
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced electrocatalytic performance of platinum nanoparticles on thiolated polyaniline-multiwalled carbon nanotubes for methanol oxidation
Zhaohong Su *a,
Chaorong Lia,
Yongbing Chenga,
Qingwen Guia,
Yuanfu Xionga,
Yueming Tanb,
Hongmei Jiang*a and
Xiaoying Liu*a
*a,
Chaorong Lia,
Yongbing Chenga,
Qingwen Guia,
Yuanfu Xionga,
Yueming Tanb,
Hongmei Jiang*a and
Xiaoying Liu*a
aCollege of Science, Hunan Agricultural University, Changsha 410128, PR China. E-mail: zhaohongsu@hunau.edu.cn; jhmndcn@hunau.edu.cn; xiaoyingliu@126.com
bCollege of Chemistry and Chemical Engineering, Hunan Normal University, Changsha 410081, China
First published on 2nd October 2018
Abstract
Pt nanoparticles (PtNPs) well-dispersed on thiolated polyaniline (TPANI)-multiwalled carbon nanotubes (MWCNTs) were prepared for enhanced electrocatalytic oxidation of methanol in acidic media. Briefly, the preparation of nanocomposites was carried out via microwave-assisted thiol–ene reaction of 2,5-dimercapto-1,3,4-thiadiazole (DMcT) with oxidized PANI, which was synthesized in the presence of MWCNTs, yielding TPANI-MWCNTs; then, PtNPs were deposited on TPANI-MWCNTs by a microwave-assisted method to obtain PtNPs/TPANI-MWCNT nanohybrids. Fourier transform infrared spectroscopy, cyclic voltammetry (CV), transmission electron microscopy (TEM), X-ray diffraction (XRD), thermogravimetric analysis (TGA) and inductively coupled plasma-atom emission spectroscopy were used to study relevant nanohybrid properties. TEM showed that PtNPs were well dispersed on TPANI-MWCNTs. TGA showed that PtNPs/TPANI-MWCNTs exhibited better thermal stability than PtNPs/TPANI-MWCNTs and PtNPs/MWCNTs. CV studies showed that PtNPs/TPANI-MWCNT-modified glassy carbon electrode (GCE) exhibited a larger electrochemically active surface area and higher electrocatalytic performance toward methanol electro-oxidation compared with those of PtNPs/PANI-MWCNTs/GCE and PtNPs/MWCNTs/GCE. Also, the PtNPs/TPANI-MWCNTs/GCE electrode possessed high stability and maintained 86% of its initial catalytic activity after 1000-cycle CV in 1.0 M CH3OH + 0.5 M H2SO4.
Introduction
Nanostructured platinum (Pt) with excellent catalytic performance is involved in a considerable number of industrial chemical reactions and fuel cells.1 However, the high cost of Pt is one of the key factors inhibiting its large-scale commercialization.2 Recently, carbon nanotubes (CNTs) including multiwalled carbon nanotubes (MWCNTs) and single-walled carbon nanotubes have been generally applied as support materials for noble-metal nanoparticles (NPs) owing to their reasonable price, outstanding chemical and electrochemical stability, high specific surface area and excellent conductive ability.3 As reported in literatures, PtNPs/CNT nanohybrids exhibit high-efficiency applications not only in fuel cells, but also in heterogeneous catalysis and biosensors.4 Unfortunately, PtNPs often undergo dissolution, aggregation and Oswald ripening during fuel cell operation.4a,5 Synthesis of PtNPs on CNTs with small particle size and high dispersion with good catalytic performance and low cost has attracted the attention of many researchers. However, depositing PtNPs with high dispersion and stability on CNTs is still a challenge.It is universally acknowledged that pristine CNTs without adequate binding sites for anchoring the precursors of metal ions or metal nanoparticles, owing to self-graphitization, exhibit inhomogeneous dispersion, uncontrollable size and easy aggregation of metal nanoparticles.3,4,6 To introduce more functional active sites on the CNT surface and obtain highly dispersed noble metal NPs, functionalization of CNTs is often conducted by decorating with polymer,6,7 modification via π-stacking,4a tethering of ionic liquid,4c chemical oxidation at defect sites of CNTs,8 etc. Meanwhile, several other approaches were studied to improve the stability of PtNPs on CNTs, and these include depositing PtNPs on thiolated CNTs5 or hydrofluoric acid-treated CNTs9 and applying silica10 or ionic liquid polymer6 as a protection layer. It is interesting that the conducting polymers such as polypyrrole, polythiophene and PANI can be functionalized with thiol by the thiol–ene reaction.11 However, functionalizing conducting polymers with thiol via the microwave-assisted thiol–ene reaction has not been reported to date.
In this study, a novel method to disperse PtNPs on functionalized MWCNTs was reported. The approach includes functionalizing MWCNTs with thiolated polyaniline (TPANI) that was synthesized via microwave-assisted thiol–ene reaction and employing TPANI-MWCNTs as a support for PtNPs (Scheme 1). TPANI is 2,5-dimercapto-1,3,4-thiadiazole (DMcT)-functionalized PANI with many S and N groups. In contrast to conventional PANI-MWCNTs and pristine MWCNTs, TPANI functionalization of MWCNTs can introduce many S and N groups on the MWCNT surface without structural damage of MWCNTs. Transmission electron microscopy (TEM) showed that PtNPs exhibited better dispersion on TPANI-MWCNTs than that on conventional PANI-MWCNTs and pristine MWCNTs, and the obtained PtNPs/TPANI-MWCNTs/GCE showed higher electrocatalytic activity and enhanced stabilization toward methanol electro-oxidation than PtNPs/PANI-MWCNTs/GCE and PtNPs/MWCNTs/GCE.
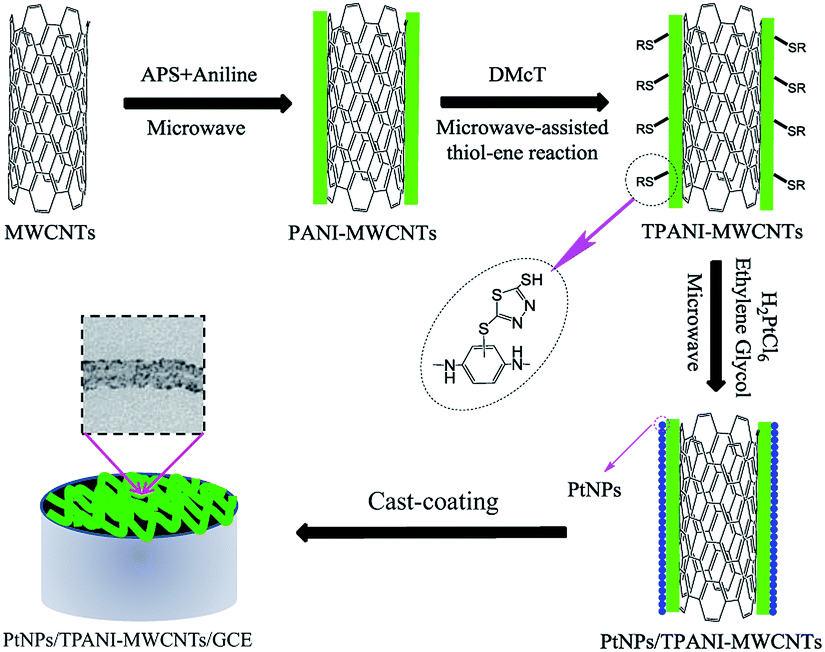 | ||
| Scheme 1 Schematic diagram for preparation of PtNPs/TPANI-MWCNTs/GCE. | ||
Experimental methods
Materials
Pristine MWCNTs with an average diameter of 20–40 nm were obtained from Shenzhen Nanotech Port Co., Ltd. (Shenzhen, China). Aniline was purchased from Shantou Xilong Chemical Engineering Factory (Guangdong, China), which was purified via double distillation and stored in a refrigerator prior to use. 2,5-Dimercapto-1,3,4-thiadiazole (DMcT), chloroplatinic acid, ammonium persulfate (APS), methanol and ethylene glycol were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Nafion (5 wt%) was purchased from Aldrich, and diluted 0.05 wt% Nafion solution was prepared with absolute ethanol. Ultra-pure N2 was used for the deaeration of the electrolytes. Other chemicals were of analytical grade and used as received.Apparatus
All electrochemical experiments were conducted on a CHI760E electrochemical workstation (Chenhua Instrument Company of Shanghai, China), and a classic three-electrode electrolytic cell was used. The working electrode was a glassy carbon electrode (GCE, 3 mm diameter), the reference electrode was a KCl-saturated calomel electrode (SCE), and the counter electrode was a platinum plate. All potentials reported here are cited versus SCE (vs. SCE). Transmission electron microscopy (TEM, JEM-3010, Jeol, Japan) was applied to characterize the morphology and particle size of PtNPs on MWCNTs. X-ray diffraction (XRD, Shimadzu XD-3A diffractometer) was applied to characterize the crystal phase structures of the nanocomposites. Thermogravimetric analysis (TGA, Mettler Toledo) was performed to characterize the thermal stability of the nanocomposites. Fourier transform infrared (FT-IR) spectra were obtained using a Nicolet Nexus 670 FT-IR spectrophotometer. The PtNPs loading masses for PtNPs/TPANI-MWCNTs, PtNPs/PANI-MWCNTs and PtNPs/MWCNTs were determined by inductively coupled plasma-atom emission spectroscopy (ICP-AES).Synthesis of TPANI-MWCNTs
Briefly, the desired amounts of MWCNTs (1 mg mL−1) and aniline (5 mmol L−1) were dispersed in HCl solution (1 mol L−1) under ultrasonication for 30 min. Then, APS (5 mmol L−1) was added to the solution under magnetic stirring and left for 5 min in a microwave reactor (600 W). The PANI-MWCNT nanohybrid was collected by centrifugation, washed with ethanol and deionized water thrice, and finally dried under vacuum at room temperature for further use. A desired amount of PANI-MWCNTs (1 mg mL−1) was dispersed in H2SO4–ethanol (v/v 1/1) solution (0.1 mol L−1) under ultrasonication for 30 min to form uniform suspension. DMcT (0.05 mg mL−1) was then added into the PANI-MWCNT suspension under magnetic stirring and left for 5 min in the microwave reactor. TPANI-MWCNTs were collected by centrifugation, washed and dried similar to that for PANI-MWCNTs.Synthesis of PtNPs/TPANI-MWCNTs
A desired amount of TPANI-MWCNTs (1 mg mL−1) was dispersed in 330 μL of chloroplatinic acid solution (7.72 mmol L−1) under ultrasonication for 30 min. The pH value of the solution was tuned to 8–9 using NaOH (0.1 mol L−1); 25 mL of ethylene glycol was added into the TPANI-MWCNT suspension under magnetic stirring and left for 10 min in a microwave reactor. Then, the obtained PtNPs/TPANI-MWCNT nanohybrid was collected by centrifugation, washed with acetone, ethanol and deionized water thrice, and finally dried under vacuum at room temperature. PtNPs/PANI-MWCNTs and PtNPs/MWCNTs were also prepared using the same strategy as contrastive samples.Preparation of modified electrode
Prior to surface modification, GCE was polished carefully with 1.0 and 0.05 mm alumina slurry sequentially and then washed ultrasonically in ethanol and water for 5 min. Then, the GCE was scanned between −0.2 and 1.0 V versus SCE in 0.5 mol L−1 H2SO4 at 50 mV s−1 for a sufficient number of cycles to obtain reproducible cyclic voltammograms. The catalyst ink was prepared by dispersing 5 mg of catalyst in 5 mL of ethanol by sonication. When a dark homogeneous dispersion was formed, 10 μL of the ink was dropped onto the GCE using a micro-syringe. After drying in air, the electrode was coated with 2 μL of 0.05 wt% Nafion ethanol solution to fix the catalyst powder.Results and discussion
Characterization of PtNPs/TPANI-MWCNTs
Surface-functionalization of MWCNTs with TPANI was characterized by FT-IR spectroscopy. FT-IR spectra of DMcT, PtNPs/PANI-MWCNTs, and PtNPs/DMcT-PANI-MWCNTs are shown in Fig. 1(A). The main peaks are explained as follows: For DMcT, the peak at 2478 cm−1 corresponds to its S–H stretching vibration; the peak at 1445 cm−1 is assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration, and that at 1048 cm−1 is derived from N–N stretching vibration. The peaks at 710 and 646 cm−1 are due to C–S–C asymmetric and symmetric stretching vibrations, respectively.12 For PtNPs/PANI-MWCNTs, the peaks at 1644 and 1409 cm−1 belong to the CC stretching of the quinoid ring and benzenoid ring, respectively; the peaks at 1123 cm−1 are due to C–N stretching of secondary aromatic amines.13 After interaction of PANI with DMcT (PtNPs/TPANI-MWCNTs), a new peak at 628 cm−1 (C–S–C symmetric stretching vibration in tethered DMcT) appears.12 Simultaneously, the peak at 2478 cm−1 (S–H stretching vibration of DMcT) does not appear. The FT-IR results indicate the occurrence of the PANI-DMcT interaction via ring-nucleophilic substitution of PANI with DMcT.
N stretching vibration, and that at 1048 cm−1 is derived from N–N stretching vibration. The peaks at 710 and 646 cm−1 are due to C–S–C asymmetric and symmetric stretching vibrations, respectively.12 For PtNPs/PANI-MWCNTs, the peaks at 1644 and 1409 cm−1 belong to the CC stretching of the quinoid ring and benzenoid ring, respectively; the peaks at 1123 cm−1 are due to C–N stretching of secondary aromatic amines.13 After interaction of PANI with DMcT (PtNPs/TPANI-MWCNTs), a new peak at 628 cm−1 (C–S–C symmetric stretching vibration in tethered DMcT) appears.12 Simultaneously, the peak at 2478 cm−1 (S–H stretching vibration of DMcT) does not appear. The FT-IR results indicate the occurrence of the PANI-DMcT interaction via ring-nucleophilic substitution of PANI with DMcT.
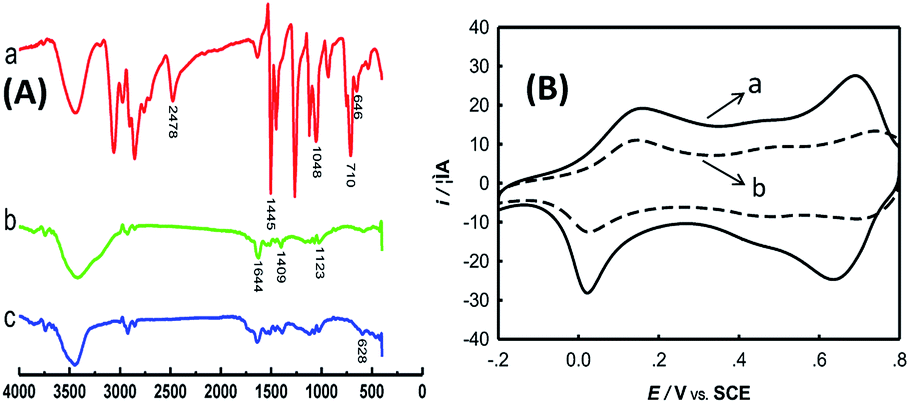 | ||
| Fig. 1 (A) FT-IR Spectra of DMcT (a), PtNPs/PANI-MWCNTs (b), PtNPs/TPANI-MWCNTs (c). (B) Cyclic voltammograms of PANI-MWCNTs/GCE (a) and TPANI-MWCNTs/GCE (b) in 0.5 M H2SO4 at a scan rate of 50 mV s−1. | ||
Surface-functionalization of MWCNTs with TPANI was also characterized by CV. As is seen in Fig. 1(B), the interaction between thiol and PANI decreased the redox peaks of PANI at 0.15/0.03 V and 0.70/0.65 V (Fig. 1(B)); this may reveal electronic and structural modification of the PANI backbones. Similar phenomena have also been reported for thio-, monoalkoxy-, and dialkoxy-substituted PANI.14 In addition, when 0.005 mg mL−1 DMcT was applied instead, a similar phenomenon was still found although the extent of current decline weakened. Thus, MWCNTs can be functionalized with thiolated polymers via microwave-assisted thiol–ene reaction with many surface functional groups (S and N groups) on MWCNTs.
Fig. 2 shows TEM images of PtNPs/TPANI-MWCNT, PtNPs/PANI-MWCNT and PtNPs/MWCNT nanohybrids. As shown in Fig. 2, TPANI-MWCNTs are successfully decorated with many well-dispersed PtNPs; although there is no clear aggregation of PtNPs, uniform dispersion of PtNPs is observed both on PANI-MWCNT and MWCNT surfaces. The average diameters of PtNPs loaded on MWCNTs, PANI-MWCNTs and TPANI-MWCNTs are 2.4 ± 0.4, 2.2 ± 0.5 and 2.8 ± 0.5 nm, respectively. The result is also proved by ICP-AES, which shows that PtNPs/TPANI-MWCNTs have the biggest Pt loading weight (Table 1). The reasons could be as follows: for pristine MWCNTs and PANI-MWCNTs, the functional groups on their surfaces not only have a limited number but also exhibit poor dispersion. When PtNPs are deposited on the surface, sufficient uniform functional groups to anchor Pt precursors and PtNPs are not present, leading to poor dispersion and extensive aggregation of PtNPs on the surface. However, for MWCNTs with TPANI modification, the TPANI layer on MWCNTs produces numerous functional groups (such as S and N) with uniform distribution. These functional groups are responsible for immobilization of Pt precursors on the MWCNT surface through the coordination effect. Therefore, high-density PtNPs with uniform distribution are observed on the surface of TPANI-MWCNTs.
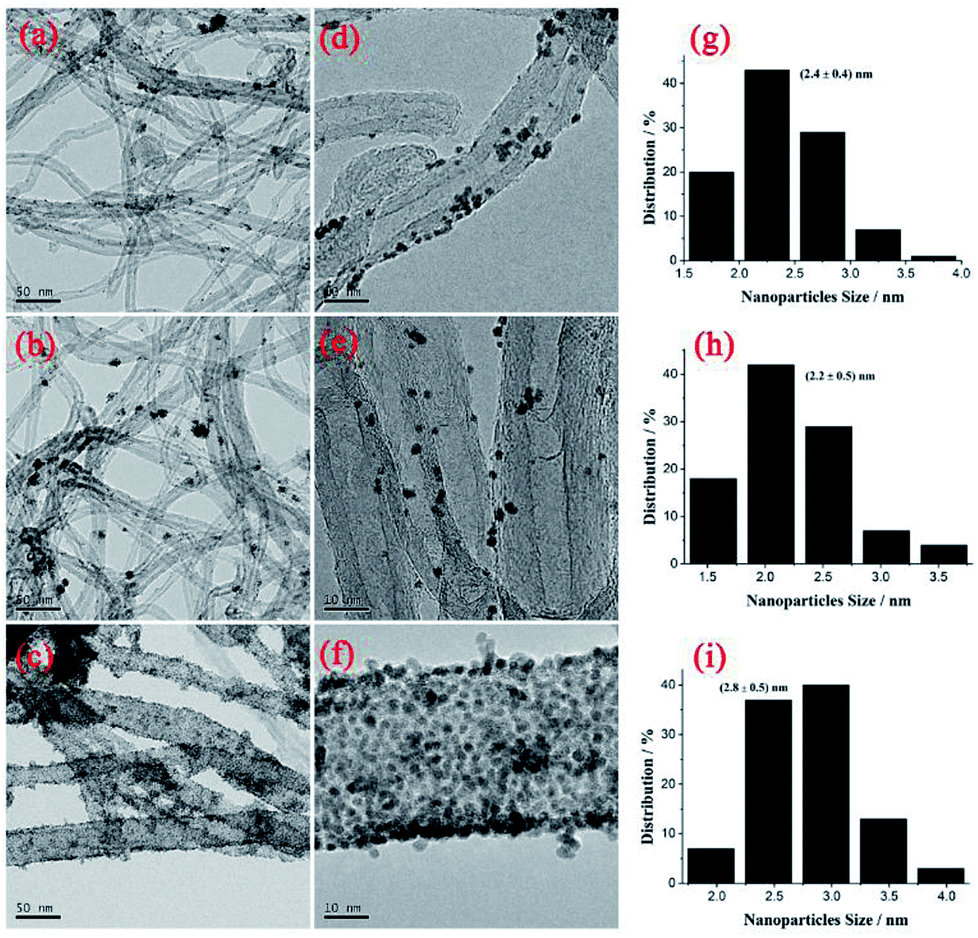 | ||
| Fig. 2 TEM (a–c), HRTEM (d–f) images and size distributions (g–i) of nanoparticles of nanohybrids. PtNPs/MWCNT (a, d and g), PtNPs/PANI-MWCNT (b, e and h), and PtNPs/TPANI-MWCNT (c, f and i) nanohybrids. | ||
| Nanohybrids | Pt (wt%) |
|---|---|
| PtNPs/TPANI-MWCNTs | 10.59 |
| PtNPs/PANI-MWCNTs | 3.16 |
| PtNPs/MWCNTs | 1.13 |
Fig. 3 shows the XRD patterns of PtNPs/TPANI-MWCNT (a), PtNPs/PANI-MWCNT (b) and PtNPs/MWCNT (c) nanohybrids. The diffraction peak at 2θ = 26.5° is assigned to the (002) crystal face of the MWCNT carrier; the peaks at 2θ = 40.3°, 46.5°, 68°, and 82° are ascribed to the face centered cubic (fcc) crystalline Pt, corresponding to the (111), (200), (220), and (311) faces.15 PtNPs/TPANI-MWCNTs exhibit higher intensity of diffraction peak for crystalline Pt compared to PtNPs/PANI-MWCNTs and PtNPs/MWCNTs; these XRD results indicate high-density PtNPs loaded on TPANI-MWCNTs.
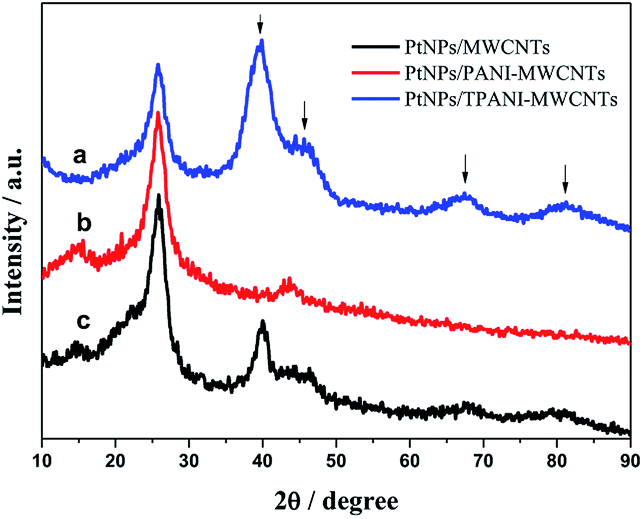 | ||
| Fig. 3 XRD patterns of the PtNPs/TPANI-MWCNT (a), PtNPs/PANI-MWCNT (b) and PtNPs/MWCNT (c) nanohybrids. | ||
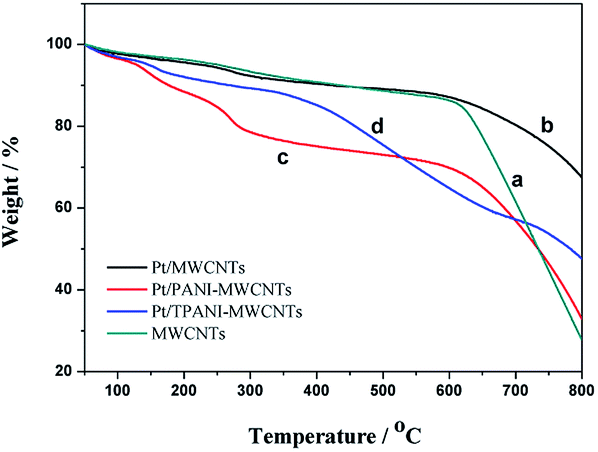
Fig. 4 shows the TGA of MWCNT (a), PtNPs/MWCNT (b), PtNPs/PANI-MWCNT (c) and PtNPs/TPANI-MWCNT (d) nanohybrids. The first weight loss observed between 40 and 250 °C can be due to the loss of water and unreacted substances.16 The second weight loss observed between 250 and 550 °C is due to the decomposition of PANI chains.16 The PANI nanocomposite decomposes completely at above 560 °C, whereas TPANI nanocomposite decomposes at 720 °C, which is due to increased thermal stabilization of incorporated functional thiol into the PANI matrix. PtNPs/TPANI-MWCNTs exhibit higher thermal stability compared to PtNPs/PANI-MWCNTs and PtNPs/MWCNTs.
 | ||
| Fig. 4 TGA of MWCNT (a), PtNPs/MWCNT (b), PtNPs/PANI-MWCNT (c) and PtNPs/TPANI-MWCNT (d) nanohybrids. | ||
The electrochemical performance of PtNPs/TPANI-MWCNTs/GCE toward electrocatalytic oxidation of methanol
The electrocatalytic activity of PtNPs/TPANI-MWCNTs as a catalyst toward methanol electro-oxidation was investigated by CV. Fig. 5 (A) shows CVs of the GCE modified with PtNPs/MWCNTs, PtNPs/PANI-MWCNTs, and PtNPs/TPANI-MWCNTs examined in N2-saturated 0.5 M H2SO4 aqueous solution. To the best of our knowledge, the electrochemical active surface area (EASA) values of PtNPs supported on TPANI-MWCNTs, PANI-MWCNTs, and MWCNTs can be obtained from the hydrogen adsorption–desorption charges. EASA's formula is given as follows:4a EASA = QH/(0.21 × [Pt]); here, QH (mC cm−2) represents the mean value between the amounts of charges exchanged during the electro-adsorption and desorption of H2 on Pt sites, [Pt] is the Pt loading (mg cm−2) on the electrode (Table 1), and 0.21 (mC cm−2) represents the charge required to oxidize a monolayer of H2 on bright Pt. The EASA value of PtNPs/TPANI-MWCNTs is about 42.53 m2 g−1 of Pt, which is remarkably higher than that of PtNPs/PANI-MWCNTs (EASA: 28.27 m2 g−1) and PtNPs/MWCNTs (EASA: 23.75 m2 g−1); this is most likely due to the better dispersion of PtNPs on TPANI-MWCNTs. This also demonstrates that PtNPs deposited on TPANI-MWCNTs are more electrochemically accessible, which is very important for the electrochemical oxidation of methanol.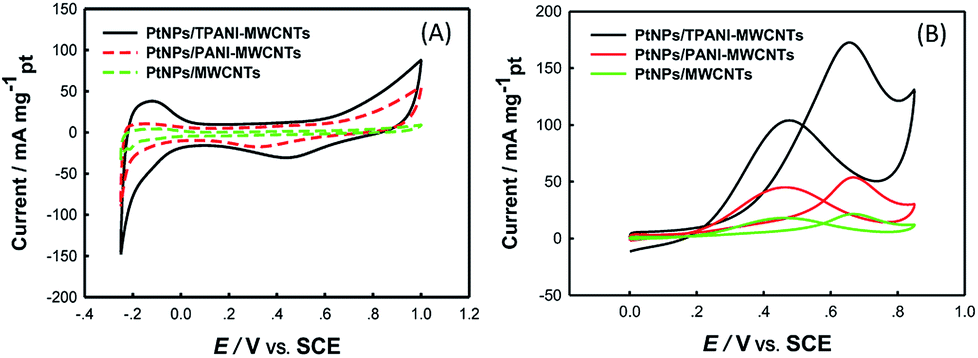 | ||
| Fig. 5 CVs of PtNPs/MWCNTs/GCE, PtNPs/PANI-MWCNTs/GCE, PtNPs/TPANI-MWCNTs/GCE in N2-saturated 0.5 M H2SO4 (A) and in N2-saturated 0.5 M H2SO4 + 1.0 M CH3OH (B) at a scan rate of 50 mV s−1. | ||
The electrochemical performances of PtNPs/TPANI-MWCNTs/GCE, PtNPs/PANI-MWCNTs/GCE and PtNPs/MWCNTs/GCE toward methanol electro-oxidation were investigated by CV in N2-saturated 0.5 M H2SO4 + 1 M CH3OH aqueous solution, as shown in Fig. 5(B). Compared with PtNPs/MWCNT and PtNPs/PANI-MWCNT catalysts, the PtNPs/TPANI-MWCNT catalyst exhibited significant enhancement of peak current and a clear negative shift of the peak potential of methanol oxidation. Notably, the forward peak current of methanol oxidation on the PtNPs/TPANI-MWCNT catalyst (173.04 mA mg−1) was higher than that on the PtNPs/MWCNT (53.44 mA mg−1) and PtNPs/PANI-MWCNT catalysts (20.77 mA mg−1); the result was comparable with or better than those of many other Pt-based catalysts reported previously toward methanol electro-oxidation in acidic media, as listed in Table 2. This result showed a noticeable feature that the PtNPs/TPANI-MWCNT catalyst exhibited higher electrocatalytic activity than PtNPs/PANI-MWCNTs and PtNPs/MWCNTs as expected by high EASA of the PtNPs/TPANI-MWCNT catalyst. This could be because of the superior features of PtNPs/TPANI-MWCNT nanohybrids over those of PtNPs/PANI-MWCNT and PtNPs/MWCNT samples: better dispersion and higher EASA of PtNPs. Numerous functional groups (such as S and N) were introduced onto TPANI-MWCNTs via microwave-assisted thiol–ene reaction, which can be used as anchors for fixing Pt precursor and PtNPs, resulting in uniform distribution of PtNPs on TPANI-MWCNTs.
| Catalyst fabrication | Investigated solution | EASA (m2 g−1) | Current density (mA mg−1) | Ref. |
|---|---|---|---|---|
| a CA: carbon aerogel; t-MWCNTs: thiolated multiwalled carbon nanotubes; N-CNT: nitrogen-doped carbon nanotubes; O-CNT: conventional acid-treated carbon nanotubes; HCSs: hollow carbonaceous composites; PDDA: poly(diallyldimethylammonium chloride); GNs: graphene nanosheets; GS: graphene sheets; SAANs: sheet-assembled alloy networks; PNCs: porous nanoclusters. | ||||
| Pt/CA | 0.5 M CH3OH + 0.5 M H2SO4 | 50.04 | — | 17 |
| Pt/t-MWCNTs | 2 M CH3OH + 0.5 M H2SO4 | — | 140.0 | 18 |
| PtRu/N-CNT | 1 M CH3OH + 0.5 M H2SO4 | 30.0 | 120.0 | 19 |
| PtRu/O-CNT | 1 M CH3OH + 0.5 M H2SO4 | 28.8 | 69.0 | 19 |
| Pt/HCSs | 0.5 M CH3OH + 0.5 M H2SO4 | 46.22 | 46.5 | 20 |
| Pt/PDDA-GNs | 1 M CH3OH + 0.5 M H2SO4 | — | 170.0 | 21 |
| PtRu/GS-MWCNTs | 1 M CH3OH + 0.5 M H2SO4 | 118.69 | 136.7 | 22 |
| PtRu/CeO2/MWCNTs | 1 M CH3OH + 0.5 M H2SO4 | 134.8 | 140.0 | 23 |
| PtPd SAANs | 0.5 M CH3OH + 0.5 M H2SO4 | — | 376.0 | 24 |
| PtAu PNCs | 0.5 M CH3OH + 0.5 M H2SO4 | — | 85.2 | 25 |
| PtNPs/TPANI-MWCNTs | 1 M CH3OH + 0.5 M H2SO4 | 42.53 | ||
| 173.0 | This work | |||
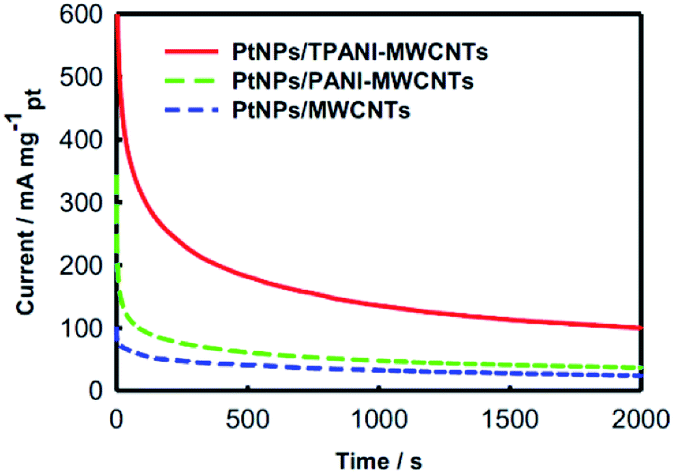
The chronoamperometric technique, which is a typical method for the evaluation of electrocatalysts in fuel cells,6 was used to further study the electrochemical performance of PtNPs supported on MWCNTs. Fig. 6 shows the chronoamperometric curves of three electrodes modified with PtNPs/TPANI-MWCNTs, PtNPs/PANI-MWCNTs and PtNPs/MWCNTs toward methanol electro-oxidation at a constant potential of 0.50 V in N2-saturated 0.5 M H2SO4 + 1.0 M CH3OH aqueous solutions at 2000 s. It is easy to find out that PtNPs/TPANI-MWCNTs possess optimal performance for electrocatalytic methanol oxidation among the three modified electrodes. This may also be caused by the superior features of PtNPs/TPANI-MWCNT nanohybrids: better dispersion and higher EASA of PtNPs. All these observations are due to sufficient functional groups (such as S and N) on the TPANI-MWCNT surface, which are used as anchors for fixing Pt precursor and PtNPs, leading to uniform distribution of PtNPs on the TPANI-MWCNT surface.
 | ||
| Fig. 6 Transient currents of PtNPs/MWCNT, PtNPs/PANI-MWCNT and PtNPs/TPAN-MWCNT catalysts for methanol electro-oxidation at 0.50 V in N2-saturated 0.5 M H2SO4 + 1.0 M CH3OH aqueous solution. | ||
The long-term cycle stability of PtNPs/TPANI-MWCNTs/GCE toward methanol electro-oxidation
The long-term cycle stabilities of PtNPs/TPANI-MWCNTs/GCE, PtNPs/PANI-MWCNTs/GCE and PtNPs/MWCNTs/GCE were studied in 0.5 M H2SO4 + 1.0 M CH3OH aqueous solution by CV (Fig. 7). For PtNPs/TPANI-MWCNTs/GCE, the peak current at 1000th cycle (IpI) was about 86% of that examined at the first cycle (IpI(1)). The decline of IpI/IpI(1) on PtNPs/TPANI-MWCNTs/GCE was only 14% after 1000 cycles, whereas for PtNPs/PANI-MWCNTs/GCE and PtNPs/MWCNTs/GCE, large decline (32% and 45%) was observed. This revealed that the long-term stability of PtNPs/TPANI-MWCNTs/GCE is much better than that of PtNPs/PANI-MWCNTs/GCE and PtNPs/MWCNTs/GCE. The above results can be due to strong interaction between PtNPs and functional thiol on TPANI-MWCNTs.5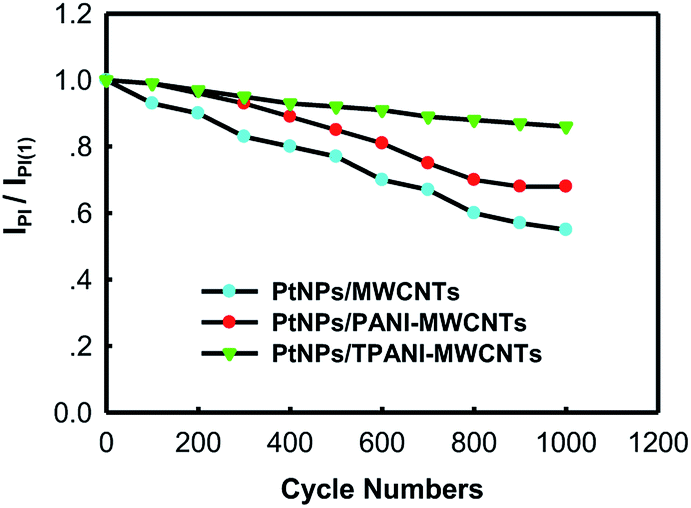 | ||
| Fig. 7 Long-term cycle stabilities of PtNPs/MWCNTs/GCE, PtNPs/PANI-MWCNTs/GCE and PtNPs/TPANI-MWCNTs/GCE in N2-saturated 0.5 M H2SO4 + 1.0 M CH3OH aqueous solution. IpI, the forward peak current; IpI(1), the forward peak current at the first cycle. | ||
Conclusions
In summary, MWCNTs were successfully functionalized with a thiolated polymer via microwave-assisted thiol–ene reaction and the as-obtained TPANI-MWCNTs were used as a support for PtNPs dispersion. Compared to PtNPs deposited on PANI-MWCNTs and MWCNTs, PtNPs deposited on TPANI-MWCNTs showed better dispersion, which endowed PtNPs/TPANI-MWCNTs/GCE with better electrocatalytic activity and stability toward methanol electro-oxidation. The as-prepared PtNPs/TPANI-MWCNTs can be promising catalysts for fuel cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21305039), the Foundation of Hunan Province (14JJ3097), the Foundation of Hunan Agricultural University for 1515 Talents, and the Foundation of Hunan Provincial Key Laboratory of Crop Germplasm Innovation and Resource Utilization.References
- (a) G. Zhang, Z. Yang, W. Zhang and Y. Wang, J. Mater. Chem. A, 2016, 4, 3316 RSC; (b) L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J. Y. Ma, D. Su and X. Huang, Science, 2016, 354, 1410 CrossRef CAS PubMed; (c) P. Strasser, Science, 2015, 349, 379 CrossRef PubMed.
- (a) J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37 CrossRef CAS PubMed; (b) H. A. Gasteiger and N. M. Marković, Science, 2009, 324, 48 CrossRef CAS PubMed.
- B. Wu, Y. Kuang, X. Zhang, J. Chen, B. Wu, Y. Kuang, X. Zhang and J. Chen, Nano Today, 2011, 6, 75 CrossRef CAS.
- (a) B. Wu, D. Hu, Y. Kuang, Y. Yu, X. Zhang and J. Chen, Chem. Commun., 2011, 47, 5253 RSC; (b) D. Wang, S. Lu and S. P. Jiang, Chem. Commun., 2010, 46, 2058 RSC; (c) S. Guo, S. Dong and E. Wang, Adv. Mater., 2010, 22, 1269 CrossRef CAS PubMed.
- S. Chen, Z. Wei, L. Guo, W. Ding, L. Dong, P. Shen, X. Qi and L. Li, Chem. Commun., 2011, 47, 10984 RSC.
- B. Wu, D. Hu, Y. Kuang, B. Liu, X. Zhang and J. Chen, Angew. Chem., Int. Ed., 2009, 48, 4751 CrossRef CAS PubMed.
- (a) M. M. Oliveira and A. J. G. Zarbin, J. Phys. Chem. C, 2008, 112, 18783 CrossRef CAS; (b) V. Selvaraj, M. Alagar and K. S. Kumar, Appl. Catal., B, 2007, 75, 129 CrossRef CAS; (c) P. Santhosh, A. Gopalan and K. P. Lee, J. Catal., 2006, 238, 177 CrossRef CAS.
- H. Xu, L. Zeng, S. Xing, G. Shi, Y. Xian and L. Jin, Electrochem. Commun., 2008, 10, 1839 CrossRef CAS.
- Y. Li, F. P. Hu, X. Wang and P. K. Shen, Electrochem. Commun., 2008, 10, 1101 CrossRef CAS.
- H. Matsumori, S. Takenaka, H. Matsune and M. Kishida, Appl. Catal., A, 2010, 373, 176 CrossRef CAS.
- (a) Z. Su, Y. Liu, Q. Xie, L. Chen, Y. Zhang, Y. Meng, Y. Li, Y. Fu, M. Ma and S. Yao, Biosens. Bioelectron., 2012, 36, 154 CrossRef CAS PubMed; (b) L. Chen, Z. Su, X. He, Y. Liu, C. Qin and Y. Zhou, Electrochem. Commun., 2012, 15, 34 CrossRef CAS; (c) C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355 RSC.
- J. M. Pope, T. Sato, E. Shoji, D. A. Buttry, T. Sotomura and N. Oyama, J. Power Sources, 1997, 68, 739 CrossRef CAS.
- (a) J. Ding, X. Li, X. Wang, J. Zhang, D. Yu and B. Qiu, Nanoscale Res. Lett., 2015, 10, 1 CrossRef CAS PubMed; (b) M. R. Gizdavic-Nikolaidis, M. M. Jevremovic, M. C. Allison, D. R. Stanisavljev, G. A. Bowmaker and Z. D. Zujovic, eXPRESS Polym. Lett., 2014, 8, 745 CrossRef CAS.
- (a) B. Bergman and T. W. Hanks, Macromolecules, 2000, 33, 8035 CrossRef CAS; (b) C. C. Han, W. D. Hseih, J. Y. Yeh and S. P. Hong, Chem. Mater., 1999, 11, 480 CrossRef CAS.
- Y. Liu, N. Chen, F. Wang, Y. Cai and H. Zhu, New J. Chem., 2017, 41, 6585 RSC.
- P. Boomi, H. G. Prabu and J. Mathiyarasu, Eur. J. Med. Chem., 2014, 72, 18 CrossRef CAS PubMed.
- Z. Guo, H. Zhu, X. Zhang, F. Wang, Y. Guo and Y. Wei, Bull. Mater. Sci., 2011, 34, 577 CrossRef CAS.
- Y. T. Kim and T. Mitani, J. Catal., 2006, 238, 394 CrossRef CAS.
- R. Chetty, S. Kundu, W. Xia, M. Bron, W. Schuhmann, V. Chirila, W. Brandl, T. Reinecke and M. Muhler, Electrochim. Acta, 2009, 54, 4208 CrossRef CAS.
- Z. Wen, Q. Wang, Q. Zhang and J. Li, Electrochem. Commun., 2007, 9, 1867 CrossRef CAS.
- B. Luo, X. Yan, S. Xu and Q. Xue, Electrochim. Acta, 2012, 59, 429 CrossRef CAS.
- Y. S. Wang, S. Y. Yang, S. M. Li, H. W. Tien, S. T. Hsiao, W. H. Liao, C. H. Liu, K. H. Chang, C. C. M. Ma and C. C. Hu, Electrochim. Acta, 2013, 87, 261 CrossRef CAS.
- Z. Sun, X. Wang, Z. Liu, H. Zhang, P. Yu and L. Mao, Langmuir, 2010, 26, 12383 CrossRef CAS PubMed.
- Y. Shi, L. Mei, A. Wang, T. Yuan, S. Chen and J. Feng, J. Colloid Interface Sci., 2017, 504, 363 CrossRef CAS PubMed.
- X. Xie, J. Lv, L. Liu, A. Wang, J. Feng and Q. Xu, Int. J. Hydrogen Energy, 2017, 42, 2104 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |