
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmphiphilic polypeptides with prolonged enzymatic stability for the preparation of self-assembled nanobiomaterials
Irina Tarasenkoa,
Natalia Zashikhinaab,
Ivan Guryanov *b,
Maria Volokitinaa,
Barbara Biondic,
Stefano Fioruccid,
Fernando Formaggioc,
Tatiana Tennikova*b and
Evgenia Korzhikova-Vlakhab
*b,
Maria Volokitinaa,
Barbara Biondic,
Stefano Fioruccid,
Fernando Formaggioc,
Tatiana Tennikova*b and
Evgenia Korzhikova-Vlakhab
aInstitute of Macromolecular Compounds, Russian Academy of Sciences, 31 Bolshoy pr., 199004 St Petersburg, Russia
bInstitute of Chemistry, St Petersburg State University, 26 Universitetskij Pr., 198504 Petrodvorets, St Petersburg, Russia. E-mail: ivan.guryanov1@gmail.com; tennikova@mail.ru
cDepartment of Clinical and Experimental Medicine, University of Perugia, 1 Piazza L. Severi, 06132 Perugia, Italy
dICB, Padova Unit, CNR, Department of Chemistry, University of Padova, 1 Via Marzolo, 35131 Padova, Italy
First published on 9th October 2018
Abstract
Due to their excellent biocompatibility and biodegradability, polypeptides have emerged as versatile bio-inspired scaffolds for the preparation of artificial biomaterials. In order to create self-assembled polypeptide nanoparticles with enhanced stability towards enzymatic degradation, we synthesized a series of random and block polypeptides based on lysine and α-aminoisobutyric acid (Aib) by the ring-opening polymerization of N-carboxyanhydrides (ROP NCA) of the corresponding amino acids. A conformational analysis carried out by means of FT-IR absorption and CD spectroscopies revealed a noticeable difference between random and block copolymers. In turn, the spatial organization of the polypeptide chains induced the formation of nanostructures of different types. The block copolymers self-assembled in vesicle-like structures, whereas polypeptides with randomly distributed monomers formed micelles. In contrast with the polymers with only natural amino acids, all nanoparticles based on Aib/Lys polypeptides showed strong resistance to proteolytic cleavage. The cytotoxicity and the kinetics of the cellular uptake of the prepared nanostructures were also studied. The results obtained could not only contribute to the understanding of long Aib polypeptide folding and self-assembling, but also pave the way to the design of nanomaterials with finely tuned properties in the fields of drug delivery and tissue engineering.
Introduction
The main goal of drug delivery systems is the safe delivery of a bioactive compound at controlled release rate under physiological conditions by various ways of administration (oral, intravenous, transdermal, etc.).1 In recent years, a number of organic, inorganic and hybrid micro- and nanomaterials has been developed.2–5 Among these, polymer nanostructures offer the widest variability in properties and morphology.6–8 They can differ in chemical and biological functionalities, biodegradation rate, optical characteristics and stimuli–response properties. However, not all the polymers, which are successfully shaped as nanoparticles, meet the requirements, such as biocompatibility and/or biodegradability, needed to consider them as biomaterials. In this regard, self-assembled polypeptide nanostructures were recently shown to be a very promising, bio-inspired class of polymers.9–13 The structural diversity of the amino acids and the possibility of their different combinations are suitable for designing and producing polypeptides endowed with the desired physical and chemical properties, such as hydrophilic/hydrophobic balance, folding motifs, recognition ability, biocompatibility and biodegradability.14 Moreover, the insertion of D-α-amino acids and other non-coded or unnatural residues into the polypeptide backbone enables the fabrication of macromolecules, which are stable to the enzymatic degradation.One of the most extensively studied, non-coded α-amino acids is α-aminoisobutyric acid. It does occur in Nature and in particular it characterizes the peptaibiotics, a peptide antibiotic class with membrane permeation abilities.15,16 In addition, its strong tendency to induce helical conformations in peptides is particularly attractive.17–19 Aib-rich peptides longer than eight residues tend to adopt the classical α-helical conformation,17–22 whereas shorter peptides preferentially form 310-helices.18,19,23–27 Since Aib is not involved in the standard ribosomal biosynthesis, Aib-containing peptides display a remarkable stability to biodegradation.21
A variety of syntheses and studies of relatively short Aib-containing peptides is currently available in the literature. Few representative examples are the peptides (Aib-Arg-Aib-Leu)n (n = 1–4),21 (Glu-Aib-Leu-Aib)n (MW approx. 2800),22 Boc-Gpn-Aib-Xaa-OMe (Boc = tert-butyloxycarbonyl, Gpn = gabapentin, Xaa = Leu or Phe, OMe = methoxy),28 Nα-thioacylated (Aib)6 (on the surface of a gold surface),29 Ser-Aib-Phe-Ser-Aib-Phe-Aib30 and Phe-Aib-Leu-Ala/Glu/Lys-Aib-Leu-Phe.26 The results from the molecular dynamics study of a series of Ac-(Aib)n-NHMe (where Ac, acetyl; n = 5–7, 10; NHMe = methylamino) were recently published as well.27 A range of publications reported the synthesis of Aib-containing peptides via solution22,31–33 or solid-phase techniques.21,34–37 Recently, Tsuchiya and Numata described also the enzymatic synthesis of hydrophobic polypeptides based on Ala and Aib.38 On the contrary to the short Aib peptides, the preparation of long polypeptides with multiple Aib residues is complicated because of the sterical hindrance of the Aib residue.
The self-assembling properties of Aib-containing peptides were the subject of many investigations.28–32,39–41 In particular, Kimura and co-workers synthesized peptide nanotubes of different size (50–200 nm in diameter and 100–600 nm in length) based on block polypeptides (Sar)m-b-(Leu-Aib)n (m = 22–25; n = 6–10)31,39 and [(Sar)26]3-b-[(His)2-(Leu-Aib)6].40 More recently, Haldar and coworkers demonstrated that Boc-Phe-Aib-Gabu-OMe and Boc-Tyr-Aib-Gabu-OMe (Gabu, γ-aminobutyric acid) are able to form polydisperse microspheres with a diameter of ca. 750 nm,32 while Boc-Leu-Aib-Ser-OMe self-assembles into supramolecular nanotubes able to encapsulate hydrophobic guests.41
Stimulated by these findings, we designed and synthesized amphiphilic, random and block copolypeptides of Lys and Aib as a novel biomaterial for various biological applications in drug delivery, tissue engineering, etc. On the contrary to the above-mentioned works, where relatively short peptides were described, we decided to explore the use of the easy and cost-effective ring-opening polymerization of N-carboxyanhydrides (ROP NCA) for the preparation of long-chain polypeptides.42 Lys was chosen to provide solubility in aqueous media and stability to the formed colloids. Furthermore, these positively charged units can bind to negatively charged membranes through electrostatic interactions with subsequent penetration into the cells by means of unspecific cellular uptake.43 In turn, being a hydrophobic amino acid, Aib might induce self-aggregation and formation of nanoparticles in aqueous media when inserted in peptides rich of hydrophilic residues. Moreover, the presence of non-coded amino acid Aib can ensure the enhanced enzymatic stability of the nanoparticles, respect to those containing only protein amino acids.
Experimental
Materials
H-Lys(Z)-OH (Z, benzyloxycarbonyl) (≥99.0%), H-Aib-OH (≥99.0%), triphosgene (98%), hexylamine (99.0%), α-pinene, trifluoromethanesulfonic acid (TFMSA), trifluoroacetic acid (TFA), Cy3 succinimidyl ester, methylthiazolyldiphenyl-tetrazolium bromide (MTT) and 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI) were purchased from Sigma-Aldrich (Germany) and used without further purification. Dimethylformamide (DMF), dimethylsulfoxide (DMSO), 1,4-dioxane, n-hexane, tetrahydrofuran (THF), ethyl acetate, 2,2,2-trifluoroethanol (TFE) and diethyl ether were purchased from Vecton Ltd. (Russia). Prior to use, all solvents were purified by distillation and dried using standard procedures. The culture media MEM and Fetal Bovine Serum (FBS) Medium 200 were purchased from Gibco (Thermo Fisher Scientific, Switzerland). HepG2 was an ATCC (USA) product.Methods
For the synthesis of the monomer, the magnetic stirrer IKA MSH-basic with controllable heating (Germany) was used. Polymerization was carried out in a thermostatic shaker Unimax1010 Incubator 100 Heidolph (Germany). For NMR analysis a Bruker 400 MHz Avance III instrument (Germany) was utilized and DMSO-d6 was used. Gel-permeation chromatography (GPC) was performed using LC-10 Shimadzu system (Japan) supplied with a RID 10-A detector and a Styragel column, HMW 6E, 15–20 μm, 7.8 i.d. × 300 mm. HPLC experiments were performed on a Shimadzu LC-20 system (Canby, USA). The data was acquired and processed with the LS Solution software (Shimadzu, Japan). The colloids were characterized by measuring the hydrodynamic diameter of the nanoobjects by means of a Malvern Zetasizer Nano-ZS DLS instrument, at a scattering angle of 173° and 25 °C. TEM images were recorded using EM 902A TEM from Philips (Netherlands). The 400 mesh copper grids were hydrophilized by treatment with nitrogen plasma during 30 min using Bal-Tec Med 020 High Vacuum Coating System. The fluorescent plate reader FLUOstar Omega BMG Labtech (Germany) was employed both for the MTT-assay and for studying the cellular uptake kinetics. Cellular uptake visualization was carried out using an Axio Observer Z1 Carl Zeiss microscope (Germany) equipped with the Axiovision MRm digital camera and the ApoTome filtering device.This polymer, hereafter called P[Lys(Z)], was used as macroinitiator for the polymerization of Aib NCA. To this end, 110 mg (850 μmol) of Aib NCA were added to 53, 70 or 105 mg (8.5, 11.3 or 16.9 μmol) of P[Lys(Z)], previously dissolved in 3.0, 3.5 or 4.0 mL, respectively, of purified and dried DMF. Thus, the ratio [Aib NCA]/P[Lys(Z)] was 100, 75 and 50, respectively. The reaction mixtures were incubated at 35 °C for 72 h. The block copolymers obtained were precipitated with diethyl ether, recrystallized twice from chloroform/diethyl ether and dried in vacuo. The block copolymer yield was 61–65%.
The self-assembly of the copolymers was carried out by phase-inversion method.52 To this aim polymer solution in DMF was placed into dialysis bag with MWCO 3500 and the bag was immersed into the glass filled with 50% DMF in water (v/v). After 2 h of dialysis with slow stirring the medium was changed to water. The dialysis against water was carried out for 24 h. During this time water was replaced twice with fresh one. Then the nanoparticles were transferred from dialysis bag to the vial and lyophilized. Before the use the samples were redispersed in 0.1 M sodium citrate buffer (pH 3.0) with ultrasound probe for 30 s. For biodegradation study and cell experiments, sodium citrate buffer was replaced with 0.01 M PBS (pH 7.4) via ultracentrifugation with VivaSpin (MWCO 3000). Dynamic light scattering (DLS) and TEM (transmission electron microscopy) were performed to determine size and morphology of the prepared particles.
Results and discussion
Synthesis of the polypeptides
Ring-opening polymerization of NCA is the most common method for the preparation of long-chain, random or block copolypeptides. Below, we illustrate the strategy for the synthesis of the random and block copolymers based on the Lys and Aib α-amino acid units (Scheme 1). | ||
| Scheme 1 Strategy for the synthesis of random (A) and block (B) copolymers based on the Lys and Aib units. | ||
A series of random polypeptides with various amounts of Lys and Aib units was obtained and characterized in terms of molecular weight and composition. The initial ratios of monomers and the characteristics of the polymers are given in Table 1.
| Sample | Initial ratio [Lys]/[Aib] | Calculated composition, mol% | Mna P[Aib-co-Lys(Z)] | Đ | [Lys]/[Aib] ratio in polypeptidesb | Determined composition,b mol% | ||
|---|---|---|---|---|---|---|---|---|
| Lys | Aib | Lys | Aib | |||||
| a GPC analysis.b Quantitative RP HPLC amino acid analysis. | ||||||||
| R1 | 8 | 89 | 11 | 138![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
1.67 | 4.5 | 82 | 18 |
| R2 | 6 | 86 | 14 | 128000 |
1.66 | 4.0 | 80 | 20 |
| R3 | 4 | 80 | 20 | 95000 |
1.56 | 3.2 | 76 | 24 |
| R4 | 2 | 67 | 33 | 70000 |
1.46 | 2.6 | 71 | 29 |
| R5 | 1 | 50 | 50 | 54000 |
1.33 | 0.7 | 42 | 58 |
The molecular weights of the random Lys(Z)/Aib copolymers were examined using gel-permeation chromatography. It was found that an increase of Aib molar concentration in the polymerization mixture led to a decrease of the polypeptide molecular weight. This result is in agreement with the molecular weights of the monomers, e.g. the molecular weight of Aib NCA is approx. 2.4 times lower than that of Lys(Z) NCA. According to the GPC analysis, also Đ (dispersity) values diminished with the increase of the Aib amount in the polymerization mixture. To determine the [Lys]/[Aib] ratio in the polypeptides, we carried out their acid hydrolyses and analyzed the amino acid content by quantitative RP-HPLC. It was found that an increase of the Aib molar concentration in the polymerization mixture favors the increase of the Aib amount in the final polypeptide (Table 1). A good correlation between calculated and experimentally determined monomer compositions was observed.
In the case of the block copolymers, the polymerization included the preparation of the [Lys(Z)]n polypeptide and its use as the macroinitiator for the synthesis of PAibm-b-[PLys(Z)]n block sequences. The GPC analysis of [Lys(Z)]n showed the formation of a quite narrow-dispersed polymer with weight average molecular weight (Mw) = 7700 and number average molecular weight (Mn) = 6200 (Đ = 1.24). The mean number of monomer units in the polypeptide chain was found to be 23. To vary the length of the hydrophobic Aibm block, the syntheses of the PAibm-b-[PLys(Z)]n polymers were carried out at the [Aib NCA]/[PLys(Z)] ratios of 50, 75 and 100 (Table 2).
On the contrary to the random Lys(Z)/Aib polymer, the block polypeptides obtained were poorly soluble in many organic solvents and had the tendency to self-aggregate and to form nanoparticles even in DMF, a widely used solvent for polypeptides. Thus, a GPC analysis could not be performed in this case. The polypeptide composition was then calculated from the quantitative RP HPLC amino acid analysis of the hydrolyzed polypeptide samples.
Conformational study
The information about the peculiarities of the polypeptide 3D-structures in the solid state was gained by using FT-IR absorption spectroscopy. The frequencies of the IR absorption bands in the amide I (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, 1600–1700 cm−1), amide II (N–H bending and C–N stretching, 1480–1575 cm−1) and amide A (N–H stretching, 3300–3500 cm−1) regions strongly depend on the overall conformation and the resulting spatial orientation of the peptide units in the backbone.45,46 Within each series, either random or block copolymers, the FT-IR absorption spectra show almost identical band patterns, with broad bands due to the presence of a number of co-existing different conformations, whereas a noticeable difference was observed between the two series (Fig. 1; the data are shown for the samples R2, R4, B2 and B3).
O stretching, 1600–1700 cm−1), amide II (N–H bending and C–N stretching, 1480–1575 cm−1) and amide A (N–H stretching, 3300–3500 cm−1) regions strongly depend on the overall conformation and the resulting spatial orientation of the peptide units in the backbone.45,46 Within each series, either random or block copolymers, the FT-IR absorption spectra show almost identical band patterns, with broad bands due to the presence of a number of co-existing different conformations, whereas a noticeable difference was observed between the two series (Fig. 1; the data are shown for the samples R2, R4, B2 and B3).
 | ||
| Fig. 1 FT-IR absorption spectra of lyophilized powders of the R2, R4, B2 and B3 copolymers ((A) from top to bottom) and amide I and II bands of R4 and B2 (B). | ||
Very broad amide A bands in the FT-IR absorption spectra of the lyophilized R2, R4, B2 and B3 copolymers are indicative of wide inter- and intramolecular H-bond networks. The detailed analysis of the amide I and II bands in the spectra of the random copolymers allowed identifying several individual components that could be assigned to various conformations. In particular, the bands centered at about 1650 and 1540 cm−1 suggest the co-existence of random coil and helical conformations, the latter due to the presence of a large amount of the strong helix promoter Aib (Fig. 1, data are shown for the sample R4).47,48 Nevertheless, due to the broadness of the FT-IR absorption bands, a noticeable amount of the β-sheet conformation can be also present in the copolymer structure. On the contrary, the analysis of the amide I structure of the block polypeptides showed the features typical for the β-sheet conformation: a strong main band at 1625 cm−1 and weaker bands at 1665 and 1690 cm−1, resembling those of spray-dried (Lys)n in the solid state.49 Apparently, the presence of Aib in the random co-polymers prevents β-sheet formation and disrupts the aggregation. This fact can also explain the higher solubility of the random copolymers with respect to the block copolymers.
The CD analysis showed noticeable differences in the copolymer secondary structures, which depended on the Lys/Aib ratio (Fig. 2).
 | ||
| Fig. 2 CD spectra of the copolymers R2, R4, B2 and B3 in water (A), HCl solution, pH 3, (B) and TFE/water mixture (3/1 v/v) (C). | ||
The lower solubility of the block copolymers, with formation of opalescent solutions, resulted also in lower intensities of the CD spectra as compared to those of the random co-polymers. In water, at neutral or low pH, all copolymers were in random conformation because of the repulsion of the positively charged amino groups.
On the contrary, in a water/TFE mixtures (the latter solvent is known to induce helix formation in peptides)50 all spectra showed the CD pattern typical for helical conformations, even in the case of the copolymers B2 and B3, forming β-sheets in lyophilized powders. However, in the spectra of B2 and B3 a noticeable contribution of β-sheet structure was evidenced by the more intense band at about 222 nm as compared to that at about 207 nm. The ratio Θ222/Θ207 decreased and the helix content increased with increasing amount of helix-inducing Aib residues in the copolymers (samples R4 and B3).51 Thus, it can be expected, that the composition of the polypeptides and their tendency to form β-sheets could influence their propensity to form aggregates and further to self-assemble into supramolecular structures.
Preparation and characterization of the nanoparticles
After removal of Z protecting group the polypeptides became amphiphilic and self-assembled into supramolecular structures in aqueous media with addition of the organic solvent. Both random and block polypeptides were used for the preparation of nanoparticles. To this aim, the gradual phase-inversion approach (from organic to aqueous medium) was used. In our previous studies on PLys-b-PLeu52 and PLys-b-PIle block copolymers we found that for Lys-based polypeptides the acidic conditions were more suitable for self-assembly.53 Apparently, formation of particles with smaller size at pH 3, associated with a more extensive protonation of the Lys side chains, imparts higher solubility to the polypeptides. The subsequent transfer of the nanospheres from the acidic pH to a PBS environment at pH 7.4 did not influence the particle size.53 Thus, a 0.1 M Na-citric buffer, pH 3.0, was chosen for the preparation of Lys/Aib block and random polypeptide nanoparticles.Obviously, the differences in the distribution of hydrophilic and hydrophobic units along the macromolecule chain may affect the morphology of the self-assembled particles. Usually, a random distribution of hydrophilic and hydrophobic residues favors the formation of micelles.54 In turn, macromolecules with block structures form polymersomes (liposome-like particles or vesicles), as well as micelles.6 Indeed, our detailed TEM study of the particles prepared from random and block polypeptides allowed us to conclude that nanospheres of different morphologies do form (Fig. 3).
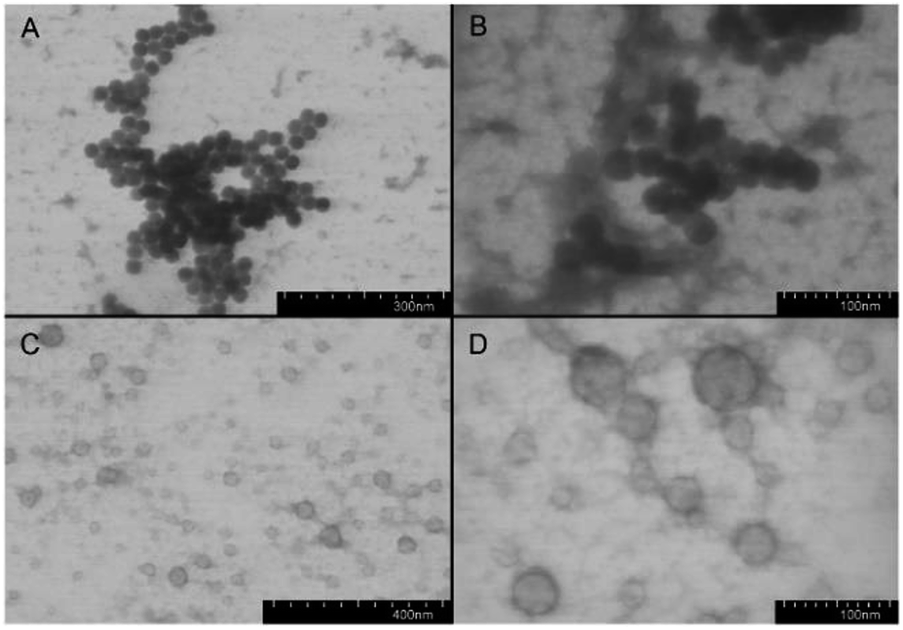 | ||
| Fig. 3 TEM images of nanoparticles formed by P(Aib-co-Lys) random copolymer ((A and B) sample R4) and PAib-b-PLys block copolymer ((C and D) sample B2). | ||
The nanospheres from random polypeptides were contrasted uniformly with uranyl acetate. On the contrary, the nanospheres from block polypeptides had a dark envelope and a bright core. Thus, in the first case, formation of micelles took place, whereas in the second case the self-assembly generated vesicles. The comparison of the CD spectra in the organic solvent/water mixtures with the morphology of the forming nanoparticles allows to conclude, that not only β-structure content, but also the distribution of β-sheets along the polypeptide chain can influence the self-assembly.
Table 3 summarizes the results of the measurements of the hydrodynamic diameter (dH) and the PDI of the nanoparticles. The nanoparticles obtained had unimodal size distribution with a mean hydrodynamic diameter from approx. 200 to 400 nm (depending on the polymer type and composition) and with PDI ranging from 0.15 to 0.24. In the case of random polypeptides, dH for R1-R3, that contained 18–24 mol% of Aib monomer, was about 300 nm. Interestingly, the increase of Aib amount up to 58 mol% for R5 induced a decrease in dH. This finding can be explained by the tight folding of the hydrophobic part (Aib stretch) of the macromolecule in aqueous environments. Another reason for this phenomenon can be the diminished repulsion among the less abundant, positively charged monomers. The discrepancy of the data acquired by TEM and DLS methods originates from the different principles of the measurement. DLS detects hydrodynamic diameter of the particles with polymeric chains exposed to the solvent, and it is obviously higher than that determined for the dried samples in TEM.55,56
| Sample | Hydrodynamic diameter, nm | PDI |
|---|---|---|
| P(Aib-co-Lys) | ||
| R1 | 294 ± 127 | 0.20 |
| R2 | 312 ± 138 | 0.20 |
| R3 | 310 ± 151 | 0.24 |
| R4 | 238 ± 101 | 0.18 |
| R5 | 185 ± 87 | 0.22 |
|
||
| PAib-b-PLys | ||
| B1 | 216 ± 84 | 0.15 |
| B2 | 238 ± 137 | 0.23 |
| B3 | 398 ± 193 | 0.24 |
While the amphiphilic random copolymers self-assembled into micelles, with a compact hydrophobic core, the amphiphilic block-copolymers formed vesicle-like structures containing a hydrophobic membrane and an aqueous interior. For the block polypeptides the increase of the hydrophobic part was accompanied by an increase of their hydrodynamic diameter. A similar result was previously observed for PGlu-b-PPhe polymersomes.57 The most probable reason for such effect is the decrease of polypeptide solubility because of its increased hydrophobicity. Therefore, the higher self-aggregation caused by the hydrophobic interactions leads to a reduction of the particle core.
Biodegradation study
In vitro biodegradation studies represent a complicated task as usually the degradation rate differs from that of the in vivo process because of the complex composition of the biological fluids containing a number of highly active enzymes. Nevertheless, the results of the in vitro degradation allow a comparative analysis of the biodegradability of different biomaterials.58To evaluate the biodegradation of the prepared nanoparticles, the representative samples R1, R4 and B2 were selected. R4 and B2 were chosen due to the similarity of the amino acid ratio and size after self-assembly. R1 sample was added for the comparison with R4 to evaluate the effect of hydrophobic to hydrophilic amino acid ratio on biodegradation of random copolymers. To assess the contribution of Aib to the biodegradation rate, PLys-b-PLeu nanoparticles were used as well. Papain was selected as a biocatalyst. This proteolytic enzyme works at physiological conditions (pH 7.4 and 37 °C) and cleaves the peptide bonds between numerous α-amino acids.
During the 35 day biodegradation experiments, the hydrodynamic diameters of the particles were monitored by DLS (Fig. 4). Except R1, containing the highest ratio of hydrophilic to hydrophobic amino acids, the degradation of the polypeptides (R4, B2 and PLys-b-PLeu) was accompanied by the increase in the particle size after the first day of degradation process and a slow decrease of the size within further two weeks. Such surge in the particle size is caused by the partial degradation of the hydrophilic Lys-based shell and the exposure of the hydrophobic part to the solvent. In turn, this latter event favored the aggregation of the particles because of the hydrophobic interactions in the aqueous medium. The highest aggregation and, consequently, the highest burst in size was observed for the polypeptide based on coded amino acids, e.g. Lys and Leu. This peptide was used as benchmark for the degradation studies. In this case, the fast degradation of the (Lys)n block led to the quick aggregation of the nanoparticles after a day of co-incubation with the enzyme. A considerable aggregation of the particles formed by Aib-containing polypeptides was observed after co-incubation with the enzyme within three weeks for PAib-b-PLys and within a month for random polypeptides.
 | ||
| Fig. 4 Changes in the hydrodynamic sizes of polypeptide particles during the biodegradation processes induced by papain (0.01 M PBS, pH 7.4, 37 °C). | ||
The HPLC amino acid analysis allowed identification of free amino acids released in the reaction media during the degradation process. On the contrary to PLys-b-PLeu degradation, where Leu was detected already after one day of biodegradation, free Aib was found after 5 days for R4 and B2 and 9 days for R1. Moreover, the analysis of free amino acid content after 14 and 35 days revealed that Lys accumulation for both PLys-b-PLeu and B2 was faster than that of the random polypeptides R1 and R4 (Fig. 5A). In case of Aib, the slowest accumulation of the amino acid was observed for B2 sample. It indicates the slowest degradation rate of hydrophobic block of B2 copolymer in comparison with PLys-b-PLeu, as well as with R1 and R4 samples (Fig. 5B). This behavior can be explained by the fast degradation of Lys stretches by papain, whereas Aib sequence can be hardly targeted by the hydrolytic enzyme.
 | ||
| Fig. 5 Accumulation of free Lys (A) and Aib/Leu (B) during the biodegradation process of random and block copolymers based on Lys and Aib, as well as PLys-b-PLeu induced by papain (0.01 M PBS, pH 7.4, 37 °C). | ||
The accumulation of free amino acids produced during the degradation of random Aib-containing polypeptides was comparable for R1- and R4-based particles, but it was slower than that observed for the benchmark polypeptide as well.
Cell experiments
The nanoparticles obtained were tested in cell experiments using the HepG2 cell line.The results on the cell viability determined by the MTT assay are presented in Fig. 6. Up to the concentration 0.125 mg mL−1, cell viability for nanoparticles based on both kinds of copolymers was higher than 80%. However, at concentration of 0.25 mg mL−1 the cytotoxicity of the random copolymers was twice higher than that of the block copolymers. The EC50 values for the tested random (R4) and block-copolymer (B2) were 0.170 and 0.245, respectively.
 | ||
| Fig. 6 Cell viability (HepG2) in the presence of P(Aib-co-Lys) (sample R4) and PAib-b-PLys (sample B2) during 48 hours. | ||
The lower cytotoxicity of the nanomaterials based on block copolymers can be related to the better burial of the hydrophobic residues inside the particles. This behavior, in turn, favors the stabilization of the particles in the culture medium. In addition, the exposed hydrophobic residues of the random polymers can favor the membrane disrupting activity of the nanostructures.
To study the cellular uptake of Lys/Aib-based nanomaterials, the fluorescent microscopy technique was applied. Samples R4 and B2 were covalently labeled by use of the Cy3 dye and incubated with HepG2 cells for 4 hours. When the incubation time was over the cell nuclei were stained with DAPI. The internalization of both kinds of nanoparticles indicated a relatively uniform distribution of Cy3 (red) in the cytoplasm around the nucleus (blue) (Fig. 7).
 | ||
| Fig. 7 Fluorescent images (×20) of HepG2 cells treated with Cy3-labeled polypeptides (60 μg mL−1): (A) P(Lys-co-Aib) (sample R4); (B) PAib-b-PLys (sample B2). Each image comprises (from left to right) Cy3, DAPI and merged fluorescence. | ||
The kinetics of internalization was studied via incubation of Cy3-labeled nanoparticles with HepG2 for 48 hours. The fluorescent signal for both kinds of nanoparticles (series R and B) considerably increased during the initial hours and reached the maximum after 24 hours of incubation (Fig. 8). The difference in fluorescence intensity can be explained by different amount of Cy3 label covalently bound to the nanoparticle surface. In case of nanoparticles formed by random polymers (R4 sample) the labeling efficiency is much higher because most hydrophilic lysine residues are exposed to aqueous surrounding and accessible for the reaction with the dye. In turn, for polymersomes (B2 sample), which have bilayer polymer membrane, about a half of lysines are buried inside the particle. Thus, for polymersomes the lower amount of lysine units is accessible for modification.
 | ||
| Fig. 8 Kinetics of cell uptake of Cy3-labeled P(Aib-co-Lys) (sample R4) and PAib-b-PLys (sample B2) nanoparticles (concentration: 60 μg mL−1). | ||
Conclusions
A series of random and block copolypeptides based on Lys and Aib was synthesized via ROP NCA. The random polypeptides, lyophilized from water, adopted both random coil and α-helical conformations, whereas the block-type polypeptides showed a high content of β-sheets in their structure. In water, at neutral or low pH, all copolymers were unordered because of the repulsion of the positively charged side chains of Lys residues. On the contrary, in TFE all spectra revealed a pattern typical for helical conformations, even in the case of block copolymers with high β-sheet propensity. As expected, the helix content increased with increasing amount of the helix-inducing Aib residue in the polypeptides. All the copolymers formed nanoparticles of different morphology, which depended on the distribution of the Aib residues (random or block) along the peptide chain. In particular, random polypeptides self-assembled into micelles, whereas the block-type polypeptides formed vesicles. At optimal conditions, the nanoparticles obtained showed an almost unimodal size distribution with mean hydrodynamic diameter of 200–400 nm. By increasing the Aib amount, the hydrodynamic diameter of the particles based on random copolymer decreased, probably, because of the increasing self-aggregation and the reduction of the particle core. These results can be important for the further understanding of the peculiarities of the long Aib polypeptides folding and the influence of the secondary structure on of the morphology of the resulting nanoparticles. Aib-containing nanoparticles proved to be more stable to enzymatic degradation than those based on only coded amino acids. They did not induce cell death up to the concentration 0.125 mg mL−1 and were internalized into the cell cytoplasm within 24 hours. Due to all these features the novel, bio-inspired polypeptide nanomaterials described herein can find various biomedical applications, such as development of improved drug delivery formulations, fillers for the scaffolds, etc.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The chemical synthesis and biological evaluation of amphiphilic block copolymers was supported by a grant of the Russian Science Foundation (#14.50.00069). The authors also thank the Chemical Analysis and Materials Research Centre of St Petersburg State University for the HPLC amino acid analysis. B. B. and F. F. are grateful to the Italian Ministry of Research for financial support (PRIN 2015, Prot. 20157WW5EH).References
- C. Ding and Z. Li, Mater. Sci. Eng., C, 2017, 76, 1440 CrossRef CAS PubMed.
- J. Li, X. Chang, X. Chen, Z. Gu, F. Zhao, Z. Chai and Y. Zhao, Biotechnol. Adv., 2014, 32, 727 CrossRef CAS PubMed.
- J. Wang, C. Gong, Y. Wang and G. Wu, RSC Adv., 2014, 4, 15856 RSC.
- M. S. Shim and Y. J. Kwon, Adv. Drug Delivery Rev., 2012, 64, 1046 CrossRef CAS PubMed.
- I. Guryanov, S. Fiorucci and T. Tennikova, Mater. Sci. Eng., C, 2016, 68, 890 CrossRef CAS PubMed.
- S. Kumar and P. Maiti, RSC Adv., 2016, 6, 67449 RSC.
- K. Letchford and H. Burt, Eur. J. Pharm. Biopharm., 2007, 65, 259 CrossRef CAS PubMed.
- J. S. Lee and J. Feijen, J. Controlled Release, 2012, 161, 473 CrossRef CAS PubMed.
- N. Habibi, N. Kamaly, A. Memic and H. Shafiee, Nano Today, 2016, 11, 41 CrossRef CAS PubMed.
- K. Matsuurua, RSC Adv., 2014, 4, 2942 RSC.
- A. V. Hubina, A. A. Pogodaev, V. V. Sharoyko, E. G. Vlakh and T. B. Tennikova, React. Funct. Polym., 2016, 100, 173 CrossRef CAS.
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589 RSC.
- Z. Song, Z. Han, S. Lv, C. Chen, L. Chen, L. Yin and J. Cheng, Chem. Soc. Rev., 2017, 46, 6570 RSC.
- V. Mikhalevich, I. Craciun, M. Kyropoulou, C. G. Palivan and W. Meier, Biomacromolecules, 2017, 18, 3471 CrossRef CAS PubMed.
- N. Stoppacher, N. K. N. Neumann, L. Burgstaller, S. Zeilinger, T. Degenkolb, H. Brückner and R. Schuhmacher, Chem. Biodivers., 2013, 10, 734 CrossRef CAS PubMed.
- Peptaibiotics, ed. C. Toniolo and H. Bruckner, Wiley-VCH, Weinheim, Germany, 2009 Search PubMed.
- M. Venanzi, E. Gatto, F. Formaggio and C. Toniolo, J. Pept. Sci., 2017, 23, 104 CrossRef CAS PubMed.
- I. L. Karle and P. Balaram, Biochemistry, 1990, 29, 6747 CrossRef CAS PubMed.
- C. Toniolo, M. Crisma, F. Formaggio and C. Peggion, Biopolymers, 2001, 60, 396 CrossRef CAS PubMed.
- E. Longo, A. Moretto, F. Formaggio and C. Toniolo, Chirality, 2011, 23, 756 CrossRef CAS PubMed.
- S. Zikou, A.-I. Koukkou, P. Mastora, M. Sakarellos-Daitsiotis, C. Sakarellos, C. Drainas and E. Panou-Pomonis, J. Pept. Sci., 2007, 13, 481 CrossRef CAS PubMed.
- K. Kono, H. Nishii and T. Takagishi, Biochim. Biophys. Acta, 1993, 1164, 81 CrossRef CAS.
- L. Garbuio, S. Antonello, I. Guryanov, Y. Li, M. Ruzzi, N. J. Turro and F. Maran, J. Am. Chem. Soc., 2012, 134, 10628 CrossRef CAS PubMed.
- R. A. G. D. Silva, S. C. Yasui, J. Kubelka, F. Formaggio, M. Crisma, C. Toniolo and T. A. Keiderling, Biopolymers, 2002, 65, 299 CrossRef PubMed.
- P. Hanson, G. Millhauser, F. Formaggio, M. Crisma and C. Toniolo, J. Am. Chem. Soc., 1996, 118, 7618 CrossRef CAS.
- S. Mondal, M. Varenik, D. N. Bloch, Y. Atsmon-Raz, G. Jacoby, L. Adler-Abramovich, L. J. W. Shimon, R. Beck, Y. Miller, O. Regev and E. Gazit, Nat. Commun., 2017, 8, 14018 CrossRef CAS PubMed.
- S. Grubičić, B. Chandramouli, V. Barone and G. Brancato, Phys. Chem. Chem. Phys., 2016, 18, 20389 RSC.
- M. Konda, S. Bhowmik, S. M. Mobin, S. Biswas and A. K. Das, ChemistrySelect, 2016, 1, 2586 CrossRef CAS.
- P. Gobbo, S. Antonello, I. Guryanov, F. Polo, A. Soldà, F. Zen and F. Maran, ChemElectroChem, 2016, 3, 2063 CrossRef CAS.
- S. Mondal, L. Adler-Abramovich, A. Lampel, Y. Bram, S. Lipstman and E. Gazit, Nat. Commun., 2015, 6, 8615 CrossRef CAS PubMed.
- M. Ueda, A. Makino, T. Imai, J. Sugiyama and S. Kimura, J. Pept. Sci., 2011, 17, 94 CrossRef CAS PubMed.
- A. Pramanik, A. Paikar, T. Das, K. Maji and D. Haldar, RSC Adv., 2016, 6, 39172 RSC.
- Synthesis of peptides and peptidomimetics, Houben-Weyl: Methods of Organic Chemistry, ed. M. Goodman, A. Felix, L. Moroder and C. Toniolo, vol. E 22, Thieme, Stuttgart, Germany, 2003 Search PubMed.
- A. Takasu, S. Horikoshi and T. Hirabayashi, Biomacromolecules, 2005, 6, 2334 CrossRef CAS PubMed.
- M. De Zotti, B. Biondi, C. Peggion, F. Formaggio, Y. Park, K.-S. Hahm and C. Toniolo, Org. Biomol. Chem., 2012, 10, 1285 RSC.
- K. B. H. Salah and N. Inguimbert, Org. Lett., 2014, 16, 1783 CrossRef PubMed.
- A. Siow, K. Hung, P. W. R. Harris and M. A. Brimble, Eur. J. Org. Chem., 2017, 350 CrossRef CAS.
- K. Tsuchiya and K. Numata, Chem. Commun., 2017, 53, 7318 RSC.
- M. Ueda, A. Makino, T. Imai, J. Sugiyama and S. Kimura, Polym. J., 2013, 45, 509 CrossRef CAS.
- M. Ueda, A. Uesaka and S. Kimura, Chem. Commun., 2015, 51, 1601 RSC.
- A. Paikar, A. Pramanik, T. Das and D. Haldar, Polym. Chem., 2017, 8, 396 RSC.
- J. Cheng and T. J. Deming, Top. Curr. Chem., 2012, 310, 1 CAS.
- E. Fröhlich, Int. J. Nanomed., 2012, 7, 5577 CrossRef PubMed.
- R. Wilder and S. Mobashery, J. Org. Chem., 1992, 57, 2755 CrossRef CAS.
- M. Jackson and H. H. Mantsch, Crit. Rev. Biochem. Mol. Biol., 1995, 30, 95 CrossRef CAS PubMed.
- A. Mirtič and J. Grdadolnik, Biophys. Chem., 2013, 175–176, 47 CrossRef PubMed.
- I. Guryanov, F. Polo, E. V Ubyvovk, E. Korzhikova-Vlakh, T. Tennikova, A. T. Rad, M.-P. Nieh and F. Maran, Chem. Sci., 2017, 8, 3228 RSC.
- S. J. Prestrelski, N. Tedeschi, T. Arakawa and J. F. Carpenter, Biophys. J., 1993, 65, 661 CrossRef CAS PubMed.
- A. Mauerer and G. Lee, Eur. J. Pharm. Biopharm., 2006, 62, 131 CrossRef CAS PubMed.
- M. Goodman, A. S. Verdini, C. Toniolo, W. D. Phillips and F. A. Bovey, Proc. Natl. Acad. Sci. U. S. A., 1969, 64, 444 CrossRef CAS.
- C. Toniolo, A. Polese, F. Formaggio, M. Crisma and J. Kamphuis, J. Am. Chem. Soc., 1996, 118, 2744 CrossRef CAS.
- N. N. Zashikhina, M. V. Volokitina, V. A. Korzhikov-Vlakh, I. I. Tarasenko, A. Lavrentieva, T. Scheper, E. Rühl, R. V. Orlova, T. B. Tennikova and E. G. Korzhikova-Vlakh, Eur. J. Pharm. Sci., 2017, 109, 1 CrossRef CAS PubMed.
- E. G. Vlakh, E. V. Grachova, D. D. Zhukovsky, A. V. Hubina, A. S. Mikhailova, J. R. Shakirova, V. V. Sharoyko, S. P. Tunik and T. B. Tennikova, Sci. Rep., 2017, 7, 41991 CrossRef CAS PubMed.
- Functional Monomers and Polymers, ed. K. Takemoto, R. M. Ottenbrite and M. Kamachi, Marcel Dekker, NY, 2nd edn, 1997 Search PubMed.
- A. O. Moughton, J. P. Patterson and R. K. O'Reilly, Chem. Commun., 2011, 47, 355 RSC.
- H. R. Marsden, L. Gabrielli and A. Kros, Polym. Chem., 2010, 1, 1512 RSC.
- E. Vlakh, A. Ananyan, N. Zashikhina, A. Hubina, A. Pogodaev, M. Volokitina, V. Sharoyko and T. Tennikova, Polymers, 2016, 8, 212 CrossRef.
- H. S. Azevedo and R. L. Reis, Understanding the enzymatic degradation of biodegradable polymers and strategies to control their degradation rate, in Biodegrad. Syst. Tissue Eng. Regen. Med., ed. R. L. Reis and J. S. Roman, CRC Press, Boca Raton, 2004, pp. 177–201 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |