuss the site occupancy of dopants into the host. In previous papers, this method has been proven; for example, the site preferential occupancy for Eu in Sr2V2O7, Sr9Gd(VO4)7 and Sr2V2O7/Sr9Gd(VO4)7, CaAl2Si2O8 phosphors,20 as well as Bi2+ in β-Ca2P2O7 crystals,21 have been solved though the bond energy method. Furthermore, this method was also used in study of bond energy and preferential occupancy of Eu3+ doped in the Ca10M(PO4)7 (M = Li, Na, K) systems.22
In our work, Eu3+ doped Ca5(BO3)3F was prepared by a high-temperature solid-state method. We firstly used the chemical bond parameters and analyzed the sites of Eu ion systematically based on the refined crystal structure parameters. Then, the site occupancy of Eu3+ was investigated by analyzing the luminescent properties and environmental factor he and the standard deviation of environmental factor (EFSD) 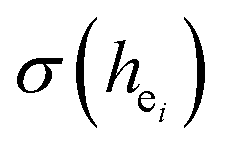 of Ca5(BO3)3F:1% Eu3+ when Eu3+ replaced the different Ca2+ sites in the matrix. Finally, the bond energy method was applied to illustrate and explain the site occupation of Eu3+ into the host lattice.
of Ca5(BO3)3F:1% Eu3+ when Eu3+ replaced the different Ca2+ sites in the matrix. Finally, the bond energy method was applied to illustrate and explain the site occupation of Eu3+ into the host lattice.
2. Experimental
2.1 Synthesis of material
Ca5(BO3)3F:1% Eu3+ phosphor was prepared by high-temperature solid-state reaction in air. The desired CaF2 (A.R., Macklin), H3BO3 (A.R., Sinopharm), CaCO3 (99.99%, Aladdin), and Eu2O3 (99.99%, Aladdin) were weighed according to the stoichiometric ratio of Ca5(BO3)3F:1% Eu3+, and the raw materials were placed in an agate mortar and ground for half an hour. The uniformly mixed raw materials were transferred to an alumina crucible, heated in a crucible calciner to 1150 °C, and then kept for 4 h. After the sample was cooled to room temperature, it was ground into a powder to obtain the desired sample.2.2 Material characterization
In this experiment, we used a Bruker D8 Advance X-ray diffractometer to analyze the crystal structure of the prepared phosphor with Cu Kα (λ = 1.54056 Å) radiation and acceleration voltage of 30 kV. The scanning range was 5 to 80 degrees. The sample was detected by FLS980 fluorescence spectrometer to obtain the excitation spectrum and emission spectrum. The excitation source was a 450 W Xe lamp with a slit width of 2 nm and a measured spectral range of 200 nm to 750 nm, with a resolution of 0.2 nm. The sample was tested at room temperature. The Rietveld structure refinement was performed using the General Structure Analysis System (GSAS)23 software in order to determine the change of crystal structure.3. Results and discussion
3.1 Phase characterization and crystal analysis
Fig. 1 is an XRD pattern of Ca5(BO3)3F:1% Eu3+ sample. Comparing the sample diffraction pattern with the standard card, it was found that the diffraction peak data were basically consistent with the Ca5(BO3)3F (ICSD-65763) card data, with a C1m1 space group and a monoclinic crystal system. There is an impurity phase, namely, Ca3B2O6. Fortunately, a very small amount of Ca3B2O6 has no effect on our system. Therefore, the substitution of Eu3+ for Ca2+ into Ca5(BO3)3F did not change the structure of the crystal.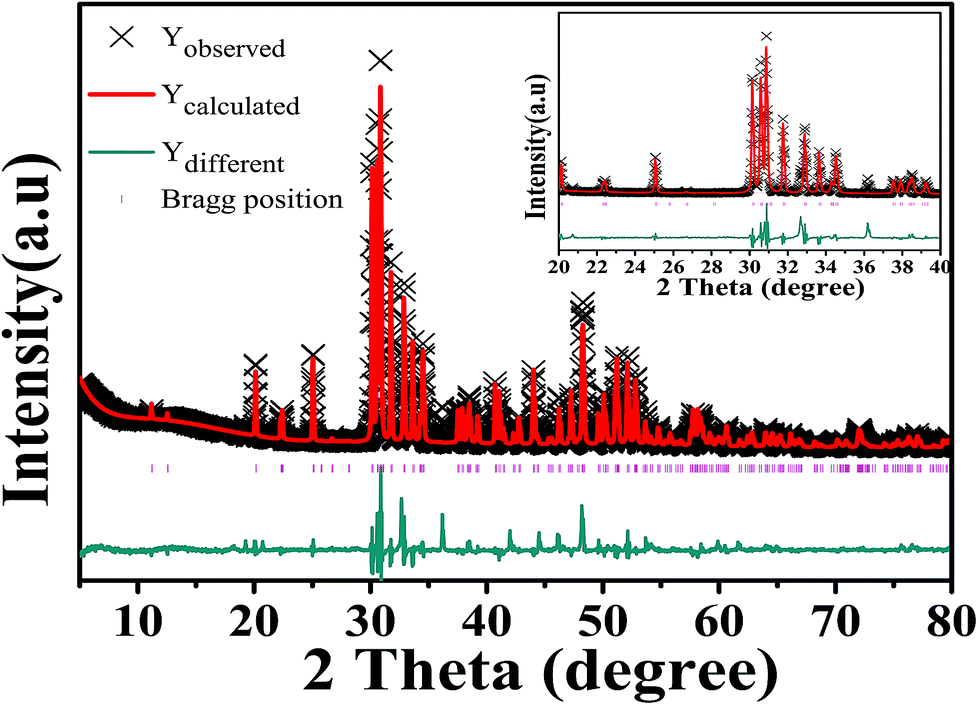 | ||
| Fig. 1 Observed, calculated and difference X-ray diffraction patterns of an Eu3+ (1%)-doped Ca5(BO3)3F phosphor (the inset gives [20–40] 2θ range). | ||
Fig. 2 shows the crystal structure of Ca5(BO3)3F. There are three Ca ion coordination environments in this structure, namely, Ca1, Ca2 and Ca3. The anionic structural group of the Ca5(BO3)3F crystal is a planar BO3 group. Both Ca and the surrounding anions form a CaO4X2 (X = F or O) octa-coordination, in which Ca(1) forms a CaO5F octahedron with five O atoms and one F atom, and Ca(2) forms a distorted octahedral structure with six O atoms. Ca(3) is coordinated with two F atoms and four O atoms. These polyhedrons are connected to the BO3 group by sharing O atoms to form a three-dimensional space structure.
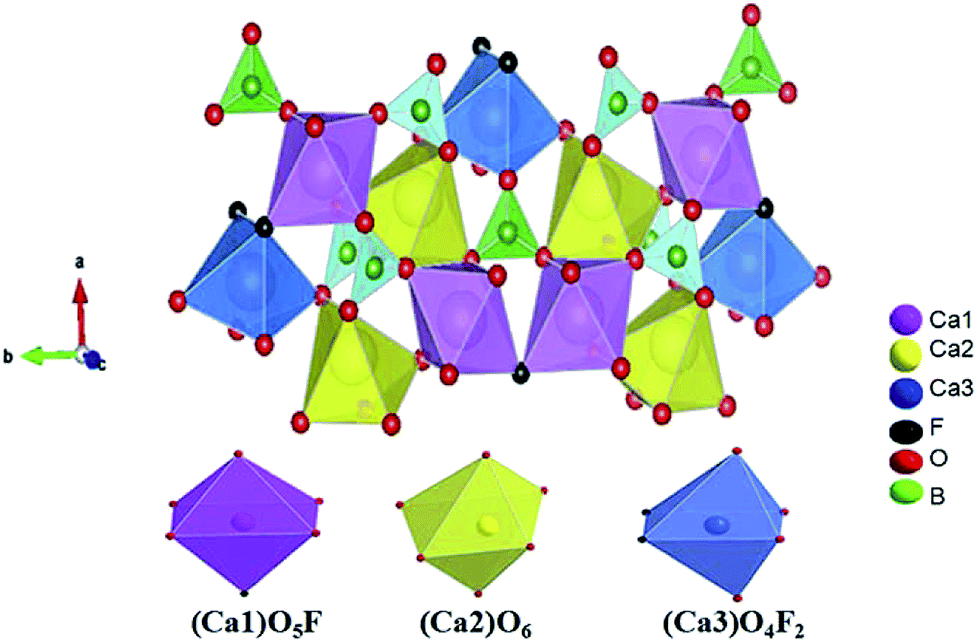 | ||
| Fig. 2 Crystal structure of Ca5(BO3)3F and the coordination environment of Ca1, Ca2, and Ca3. | ||
The crystal structure of Ca5(BO3)3F (ICSD-65763) was used as the starting model for structure refinement. The Rietveld method refers to the point-by-point comparison of the calculated and measured values of the diffraction intensity of a crystal using a computer program, and the least squares method is used to adjust the parameters of the structural atoms and of the peak shape, so that the calculated peak shape is consistent with the measured peak shape. In the structural refinement of this paper, based on the XRD peak of the existing Ca5(BO3)3F:1% Eu3+ crystal and Ca5(BO3)3F standard card as the background, the set function is type 4, and about 30 terms were refined. Fig. 1 shows the observed, calculated and difference results for the Rietveld refinement of Ca5(BO3)3F:1% Eu3+ phosphor. The peak intensities and positions exhibited few differences between the experimental and calculated patterns. Crystallographic and refinement parameters are summarized in Table 1. The results show that almost all diffraction peaks can be directed to Ca5(BO3)3F with a monoclinic unit cell (C1m1). The cell parameters a, b, c, and β, etc., are close to those of Ca5(BO3)3F (ICSD-65763). The atomic coordinates and isotropic displacement parameters of Ca5(BO3)3F:1% Eu3+ phosphor are listed in Table 2. The above results show that the crystal structure data of Ca5(BO3)3F:1% Eu3+ simulated by refinement can be well matched with its experimental data.
| Formula | Ca5(BO3)3F | E |
| Space-group | C1m1 | |
| a/Å | 8.132 | 0.003 |
| b/Å | 16.054 | 0.003 |
| c/Å | 3.542 | 0.003 |
| α = γ | 90° | 0 |
| β | 100.946° | 0 |
| Rwp | 5.30% | |
| Rp | 9.35% | |
| χ2 | 6.806 |
| Name | x | y | z | Eposition | Mult | Occ | Uiso | EUiso |
|---|---|---|---|---|---|---|---|---|
| Ca1 | 0.6501 | 0.1177 | 0.7786 | 0.013 | 4 | 1 | 0.0231 | 0.001 |
| Ca2 | 0.0278 | 0.1796 | 0.4421 | 0.02 | 4 | 1 | 0.0145 | 0.002 |
| Ca3 | 0.2583 | 0 | 0.0769 | 0.04 | 2 | 1 | 0.0283 | 0.02 |
| F1 | 0.4691 | 0 | −0.3287 | 0.03 | 2 | 1 | 0.0737 | 0.01 |
| O1 | 0.8282 | 0.0743 | 0.3639 | 0.01 | 4 | 1 | 0.0603 | 0.002 |
| O2 | −0.001 | 0.3279 | 0.2448 | 0.018 | 4 | 1 | 0.0504 | 0.001 |
| O3 | 0.0722 | 0 | 0.5116 | 0.024 | 2 | 1 | 0.0066 | 0.002 |
| O4 | 0.829 | 0.2254 | 0.8459 | 0.049 | 4 | 1 | 0.0307 | 0.02 |
| O5 | 0.2071 | 0.143 | 0.0125 | 0.02 | 4 | 1 | 0.0134 | 0.001 |
| B1 | 0.8626 | 0.307 | 0.0036 | 0.014 | 4 | 1 | 0.0594 | 0.011 |
| B2 | 0.9239 | 0 | 0.3499 | 0.006 | 2 | 1 | −0.0113 | 0.003 |
3.2 Photoluminescence properties
Fig. 3(b) is an excitation spectrum of the Ca5(BO3)3F:1% Eu3+ sample at a monitoring wavelength of 633 nm, which is composed of a broad excitation band of 225–350 nm, derived from the charge transfer transition of O2− to Eu3+, where the strongest absorption peak is at 266 nm. The emission spectrum at an excitation wavelength of 266 nm is shown in Fig. 3(d), consisting of emission peaks at around 576, 592, 612, 652 and 708 nm, corresponding to 5D0 → 7F0, 5D0 → 7F1, 5D0 → 7F2, 5D0 → 7F3, and 5D0 → 7F4 transitions of Eu3+ ions, respectively.24,25 We can clearly see that the charge transfer transition peaks of samples O to Eu have a significant shift when the monitoring wavelengths are 576, 612, 619 and 633 nm, as shown in Fig. 3(a) and (b). The two broadband peaks (O2− → Eu3+) are located at 266 and 283 nm, respectively, which means that the Eu3+ ions exhibit two lattice environments in the Ca5(BO3)3F matrix. However, the excitation and emission spectra of Eu-doped Ca3B2O6 are completely different with our luminescence properties of Eu-doped Ca5(BO3)3F. The former shows an emission band peaking at 422 nm and excitation peaks at 362, 380 and 394 nm (λem = 616 nm). These prove that the different luminescence of Eu is due to Eu occupying the different sites in the Ca5(BO3)3F.26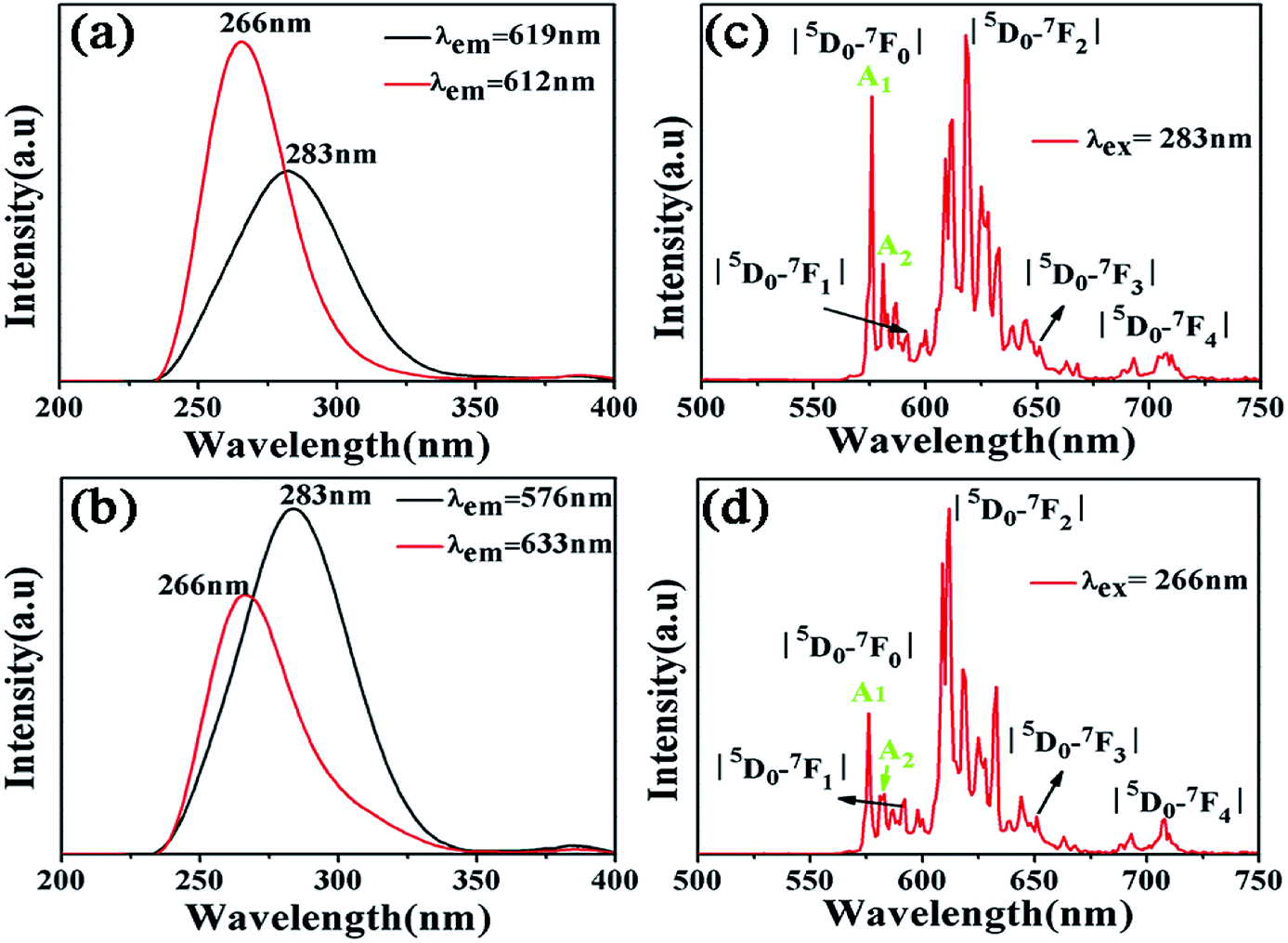 | ||
| Fig. 3 (a) and (b) Excitation spectra under different monitoring wavelengths of Ca5(BO3)3F:1% Eu3+; (c) and (d) emission spectra of Ca5(BO3)3F:1% Eu3+ under 266 and 283 nm excitation. | ||
When the Eu3+ ion deviates from the center of the inversion, due to the opposite parity configuration in the 4f configuration, the parity selection in the crystal is relaxed and the 5D0 → 7F2 electric dipole transition will occur. If the Eu3+ ion is located in the non-inversion center, its emission spectrum will be dominated by the 5D0 → 7F2 electric dipole transition, and the emission spectrum will be around 610 nm. For the electronic transition of Eu3+ ion, the 5D0 → 7F0 transition originally belongs to the forbidden transition. However, when it is in the ten symmetry positions of Cs, C1, C2, C3, C4, C6, C2V, C3V, C4V and C6V, 5D0 → 7F0 transition emission will occur, and the emission spectrum peak will be around 580 nm. A 5D0 → 7F0 transition peak appears in each site.27 Therefore, based on the number of the peaks, the number of occupied sites of the Eu3+ ion crystals can be judged. Fig. 3(c) and (d) show the emission spectra of Ca5(BO3)3F:1% Eu3+ upon excitation at 266 nm and 283 nm, respectively. Their peaks locate at 576 nm, which is due to 5D0 → 7F0 transition of Eu3+ ions. Each 5D0 → 7F0 transition peak corresponds to a lattice. Two 5D0 → 7F0 peaks, A1 and A2, were found in Fig. 3(c) and (d), which indicate that Eu3+ doping in Ca5(BO3)3F has two sites.28–30 The data are shown in Table 3 for the luminescent levels of Ca5(BO3)3F:1% Eu3+ upon 266 and 283 nm excitation.
| Energy level transition | The location of the peak (nm) (λex = 266 nm) | Intensity | The location of the peak (nm) (λex = 283 nm) | Intensity |
|---|---|---|---|---|
| 5D0 → 7F0 | 576 | 576 | ||
| 583 | 581 | |||
| 5D0 → 7F1 | 592 | 233![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 668.172 668.172 |
592 | 149367.984 |
| 5D0 → 7F2 | 612 | 685254.813 |
618 | 1102500 |
| 5D0 → 7F3 | 652 | 651 | ||
| 5D0 → 7F4 | 708 | 708 | ||
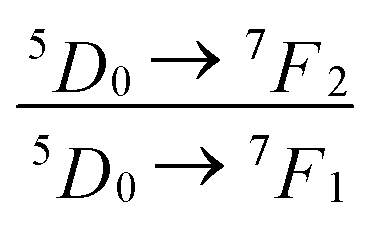 |
2.933 | 7.381 |
Since CT energy is susceptible to the central ionic environment, it can be quantitatively expressed by using environmental factors (he). he consists of four chemical bond parameters: the bond volume polarization 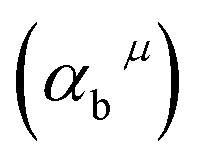 , the covalency
, the covalency 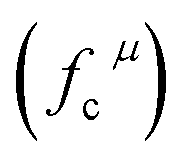 , the coordination number (C. N.) and the presented charge of the ligand
, the coordination number (C. N.) and the presented charge of the ligand 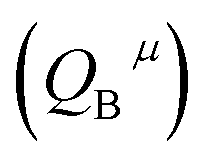 . Its calculation formula is as follows:31
. Its calculation formula is as follows:31
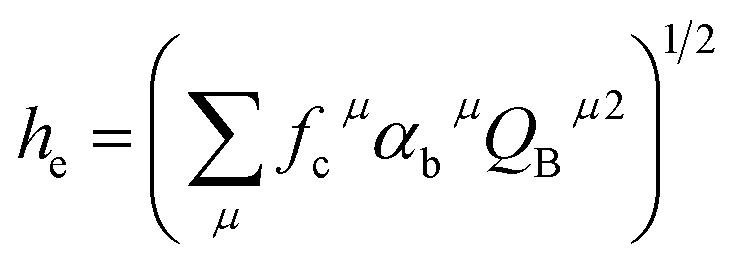 | (1) |
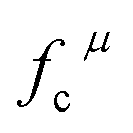 represents the covalent value of μ type bond,
represents the covalent value of μ type bond, 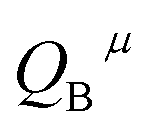 is the charge exhibited by the nearest anion and
is the charge exhibited by the nearest anion and 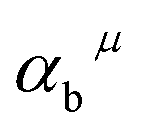 stands for the polarizability of the μ-type chemical bond volume.
stands for the polarizability of the μ-type chemical bond volume.
The four chemical bond parameters, with any change, will cause a shift in the CT bands. As he increases, the CT energy decreases, which means that the CT bands will produce a red shift. In Table 4, we can see that the he values of Ca1, Ca2, and Ca3 are 0.6397, 0.7439, and 0.3514, respectively. Therefore, it can be known that Eu will occupy the Ca1 and Ca2 sites, and at the same time, the O–Eu1 CT band at the Ca1 site corresponds to the peak at 266 nm and the O–Eu2 CT band at Ca2 corresponds to the position at 283 nm.
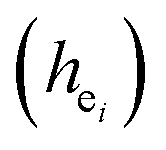 and their standard deviation of the Ca–O environmental factors
and their standard deviation of the Ca–O environmental factors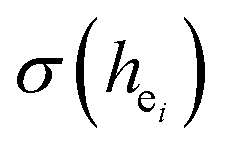
| Central ion | Bond type | Distance (Å) |
|
|
|
C.N. | he |
|
|
O–Eu charge transfer peak |
|---|---|---|---|---|---|---|---|---|---|---|
| Ca1/Eu | Ca1–O1 | 2.3556 | 0.1160 | 0.4491 | 1.3333 | 1 | 0.6397 | 0.3043 | 0.1607 | 266 nm |
| Ca1–O1 | 2.3960 | 0.1151 | 0.4819 | 1.3333 | 1 | 0.3140 | ||||
| Ca1–O2 | 2.2283 | 0.1191 | 0.3569 | 1.3333 | 1 | 0.2748 | ||||
| Ca1–O2 | 2.4020 | 0.1150 | 0.4870 | 1.3333 | 1 | 0.3155 | ||||
| Ca1–O4 | 2.2428 | 0.1187 | 0.3666 | 1.3333 | 1 | 0.2781 | ||||
| Ca1–F | 2.3810 | 0.0258 | 0.2053 | 1.3333 | 1 | 0.0970 | ||||
| Ca2/Eu | Ca2–O1 | 2.3237 | 0.1167 | 0.4244 | 1.3333 | 1 | 0.7439 | 0.2967 | 0.0184 | 283 nm |
| Ca2–O2 | 2.4795 | 0.1135 | 0.5557 | 1.3333 | 1 | 0.3348 | ||||
| Ca2–O4 | 2.4653 | 0.1137 | 0.5426 | 1.3333 | 1 | 0.3312 | ||||
| Ca2–O4 | 2.5136 | 0.1129 | 0.5882 | 1.3333 | 1 | 0.3436 | ||||
| Ca2–O5 | 2.3311 | 0.1165 | 0.4300 | 1.3333 | 1 | 0.2984 | ||||
| Ca2–O5 | 2.3726 | 0.1156 | 0.4627 | 1.3333 | 1 | 0.3083 | ||||
| Ca3/Eu | Ca3–O3 | 2.2691 | 0.1050 | 0.3493 | 1.0000 | 1 | 0.3514 | 0.1915 | 0.0520 | |
| Ca3–O3 | 2.3551 | 0.1031 | 0.4080 | 1.0000 | 1 | 0.2051 | ||||
| Ca3–O5 | 2.3363 | 0.1056 | 0.2512 | 0.6667 | 1 | 0.1086 | ||||
| Ca3–O5 | 2.3371 | 0.1056 | 0.2515 | 1.0000 | 1 | 0.1630 | ||||
| Ca3–F | 2.4366 | 0.0257 | 0.2289 | 1.0000 | 1 | 0.0767 | ||||
| Ca3–F | 2.4497 | 0.0257 | 0.2347 | 1.0000 | 1 | 0.0777 |
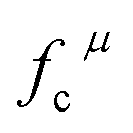
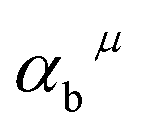
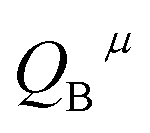
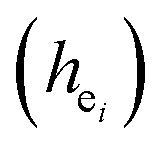
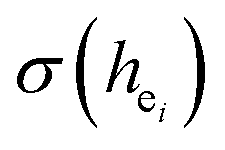
Obviously, the intensity of f–f transitions is affected by the crystal environment because of the different symmetry of the doping ions occupying the host sites. When the Eu3+ is doped into Ca5(BO3)3F, the Eu ions occupy the Ca2+ sites, wherefore, the f–f transition relative intensity is mainly affected by the Ca2+ symmetry. The different 5D0 → 7FJ transition intensities of Eu3+ depend on the local symmetry of the Eu3+ ion crystal field. The 5D0 → 7F2 transition is sensitive, while the 5D0 → 7F1 transition is stable to the crystal field environment. For example, when the Eu3+ ion is in a site with a strict inversion center, it will be dominated by the allowable 5D0 → 7F1 magnetic dipole transition, and the emission spectrum is around 590 nm, which is orange light. When the Eu3+ ion is in the site away from the inversion center, the parity selection in the crystal is relaxed, and a 5D0 → 7F2 electric dipole transition will occur; the emission spectrum is around 610 nm, emitting red light. If the intensity of the 5D0 → 7F2 transition is much higher than the intensity of 5D0 → 7F1, the Eu3+ ion mainly occupies the non-inversion symmetry of the lattice. It is known that the PL intensity ratio between 5D0 → 7F2 and 5D0 → 7F1 transition of Eu3+ increases as the crystal distortion increases. The degree of distortion can be calculated by using the standard deviation of environmental factor (EFSD)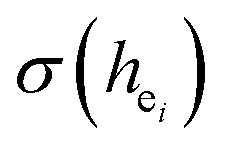 ,22,32 which can be calculated as below:
,22,32 which can be calculated as below:
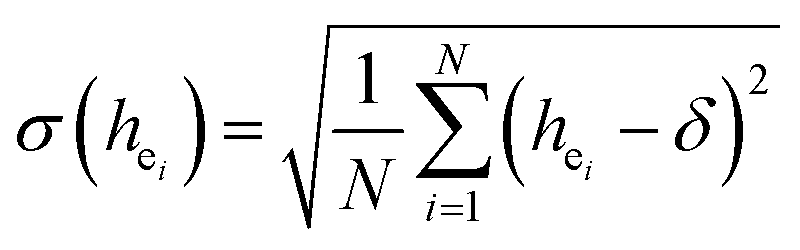 | (2) |
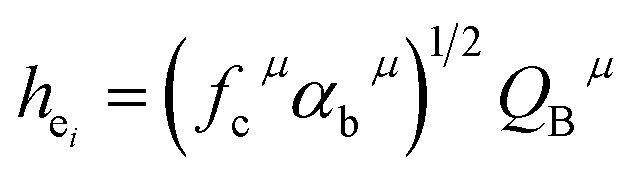 | (3) |
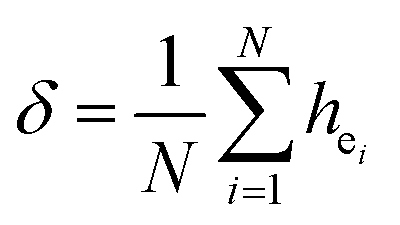 | (4) |
The related chemical parameters of the covalency 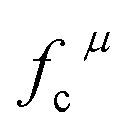 , the present charge of the ligand in the binary crystals, and the polarizability of the chemical bond volume
, the present charge of the ligand in the binary crystals, and the polarizability of the chemical bond volume 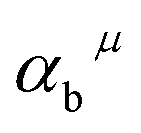 are shown in Table 4. On the basis of the eqn (2)–(4), their standard deviation for the six Ca–O environmental factors
are shown in Table 4. On the basis of the eqn (2)–(4), their standard deviation for the six Ca–O environmental factors 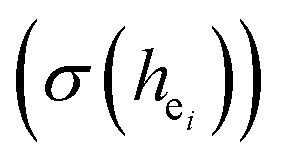 of Ca1O5F, Ca2O4F2, and Ca3O6 polyhedrons in Ca5(BO3)3F:1% Eu can be calculated to be 0.1607, 0.0184 and 0.052, respectively. Generally, the I(5D0/7F2)/I(5D0/7F1) value of Eu3+ increases with increasing
of Ca1O5F, Ca2O4F2, and Ca3O6 polyhedrons in Ca5(BO3)3F:1% Eu can be calculated to be 0.1607, 0.0184 and 0.052, respectively. Generally, the I(5D0/7F2)/I(5D0/7F1) value of Eu3+ increases with increasing 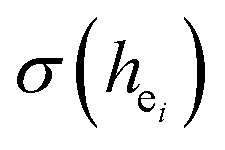 . The
. The 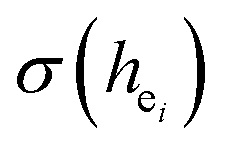 value of Ca1O5F in Ca5(BO3)3F:1% Eu is larger than that of Ca2O4F2. By comparison, the intensity ratio of 5D0 → 7F2 transition to the 5D0 → 7F1 transition of Eu3+ at Ca2 (7.381) is 2.5 times stronger than that of Eu3+ at Ca1 site (2.933). Therefore, the I(5D0 → 7F2)/I(5D0 → 7F1) of Eu3+ in the Ca1O5F site is stronger than that in the Ca2O4F2 site. Ca1 and Ca2 correspond to excitations at 283 nm and 266 nm, respectively. However, this is contrary to the conclusion drawn in Table 4. In ideal state, the order of
value of Ca1O5F in Ca5(BO3)3F:1% Eu is larger than that of Ca2O4F2. By comparison, the intensity ratio of 5D0 → 7F2 transition to the 5D0 → 7F1 transition of Eu3+ at Ca2 (7.381) is 2.5 times stronger than that of Eu3+ at Ca1 site (2.933). Therefore, the I(5D0 → 7F2)/I(5D0 → 7F1) of Eu3+ in the Ca1O5F site is stronger than that in the Ca2O4F2 site. Ca1 and Ca2 correspond to excitations at 283 nm and 266 nm, respectively. However, this is contrary to the conclusion drawn in Table 4. In ideal state, the order of 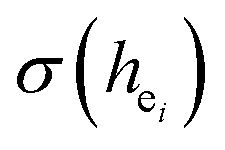 is in Fig. 4. When Eu3+ is doped into Ca5(BO3)3F, part of the Ca2+ ions must be occupied by Eu3+, so the local environment of Eu3+ must be changed in order to keep the conservation of charge in Fig. 4(a) and (b). At this time, one F of the sub-stationary [(Eu1)O5F]* and [(Eu3)O4F2]* will be replaced by an O to maintain their own stability; [(Eu2)O6]* will receive an F, and it will eventually become a 7-coordinated environment in Fig. 4(c). In summary, we can conclude that the Ca2 distortion degree should be greater than that of Ca1. Therefore, Ca1 and Ca2 correspond to excitations at 266 nm and 283 nm, respectively, consistent with the results obtained in Table 4.
is in Fig. 4. When Eu3+ is doped into Ca5(BO3)3F, part of the Ca2+ ions must be occupied by Eu3+, so the local environment of Eu3+ must be changed in order to keep the conservation of charge in Fig. 4(a) and (b). At this time, one F of the sub-stationary [(Eu1)O5F]* and [(Eu3)O4F2]* will be replaced by an O to maintain their own stability; [(Eu2)O6]* will receive an F, and it will eventually become a 7-coordinated environment in Fig. 4(c). In summary, we can conclude that the Ca2 distortion degree should be greater than that of Ca1. Therefore, Ca1 and Ca2 correspond to excitations at 266 nm and 283 nm, respectively, consistent with the results obtained in Table 4.
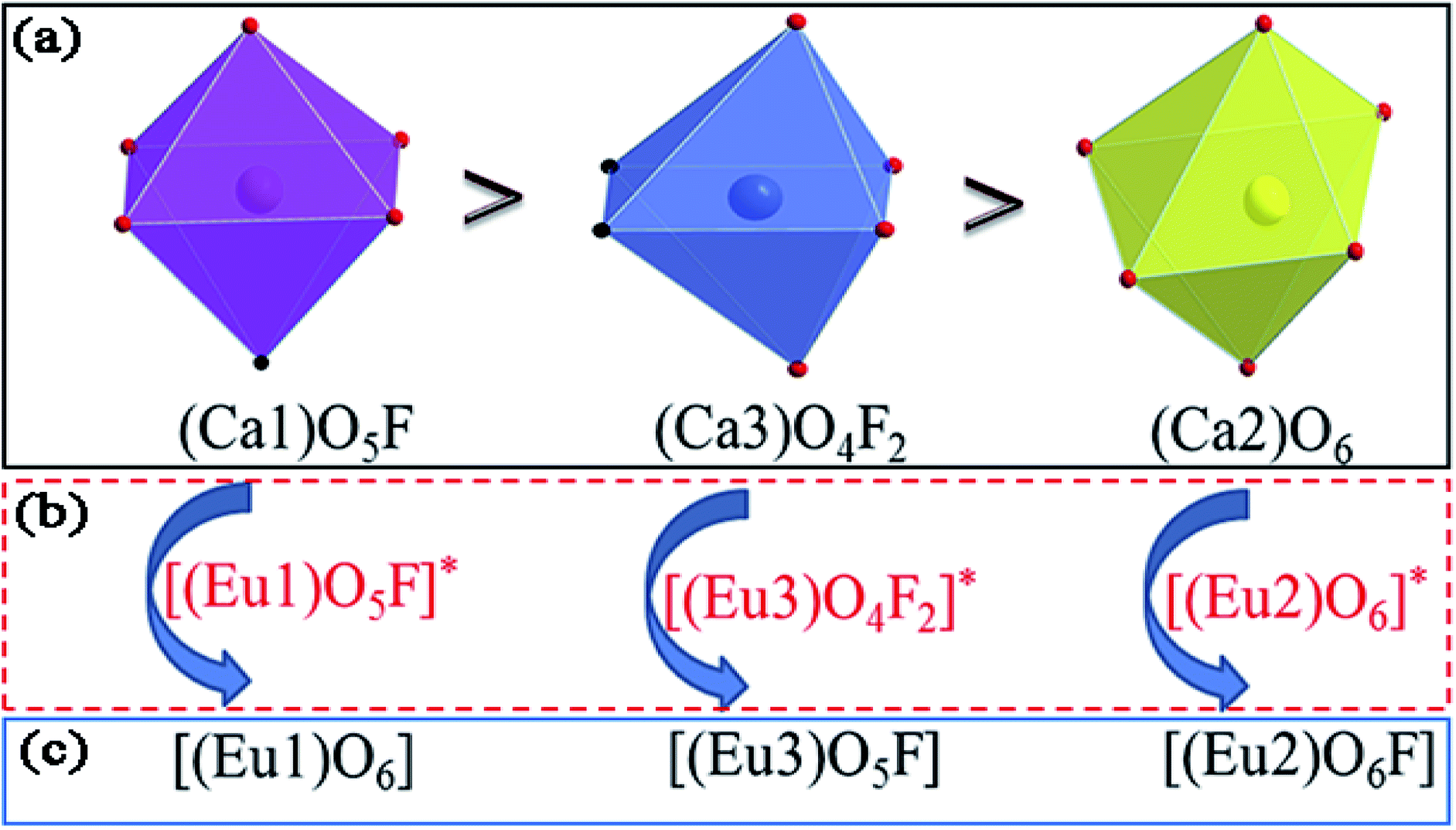 | ||
| Fig. 4 The coordination environment mechanism of Ca1 (or Eu1), Ca2 (or Eu2), and Ca3 (or Eu3) in Ca5(BO3)3F:1% Eu. | ||
3.3 Chemical bond energy calculation
From the point of view of matched valence, the bond energy of Eu3+ into the Ca5(BO3)3F phosphor can be estimated by the following equation,20,33
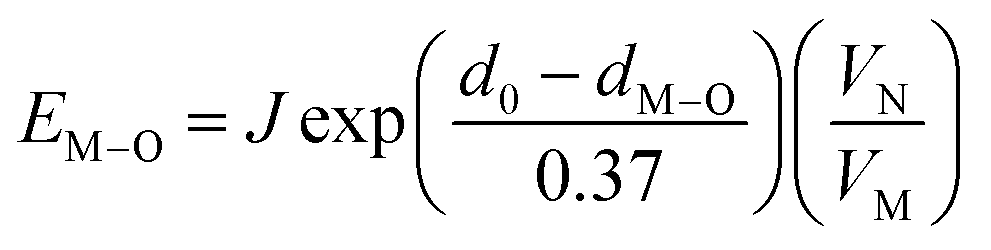 | (5) |
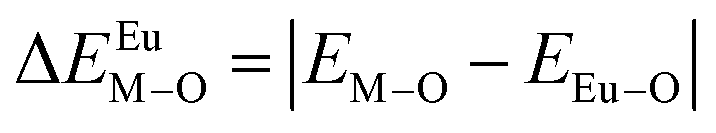 | (6) |
| Ions | d0 (Å) | J (kcal mol−1) |
|---|---|---|
| Ca2+–O2− | 1.967 | 126.60 |
| Ca2+–F− | 1.842 | 187.03 |
| B3+–O2− | 1.371 | 126.30 |
| Eu3+–F− | 2.056 | 163.35 |
| Eu3+–O2− | 2.074 | 109.40 |
Here, 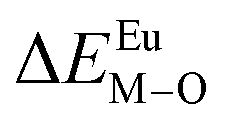 is the bond energy difference when the Eu ion locates at the site of Ca2+. As previously reported, we can know that the dopants preferentially occupy a site where the bond energy difference is small
is the bond energy difference when the Eu ion locates at the site of Ca2+. As previously reported, we can know that the dopants preferentially occupy a site where the bond energy difference is small 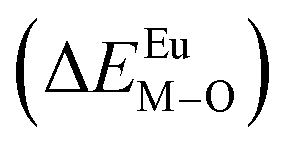 , that is, a site having a smaller absolute value of
, that is, a site having a smaller absolute value of 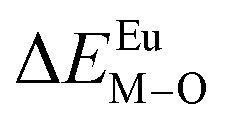 .20
.20
According to the detailed crystallographic data from a pure Ca5(BO3)3F phosphor (longer Ca–O bonds of 1.967 Å; shorter B–O bonds of 1.371 Å; longer Ca–F bonds of 1.842 Å), all calculated EM–O and ΔEM–O values of Eu3+ on both Ca2+ and B3+ sites are shown in Table 6. The corresponding occupancies of Eu3+ are summarized, according to the calculated 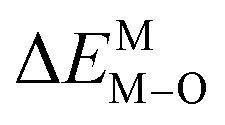 values. Dopants preferentially occupy the sites where the bond energy difference
values. Dopants preferentially occupy the sites where the bond energy difference 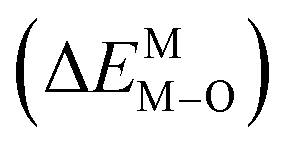 is smaller; that is, Eu3+ ions preferentially occupy Ca2+ sites if
is smaller; that is, Eu3+ ions preferentially occupy Ca2+ sites if 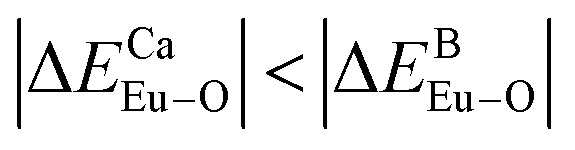 ; they preferentially occupy B3+ sites if
; they preferentially occupy B3+ sites if 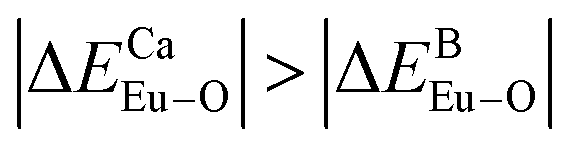 , otherwise.
, otherwise.
| Central atom | Coordination atom | Count | d (Å) | EM–O |
|
|
|---|---|---|---|---|---|---|
| Ca1 | O2 | 1× | 2.2283 | 62.4781 | 48.0633 | 11.0351 |
| O4 | 1× | 2.2428 | 60.0770 | 46.2162 | ||
| O1 | 1× | 2.3556 | 44.2901 | 34.0716 | ||
| O1 | 1× | 2.3960 | 39.7088 | 30.5473 | ||
| O2 | 1× | 2.4020 | 39.0701 | 30.0559 | ||
| F1 | 1× | 2.3810 | 41.3517 | 31.8111 | ||
| Ca2 | O1 | 1× | 2.3237 | 48.2781 | 48.0632 | 1.7744 |
| O2 | 1× | 2.4795 | 31.6867 | 46.2162 | ||
| O4 | 1× | 2.4653 | 32.9264 | 34.0716 | ||
| O4 | 1× | 2.5136 | 28.8969 | 30.5473 | ||
| O5 | 1× | 2.3311 | 47.3221 | 30.0559 | ||
| O5 | 1× | 2.3726 | 42.3012 | 31.8111 | ||
| Ca3 | O3 | 1× | 2.2691 | 55.9549 | 37.1394 | 14.2389 |
| O3 | 1× | 2.3551 | 44.3500 | 24.3760 | ||
| O5 | 1× | 2.3363 | 46.6617 | 25.3297 | ||
| O5 | 1× | 2.3371 | 46.5609 | 22.2299 | ||
| F1 | 1× | 2.4366 | 35.5821 | 36.4041 | ||
| F1 | 1× | 2.4497 | 34.3444 | 32.5416 | ||
| B1 | O4 | 1× | 1.4296 | 107.8004 | 734.209 | 551.7228 |
| O5 | 1× | 1.5032 | 88.35499 | 707.712 | ||
| O2 | 1× | 1.3091 | 149.2999 | 687.725 | ||
| B2 | O1 | 1× | 1.4297 | 107.7712 | 624.1322 | 636.8898 |
| O1 | 1× | 1.4304 | 107.5676 | 622.9525 | ||
| O3 | 1× | 1.2329 | 183.4429 | 1062.3664 |
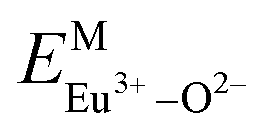
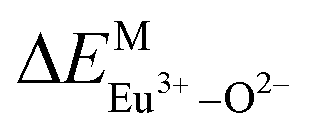
There are three Ca2+ sites and two B3+ sites in the structure of Ca5(BO3)3F. On the basis of the bond energy method, the values of 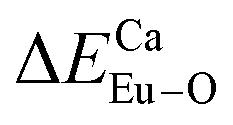 and
and 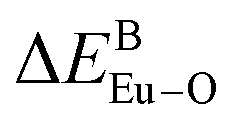 have been listed in Table 6. The order of difference of bond energy that Ca2+and B3+ ions are replaced by Eu3+ is
have been listed in Table 6. The order of difference of bond energy that Ca2+and B3+ ions are replaced by Eu3+ is 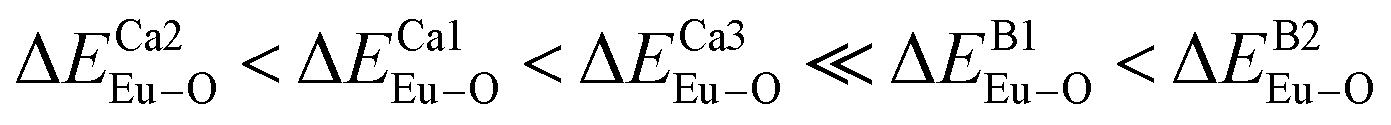 , which means that Eu3+ preferentially replaces Ca2 and Ca1. According to our calculation, Eu3+ preferentially occupies Ca2 and Ca1 sites, which is consistent with the environmental factor, the
, which means that Eu3+ preferentially replaces Ca2 and Ca1. According to our calculation, Eu3+ preferentially occupies Ca2 and Ca1 sites, which is consistent with the environmental factor, the 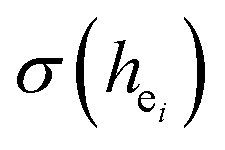 analysis and PL spectrum.
analysis and PL spectrum.
4. Conclusions
The photoluminescence of Ca5(BO3)3F:1% Eu3+ shows that the excitation spectrum consists of some broadband from the O2− → Eu3+ charge transfer (CT) band and some sharp emission peaks derived from the f–f transition of Eu3+. Two different O2− → Eu3+ CT broad bands with the peaks at 266 and 283 nm in Ca5(BO3)3F:1% Eu3+ can be assigned to the Eu3+ sites occupying Ca1 and Ca2, respectively. Two 5D0 → 7F0 peaks, A1 and A2, were found, which implies that Eu3+ doped in Ca5(BO3)3F has two sites. According to the dielectric theory of the crystal, the important chemical bonds such as the polarizability, the covalency and the environmental factor were quantitatively calculated. When Eu3+ ions occupy the Ca1, Ca2 and Ca3 sites of Ca5(BO3)3F, their environmental factors are 0.6397, 0.7439 and 0.3514, respectively. The intensity ratio of 5D0 → 7F2 transition to the 5D0 → 7F1 transition of Eu3+ at Ca2 (7.381) is 2.5 times stronger than that of Eu3+ at Ca1 site (2.933). The calculated ideal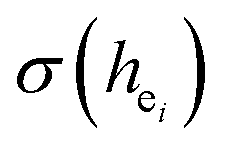 showed that the I(5D0/7F2)/I(5D0/7F1) value at Eu1 is larger than that at Eu2 site. The local nonequivalence substitution distortion model was proposed to explain the result. The smaller deviation value of the bond energy method of
showed that the I(5D0/7F2)/I(5D0/7F1) value at Eu1 is larger than that at Eu2 site. The local nonequivalence substitution distortion model was proposed to explain the result. The smaller deviation value of the bond energy method of 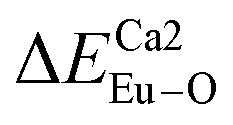 and
and 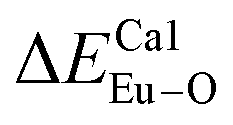 showed that the preferential sites of Eu3+ ion occupancy in Ca5(BO3)3F are Ca2 and Ca1. All of results are consistent with each other. The three theoretical methods provide us a new strategy to study the occupancy of Eu3+ in Eu3+-doped inorganic compounds.
showed that the preferential sites of Eu3+ ion occupancy in Ca5(BO3)3F are Ca2 and Ca1. All of results are consistent with each other. The three theoretical methods provide us a new strategy to study the occupancy of Eu3+ in Eu3+-doped inorganic compounds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundations of China (Grant no. 21301053 and 21571165) and Hubei Natural Science Foundations from Science and Technology Department of Hubei Province (2018CFB517).Notes and references
- D. Liu, Y. Jin, Y. Lv, G. Ju, C. Wang, L. Chen, W. Luo and Y. Hu, J. Am. Ceram. Soc., 2018 DOI:10.1111/jace.15877 , ahead of print.
- H.-W. Wei, X.-M. Wang, H. Jiao and X.-P. Jing, J. Alloys Compd., 2017, 726, 22–29 CrossRef.
- H. Xu, L. Wang, L. Tan, D. Wang, C. Wang and J. Shi, J. Am. Ceram. Soc., 2018, 101, 3414–3423 CrossRef.
- R. Wei, L. Wang, F. Hu, X. Li, X. Peng, Y. Shi, H. Guo and J. Qiu, J. Lumin., 2018, 197, 291–296 CrossRef.
- A. K. Pal, S. Som and C.-H. Lu, Ceram. Int., 2018, 44, 18256–18263 CrossRef.
- Y. Miao, K. Wang, L. Gao, B. Zhao, H. Wang, F. Zhu, B. Xu and D. Ma, J. Mater. Chem. C, 2018, 6, 8122–8134 RSC.
- P. Chen, D. Yang, W. Hu, J. Zhang and Y. Wu, Chem. Phys. Lett., 2017, 689, 169–173 CrossRef.
- Z. Xie, W. Zhou, W. Zhao, H. Zhang, Q. Hu and X. Xu, Chem. Phys. Lett., 2017, 685, 177–184 CrossRef.
- T. S. Sreena, P. P. Rao, A. K. V. Raj and T. R. A. Thara, J. Alloys Compd., 2018, 751, 148–158 CrossRef.
- G. Li, Y. Wei, W. Long and G. Xu, Mater. Res. Bull., 2017, 95, 86–94 CrossRef.
- A. Kruopyte, R. Giraitis, R. Juskenas, D. Enseling, T. Justel and A. Katelnikovas, J. Lumin., 2017, 192, 520–526 CrossRef.
- D. Kumar, B. P. Singh, M. Srivastava, A. Srivastava, P. singh, A. Srivastava and S. K. Srivastava, J. Lumin., 2018, 203, 507–514 CrossRef.
- K. Das, A. Marathe, X. Zhang, Z. Zhao and J. Chaudhuri, RSC Adv., 2016, 6, 95055–95061 RSC.
- A. Huang, Z. Yang, C. Yu, Z. Chai, J. Qiu and Z. Song, Mater. Lett., 2016, 185, 440–442 CrossRef.
- P. Chen, F. Mo, A. Guan, R. Wang, G. Wang, S. Xia and L. Zhou, Appl. Radiat. Isot., 2016, 108, 148–153 CrossRef PubMed.
- G. Chen, Y. Wu and P. Fu, J. Cryst. Growth, 2006, 292, 449–453 CrossRef.
- N. Kozhaya, M. Ferriol, M. Cochez, M. Aillerie and A. Maillard, Opt. Mater., 2011, 33, 1621–1625 CrossRef.
- X. Li, P. Li, Z. Wang, S. Liu, Q. Bao, X. Meng, K. Qiu, Y. Li, Z. Li and Z. Yang, Chem. Mater., 2017, 29, 8792–8803 CrossRef.
- L. Yi, J. Zhang, Z. Qiu, W. Zhou, L. Yu and S. Lian, RSC Adv., 2015, 5, 67125–67133 RSC.
- L. Li, W. Wang, Y. Pan, Y. Zhu, X. Liu, H. M. Noh, B. K. Moon, B. C. Choi and J. H. Jeong, RSC Adv., 2018, 8, 1191–1202 RSC.
- L. Li, J. Cao, B. Viana, S. Xu and M. Peng, Inorg. Chem., 2017, 56, 6499–6506 CrossRef PubMed.
- W. Wang, Y. Pan, Y. Zhu, H. Xu, L. Zhou, X. Liu, L. Li, H. M. Noh and J. H. Jeong, Dalton Trans., 2018, 47, 6507–6518 RSC.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef.
- Y. Zhang, A. Abraha, R. Zhang, T. Shahbazyan, M. Fadavi, E. Heydari and Q. Dai, Opt. Mater., 2018, 84, 115–122 CrossRef.
- W. You, Z. Xiao, F. Lai, X. Ye, C. Wang and H. Jiang, Zhongguo Xitu Xuebao, 2016, 34, 11–16 Search PubMed.
- M. Shi, C. Zhu, M. Lu, X. Meng and M. Wei, J. Am. Ceram. Soc., 2018 DOI:10.1111/jace.15782 , ahead of print.
- J. B. Gruber, U. Vetter, T. Taniguchi, G. W. Burdick, H. Hofsaess, S. Chandra and D. K. Sardar, J. Appl. Phys., 2011, 110, 023104 CrossRef.
- L. Zhou, J. Shi and M. Gong, J. Lumin., 2005, 113, 285–290 CrossRef.
- P. A. Tanner, Y. Y. Yeung and L. Ning, J. Phys. Chem. A, 2013, 117, 2771–2781 CrossRef PubMed.
- Y. Zhang, Q. Xiao, H. He, J. Zhang, G. Dong, J. Han and J. Qiu, J. Mater. Chem. C, 2015, 3, 10140–10145 RSC.
- L. Li, X. Liu, H. M. Noh, S. H. Park, J. H. Jeong and K. H. Kim, J. Alloys Compd., 2015, 620, 324–328 CrossRef.
- L. Li, Y. Pan, W. Wang, W. Zhang, Z. Wen, X. Leng, Q. Wang, L. Zhou, H. Xu, Q. Xia, L. Liu, H. Xiang and X. Liu, J. Alloys Compd., 2017, 726, 121–131 CrossRef.
- Y. He and D. Xue, J. Phys. Chem. C, 2007, 111, 13238–13243 CrossRef.
- L. Li, Y. Pan, W. Wang, Y. Zhu, W. Zhang, H. Xu, L. Zhou and X. Liu, J. Alloys Compd., 2018, 731, 496–503 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |