DOI:
10.1039/C8RA06240D
(Paper)
RSC Adv., 2018,
8, 29513-29525
Experimental and theoretical interpretation of the magnetic behavior of two Dy(III) single-ion magnets constructed through β-diketonate ligands with different substituent groups (–Cl/–OCH3)†
Received
23rd July 2018
, Accepted 6th August 2018
First published on 20th August 2018
Abstract
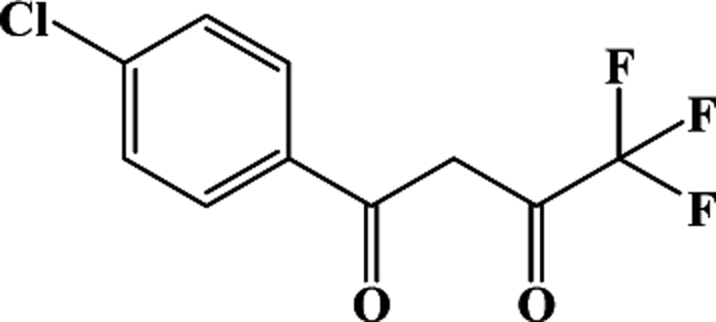
Two Dy(III) single-ion magnets, formulated as [Dy(Phen)(Cl-tcpb)3] (Cl-1) and [Dy(Phen)(CH3O-tmpd)3] (CH3O-2) were obtained through β-diketonate ligands (Cl-tcpb = 1-(4-chlorophenyl)-4,4,4-trifluoro-1,3-butanedione and CH3O-tmpd = 4,4,4-trifluoro-1-(4-methoxyphenyl)-1,3-butanedione) with different substituent groups (–Cl/–OCH3) and auxiliary ligand, 1,10-phenanthroline (Phen). The Dy(III) ions in Cl-1 and CH3O-2 are eight-coordinate, with an approximately square antiprismatic (SAP, D4d) and trigonal dodecahedron (D2d) N2O6 coordination environment, respectively, in the first coordination sphere. Under zero direct-current (dc) field, magnetic investigations demonstrate that both Cl-1 and CH3O-2 display dynamic magnetic relaxation of single-molecule magnet (SMM) behavior with different effective barriers (Ueff) of 105.4 cm−1 (151.1 K) for Cl-1 and 132.5 cm−1 (190.7 K) for CH3O-2, respectively. As noted, compound CH3O-2 possesses a higher effective barrier than Cl-1. From ab initio calculations, the energies of the first excited state (KD1) are indeed close to the experimental Ueff as 126.7 cm−1 vs. 105.4 cm−1 for Cl-1 and 152.8 cm−1 vs. 132.5 cm−1 for CH3O-2. The order of the calculated energies of KD1 is same as that of the experimental Ueff. The superior SIM properties of CH3O-2 could have originated from the larger axial electrostatic potential (ESP(ax)) felt by the central Dy(III) ion when compared with Cl-1. The larger ESP(ax) of CH3O-2 arises from synergic effects of the more negative charge and shorter Dy–O distances of the axial O atoms of the first sphere. These charges and distances could be influenced by functional groups outside the first sphere, e.g., –Cl and –OCH3.
1. Introduction
Since the first single-molecule magnet (SMM), Mn12Ac, was discovered in the 1990s,1 the search for improved properties, in particular the enhanced relaxation barrier (Ueff) and blocking temperature (TB), has advanced the development of new compounds with magnetic properties customized by the coordination environment.2 The relevant explorations focus on mononuclear lanthanide compounds, especially single Dy-center systems, owing to the intrinsic strong spin–orbit coupling and large magnetic anisotropy of lanthanide ions.3 Furthermore, the relatively simple structure is convenient for chemists to improve the understanding of the magneto-structural correlation by combining with ab initio calculations.4 The number of mononuclear lanthanide SMMs or single-ion magnets (SIMs) derived from the above approach, following significantly slow relaxations of the magnetization, has grown significantly. An equatorially coordinated triangular geometry for C3,5 square antiprismatic systems for D4d,6 sandwich-type systems for D8d or D8h,7 the pentagonal bipyramidal local geometry for D5h,4a,4b,8 linear 2-coordinated systems for D∞h9 and some systems possessing high charge distribution symmetry,10 are given different priorities in lanthanide SIM construction. The research reveals that the single-ion magnetic anisotropy of lanthanide ions is extremely sensitive to the subtle changes in the ligand and the local geometrical symmetry. The electrostatic potential distribution around the magnetic center can be regulated by the amount of solvent,11 the anion ligands,12 the lattice/coordinated solvents,13 the pH values of the solution systems,14 or the counter ions,15 on the basis of weakening or strengthening the electron density.16 Interestingly, other functional groups from ligands outside the first sphere could also effectively influence the electron density of the first-sphere atoms and thus affect the SIM properties, which should also be considered in the rational design of promising molecular magnets.4d
Finding a feasible strategy to regulate and control the electrostatic environment around the metal centers and the single-ion magnetic anisotropy in SIMs would provide an active direction for understanding the magneto-structural correlation in depth and developing magnetic materials with high Ueff and blocking temperature (TB). Herein, two β-diketonate ligands with different functional groups were chosen for the following reasons: the classical β-diketonate ligands with different functional groups usually coordinate with metal ions in bidentate chelating modes in SIMs, which is beneficial for discussing the influence of the electron density.8 Fortunately, with the introduction of a capping ligand, two mononuclear compounds, [Dy(Phen)(Cl-tcpb)3] (Phen = 1,10-phenanthroline, Cl-tcpb = 1-(4-chlorophenyl)-4,4,4-trifluoro-1,3-butanedione, Cl-1) and [Dy(Phen)(CH3O-tmpd)3] (CH3O-tmpd = 4,4,4-trifluoro-1-(4-methoxyphenyl)-1,3-butanedione, CH3O-2), were obtained through solution reaction. The Dy(III) ion in compound Cl-1 has an approximately square antiprismatic (SAP) N2O6 coordination environment, while CH3O-2 has a trigonal dodecahedron (D2d) configuration. Magnetic characterization indicates that the subtle changes in the different substituent groups from the β-diketonate ligand results in great differences in the coordination environment and dramatically alters the relaxation behaviors of Cl-1 and CH3O-2. To further understand the different magnetic behaviors of Cl-1 and CH3O-2, ab initio calculations were also performed to explore the magnetic anisotropies of the central Dy(III) ions. A preliminary analysis on the electrostatic potential felt by the Dy(III) ion is utilized to identify the effect of ligands on its magnetic anisotropy. Expectedly, other functional groups outside the first sphere could also effectively influence the electron density of the first sphere atoms and thus affect the SIM properties.
2. Experimental
2.1 Materials and instruction
All the materials and reagents were obtained commercially without further purification. The FT-IR spectra were recorded in the range of 400–4000 cm−1 using KBr pellets on an EQUINOX55 FT/IR spectrophotometer. Elemental analysis (C, H, N) was implemented on a Perkin-Elmer 2400 CHN elemental analyzer. The phase purity of the bulk or polycrystalline samples were confirmed by powder X-ray diffraction (PXRD) measurements executed on a Rigaku RU200 diffractometer at 60 kV, 300 mA, and Cu Kα radiation (λ = 1.5406 Å), with a scan speed of 5° min−1 and a step size of 0.02° in 2θ. Diffuse reflectance spectra were obtained by a U-41000 spectrophotometer applying BaSO4 powder as a 100% reflectance reference. Magnetic measurements were performed in the temperature range of 1.8–300 K with an applied field of 1000 Oe, using a Quantum Design MPMS-XL-7 SQUID magnetometer on polycrystalline samples. The diamagnetic corrections for the compounds were estimated using Pascal's constants. Alternating current (ac) susceptibility experiments were performed using an oscillating ac field of 2.0 Oe at ac frequencies ranging from 1 to 1000 Hz. The magnetization was measured in the field range 0–7 T.
2.2 Synthesis and characterization of the lanthanide compounds
All chemicals were obtained from commercial sources and were used as received without further purification.
Synthesis of [Dy(Phen)(Cl-tcpb)3] (Cl-1). A methanol solution (10 mL) of Dy(NO3)3·6H2O (0.1 mmol, 0.0456 g) and Phen (0.1 mmol, 0.0200 g) was added to a solution of Cl-tcpb (0.3 mmol, 0.0753 g) and KOH (0.3 mmol, 0.0168 g) in 10 mL of dichloromethane under stirring. The resultant solution was filtered and allowed to stand undisturbed at room temperature for three weeks. Block crystals of Cl-1 were obtained in 37% yield (based on the Dy(III) salt). Anal. Calcd for C45H32DyF9N2O6: C, 52.42; H, 3.11; N, 2.72. Found: C, 52.77; H, 3.32; N, 2.53. IR (KBr): 3077 (w), 1645 (s), 1623 (m), 1553 (s), 1564 (m), 1453 (w), 1401 (m), 1334 (w), 1299 (s), 1168 (m), 1111 (s), 1043 (w), 1009 (m), 923 (m), 833 (m), 801 (w), 775 (w), 722 (w), 677 (w), 611 (m), 575 (w), 487 (m), 439 cm−1 (w).
Synthesis of [Dy(Phen)(CH3O-tmpd)3] (CH3O-2). A similar synthetic procedure to that for Cl-1 was used to synthesize CH3O-2, except that Cl-tcpb (0.3 mmol, 0.0753 g) was replaced by CH3O-tmpd (0.3 mmol, 0.0738 g). Finally, pink block crystals were obtained after three weeks in 51% yield (based on the Dy(III) salt). Anal. Calcd for C47H36DyF9N2O7: C, 52.50; H, 3.35; N, 2.61. Found: C, 52.38; H, 3.52; N, 2.82. IR (KBr): 3092 (w), 1621 (s), 1589 (s), 1567 (s), 1533 (s), 1462 (m), 1401 (m), 1343 (w), 1302 (m), 1166 (m), 1145 (m), 1054 (w), 1022 (m), 921 (w), 846 (m), 766 (m), 755 (w), 732 (w), 681 (m), 587 (m), 572 (m), 481 (w), 443 (w), 409 (m) cm−1.
2.3 X-ray single-crystal diffraction analysis
The single crystal X-ray experiment was performed on an Agilent Xcalibur Eos Gemini diffractometer using graphite-monochromatized Cu Kα radiation (λ = 1.5418 Å). The data integration and reduction were processed with the CrysAlisPro software. Absorption correction based on multi-scans was performed using the SADABS program.17 The structures were solved by the direct method and refined by means of full-matrix least-squares procedures on F2 with the SHELXL program.18 All non-hydrogen atoms were refined anisotropically. Other details of crystal data, data collection parameters, and refinement statistics are given in Table S1.† The selected bond lengths and angles are listed in Table S2.†
2.4 Theoretical methods and computational details
Multiconfigurational ab initio calculations, including spin–orbit coupling (SOC), were performed on the experimental structures of 1 and 2 to explore their magnetic anisotropy. This type of calculation includes two steps:19 (1) a set of spin eigenstates, were obtained by the state-averaged (SA) CASSCF method;20 (2) the low-lying SOC states, i.e., Kramers doublets (KD) herein, were obtained by state interaction, which is the diagonalization of the SOC matrix in the space spanned by the spin eigenstates from the first step. In the CASSCF step, the active space consisted of 9 electrons in 7 orbitals and all the spin eigenstates of 21 sextets were included. Due to the hardware limitations, other highly excited quartets and doublets were not considered. The state interaction step was performed by the RASSI-SO module21 with the SOC integrals from the AMFI method.22 The ANO-RCC basis sets,23–25 including VTZP for Dy, VDZ for C and H as well as VDZP for other atoms, were used. All the calculations were carried out with the MOLCAS@UU, a version of MOLCAS 8.0 (ref. 26 and 27) which is freely distributed for academic users. The SINGLE_ANISO module,28,29 developed by Chibotaru et al., was used to obtain the g-tensors, transition magnetic moments and other parameters characterizing the magnetic anisotropy.
3. Results and discussion
3.1 Crystal structures
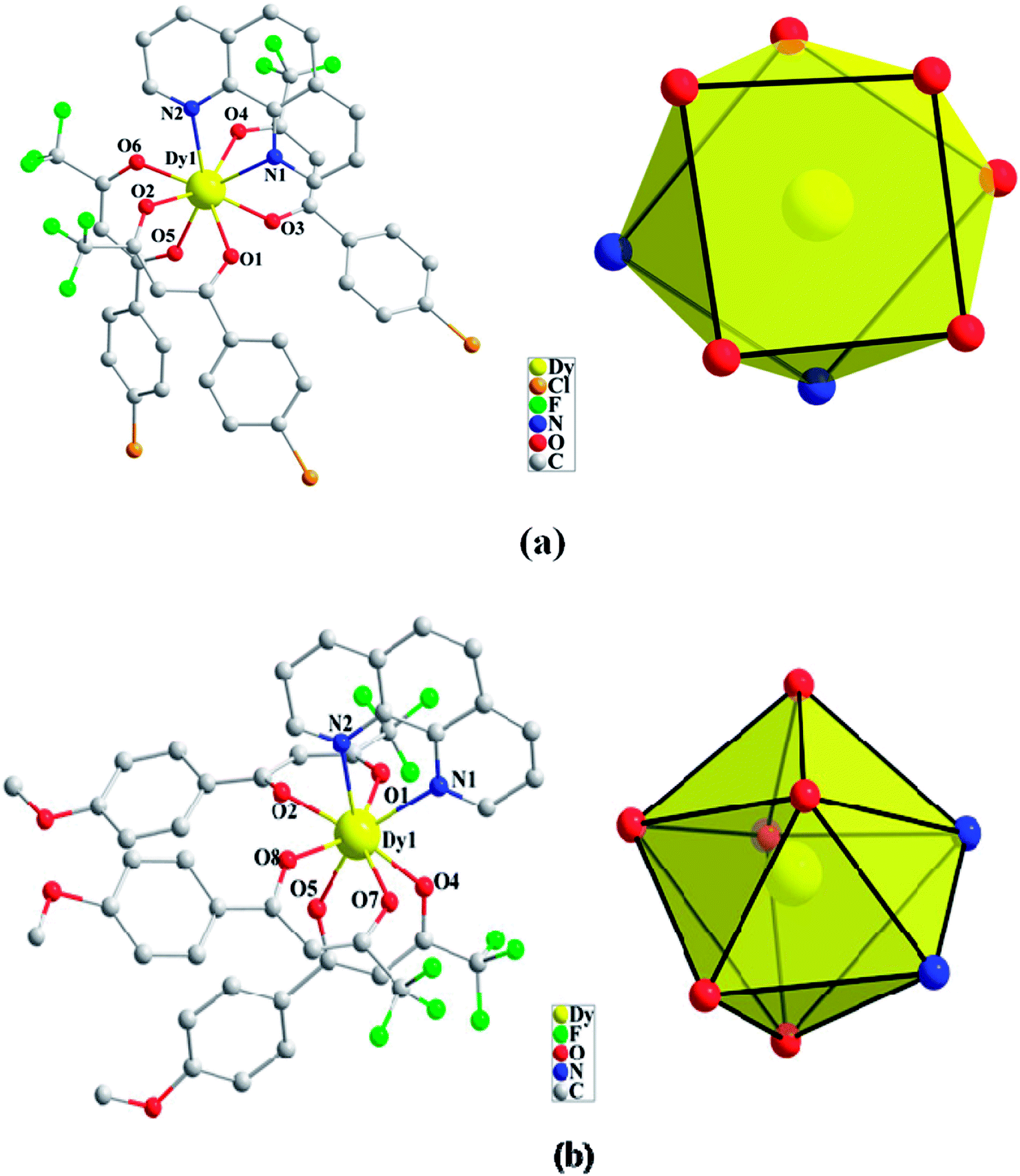
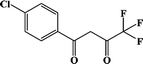
Compounds Cl-1 and CH3O-2 are crystallized in the triclinic space group P-1. Cl-1 and CH3O-2 have N2O6 coordination environments. Each Dy(III) ion is surrounded by three negative-ion ligands and a neutral capping ligand (Phen), as shown in Fig. 1. The Dy–O distances are from 2.319 (7) to 2.369 (6) Å in Cl-1 and from 2.305 (7) to 2.350 (5) Å in CH3O-2. Additionally, the Dy–N distances are 2.544 (8) Å and 2.586 (8) Å in Cl-1 as well as 2.554 (6) Å and 2.571 (6) Å in CH3O-2. By using the SHAPE 2.1 software, the configurations of Dy(III) ions in Cl-1 and CH3O-2 were calculated (Table S2†), indicating that the compounds Cl-1 and CH3O-2 belong to an approximately square-antiprismatic (SAP, D4d) and trigonal dodecahedron (D2d) configuration, respectively.30 The shortest interdinuclear Dy(III)⋯Dy(III) distances are 7.911 (6) Å in Cl-1 and 10.742 (18) Å in CH3O-2, respectively.
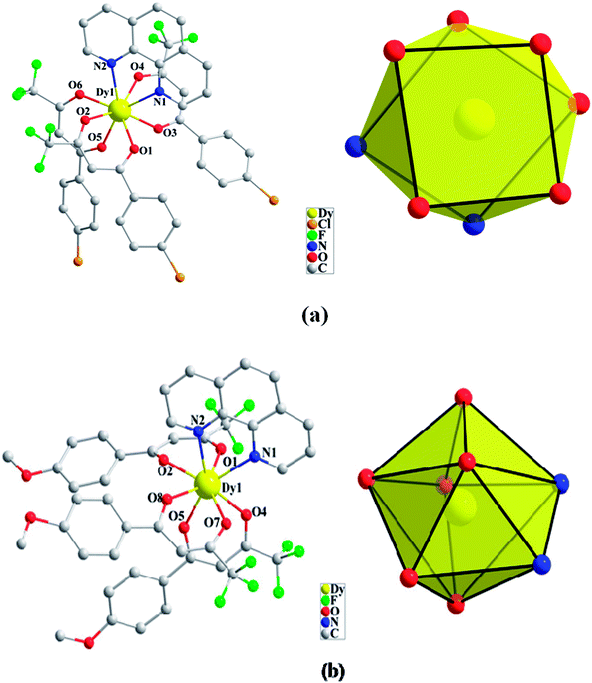 |
| | Fig. 1 Coordination environments of Cl-1 (a) and CH3O-2 (b) and the local coordination geometries of the Dy(III) ions of Cl-1 (a) and CH3O-2 (b); hydrogen atoms were omitted for clarity. | |
For CH3O-2, the neutral molecules are assisted by weak ð–ð stacking between the parallel interlayer (Fig. 2) to generate a 1D supramolecular chain, and the centroid distance is 3.597 (5) Å, from a slipped stacking and leading to the Dy(III)⋯Dy(III) distance of 10.742 (18) Å. However, there is no obvious ð–ð stacking or hydrogen bonding interaction in compound Cl-1.
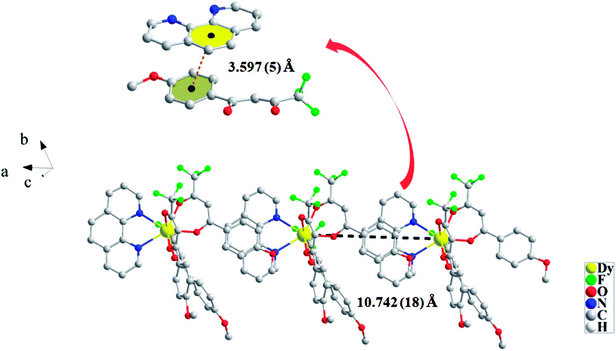 |
| | Fig. 2 Packing arrangement between two neutral molecules to form a 1D supramolecular chain in CH3O-2. | |
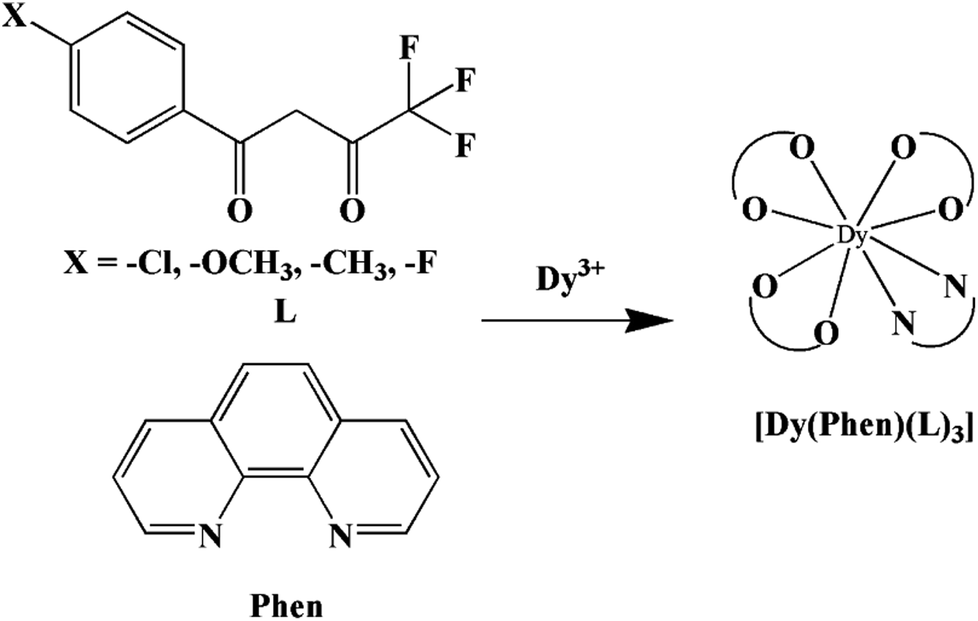
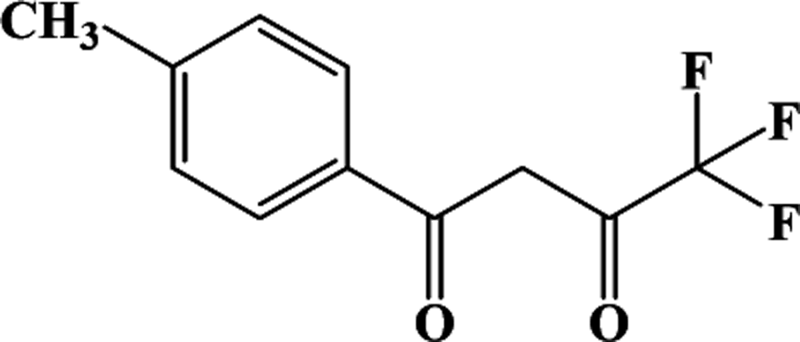
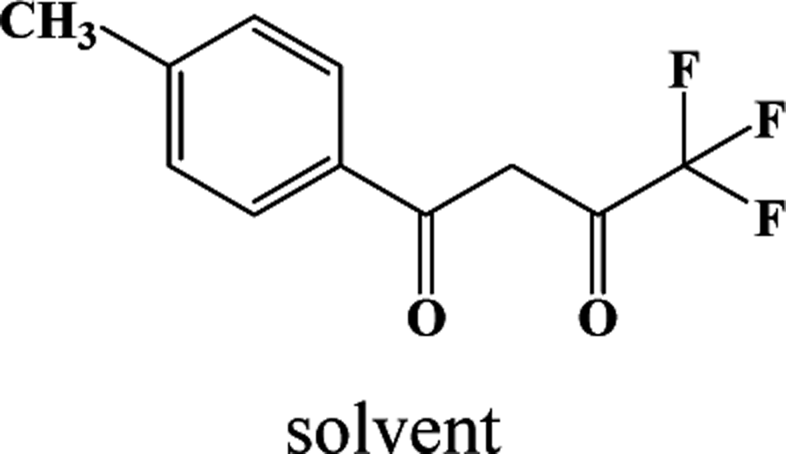
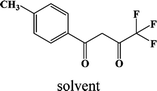
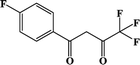
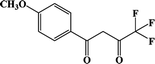
In our previous works, the β-diketonate ligands with different substituent groups (–F/–CH3) were employed to obtain a series of mononuclear Dy(III) compounds (Scheme 1). For CH3-4 (solvent) and CH3-3, the latter has a trigonal dodecahedron (D2d) configuration of Dy(III) ions, while CH3-4 (solvent) shows an approximately square antiprismatic (SAP, D4d) N2O6 coordination environment of Dy(III) ions. The uncoordinated 1,4-dioxane molecules exist in CH3-4 (solvent). Interestingly, the compounds above have weak interactions between the neutral molecules. In CH3-3, the neutral molecules are assisted by weak π⋯π stacking between the parallel interlayer, and the centroid distance is 3.767 (6) Å, belonging to a slipped stacking and leading to the Dy(III)⋯Dy(III) distance of 9.193 (5) Å. In CH3-4 (solvent), the neutral molecules are connected by weak C(171)–H(117)⋯O(7) interactions, leading to the Dy(III)⋯Dy(III) distance of 18.853 (2) Å. F-5 belongs to an approximately SAP configuration. For Cl-1, CH3-4 (solvent) and F-5, the CH3-4 (solvent) is more inclined toward the SAP configuration, calculated by utilizing the SHAPE 2.1 software. Cl-1 has the closest distance between the neutral molecules. In CH3O-2 and CH3-3, the former has the smaller deviation relative to a trigonal dodecahedron (D2d) configuration. The longer distance between the neutral molecules can be observed in CH3O-2. It is a remarkable fact that the maximum average Dy–N bond length is 2.563 Å (2) in CH3O-2. F-5 and CH3O-2 show similar average Dy–O bond lengths, which are smaller than Cl-1, CH3-3 and CH3-4 (solvent). The different types of weak interactions between the neutral molecules, configurations and bond lengths would result in different magnetic behaviors.
 |
| | Scheme 1 The synthetic process for compounds with molecular formula Dy(Phen)(L)3. | |
3.2 Magnetic properties
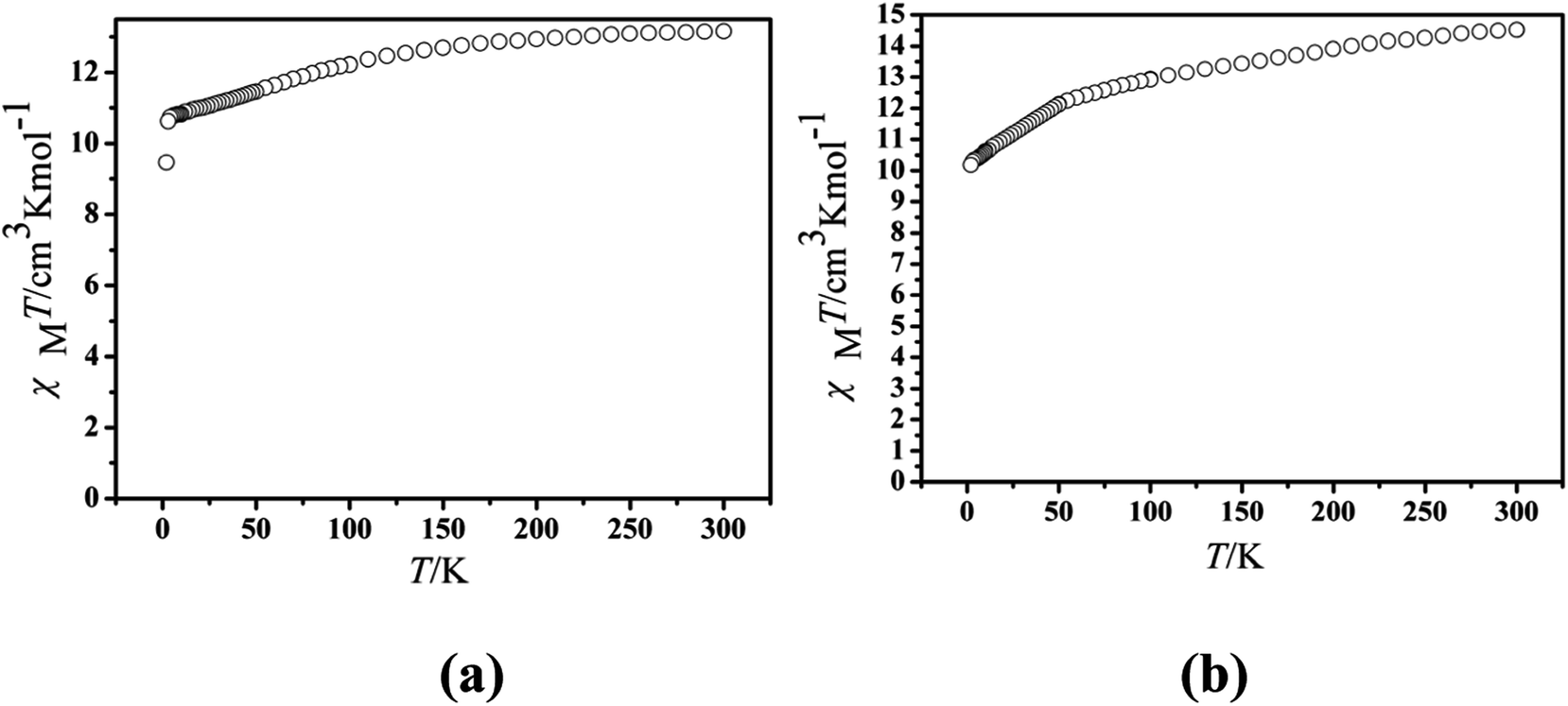
The magnetic experiments of Cl-1 and CH3O-2 were performed on polycrystalline samples. PXRD results of Cl-1 and CH3O-2 support the pure state of the bulk materials (Fig. S1†). The values of χMT of Cl-1 and CH3O-2 are 13.16 cm3 mol−1 K and 14.51 cm3 mol−1 K at room temperature, respectively, which are close to the free-ion value of 14.17 cm3 mol−1 K for a single Dy(III) ion (6H15/2, S = 5/2, L = 5, J = 15/2, g = 4/3) (Fig. 3).13 When cooled, the χMT curves for compound Cl-1 decreased slowly in the range from 300 to 100 K. Subsequently, the χMT products decreased sharply below 100 K to the minimum of 9.46 cm3 mol−1 K for Cl-1 and 11.75 cm3 mol−1 K for CH3O-2 at 1.8 K. In CH3O-2, on lowering the temperature, the χMT product decreased gradually and more rapidly below 50 K. These behaviors could be ascribed to crystal field splitting, particularly the progressive quenching of excited Dy(III) Stark sublevels and/or weak intermolecular dipole–dipole effects.32
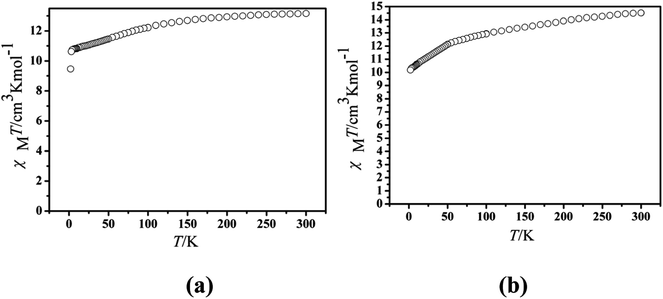 |
| | Fig. 3 Temperature dependence of the χMT product at 1000 Oe for compounds Cl-1 (a) and CH3O-2 (b). | |
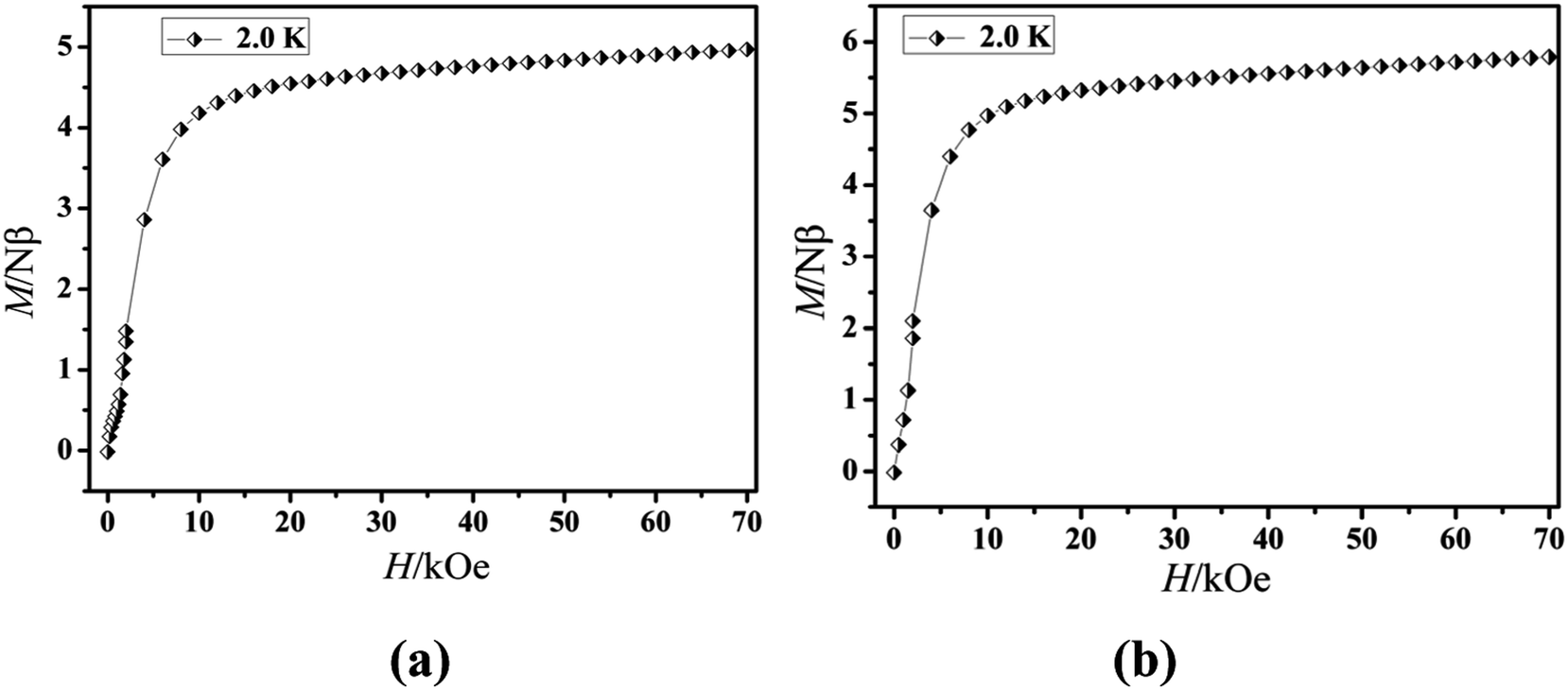
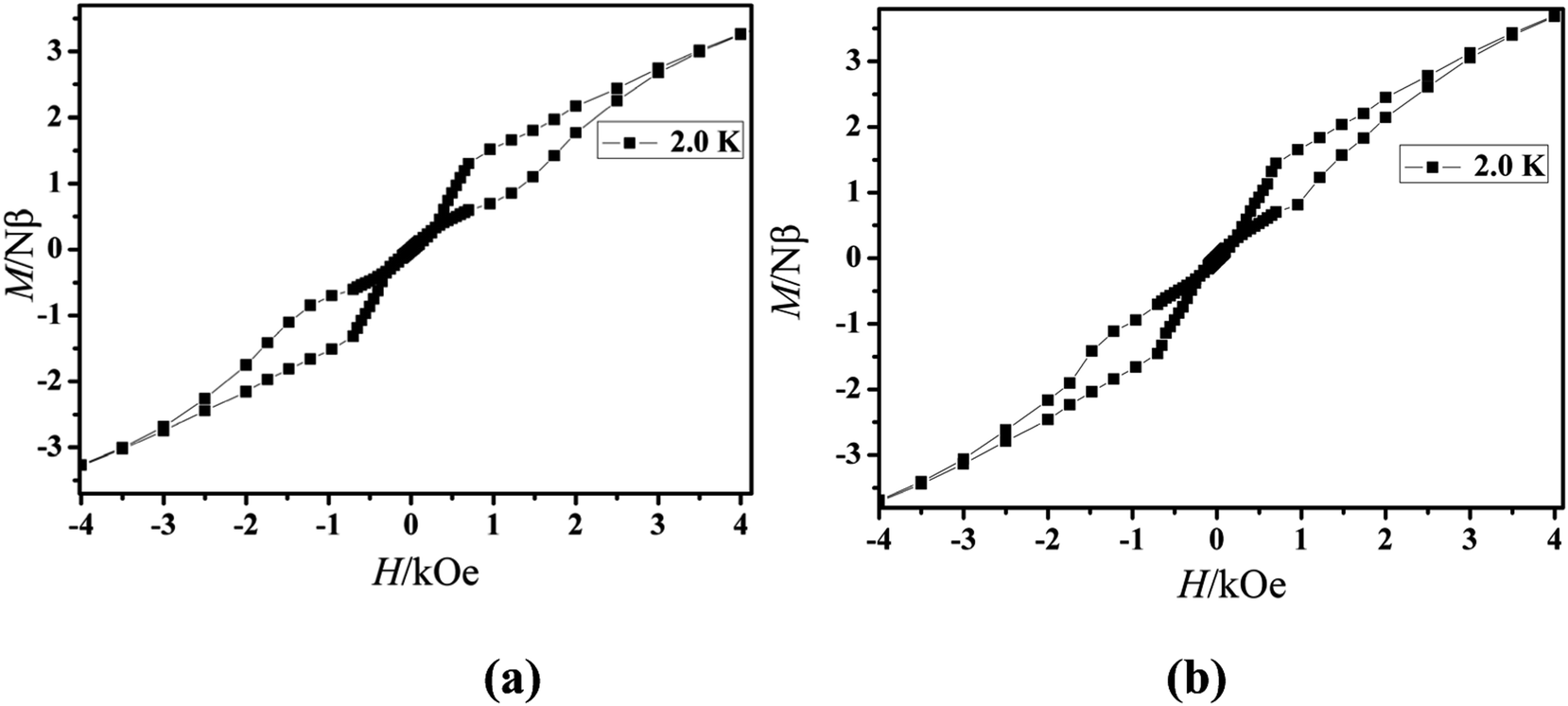
The magnetization of the two compounds from zero dc field to 70 kOe at different temperatures is shown in Fig. 4. The magnetization of Cl-1 and CH3O-2 at 2 K increased upon application of an external field to a maximum of 4.97 Nβ and 5.79 Nβ. The maximum values in Cl-1 and CH3O-2 at 7 T largely deviate from the expected saturation point of 10 Nβ, consisting of the magnetic anisotropy and crystal field effects at the dysprosium center, which dispel the 16-fold degeneration of the 6H15/2 ground state.33 The M versus H data exhibit obvious butterfly-shaped hysteresis loops at 2 K for Cl-1 and CH3O-2 (Fig. 5), indicating the fast zero-field relaxation between the two ground states.
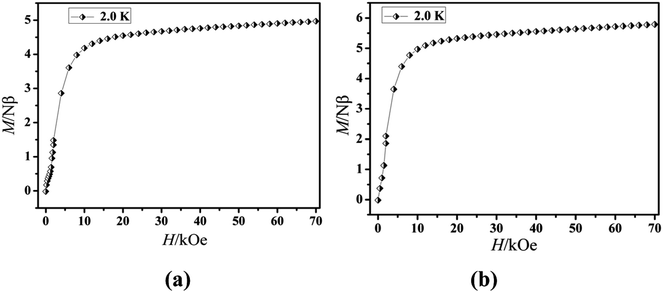 |
| | Fig. 4 M vs. H plots at 2.0 K for compounds Cl-1 (a) and CH3O-2 (b). | |
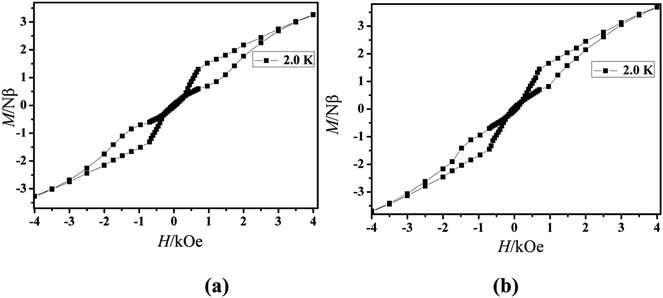 |
| | Fig. 5 M versus H data of Cl-1 (a) and CH3O-2 (b) at 2.0 K. | |
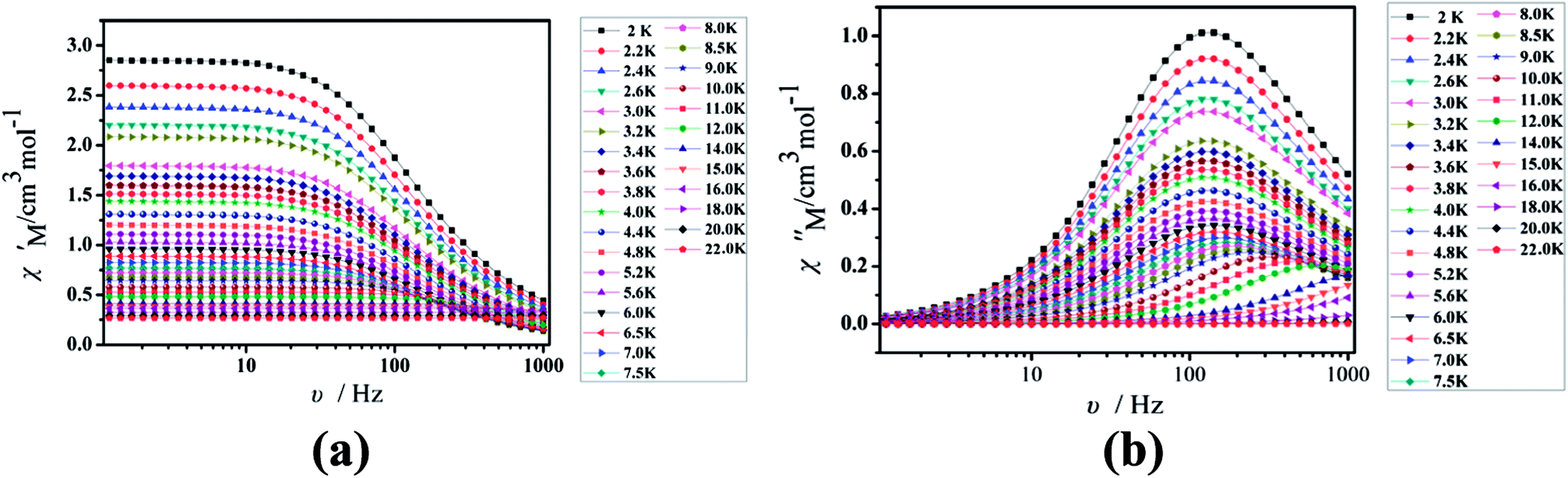
Under the oscillating field of 3.5 Oe, the zero-field AC susceptibility experiments were determined in the range of 1.8–20 K and at frequencies of 1, 10, 100, 333, 500, 800 and 1000 Hz in Cl-1. However, for CH3O-2, zero-field AC susceptibilities were measured in the range of 2–18 K and at frequencies of 1, 10, 100, 300, 500, 800, 900 and 1000 Hz. Both in-phase (χ′) and out-of-phase (χ′′) susceptibilities in compound Cl-1 and CH3O-2 showed significant temperature dependence peaks at a relatively high-temperature range (Fig. 6 and 7), which clearly indicates the slow relaxation of magnetization. When cooled, χ′ and χ′′ increased again at lower temperatures; such a situation could be due to the emergence of quantum tunneling of magnetization (QTM) without an extra dc field, which often occurs in Ln(III)-based SMMs or SIMs (Table 1).34
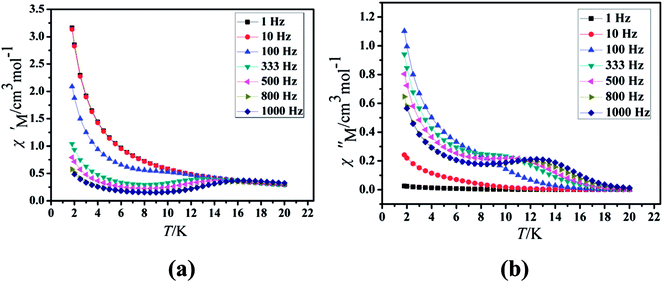 |
| | Fig. 6 Temperature dependence of the in-phase (χ′, a) and out-of-phase (χ′′, b), respectively, of the ac susceptibility for Cl-1 under the zero-dc field. | |
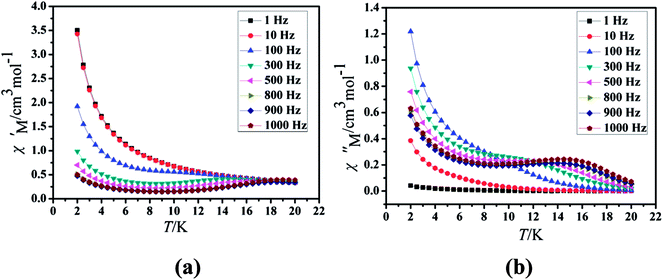 |
| | Fig. 7 Temperature dependence of the in-phase (χ′, a) and out-of-phase (χ′′, b), respectively, of the ac susceptibility for CH3O-2 under the zero-dc field. | |
Table 1 The notable examples of DyN2O6 mononuclear dysprosium compounds, namely [Dy(Phen)(L)3], based on the β-diketonate ligands with different substituent groups (–CI, –CH3,–CH3O,–F) at the 4-position
| L |
dDy–O |
dDy–N |
dDy⋯Dy |
Symmetry |
Ueff (K) |
Ref. |
Name |
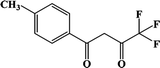 |
2.337 (2) |
2.538 (3) |
9.355 (11) |
D2d |
63.56 K/67.05 K |
13b |
CH3-3 |
| (0 Oe) |
| 118.50 K (1200 Oe) |
 |
2.329 (4) |
2.553 (5) |
9.193 (10) |
D4d |
102.82 K/95.88 K |
13b |
CH3-4 (solvent) |
| (0 Oe) |
| 164.55 (1200 Oe) |
 |
2.311 (3) |
2.579 (3) |
9.679 (8) |
D4d |
91.70 K (1200 Oe) |
31 |
F-5 |
 |
2.339 (6) |
2.562 (8) |
7.911 (6) |
D4d |
151 (0 Oe) |
Here |
Cl-1 |
 |
2.322 (5) |
2.563 (6) |
10.742 (13) |
D2d |
190.7 (0 Oe) |
Here |
CH3O-2 |
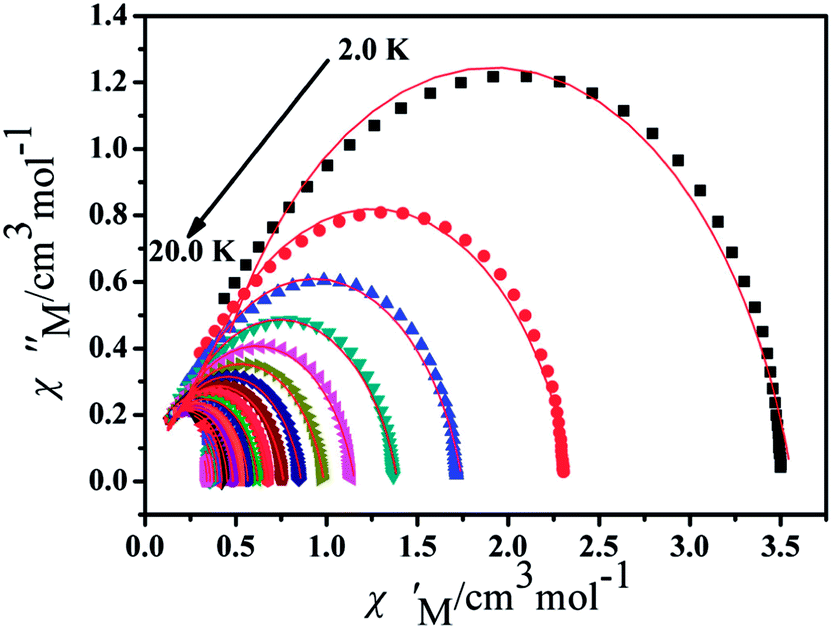
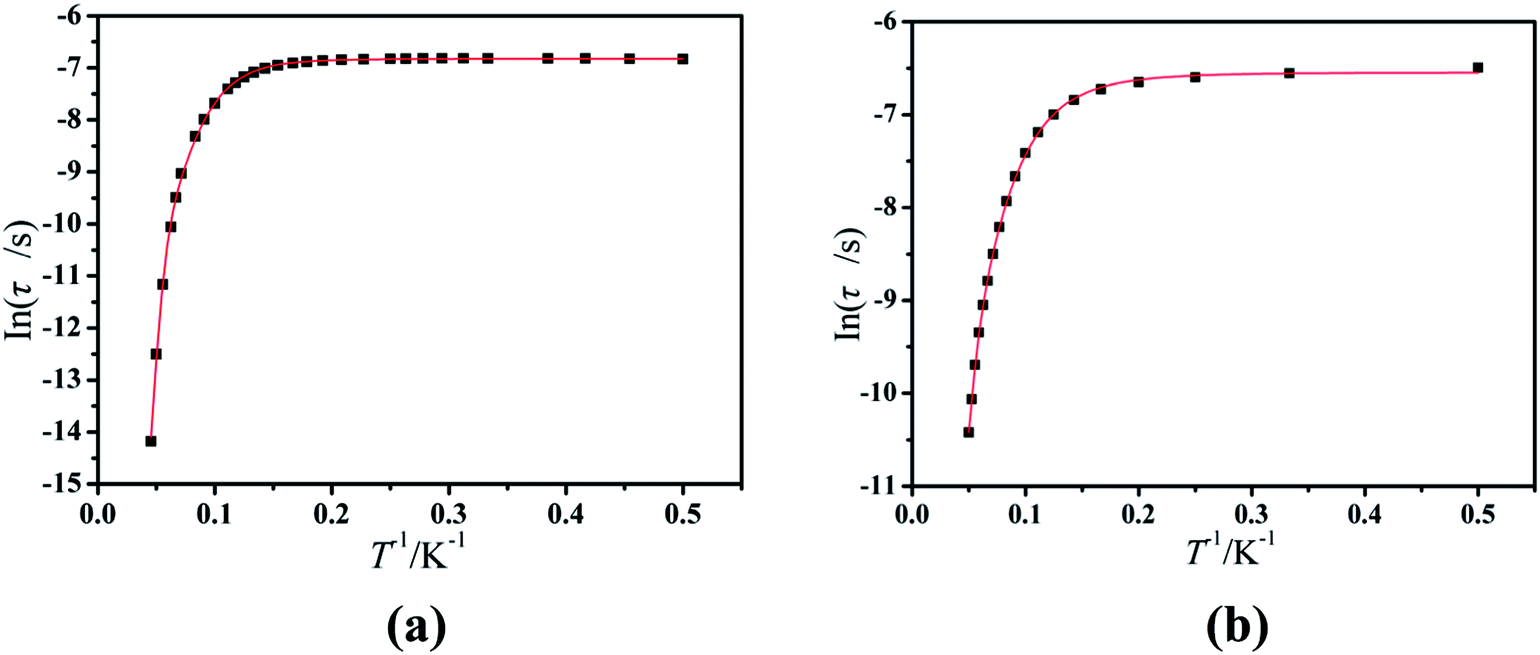
Furthermore, the frequency-dependent ac data for compounds Cl-1 and CH3O-2 were characterized in the absence of a dc field at various temperatures; the peaks of the χ′′ plots gradually shifted with the frequency sequence from middle to high, indicating that the χ′′ of compounds Cl-1 and CH3O-2 manifested frequency dependence in the selected temperature range (Fig. 8 and 9). The fitting of the Cole–Cole plots (χ′ M vs. χ′′) for Cl-1 and CH3O-2 (Fig. 10 and 11) with the Debye model13 presented a non-symmetric semicircle, which indicates the presence of a relatively moderate distribution of relaxation time (0.011 < α < 0.181 for Cl-1 and 0.007 < α < 0.161 for CH3O-2) (Table 3 and 4). For the relaxation time products under 0 Oe, the direct process can be neglected. The ln(τ) versus 1/T plots for compound Cl-1 and CH3O-2 presented some curvature (Fig. 12), indicating that the dynamics cannot be properly modelled by assuming a simple Orbach mechanism. Therefore, the total relaxation rates mainly reflect the Orbach process, Raman process and QTM process, using the following equation (eqn (1)):
| |
τ−1 = τQTM−1 + CTn + τ0−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ueff/kT) exp(−Ueff/kT)
| (1) |
where
τ is the inverse of the ac frequency,
T is the temperature of the maximum in the ac signal,
Ueff is the effective energy barrier,
k is Boltzmann's constant.
τQTM,
C, and
τ0 are the fitting parameters of the different relaxation mechanisms. In the absence of a static field, the independence of the relaxation time at low temperatures for compounds
Cl-1 and
CH3O-2 are indicative of QTM relaxation processes. The fit in the temperature range
T = 2.0–22.0 K for compound
Cl-1 by
eqn (1) resulted in
τQTM = 0.001 s,
n = 4.97,
C = 1.33 × 10
−3 s
−1 K
−4.97,
τ0 = 1.44 × 10
−8 s, and an effective energy barrier of
Ueff = 105.431 cm
−1 (151.1 K). The fit in the temperature range
T = 2.0–20.0 K for compound
CH3O-2 by
eqn (1) resulted in
τQTM = 0.001 s,
n = 4.06,
C = 8.58 × 10
−2 s
−1 K
−4.06,
τ0 = 5.08 × 10
−9 s, and an effective energy barrier of
Ueff = 132.5 cm
−1 (190.7 K).
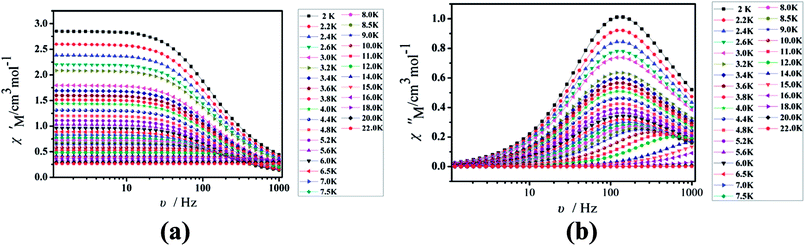 |
| | Fig. 8 Frequency dependence of the in-phase (χ′, a) and out-of-phase (χ′′, b) of the ac susceptibility for Cl-1 under the zero-dc field. | |
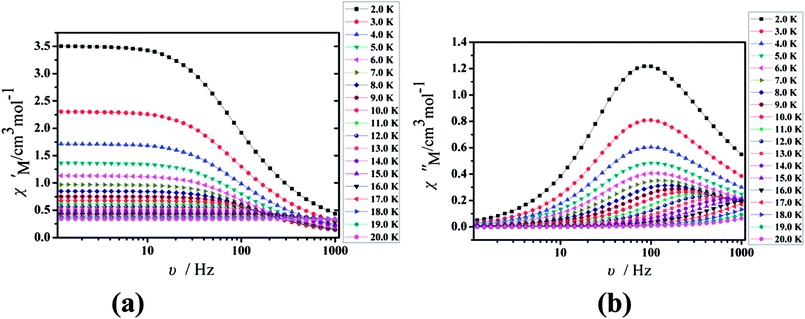 |
| | Fig. 9 Frequency dependence of the in-phase (χ′, a) and out-of-phase (χ′′, b) of the ac susceptibility for CH3O-2 under the zero-dc field. | |
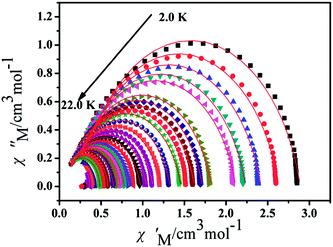 |
| | Fig. 10 Cole–Cole plots for Cl-1 from 2.0 K to 22.0 K using the ac susceptibility data. The solid lines are the best fits for the generalized Debye model between 2.0 K and 22.0 K. | |
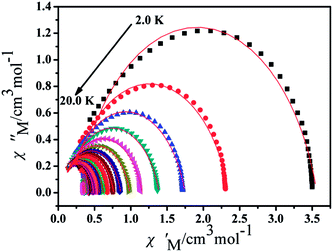 |
| | Fig. 11 Cole–Cole plots for CH3O-2 from 2.0 K to 20.0 K using the ac susceptibility data. The solid lines are the best fits for the generalized Debye model between 2.0 K and 20.0 K. | |
Table 2 Ab initio computed relative energies (in cm−1), principal values of the g-tensors and averaged transition magnetic moments μQTM (in β) of the four lowest KDs of the compounds studied in this work
| |
Cl-1 |
CH3O-2 |
| Experimental Ueff is shown in parentheses. |
| KD0 |
E |
0.000 |
0.000 |
| gZ |
19.5623 |
19.5118 |
| gX |
0.8671 × 10−2 |
0.1739 × 10−2 |
| gY |
0.1788 × 10−1 |
0.5135 × 10−2 |
| gXY |
0.1987 × 10−1 |
0.5421 × 10−2 |
| gXY/gZ |
0.1016 × 10−2 |
0.2779 × 10−3 |
| μQTM |
0.4425 × 10−2 |
0.1146 × 10−2 |
| KD1 |
E |
126.712 (105.431)a |
152.849 (132.536) |
| gZ |
15.4850 |
15.1789 |
| gX |
0.5304 × 10+00 |
0.8762 × 10+00 |
| gY |
0.1117 × 10+01 |
0.1462 × 10+01 |
| gXY |
0.1237 × 10+01 |
0.1705 × 10+01 |
| gXY/gZ |
0.7988 × 10−1 |
0.1123 × 10+00 |
| μQTM |
0.2746 × 10+00 |
0.3898 × 10+00 |
| KD2 |
E |
151.045 |
185.945 |
| gZ |
17.6519 |
16.4613 |
| gX |
0.4178 × 10+00 |
0.4877 × 10+00 |
| gY |
0.1481 × 10+01 |
0.2830 × 10+01 |
| gXY |
0.1538 × 10+01 |
0.2872 × 10+01 |
| gXY/gZ |
0.8716 × 10−1 |
0.1744 × 10+01 |
| μQTM |
0.3164 × 10+00 |
0.5530 × 10+00 |
| KD3 |
E |
177.793 |
251.832 |
| gZ |
13.8539 |
2.5445 |
| gX |
0.3264 × 10+01 |
0.8816 × 10+01 |
| gY |
0.3822 × 10+01 |
0.7151 × 10+01 |
| gXY |
0.5026 × 10+01 |
0.1135 × 10+02 |
| gXY/gZ |
0.3628 × 10+00 |
0.4461 × 10+01 |
| μQTM |
0.1181 × 10+01 |
0.2661 × 10+01 |
Table 3 The results of preliminary ESP (in a.u.) analysis
| |
Cl-1 |
CH3O-2 |
| Contributions from the equatorial N atoms. Contributions from the equatorial O atoms. |
| ESP(equ)/ESP(ax) |
0.723 |
0.701 |
| ESP(ax) |
1.082 |
1.162 |
| ESP(equ) |
0.783 |
0.814 |
| ESP(equ_N)a |
0.225 |
0.220 |
| ESP(equ_O)b |
0.557 |
0.594 |
Table 4 The negative charges (in |e|) from ab initio calculations and the related Dy–O/N bond lengths (in Å) of the atoms in the first spherea
| Cl-1 |
O3-ax |
O5-ax |
O2-ax |
O4-ax |
O6-equ |
O7-equ |
N8-equ |
N9-equ |
| “-ax” indicates the atoms at the axial positions and “-equ” means the atoms at equatorial positions. |
| Charge |
0.696 |
0.679 |
0.690 |
0.687 |
0.698 |
0.699 |
0.339 |
0.326 |
| Dy–O/N |
2.319 (7) |
2.352 (6) |
2.369 (6) |
2.339 (6) |
2.330 (6) |
2.321 (6) |
2.544 (8) |
2.586 (8) |
| CH3O-2 |
O2-ax |
O7-ax |
O3-ax |
O5-ax |
O4-equ |
O6-equ |
N8-equ |
N9-equ |
| Charge |
0.722 |
0.764 |
0.704 |
0.710 |
0.764 |
0.697 |
0.332 |
0.304 |
| Dy–O/N |
2.350 (5) |
2.307 (5) |
2.318 (5) |
2.305 (7) |
2.337 (6) |
2.312 (6) |
2.554 (6) |
2.571 (6) |
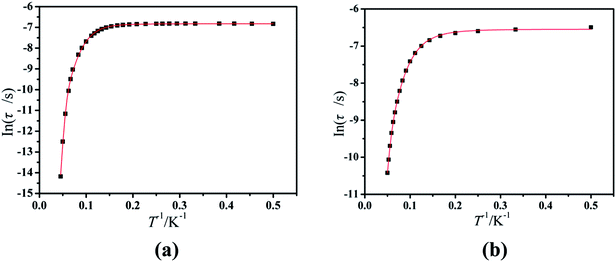 |
| | Fig. 12 Magnetization relaxation time (ln(τ)) versus T−1 plots under a zero dc field for Cl-1 (a) and CH3O-2 (b). The solid red line represents the best fitting to the multiple relaxation processes (eqn (1), see text for parameters). | |
The different local symmetries and the bond distances contribute to differing ligand fields, further resulting in diverse dynamic magnetic behaviors.13b,35 The CH3-4 (solvent) with an approximately square antiprismatic (SAP, D4d) N2O6 coordination environment exhibited more excellent properties than CH3-3 with a trigonal dodecahedron (D2d) configuration.35 However, the results in the present cases are in contrast to the explanation above, which verifies that the relaxation magnetism incompletely depends on the coordination symmetry of the Dy(III) centers. Accordingly, the magnetism of dysprosium-based SMMs might be simultaneously dominated by complicated factors including local symmetry, electrostatics, etc.10b,36
In our recent work, a similar phenomenon was observed in the two β-diketone mononuclear Dy(III) compounds, formulated as Dy(BTFA)3(H2O)2 (D2d) and Dy(BTFA)3(bpy) (D4d) (BTFA = 3-benzoyl-1,1,1-trifluoroacetone, bpy = 2,2′-bipyridine).10b As noticed, compound Dy(BTFA)3(H2O)2 possesses a higher effective barrier than Dy(BTFA)3(bpy), despite Dy(BTFA)3(H2O)2 exhibiting a lower geometrical symmetry of the Dy(III) ion. This is likely attributable to different charge distributions around the Dy(III) ions in both compounds, which compensate for the discrepancy of the geometrical symmetries and is responsible for the disparities in magnetic anisotropy, as well as energy barrier and slow relaxation behavior between the two compounds. However, the conjecture above urgently needs studies for an in-depth understanding of the significative magneto-structural correlation.
According to the Dy(III) coordination spheres, the compounds are slightly distorted, with the following order: F-5 > Cl-1 > CH3-4 (solvent); CH3-3 > CH3O-2. The shortest intermolecular distance between Dy(III) ions is 7.911 (6) Å in Cl-1. However, there are no obvious ð–ð stacking or hydrogen bonding interactions in Cl-1, probably resulting in the weakening of the QTM relaxation process from the intermolecular interactions. CH3O-2 shows shorter bond lengths for Dy–N and Dy–O than CH3-3 and a smaller degree of distortion, indicating the strong charge density around the metal ions and further generating enhanced uniaxial magnetic anisotropy. Finally, these different effective energy barriers (ΔE/kB) have the following order: CH3O-2 > Cl-1 > CH3-4 (solvent) > CH3-3 > F-5.
3.3 Theoretical analysis
The effective energy barrier for the reversal of magnetization, Ueff, is a popular parameter that is used to characterize the SMM properties of the compounds. However, only within the Orbach process is Ueff clearly defined in principle. In the early stages of the SMM, the compounds were usually polynuclear transition metal structures where the Orbach process dominated the magnetic relaxation; since then, Ueff has become popular in the field of SMM. However, in the case of mononuclear SIMs, several relaxation processes, including both Orbach and others of QTM, direct as well as Raman, exist simultaneously.37–39 Thus, the magnetic relaxation in Ln-based SIMs is not naturally dominated by the Orbach process and the necessary condition for the observation of SMM behavior is the effective suppression of all the fast relaxation process.38,39 Among all the fast relaxation processes, the quantum tunnelling of magnetization (QTM) within the ground state is the most effective and thus its suppression is the first target. Irrespective of various sources, the rate of QTM scales as the square of the so-called tunnel splitting Δtun.37–40 For Kramers ions, e.g., Dy(III), Dtun is forced to be zero under the strict absence of a magnetic field due to time-reversal symmetry; however, small internal magnetic fields actually exist with different sources.38 Therefore Δtun in Kramers systems is induced via the Zeeman interaction (eqn (1a)) between the transversal fields (HX and HY) and the corresponding components of the magnetic moments ![[small mu, Greek, vector]](https://www.rsc.org/images/entities/char_e0e9.gif) of the same directions (μX and μY).37–41
of the same directions (μX and μY).37–41| |
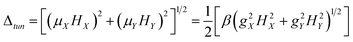 | (1a) |
| |
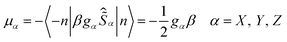 | (1b) |
| |
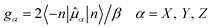 | (1c) |
Theoretically, each Kramers doublet (KD) could be associated with an effective spin (pseudospin) S = 1/2.28,37,38 The magnetic moment of such pseudospin is determined by its principal values of the g-tensors as shown in eqn (1b).37,38 Clearly, small values of the transversal gX and gY (eqn (1c)) of the ground KD, i.e., KD0, will lead to a low magnitude of Δtun and thus zero-field SIM behavior could exist if the value of gXY for KD0 is small enough.42 Besides the principal g values of each KD, ab initio calculations also provide the averaged absolute value of the transversal magnetic moments, μQTM, which could also be used to measure the strength of QTM. As shown in Table 2, the gXY values of KD0 are 0.1987 × 10−1 and 0.5421 × 10−02 for Cl-1 and CH3O-2, respectively. According to previous results from Ruiz et al.,17 zero-field SIM behavior could occur if the gXY of KD0 is smaller than 0.15 × 10−01 for mononuclear Dy(III) compounds. Clearly, this criterion is fulfilled in the case of CH3O-2 and, although a little bit larger, the gXY of KD0 of Cl-1 is also quite close to this value. Therefore our ab initio results do suggest the existence of zero-field SIM properties in these two compounds, which is consistent with the experimental observation based on ac susceptibility measurements.
Due to the smaller value of gXY of KD0, the SIM property of CH3O-2 is theoretically predicted to be superior to that of Cl-1. This theoretical prediction is also in line with the higher Ueff of CH3O-2 obtained from the fitting of the experimental data. In many cases of Ln-SIMs, the energy of the first excited KD, i.e., KD1, is closely related to the Ueff. In the two compounds here, the energies of KD1 are indeed close to the experimental Ueff: 126.7 cm−1 vs. 105.4 cm−1 for Cl-1 and 152.8 cm−1 vs. 132.5 cm−1 for CH3O-2. Moreover, the energy of KD1 for CH3O-2 is also higher than that of Cl-1. Thus, the reliability of our ab initio results is verified again in terms of energies of KD1.
As shown in our previous results,14,18,19 the desired electronic structure, which is suitable for the ideal SIM properties of Dy(III) systems, could be approached via an electrostatic route due to the oblate electron density of the Dy(III) ion; i.e., the axial electrostatic potential (ESP) should exceed the equatorial one as much as possible.6g,31,40,43 According to the orientation of the magnetic easy axis (Fig. 13), the eight atoms of the first sphere could be collected into two groups: (1) axial atoms consisting of the four oxygen atoms (O2, O3, O4 and O5 for Cl-1) that lie along the axial direction; (2) equatorial atoms consisting of the two nitrogen atoms and the residual two oxygen atoms (N8, N9, O6 and O7 for Cl-1). With ab initio atomic charge, we could approximate the axial ESP felt by the Dy(III) ion, i.e., ESP(ax), with the sum of the contribution from the four axial atoms. Similarly, the equatorial ESP, i.e., ESP(equ), could be approximated as the collection of the contribution from the four equatorial atoms. As shown before,31,40,43 the lower value of the ratio ESP(equ)/ESP(ax) indicates the higher degree of the excess of the axial ESP over the equatorial, and thus it should lead to the electronic structure, which is more suitable for the ideal SIM properties.
 |
| | Fig. 13 Direction of the ab initio magnetic easy axis of the ground KDs of the compounds (only the central Dy(III) and first sphere are shown for the sake of clarity, the equatorial atoms are shaded). | |
As shown in Table 3, the ESP(equ)/ESP(ax) ratio of CH3O-2 is 0.701, clearly lower than that of Cl-1 (0.723). Thus, the superior SIM property of CH3O-2 should originate from the more suitable ESP felt by the central Dy(III) ion when compared with 1. In detail, the difference in ESP(equ) of these two compounds is 0.03 a.u., which is clearly smaller than the corresponding difference in ESP(ax) (0.08 a.u.). In other words, the different amounts of ESP(ax) of these two compounds should play the central role in their differences in terms of SIM properties. When making a further analysis of the charges and distances to the central ion (Table 4), the averaged charges and distances to the central Dy(III) ion are 0.725 |e| and 2.320 Å, respectively, for CH3O-2. In the case of Cl-1, the averaged charges and distances are 0.688 |e| and 2.345 Å, respectively. Therefore the larger amount of ESP(ax) for CH3O-2 arises from the synergic effect of the more negative charge and shorter Dy–O distances of the axial O atoms of the first sphere. Of course, these charges and distances could be influenced by functional groups outside the first sphere, e.g., –Cl and –OCH3.
4. Conclusion
Two mononuclear compounds, [Dy(Phen)(Cl-tcpb)3] (Cl-1) and [Dy(Phen)(CH3O-tmpd)3] (CH3O-2), were synthesized based on β-diketonate ligands (Cl-tcpb = 1-(4-chlorophenyl)-4,4,4-trifluoro-1,3-butanedione and CH3O-tmpd = 4,4,4-trifluoro-1-(4-methoxyphenyl)-1,3-butanedione) with different substituent groups (–Cl/–OCH3) and auxiliary ligand 1,10-phenanthroline (Phen). The Dy(III) ions in Cl-1 have approximately square-antiprismatic (SAP, D4d) N2O6 coordination environments. The coordination geometry of Dy(III) ions in CH3O-2 can be best described as a trigonal dodecahedron (D2d). The dynamic magnetic investigations showed that both compounds exhibited SMM behavior in zero dc field, while the effective magnetization relaxation barriers increased progressively from 105.4 cm−1 (151.1 K) for Cl-1 to 132.5 cm−1 (190.7 K) for CH3O-2. CH3O-2 possessed a higher effective barrier than Cl-1, despite Cl-1 exhibiting a higher geometrical symmetry of the Dy(III) ion. Moreover, the energy of KD1 of CH3O-2 was also higher than that of Cl-1. The zero-field SIM behaviors, as well as the difference in Ueff, of these two compounds were reproduced by ab initio calculations. Further studies from the viewpoint of electrostatic potential demonstrated that the larger axial electrostatic potential (ESP) felt by the central Dy(III) ion of CH3O-2 is responsible for its better SIM properties when compared with Cl-1. The larger amount of ESP(ax) of CH3O-2 arises from the synergic effect of the more negative charge and shorter Dy–O distances of the axial O atoms of the first sphere. Beyond all doubt, these charges and distances could be affected by functional groups outside the first sphere, e.g., –Cl and –OCH3.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Nos. 21703002, 21103137), the 61th China Postdoctoral Science Foundation funded project (2017M610646), the Doctoral Scientific Research Starting Foundation of Baoji University of Arts and Science (No. ZK2017024) and the Fund of Northwest University (334100048).
Notes and references
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef.
-
(a) D. Gatteschi, R. Sessoli, J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, 2006 CrossRef;
(b) D. G. C. Benelli, Introduction to Molecular Magnetism. From Transition Metals to Lanthanide, Wiley-VCH, Weinheim, 2015 Search PubMed;
(c) G. A. Timco, S. Carretta, F. Troiani, F. Tuna, R. J. Pritchard, C. A. Muryn, E. J. L. McInnes, A. Ghirri, A. Candini, P. Santini, G. Amoretti, M. Affronte and R. E. P. Winpenny, Nat. Nanotechnol., 2009, 4, 173–178 CrossRef PubMed;
(d) S. G. McAdams, A. M. Ariciu, A. K. Kostopoulos, J. P. S. Walsh and F. Tuna, Coord. Chem. Rev., 2017, 346, 216–239 CrossRef;
(e) S. T. Liddle and J. van Slageren, Chem. Soc. Rev., 2015, 44, 6655–6669 RSC;
(f) F. Pointillart, O. Cador, B. Le Guennic and L. Ouahab, Coord. Chem. Rev., 2017, 346, 150–175 CrossRef;
(g) S. Demir, I. R. Jeon, J. R. Long and T. D. Harris, Coord. Chem. Rev., 2015, 289, 149–176 CrossRef;
(h) Y.-S. Meng, S.-D. Jiang, B.-W. Wang and S. Gao, Acc. Chem. Res., 2016, 49, 2381–2389 CrossRef PubMed.
-
(a) C. Benelli and D. Gatteschi, Chem. Rev., 2002, 102, 2369–2388 CrossRef PubMed;
(b) R. Skomski, Simple models of magnetism. Oxford University Press, New York 2008 CrossRef;
(c) D. N. Woodruff, R. E. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef PubMed;
(d) J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC;
(e) P. Zhang, Y.-N. Guo and J.-K. Tang, Coord. Chem. Rev., 2013, 257, 1728–1763 CrossRef.
-
(a) J. Liu, Y.-C. Chen, J.-L. Liu, V. Vieru, L. Ungur, J.-H. Jia, L. F. Chibotaru, Y. Lan, W. Wernsdorfer, S. Gao, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef PubMed;
(b) Y.-C. Chen, J.-L. Liu, L. Ungur, J. Liu, Q.-W. Li, L.-F. Wang, Z.-P. Ni, L. F. Chibotaru, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 2829–2837 CrossRef PubMed;
(c) J. J. Le Roy, L. Ungur, I. Korobkov, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2014, 136, 8003–8010 CrossRef PubMed;
(d) M. Li, H.-P. Wu, Y. Qi, H.-S. Ke, B. Yin, Q. Shi, W.-Y. Wang, Q. Wei, G. Xie and S.-P. Chen, Chem.–Eur. J., 2017, 23, 17775–17787 CrossRef PubMed.
- P. Zhang, L. Zhang, C. Wang, S.-F. Xue, S.-Y. Lin and J.-K. Tang, J. Am. Chem. Soc., 2014, 136, 4484–4487 CrossRef PubMed.
-
(a) S.-D. Jiang, B.-W. Wang, G. Su, Z.-M. Wang and S. Gao, Angew. Chem., Int. Ed., 2010, 49, 7448–7451 CrossRef PubMed;
(b) N. F. Chilton, S. K. Langley, B. Moubaraki, A. Soncini, S. R. Batten and K. S. Murray, Chem. Sci., 2013, 4, 1719–1730 RSC;
(c) T. T. da Cunha, J. Jung, M. E. Boulon, G. Campo, F. Pointillart, C. L. Pereira, B. Le Guennic, O. Cador, K. Bernot, F. Pineider, S. Golhen and L. Ouahab, J. Am. Chem. Soc., 2013, 135, 16332–16335 CrossRef PubMed;
(d) F. Pointillart, K. Bernot, S. Golhen, B. Le Guennic, T. Guizouarn, L. Ouahab and O. Cador, Angew. Chem., Int. Ed., 2015, 54, 1504–1507 CrossRef PubMed;
(e) Y. Bi, Y.-N. Guo, L. Zhao, Y. Guo, S.-Y. Lin, S.-D. Jiang, J. Tang, B.-W. Wang and S. Gao, Chem. - Eur. J., 2011, 17, 12476–12481 CrossRef PubMed;
(f) J. Wu, O. Cador, X. L. Li, L. Zhao, B. Le Guennic and J. K. Tang, Inorg. Chem., 2017, 56, 11211–11219 CrossRef PubMed;
(g) M. Guo, J.-F. Wu, O. Cador, J.-J. Lu, B. Yin, B. L. Guennic and J.-K. Tang, Inorg. Chem., 2018, 57, 4534–4542 CrossRef PubMed.
-
(a) K. R. Meihaus and J. R. Long, J. Am. Chem. Soc., 2013, 135, 17952–17957 CrossRef PubMed;
(b) L. Ungur, J. J. Le Roy, I. Korobkov, M. Murugesu and L. F. Chibotaru, Angew. Chem., Int. Ed., 2014, 53, 4413–4417 CrossRef PubMed;
(c) K. L. Harriman and M. Murugesu, Acc. Chem. Res., 2016, 49, 1158–1167 CrossRef PubMed;
(d) F. Gendron, B. Pritchard, H. Bolvin and J. Autschbach, Dalton Trans., 2015, 44, 19886–19900 RSC.
-
(a) Y.-S. Ding, N. F. Chilton, R. E. P. Winpenny and Y.-Z. Zheng, Angew. Chem., Int. Ed., 2016, 55, 16071–16074 CrossRef PubMed;
(b) J.-L. Liu, Y.-C. Chen, Y.-Z. Zheng, W.-Q. Lin, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M.-L. Tong, Chem. Sci., 2013, 4, 3310–3316 RSC;
(c) S. K. Gupta, T. Rajeshkumar, G. Rajaraman and R. Murugavel, Chem. Sci., 2016, 7, 5181–5191 RSC;
(d) Y.-C. Chen, J.-L. Liu, Y. Lan, Z.-Q. Zhong, A. Mansikkam-ki, L. Ungur, Q.-W. Li, J.-H. Jia, L. F. Chibotaru, J.-B. Han, W. Wernsdorfer, X.-M. Chen and M.-L. Tong, Chem. - Eur. J., 2017, 23, 5708–5715 CrossRef PubMed.
- N. F. Chilton, C. A. Goodwin, D. P. Mills and R. E. Winpenny, Chem. Commun., 2015, 51, 101–103 RSC.
-
(a) W.-B. Sun, P.-F. Yan, S.-D. Jiang, B.-W. Wang, Y.-Q. Zhang, H.-F. Li, P. Chen, Z.-M. Wang and S. Gao, Chem. Sci., 2016, 7, 684–691 RSC;
(b) P.-P. Cen, S. Zhang, X.-Y. Liu, W.-M. Song, Y.-Q. Zhang, G. Xie and S.-P. Chen, Inorg. Chem., 2017, 56, 3644–3656 CrossRef PubMed.
- G.-J. Chen, C.-Y. Gao, J.-L. Tian, J.-K. Tang, W. Gu, X. Liu, S.-P. Yan, D.-Z. Liao and P. Cheng, Dalton Trans., 2011, 40, 5579–5583 RSC.
-
(a) S.-Y. Lin, Y.-N. Guo, Y. Guo, L. Zhao, P. Zhang, H.-S. Ke and J.-K. Tang, Chem. Commun., 2012, 48, 6924–6926 RSC;
(b) X.-L. Li, H. Li, D.-M. Chen, C. Wang, J. Wu, J.-K. Tang, W. Shi and P. Cheng, Dalton Trans., 2015, 44, 20316–20320 RSC;
(c) L. Zhang, P. Zhang, L. Zhao, J.-F. Wu, M. Guo and J.-K. Tang, Inorg. Chem., 2015, 54, 5571–5578 CrossRef PubMed.
-
(a) W. B. Sun, B. Yan, L.-H. Jia, B.-W. Wang, Q. Yang, X. Cheng, H.-F. Li, P. Chen, Z.-M. Wang and S. Gao, Dalton Trans., 2016, 45, 8790–8794 RSC;
(b) S. Zhang, H.-S. Ke, L. Sun, X. Li, Q. Shi, G. Xie, Q. Wei, D.-S. Yang, W.-Y. Wang and S.-P. Chen, Inorg. Chem., 2016, 55, 3865–3871 CrossRef PubMed;
(c) X.-J. Zhang, V. Vieru, X.-W. Feng, J.-L. Liu, Z.-J. Zhang, B. Na, W. Shi, B.-W. Wang, A. K. Powell, L. F. Chibotaru, S. Gao, P. Cheng and J. R. Long, Angew. Chem., Int. Ed., 2015, 54, 9861–9865 CrossRef PubMed.
- Y.-N. Guo, X.-H. Chen, S. F. Xue and J.-K. Tang, Inorg. Chem., 2011, 50, 9705–9713 CrossRef PubMed.
-
(a) M. Gregson, N. F. Chilton, A. M. Ariciu, F. Tuna, I. F. Crowe, W. Lewis, A. J. Blake, D. Collison, E. J. L. McInnes, R. E. P. Winpenny and S. T. Liddle, Chem. Sci., 2016, 7, 155–165 RSC;
(b) K. S. Lim, J. J. Baldoví, W. R. Lee, J. H. Song, S. W. Yoon, B. J. Suh, E. Coronado, A. Gaita-Arinõ and C. S. Hong, Inorg. Chem., 2016, 55, 5398–5404 CrossRef PubMed;
(c) J.-F. Wu, J.-L. Jung, P. Zhang, H.-X. Zhang, J.-K. Tang and B. Le Guennic, Chem. Sci., 2016, 7, 3632–3639 RSC.
-
(a) G.-J. Chen, Y.-N. Guo, J.-L. Tian, J.-K. Tang, W. Gu, X. Liu, S.-P. Yan, P. Cheng and D.-Z. Liao, Chem.–Eur. J., 2012, 18, 2484–2487 CrossRef PubMed;
(b) F. Habib, G. Brunet, V. Vieru, I. Korobkov, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2013, 135, 13242–13245 CrossRef PubMed;
(c) J. Zhu, C.-Z. Wang, F. Luan, T.-Q. Liu, P.-F. Yan and G.-M. Li, Inorg. Chem., 2014, 53, 8895–8910 CrossRef PubMed;
(d) T. Pugh, F. Tuna, L. Ungur, D. Collison, E. J. McInnes, L. F. Chibotaru and R. A. Layfield, Nat. Commun., 2015, 6, 7492 CrossRef PubMed;
(e) W. Cao, C. Gao, Y. Q. Zhang, D. Qi, T. Liu, K. Wang, C. Duan, S. Gao and J. Z. Jiang, Chem. Sci., 2015, 6, 5947–5954 RSC;
(f) T. Pugh, V. Vieru, L. F. Chibotaru and R. Layfield, Chem. Sci., 2016, 7, 2128–2137 RSC.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
- J. Luzon and R. Sessoli, Dalton Trans., 2012, 41, 13556–13567 RSC.
- B. O. Roos, P. R. Taylor and P. E. M. Siegbahn, Chem. Phys., 1980, 48, 157–173 CrossRef.
- F. Aquilante, L. De Vico, N. Ferre, G. Ghigo, P. A. Malmqvist, P. Neogrady, T. B. Pedersen, M. Pitonak, M. Reiher, B. O. Roos, L. Serrano-Andres, M. Urban, V. Veryazov and R. Lindh, J. Comput. Chem., 2010, 31, 224–247 CrossRef PubMed.
- F. Aquilante, J. Autschbach, R. K. Carlson, L. F. Chibotaru, M. G. Delcey, L. De Vico, I. F. Galvan, N. Ferre, L. M. Frutos, L. Gagliardi, M. Garavelli, A. Giussani, C. E. Hoyer, G. Li Manni, H. Lischka, D. Ma, P. A. Malmqvist, T. Muller, A. Nenov, M. Olivucci, T. B. Pedersen, D. Peng, F. Plasser, B. Pritchard, M. Reiher, I. Rivalta, I. Schapiro, J. Segarra-Marti, M. Stenrup, D. G. Truhlar, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, V. P. Vysotskiy, O. Weingart, F. Zapata and R. Lindh, J. Comput. Chem., 2016, 37, 506–541 CrossRef PubMed.
- P. Malmqvist, B. O. Roos and B. Schimmelpfennig, Chem. Phys. Lett., 2002, 357, 230–240 CrossRef.
- B. A. Hess, C. M. Marian, U. Wahlgren and O. Gropen, Chem. Phys. Lett., 1996, 251, 365–371 CrossRef.
- B. O. Roos, R. Lindh, P. Malmqvist, V. Veryazov and P. Widmark, J. Phys. Chem. A, 2004, 108, 2851–2858 CrossRef.
- B. O. Roos, R. Lindh, P. Malmqvist, V. Veryazov and P. Widmark, J. Phys. Chem. A, 2005, 109, 6575–6579 CrossRef PubMed.
- B. O. Roos, R. Lindh, P. Malmqvist, V. Veryazov, P. Widmark and A. C. Borin, J. Phys. Chem. A, 2008, 112, 11431–11435 CrossRef PubMed.
- L. F. Chibotaru and L. Ungur, J. Chem. Phys., 2012, 137, 064112 CrossRef PubMed.
- L. F. Chibotaru, Adv. Chem. Phys., 2013, 153, 397–519 CrossRef.
- M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE, Universitat de Barcelona, Barcelona, 2013, vol. 2.1 Search PubMed.
- S. Zhang, H. P. Wu, L. Sun, H. S. Ke, S. P. Chen, B. Yin, Q. Wei, D. S. Yang and S. L. Gao, J. Mater. Chem. C, 2017, 5, 1369–1382 RSC.
-
(a) H. S. Ke, S. Zhang, X. Li, Q. Wei, G. Xie, W.-Y. Wang and S.-P. Chen, Dalton Trans., 2015, 44, 21025–21031 RSC;
(b) L. Zhang, P. Zhang, L. Zhao, S.-Y. Lin, S.-F. Xue, J.-K. Tang and Z.-L. Liu, Eur. J. Inorg. Chem., 2013, 2013, 1351–1357 CrossRef;
(c) B. Joarder, A. K. Chaudhari, G. Rogez and S. K. Ghosh, Dalton Trans., 2012, 41, 7695–7699 RSC;
(d) L. Liang, G. Peng, G.-Z. Li, Y.-H. Lan, A. K. Powell and H. Deng, Dalton Trans., 2012, 41, 5816–5823 RSC.
- S. Osa, T. Kido, N. Matsumoto, N. Re, A. Pochaba and J. Mrozinski, J. Am. Chem. Soc., 2004, 126, 420–421 CrossRef PubMed.
-
(a) A. Yamashita, A. Watanabe, S. Akine, T. Nabeshima, M. Nakano, T. Yamamura and T. Kajiwara, Angew. Chem., Int. Ed., 2011, 123, 4102–4105 CrossRef;
(b) H. L. C. Feltham, Y. Lan, F. Klöwer, L. Ungur, L. F. Chibotaru, A. K. Powell and S. Brooker, Chem.–Eur. J., 2011, 17, 4362–4365 CrossRef PubMed;
(c) S.-D. Jiang, B.-W. Wang, H.-L. Sun, Z.-M. Wang and S. Gao, J. Am. Chem. Soc., 2011, 133, 4730–4733 CrossRef PubMed.
-
(a) X. C. Huang, M. Zhang, D. Y. Wu, D. Shao, X.-H. Zhao, W. Huang and X.-Y. Wang, Dalton Trans., 2015, 44, 20834–20838 RSC;
(b) Y.-Z. Tong, C. Gao, Q.-L. Wang, B.-W. Wang, S. Gao, P. Cheng and D.-Z. Liao, Dalton Trans., 2015, 44, 9020–9026 RSC.
-
(a) L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092–3104 RSC;
(b) L. Ungur, S.-Y. Lin, J.-K. Tang and L. F. Chibotaru, Chem. Soc. Rev., 2014, 43, 6894–6905 RSC.
- L. F. Chibotaru, Struct. Bonding, 2015, 164, 185–230 CrossRef.
- L. Ungur and L. F. Chibotaru, Inorg. Chem., 2016, 55, 10043–10056 CrossRef PubMed.
- M. Li, H. P. Wu, Q. Yang, H.-S. Ke, B. Yin, Q. Shi, W.-Y. Wang, Q. Wei, G. Xie and S. P. Chen, Chem.–Eur. J., 2017, 23, 17775–17787 CrossRef PubMed.
- L. Ungur and L. F. Chibotaru, Phys. Chem. Chem. Phys., 2011, 13, 20086–20090 RSC.
- L. Ungur and L. F. Chibotaru, Computational Modelling of the Magnetic Properties of Lanthanide Compounds. in, Lanthanides and Actinides in Molecular Magnetism, ed. R. A.Layfield and M.Murugesu, Wiley-VCH Verlag GmbH & Co. KGaA, 1st edn, 2015, chp. 6, pp. 153–184 Search PubMed.
- D. Aravena and E. Ruiz, Inorg. Chem., 2013, 52, 13770–13778 CrossRef PubMed.
- Z. J. Jiang, L. Sun, Q. Yang, B. Yin, H. S. Ke, J. Han, Q. Wei, G. Xie and S. P. Chen, J. Mater. Chem. C, 2018, 6, 4273–4280 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1815605 and 1815606. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra06240d |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ac,
Wenjiao Moa,
Jiangwei Zhang
*ac,
Wenjiao Moa,
Jiangwei Zhang