
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAbrasive treatment of microtiter plates improves the reproducibility of bacterial biofilm assays†
Emily Bordeleau‡
,
Sina Atrin Mazinani‡,
David Nguyen,
Frank Betancourt and
Hongbin Yan *
*
Department of Chemistry and Centre for Biotechnology, Brock University, 1812 Sir Isaac Brock Way, St. Catharines, Ontario, Canada L2S 3A1. E-mail: tyan@brocku.ca
First published on 19th September 2018
Abstract
Microtiter plate-based bacterial biofilm assay is frequently used to study bacterial biofilm development and growth. While this assay is simple and relatively high-throughput, it frequently shows difficulty in establishing robust biofilm attachment in the wells. We report that the consistency of bacterial biofilm assays carried out in microtiter plates subjected to abrasive treatment, by sandblasting or drill press grinding, is significantly improved in a Pseudomonas fluorescens Pf0-1 model. Scanning electron microscopy imaging suggests that the treated surfaces could provide points of attachment to facilitate the recruitment of bacteria in the initial phase of biofilm colony establishment. The sandblast treated polypropylene, but not polystyrene, plates were found suitable in studying the impact of flavonoid quercetin on the biofilm formation in Bacillus subtilis FB17. Further investigation revealed that due to the hydrophobicity of the polystyrene surfaces, a greater amount of quercetin was adsorbed on the plate surface, effectively lowering the concentration of the flavonoid in solution.
Introduction
Biofilm formation, that is, planktonic–sessile phenotypic transition, is recognized as an important mechanism for bacterial survival under environmental stresses.1 Various systems have been developed to investigate the biofilm formation process, such as continuous-flow, chemostat, and static systems.2–4 Although the batch-grown static conditions do not readily support the formation of mature biofilms obtained in flow cell systems, largely due to limitations in nutrients and difficulty in aeration, certain static platforms, such as the crystal violet-based microtiter plate static biofilm assay, allow for relatively high-throughput screenings of multiple bacterial strains under different growth conditions on abiotic surfaces.5 This assay, however, is often associated with poor reproducibility. In this respect, small changes that occur in a biofilm assay protocol can result in the reduction, or complete inability, of a biofilm to form. Although the addition of auxiliary chemicals, such as dimethyl sulfoxide (DMSO), methanol and ethanol, to the biofilm cultures tends to stimulate biofilm formation,6–8 a robust biofilm assay protocol that does not require additional reagents will be of significance in order to avoid potential influence of auxiliaries on bacterial physiology.While successful biofilm studies were previously performed in this lab by utilizing 96-well polystyrene plates with Pseudomonas fluorescens Pf0-1 as a model microorganism, subsequent attempts to culture P. fluorescens Pf0-1 with this assay resulted in the formation of only insignificant amount of biofilm under a wide range of conditions. A rather drastic difference in the reproducibility in microtiter plates of different surface chemistry from various suppliers, or same brands of plates from different lots, was observed, similar to what was previously reported for enzyme-linked immunosorbent assays.9,10 Our effort to resolve this inconsistency in initiating biofilm development led to the investigation of mechanically altering the surface of microtiter plate wells in order to create a surface that supports consistent static biofilm assays. Finally, the utility of the surface-treated microtiter plates in the biofilm formation of a Gram-positive bacterium Bacillus subtilis FB17 in the presence of flavonoids was investigated.
Results and discussion
Topographical changes in polystyrene surfaces post-abrasive treatment with aluminum oxide
The results of microtiter plate-based static biofilm assays can be affected by a number of factors. Plates from different suppliers or even different lots can lead to difference in the level of biofilm formation.11 Thus, a polystyrene surface with a topography that could support higher, consistent, levels of biofilm would be desirable. To this end, microtiter plates were bombarded with 80-grit aluminum oxide (sand) particles in order to provide a greater surface area for the bacterial cells to adhere to. While rough surfaces have been shown to increase the adhesion of Staphylococcus epidermidis on titanium surfaces12 as well as silicone,13 microtiter plates treated abrasively have not been evaluated for their application in bacterial biofilm assays.The area near the center of the wall of the microtiter plate wells was analyzed by scanning electron microscopy (SEM) to provide the most accurate representation of the topography encountered by the bacterial cells once reaching the air–liquid interface. In backscattered electron (BSE) imaging, contrast in images is generated due to the differences in the average atomic number of the material where electron scattering occurs. Areas where heavier elements are present create brighter images as electrons are more effectively scattered out of the sample.14,15 The BSE-SEM image for the polystyrene post-abrasive treatment displayed a high amount of heavier element particles of varying sizes scattered throughout the surface (Fig. 1a). These particles are most likely the aluminum oxide particles that remained impregnated on the surface after washing. The BSE-SEM image for the untreated polystyrene surface also displayed unknown heavier elements scattered over the polystyrene surface, but in much lower quantities (Fig. 1c), possibly due to contamination.
 | ||
| Fig. 1 SEM images of sandblast-treated wells. (a–d) Comparison of polystyrene surface post-abrasive treatment with the unmodified polystyrene surface. SEM analysis was carried out at 15.0 kV at 500 × magnification. (a). BSE-SEM and (b). SEI−SEM for wells post-abrasive treatment; (c). BSE-SEM and (d). SEI-SEM for unmodified wells. (e). SEM image of polystyrene post abrasive treatment at 2.0 kV and 1000× magnification. | ||
Images of the same surface area at higher resolutions were obtained with low energy secondary electron scanning (SE), providing details about the surface topography. In this mode, structures protruding from the surface appear brighter, as more electrons are scattered at these areas. On the other hand, surface areas hidden from topographical features appear darker as fewer electrons are scattered from these surfaces.16
Various structures of random spatial orientations were seen for the sandblasted polystyrene wells (Fig. 1b). In order to enhance the surface details in the SEM images, secondary electron images (SEI) (Fig. 1e) were obtained by reducing the beam energy from 15.0 to 2.0 kV. This image also represents a 1000× magnification of the same surface area seen in Fig. 1b. The BSE and SE images suggest that the surface treatment by sandblast leads to the formation of a unique topographical profile, possibly providing points of attachment during the initial phases of biofilm formation.
Biofilm formation on sandblasted polystyrene microtiter plates
Biofilm formation assays were carried out with P. fluorescens Pf0-1 in two polystyrene microtiter plates (Corning, Cat. # 351172), one treated by sandblasting, and another untreated as comparison. As shown in Fig. 2a, the level of biofilm formed on the treated plates more than doubled. Furthermore, the coefficient of variation (CV) in the sandblasted plates was quite comparable to that of untreated ones (18 vs. 23%).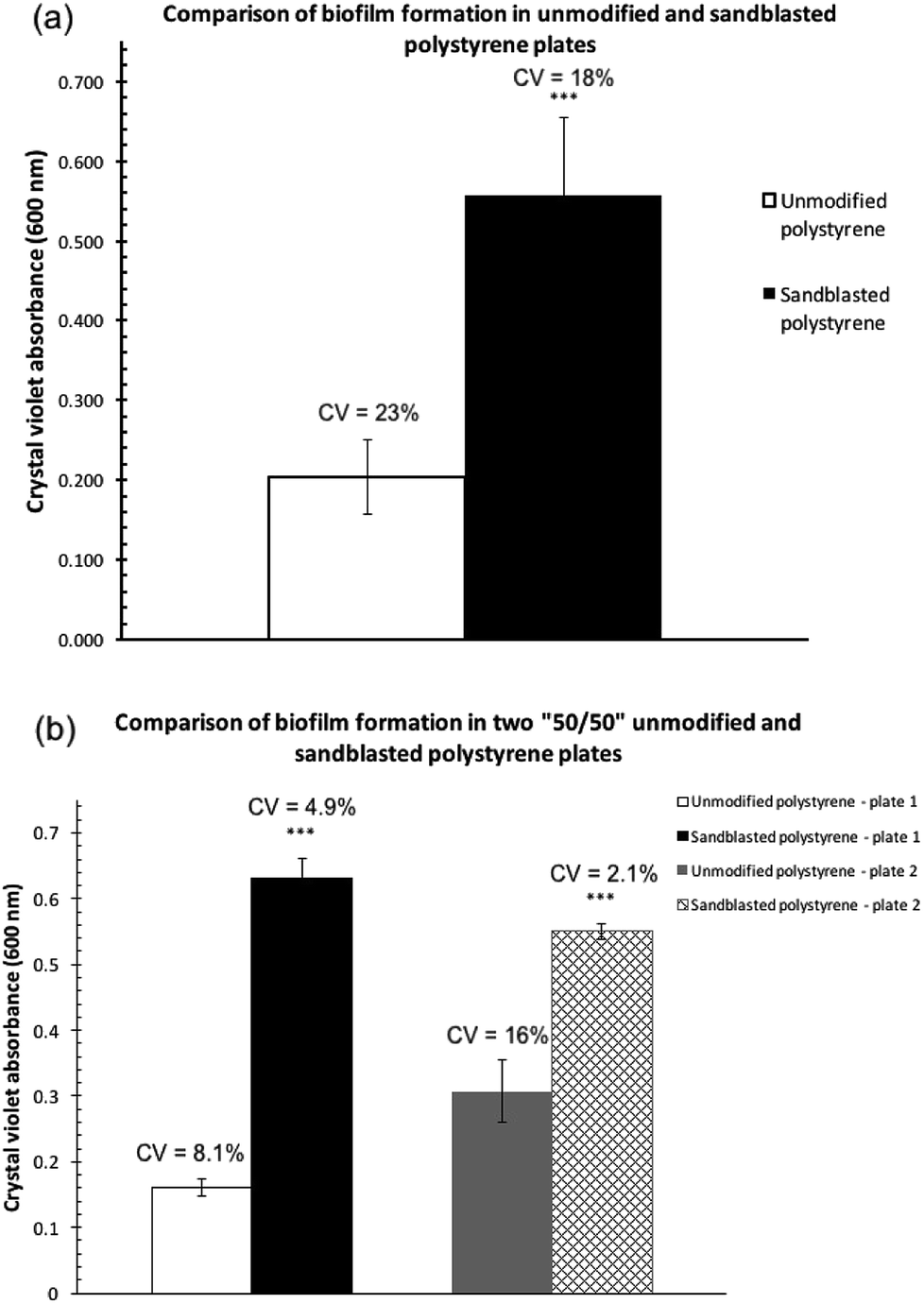 | ||
| Fig. 2 Comparisons of the quantities of P. fluorescens Pf0-1 biofilm formation as determined by measuring the absorbance at 600 nm after crystal violet bound to the biofilm was solubilized in 30% acetic acid. The bars represent the average amount of biofilm formed. (a) On an unmodified polystyrene plate and a polystyrene plate treated by sandblasting. Error bars represent the mean ± standard deviation (n = 12). ***P < 0.001 vs. unmodified plates. (b) On two “50/50” sandblast-treated polystyrene microtiter plates, where half of each plate was sandblasted but not the other. Error bars represent the mean ± standard deviation (n = 6). ***P < 0.001 vs. the unmodified side of the plates. The coefficients of variation (CV) are indicated above the bars. | ||
In order to minimize plate-to-plate variation in the comparison, subsequent biofilm assay experiments were carried out in a microtiter plate where half of the plate wells were sandblasted while the other half were not (“50/50”, See Fig. S1 in the ESI† for an image of the treated plate). This plate was produced by covering half of the plate during the sandblasting process. To eliminate edge effects of microtiter plates,17–19 the perimeter of the treated and non-treated areas was not used to cultivate biofilm cultures.
Two replicate experiments were carried out in the “50/50” sand-treated microtiter plates. In both assays, the level of biofilm formation in the treated wells was statistically significantly higher than that found in untreated wells (Fig. 2b). Each experiment, although performed at a different time and thus two separate cultures, produced quantities of biofilm on the modified polystyrene surface that were not statistically significantly different from one another. The amount of biofilm formed in the unmodified polystyrene surface in the two replicates, however, showed a much greater variation.
Biofilm formation on polystyrene microtiter plates treated with a drill press
The increase in biofilm formation observed could be attributed to not only the altered surface topography but also the presence of the aluminum oxide particulates impregnated on the surface. To eliminate the potential effects from the aluminum oxide impregnation, another set of microtiter plates were modified using a drill press.Two plates (Corning, Cat. # 351172) where only half of the wells were treated by drill press grinding were subsequently inoculated with P. fluorescens Pf0-1 to assess biofilm establishment in M63 as well as LB broth. As expected, very little biofilm was formed on both surfaces (Fig. S2†) in LB, as LB is a nutrient-rich medium.20 In M63 broth, however, drill press treatment of wells resulted in significant increases in biofilm formed across both plates (Fig. 3). The quantity of biofilm formed in M63 medium was highly reproducible, while the quantity of biofilms cultivated in the unmodified wells was significantly less.
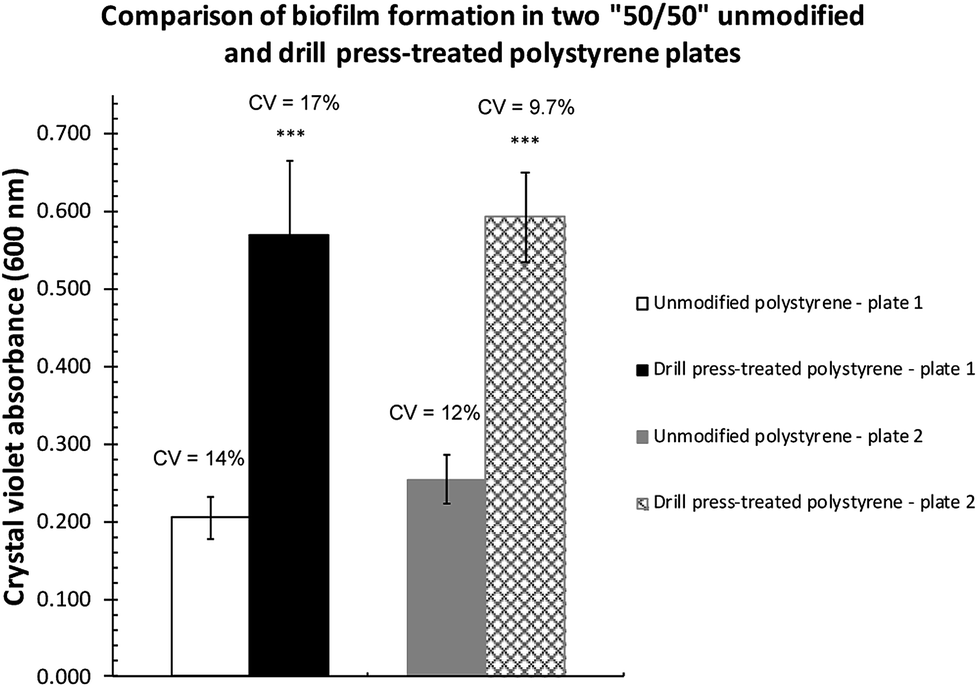 | ||
| Fig. 3 Comparisons of P. fluorescens Pf0-1 biofilm formed on unmodified and drill-modified polystyrene plates. The bars represent the average amount of biofilm formed. Error bars represent the mean ± standard deviation (n = 6). ***P < 0.001 vs. unmodified side of the plates. The coefficients of variation (CV) are indicated above the bars. | ||
Influence of microtiter plate material on the adsorption of flavonoids
Biofilm assays for P. fluorescens Pf0-1 supplemented with quercetin at 50 and 100 μg mL−1 that were carried out in sandblasted polypropylene (Norgen, Cat. # 24310) and polystyrene (Falcon, VWR Cat. # CA15705-066) gave rather different results. As Table 1 shows, a robust reduction in biofilm formation in polypropylene plates was seen at both concentrations of quercetin, while only very small differences were noted in those carried out in polystyrene plates. While it can be argued that polypropylene surface might be more suitable for P. fluorescens Pf0-1 adhesion, it is plausible that due to the aromatic nature of quercetin, adsorption of this flavonoid onto polystyrene surface is much greater than polypropylene, leaving a lower effective concentration of quercetin in the culture medium, therefore a reduced effect on biofilm formation.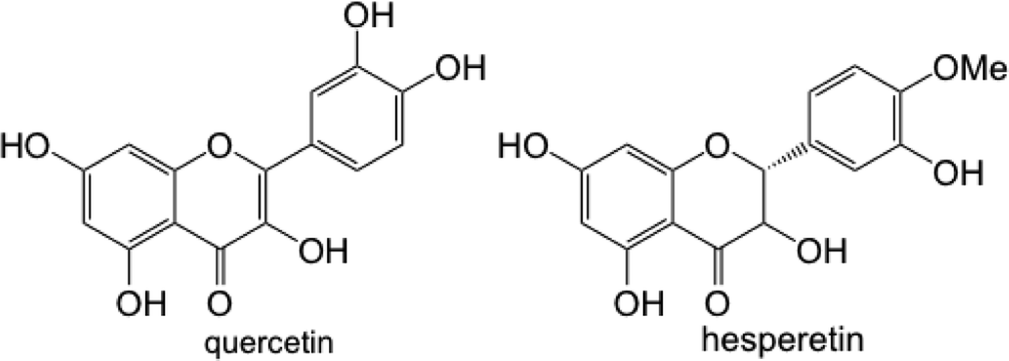
| Quercetin | Average percentage reduction in biofilm formation | |
|---|---|---|
| Norgen plates | 50 μg mL−1 | 29 |
| 100 μg mL−1 | 27 | |
| Falcon plates | 50 μg mL−1 | 7 |
| 100 μg mL−1 | 4 |
In this respect, adsorption of flavonoids to solid surfaces through non-covalent interactions has been documented.21 We confirmed through UV spectrophotometry that when a solution of quercetin in M63 medium containing 0.1% DMSO was incubated in sandblasted polypropylene and polystyrene plates, a greater reduction in absorbance representing quercetin was seen in polystyrene plates (see Fig. S3 in the ESI†), likely owing to the similar hydrophobic properties of quercetin and polystyrene surfaces.
Biofilm formation of B. subtilis FB17 in the presence of quercetin and hesperetin
To demonstrate the utility of the sandblasted microtiter plates in studying interspecies interactions in the rhizosphere between plants and plant-associated bacteria, Bacillus subtilis FB17 was chosen, as this bacterium has been shown to colonize Arabidopsis thaliana roots,22–25 likely through a chemotaxis phenomenon. As A. thaliana has also been well characterized for the production of flavonoids,26–29 including quercetin, we decided to examine the influence of supplementation of quercetin and hesperetin on B. subtilis FB17 biofilm formation in Norgen brand microtiter plates (polypropylene). It was noted, however, that B. subtilis FB17 forms submerged biofilm30 as well as floating pellicles at the air–liquid interface,31 making the quantification of submerged biofilm challenging. In the present study, pellicles were removed by washing with milli-Q water prior to staining submerged biofilm. As quercetin and hesperetin have limited solubility in water, solutions in DMSO were used to a maximal final concentration of DMSO (0.5%) in the assay cultures.It was found that while the absolute readouts from crystal violet staining across the experiments vary, the percentages of reduction in the absorbance of crystal violet staining between control cultures containing 0.5% DMSO and cultures treated with quercetin and hesperetin at 500 μg mL−1 were rather consistent. In this respect, reductions of biofilm formation by 84 and 74% (coefficient of variation 8 and 7%, respectively) in the presence of quercetin and hesperetin at 500 μg mL−1, respectively, were seen. While additional experiments are necessary in order to understand the roles of flavonoids such as quercetin and hesperetin in the interspecies communications between A. thaliana and B. subtilis FB17, the sandblast-treated microtiter plates provided a rather robust platform for these future investigations.
Materials and methods
P. fluorescens Pf0-1 growth conditions
P. fluorescens wild-type strain Pf0-1 (gift from Professor George O'Toole at Dartmouth College) was stored as 15–25% (v/v) glycerol stocks at −80 °C. Cultures were revived by streaking the stock onto LB-Miller agar followed by overnight static incubation at 30 °C. Single colonies were picked to inoculate pre-cultures in LB broth, and incubated at 30 °C and 180 revolutions per minute (rpm) until the required optical density (typically OD600 ca. 0.8) was reached, usually after 15–16 hours.LB medium
Bacto tryptone (10.00 g, Fisher Scientific, Cat.# BP1421), yeast extract (5.00 g, Sigma-Aldrich, Cat.# 70161) and NaCl (10.00 g, ACP Chemicals, Cat. # S-2830) were added to milli-Q water (950 mL). The pH of the solution was adjusted to 7.00 ± 0.10 with aqueous NaOH and diluted to 1 L with milli-Q water. Medium was autoclaved at 121 °C with a 30 min sterilization cycle, and then stored in the dark at room temperature until use.M63 minimal medium32
To milli-Q water (1 L) were added KH2PO4 (13.60 g, Sigma-Aldrich, Cat.#P5655), (NH4)2SO4 (2.00 g, Sigma-Aldrich Cat.# A4418) and glucose (2.00 g, Sigma-Aldrich Cat.#G7021), and the pH of the solution was adjusted to 7.00 ± 0.09 with KOH. The medium was then autoclaved at 121 °C with a 30 min sterilization cycle. After the solution cooled down to room temperature, syringe filter-sterilized (0.22 μm nitrocellulose syringe filters) solutions of MgSO4 (1 mL, 0.2 g mL−1 of MgSO4·7H2O, Sigma-Aldrich Cat. #M2773) and FeSO4 (500 μL, 1.0 mg mL−1 of FeSO4·7H2O, Sigma-Aldrich Cat.#F8633) were added to the medium broth. The medium was stored in the dark at room temperature until use.E-medium33
To prepare E-medium, L-glutamic acid (20.00 g, Sigma Aldrich Cat.# G-1626), citric acid (12.00 g, BDH, AnalaR grade), glycerol (80.00 g, Bioshop, Cat.# GLY002.1), NH4Cl (7.00 g), MgSO4·7H2O (0.50 g, Sigma-Aldrich Cat.#M2773), K2HPO4 (0.50 g, Sigma-Aldrich, Cat.#P5655), CaCl2·2H2O (0.15 g, Sigma-Aldrich, Cat. #C7902), MnSO4·H2O (0.15 g, Bioshop, Cat.# MAN111) and FeCl3·6H2O (40 mg, Sigma-Aldrich, Cat. # 157740) were dissolved in milli-Q water (1.00 L). The pH of the solution was adjusted to 7.4 with NaOH. The medium was then autoclaved at 121 °C with a 30 min sterilization cycle and stored in the dark at room temperature until use.UV/vis spectrophotometric measurements
Bacterial culture growth was monitored through UV spectrophotometric measurements carried out on a ThermoScientific Genesys 10S UV/vis spectrophotometer. A Biotek PowerWave XS microtiter plate reader was used to quantify the biomass of biofilm in 96-well microtiter plates by recording absorbance of crystal violet solutions at 600 nm. All measurements in the plate reader were made with the lid off microtiter plates.Statistical analysis
2-Tailed t-tests were performed in Excel to compare the significance in the quantity of biofilm formed. Experiments with p values not greater than 0.05 are considered as statistically significant.Sandblast treatment of microtiter plates
Microtiter plates were held six inches away from a SB4 Uni-blaster (Inland Manufacturing Co. Omaha, Nebraska, USA) and 80 grit brown aluminum oxide (Cyclone Manufacturing, part # 5007) was sprayed at plate surfaces at a pressure of 80 psi. The Uni-blaster was moved across each row, working left to right at a constant speed to ensure equal coverage. The plates were treated 5 times, each in a different position; first so that sand was sprayed onto the bottom of the wells directly, then angled so that each of the 4 sides of the microtiter well-walls were exposed. For experiments where only half of the microtiter plate was treated, the untreated control side was covered with a metal plate prior to sand blast treatment. For plates where the bottoms of the wells are not treated, polytetrafluoroethylene (PTFE) discs were inserted into each well. After treatment, plates were rinsed twice with 70% ethanol and then twice with distilled water. Prior to use in a static biofilm assay the plates were sterilized under germicidal UV light for 20 min.Drill press modification of microtiter plates
A drill press was used to insert a straight drill bit (1/4 inch, see Fig. S3 in the ESI† for images of the drill bit), in and out of the microtiter plate wells once to create uniform creveses on the surface. Plates were then rinsed twice with 70% ethanol and then twice with distilled water. Prior to use in a static biofilm assay the plates were sterilized under germicidal UV light for 20 min.Static biofilm assay and quantification
P. fluorescens Pf0-1 was revived from −80 °C storage by streaking directly from a glycerol stock onto an LB agar plate for static incubation at 30 °C. Upon the formation of single colonies, pre-cultures were prepared by inoculating 9 mL of LB broth with a P. fluorescens Pf0-1 single colony. The pre-cultures were incubated at 30 °C at 180 rpm until mid-exponential phase was reached after 15–16 hours. Inoculum was prepared by diluting the pre-cultures 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 into M63 broth. For each diluted culture, a minimum of six microtiter plate wells were then inoculated using 100 μL per well with blank medium as controls. Plates were then incubated statically at 30 °C for 8 hours, with the plate covered either with a lid, when provided by the manufacturer, or sterile rayon AeraSeal™ film (Sigma-Aldrich, Cat. # A9224). After incubation, the liquid culture was discarded from the wells by shaking the inverted plate over a waste bucket. The wells were then rinsed with distilled water using an 8-prong wash bottle shaking contents over a waste bucket. Biofilm was then stained with 125 μL of a 0.1% crystal violet (Sigma Aldrich, Cat.#C6158) solution for 15 min. After staining was complete, liquid in the wells was discarded and the wells were washed with distilled water and left to dry in the dark. To quantify the amount of crystal violet bound to biofilm, a 30% acetic acid solution (200 μL) was added to the wells, and the plates were incubated at room temperature for 15 min and the contents were mixed by pipette aspiration. The solutions (125 μL) were transferred to a new flat well microtiter plate, and the absorbance of the crystal violet–acetic acid solution was measured at 600 nm using 30% acetic acid as a blank.
:100 into M63 broth. For each diluted culture, a minimum of six microtiter plate wells were then inoculated using 100 μL per well with blank medium as controls. Plates were then incubated statically at 30 °C for 8 hours, with the plate covered either with a lid, when provided by the manufacturer, or sterile rayon AeraSeal™ film (Sigma-Aldrich, Cat. # A9224). After incubation, the liquid culture was discarded from the wells by shaking the inverted plate over a waste bucket. The wells were then rinsed with distilled water using an 8-prong wash bottle shaking contents over a waste bucket. Biofilm was then stained with 125 μL of a 0.1% crystal violet (Sigma Aldrich, Cat.#C6158) solution for 15 min. After staining was complete, liquid in the wells was discarded and the wells were washed with distilled water and left to dry in the dark. To quantify the amount of crystal violet bound to biofilm, a 30% acetic acid solution (200 μL) was added to the wells, and the plates were incubated at room temperature for 15 min and the contents were mixed by pipette aspiration. The solutions (125 μL) were transferred to a new flat well microtiter plate, and the absorbance of the crystal violet–acetic acid solution was measured at 600 nm using 30% acetic acid as a blank.
Scanning electron microscopy (SEM)
Half of a Corning polystyrene microtiter plate with flat bottom (Cat. # 351172) was treated by sandblasting, while the other half was untreated. The surfaces of two wells, one untreated and another treated with sandblasting, were examined with a JEOL JSM-7000F Field Emission Scanning Electron Microscope (McMaster University, Canada). Each well was originally positioned in the middle of their respective side of the microtiter plate to eliminate edge-well discrepancies. Images were taken at 200×, 500× and 1000× magnification in the COMPO mode using backscattered electrons (BSE) at 2.0 kV to obtain information on the surface composition, and in the secondary electron mode (SEI) at 15.0 kV to give surface topography, respectively.Biofilm assay-FB17
Frozen stocks of B. subtilis FB17 (ATCC PTA-11857) were streaked on LB agar plates. The plates were then incubated in a static incubator at 37 °C until single colonies of the bacteria were obtained. A single colony was then used to inoculate a 9 mL LB medium tube. The tube was incubated in a shaking incubator at 37 °C and 260 rpm until an OD600 in the range of 0.78–0.81 was achieved. An inoculum was taken from the tube and was diluted 1:50 in E-medium. Quercetin (Cayman Chemicals, Item # 10005169) and hesperetin (Cayman Chemicals, Item # 10006084) solutions were prepared at concentrations of 1000, 200 and 100 μg mL−1 in E-medium with DMSO concentrations of 1, 0.2 and 0.1% (v/v), respectively. Equal volumes of quercetin (or hesperetin) solutions and the diluted bacterial suspension (1:50) were mixed to obtain a final bacterial dilution of 1:100 in E-medium with quercetin (or hesperetin) at 500, 100 or 50 μg mL−1. Aliquots of 100 μL were transferred to microtiter plates (sandblasted Norgen and Falcon plates). The microtiter plates were incubated in a static incubator at 37 °C for 14 hours for biofilms to form. The supernatant was then aspirated from each well. The wells were washed with milli-Q water three times, and the plates were left to air dry for 20 minutes. Crystal violet solution (0.1% (w/v), 125 μL) was then added to each well and incubated at room temperature for 20 minutes. The supernatants were removed by pipetting and the wells were washed with milli-Q water three times to remove the unbound crystal violet. The plates were then left to dry overnight in the dark. Acetic acid (30% (v/v), 200 μL) was added to each well to dissolve the bound crystal violet followed by 20 minutes incubation at room temperature. Aliquots (125 μL) were then transferred to a new microtiter plate (Thermofisher) and the absorbance was read at 600 nm using a microplate absorbance reader (BioTek, PowerWave XS).
Comparison of adsorption of quercetin on sandblasted Norgen and Falcon microtiter plates
Quercetin solutions were prepared at a concentration of 50 μg mL−1 in M63 medium containing 0.1% (v/v) DMSO. Aliquots of 100 μL were transferred to microtiter plates (sandblasted Norgen and sandblasted Falcon). The microtiter plates were incubated in a static incubator at 30 °C for 8 hours. A similar process was repeated for the blank (M63 medium). From two wells in each plate, two samples of 75 μL were taken. The absorbance spectrum of the combined 150 μL sample was recorded with a Carry 4000 UV-vis spectrophotometer, using M63 medium (as treated above) as the reference.Conclusions
The present work was motivated by the challenges in the consistency of the microtiter plate-based bacterial biofilm assays. It was found that abrasive treatment of microtiter plate wells, either by sandblasting or drill press grinding, led to drastically improved reproducibility in biofilm formation in this assay. SEM microscopy revealed more rough surface morphology of the treated wells, which likely provide points of attachment for more effective cell recruitment during initial bacterial attachment phases, and increases in structural components to hold the cells together on the different topography.While the abrasive treatment described in the present work significantly improves the consistency of the biofilm assays, it is nevertheless rather tedious to treat the microtiter plate wells in a consistent manner. From practical point of view, treatment of microtiter plates by a drill press can be readily automated and will provide greater levels of reproducibility in the assay results.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Natural Science and Engineering Research Council of Canada. The authors wish to thank Professor George O'Toole for a gift of P. fluorescens Pf0-1 stock sample. The authors would also like to thank Steve Crumb and Steve Renda for their assistance with abrasive treatment of microtiter plates.Notes and references
- G. A. O'Toole, H. B. Kaplan and R. Kolter, Annu. Rev. Microbiol., 2000, 54, 49 CrossRef PubMed.
- J. H. Merritt, D. E. Kadouri, and G. A. O'Toole, Growing and analyzing static biofilms, Curr. Protoc. Microbiol., John Wiley, New York, 2005, ch. 1, Unit–1B.1. DOI:10.1002/9780471729259.mc01b01s00.
- B. E. Ramey and M. R. Parsek, Growing and analyzing biofilms in fermenters, Curr. Protoc. Microbiol., John Wiley, New York, 2005, ch. 1, Unit 1B.3, DOI:10.1002/9780471729259.mc01b03s00.
- T. Tolker-Nielsen and C. Sternberg, Growing and analyzing biofilms in flow chambers, Curr. Protoc. Microbiol., John Wiley, New York, 2005, ch. 1, Unit 1B.2, DOI:10.1002/9780471729259.mc01b02s21.
- G. A. O'Toole, J. Visualized Exp., 2011, 47, 2437 Search PubMed.
- J. Y. Lim, J. M. May and L. Cegelski, Appl. Environ. Microbiol., 2012, 78, 3369 CrossRef PubMed.
- Y. Tashiro, A. Inagaki, K. Ono, T. Inaba, Y. Yawata, H. Uchiyam and N. Nomura, Biosci., Biotechnol., Biochem., 2014, 78, 178 CrossRef PubMed.
- X. Wu, R. R Santos and J. Fink-Gremmels, J. Microbiol. Methods, 2014, 101, 63 CrossRef PubMed.
- I. C. Shekarchi, J. L. Sever, Y. J. Lee, G. Castellano and D. L. Madden, J. Clin. Microbiol., 1984, 19, 89 Search PubMed.
- S. Lilyanna, E. M. W. Ng, S. Moriguchi, S. P. Chan, R. Kokawa, S. H. Huynh, P. C. J. Chong, Y. X. Ng, A. M. Richards, T. W. Ng and O. W. Liew, J. Appl. Lab. Med., 2017 DOI:10.1373/jalm.2017.023952.
- Y. H. An and R. J. Friedman, Handbook of bacterial adhesion: principles, methods, and applications, Springer Science & Business Medium, 2000 Search PubMed.
- Y. Wu, J. P. Zitelli, K. S. TenHuisen, X. Yu and M. R. Libera, Biomaterials, 2011, 32, 951 CrossRef PubMed.
- H. Tang, T. Tao, X. Liang, A. Wang, S. O. Salley, J. McAllister and K. Ng, J. Biomed. Mater. Res., Part A, 2009, 88, 454 CrossRef PubMed.
- A. Crewe and P. Lin, Ultramicroscopy, 1976, 1, 231 CrossRef PubMed.
- D. Kaczmarek, Scanning, 1997, 19, 310 CrossRef.
- H. Seiler, J. Appl. Phys., 1983, 54, R1 CrossRef.
- S. M. Burt, T. J. N. Carter and L. J. Kricka, J. Immunol. Methods, 1979, 31, 231 CrossRef PubMed.
- L. J. Kricka, T. J. N. Carter, S. M. Burt, J. H. Kennedy, R. L. Holder, M. I. Halliday, M. E. Telford and G. B. Wisdom, Clin. Chem., 1980, 26, 741 Search PubMed.
- D. G. Oliver, A. H. Sanders, R. D. Hogg and J. W. Hellman, J. Immunol. Methods, 1981, 42, 195 CrossRef PubMed.
- P. D. Newell, R. D. Monds and G. A. O'Toole, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3461 CrossRef PubMed.
- H. Ye, Z. Chen, Y. Liu, S. Lou and D. Di, J. Appl. Polym. Sci., 2014, 131, 40188 Search PubMed.
- T. Rudrappa, K. J. Czymmek, P. W. Paré and H. P. Bais, Plant Physiol., 2008, 148, 1547 CrossRef PubMed.
- T. Rudrappa, M. L. Biedrzycki, S. G. Kunjeti, N. M. Donofrio, K. J. Czymmek, P. W. Paré and H. P. Bais, Commun. Integr. Biol., 2010, 3, 130 CrossRef PubMed.
- V. Lakshmanan, S. L. Kitto, J. L. Caplan, Y. H. Hsueh, D. B. Kearns, Y. S. Wu and H. P. Bais, Plant Physiol., 2012, 160, 1642 CrossRef PubMed.
- R. Allard-Massicotte, L. Tessier, F. Lécuyer, V. Lakshmanan, J. F. Lucier, D. Garneau, L. Caudwell, H. Vlamakis, H. P. Bais and P. B. Beauregard, mBio, 2016, 7, e01664 CrossRef PubMed.
- J. M. Routaboul, L. Kerhoas, I. Debeaujon, L. Pourcel, M. Caboche, J. Einhorn and L. Lepiniec, Planta, 2006, 224, 96 CrossRef PubMed.
- C. S. Buer, G. K. Muday and M. A. Djordjevic, Plant Physiol., 2007, 145, 478 CrossRef PubMed.
- M. Veit and G. F. Pauli, J. Nat. Prod., 1999, 62, 1301 CrossRef.
- L. Kerhoas, D. Aouak, A. Cingöz, J. M. Rountaboul, L. Lepiniec, J. Einhorn and N. Birlirakis, J. Agric. Food Chem., 2006, 54, 6603 CrossRef PubMed.
- M. A. Hamon and B. A. Lazazzera, Mol. Microbiol., 2001, 42, 1199 CrossRef PubMed.
- D. B. Kearns, F. Chu, S. S. Branda, R. Kolter and R. Losick, Mol. Microbiol., 2005, 55, 739 CrossRef PubMed.
- A. B. Pardee, F. Jacob and J. Monod, J. Mol. Biol., 1959, 1, 165 CrossRef.
- A. M. Cromwick and R. A. Gross, Int. J. Biol. Macromol., 1995, 17, 259 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06352d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |