
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Schwertmannite: occurrence, properties, synthesis and application in environmental remediation
Zhuo Zhanga,
Xue Bi *b,
Xintong Lib,
Qiancheng Zhaob and
Honghan Chena
*b,
Xintong Lib,
Qiancheng Zhaob and
Honghan Chena
aBeijing Key Laboratory of Water Resources & Environmental Engineering, School of Water Resources and Environment, China University of Geosciences, Beijing 100083, China
bBeijing Junmei Environmental Technology Co., Ltd., Room 1505. Tower B, New Logo International Tower, No. 18A, Zhongguancun South Street, Handian District, Beijing, 100081, China. E-mail: bixue@bjjunmei.com.
First published on 1st October 2018
Abstract
Schwertmannite is a typical iron-derived mineral, which was originally discovered in acid mine drainings and subsequently synthesized in the laboratory. Increasingly, it is seen as having considerable potential as an adsorbent material, which could be used for environmental remediation (such as the treatment/remediation of arsenic, chromium, antimony, fluoride, and organic contaminants). This study reviews current developments, mainly in the preparation, structure, and water treatment of Schwertmannite. Several key issues are discussed in detail, such as synthetic strategy, the structure–property relationships, potential environmental applications, and related mechanisms. Soil remediation by schwertmannite is compared to water treatment, and its application is further evaluated. Finally, the methodologies for water treatment and soil remediation using schwertmannite are also taken into consideration from an environmental point of view.
1. Introduction
Schwertmannite (Sch) is a secondary iron (Fe)-hydroxylsulfate mineral, which is found in acidic sulfate-rich environments. It could potentially be used to treat multiple heavy metals in contaminated water or soils. It is probably one of the most frequently observed minerals that precipitate from ferriferous aqueous systems, such as mine drainages, soil solutions, and lake water,1 similar to ferrihydrite.1.1 Discovery
In natural environments, the Fe–sulfur (S) system automatically cycles through a reduction path based on Fe sulfide and an oxidation path based on Fe sulfate. The Fe sulfates are commonly found in natural marine environments or lakes, etc., which often also contain Sch.2 Natural sources of Sch can be acid mine drainings (AMD), acid sulfate soils (ASS),3 solid minerals, such as ochreous sediments,4 natural Fe and S oxidation by-products in coal,5 and some types of industrial liquid/solid waste produced during human activities, such as copper heap leachate solutions.6Early studies on Sch were undertaken by Brady et al.7 and Murad8 before it was recognized as a discrete mineral. There was no chemical analysis. Therefore, Sch was tentatively identified as a “well” crystallized ferrihydrite phase. In 1990, Bigham et al.9 discovered a poorly crystallized oxyhydroxy-sulfate of Fe in sediment samples collected from mine effluents in the eastern Ohio coalfield and drainage exiting an abandoned copper–arsenic mine in Finland, which was identified as the primary component of ochreous precipitates from sulfate-rich mine waters in the pH range of 2.5–4.0. This material was approved as an independent mineral species by the Commission on New Materials and Mineral Names of the International Mineralogical Association in 1992 (90-006).10 Two years later, Bigham et al.10 reported its occurrence at the Pyhäsalmi sulfide mine, Province of Oulu, Finland. In the same year, this mineral was named in honour of Udo Schwertmann, Professor of Soil Science from the Technical University of Munich, who published the first description of Sch.9 In 1995, after being frequently identified in precipitates from acid mine waters, a fully natural Sch was found in a small stream draining a pyritic Schist at PfitScher Joch, Zillertaler Alps, Austria.11 In Japan, Sch was discovered in 2003, based on a phenomenon of naturally attenuated arsenic (As) concentration form abandoned As mine in Nishinomaki,12 and then the first patent about Sch for absorbent was published.13 In Chinese, Sch is called “shī weī tè màn shí”,14 and was first found in AMD sediments from the Malan coal mine, Shanxi Province, China, in 2006,15 and afterwards at the Dabaoshan mine in South China.16–19 The research development of Sch and the primary contributions of researchers or groups are listed in Table 1. The table shows that natural Sch was discovered, identified, and structurally classified within the first few years of its discovery. After this, related theoretical studies (over nearly 20 years) about its properties and further application, especially for environmental protection, began to attract more attention.
| Starting years | Authors | Groups | Contribution | Ref. |
|---|---|---|---|---|
| 1990 | J. M. Bigham | J. M. Bigham | Discovered Sch in acid mine draining and studied on the Sch's structure | 9 |
| 1994 | J. M. Bigham | E. Murad | The occurrence in Pyhäsalmi sulfide mine, Province of Oulu, Finland. | 10 |
| 1994 | IMA&CNMMN | — | Official approval | 10 |
| 1995 | U. Schwertmann | J. M. Bigham | A fully occurrence in a stream draining of Austria | 11 |
| 1996 | G. A. Waychunas | G. A. Waychunas | AsO43− and SeO43− substituted Sch | 49 |
| 2003 | F. Keisuke | F. Keisuke | The discovery of Sch in As mine | 12 |
| 2004 | Oishi | — | The first patent about Sch for absorbent | 13 |
| 2004 | S. Regenspurg | S. Regenspurg | Chemical oxidation for Sch's preparation | 20 |
| 2005 | S. Regenspury | S. Regenspurg | AsO43− and CrO42− incorporation in Sch | 29 |
| 2006 | H. F. Sun | F. H. Zhao | Found in Shanxi Province, China | 15 |
| 2006 | P. Acero | P. Acero | Behavior of trace elements during Sch precipitation | 81 |
| 2009 | E. D. Burton | E. D. Burton | Sorption of As(V) and As(III) by Sch | 32 |
| 2011 | S. Paikaray | S. Paikaray | Removal of As(III) using Sch upon various synthesis | 38 |
| 2013 | E. D. Burton | E. D. Burton | Microbially mediated reductive transformation of Sch for As removal | 65 |
| 2013 | W. M. Wang | X. Han | Sch as a new Fenton-like catalyst for oxidation of phenol | 62 |
| 2014 | C. L. Vithana | C. L. Vithana | Effect of fulvic acid during the arsenic removal by Sch | 78 |
| 2015 | F. W. Liu | W. H. Fan | Ferrous ion chemical oxidation: different H2O2 supply rates | 31 |
| 2015 | M. Q. Chen | G. N. Lu & Z. Dang | Sch's role during the natural sulfate migration | 16 |
1.2 Chemical composition
The groups led by Bigham9,10 and Schwertmann11 proved experimentally that this type of mineral can be expressed as Fe8O8(OH)8−2x(SO4)x (1 ≤ x ≤ 1.75), with a molar ratio for Fe/S of between 4.6–8.0, and with a mass percentage for SO42− of between 12.5%–20.5% in the theoretical chemical formula when crystal water is not considered. However, differences between operators, methods, and conditions mean that the mass percentage for SO42− can range widely from 5.3% (ref. 4) to 32%.20The Fe species was proven to be entirely Fe3+ by the Murad group and others after using Mössbauer spectroscopy.9,21–24 This stable Fe chemical state gives this material a unique structure and could potentially be used in a number of different fields.
1.3 Microstructure
Characterization by X-ray diffraction (XRD) has suggested that Sch has poorer crystallinity characteristics than jarosite and goethite, but it is quite similar to ferrihydrite.25 However, Loan et al.26 demonstrated that most Sch whiskers have a structure that is consistent with maghemite, which is similar to some ferrihydrite components, whereas some sections also contain highly disordered ferrihydrite structural components and more amorphous regions. This description is mainly based on XRD data and some other direct images about the surface morphology of Sch. The Sch particles are transformed into crystal cubic and hexagonal phases, during which, sulfate and OH/H2O are ejected from the structure and sulfate is structurally incorporated. The microstructure of Sch is shown in Fig. 1A.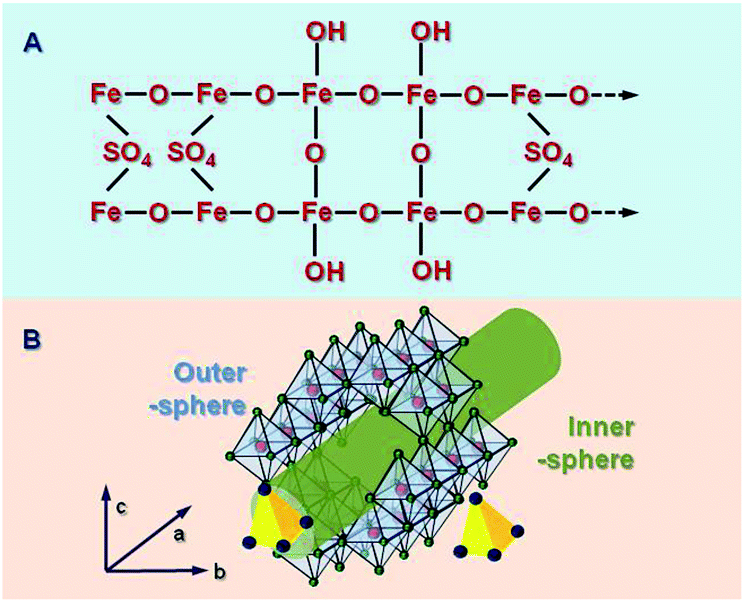 | ||
| Fig. 1 Chemical (A) and channel (B) structure of schwertmannite. | ||
Schwertmannite is a novel mineral and possesses a 3D structure with ordered channels. Fukushi et al.27 studied previous reports and gave a comprehensive description of possible coordination modes, which involved a bidentate bridge of SO42− within an akaganéite-like structure. Sch has dominant crystal planes at {100},9 which feature double sequences of surface O atoms that are arranged in six rows, are coordinated with Fe, and oriented parallel to c, as in the following: Fe3O, Fe1O, Fe3OH, Fe3O, Fe3OH, and Fe2O. Four surface O atoms and –OH groups are present in the 1st, 2nd, and 5th rows. In the 3rd and 5th rows, SO4 structures replace one –OH group to form a bridging bidentate (Fe3)SO4 surface group. Fernandez-Martinez et al.28 showed a deformed frame of an Fe octahedral that was similar to that of akaganéite. This was based on pair distribution functional data, XRD analysis, and density functional theory calculations. The simulations suggested that two sulfate molecules per unit cell were present in the structure, which formed one outer sphere and one inner sphere complex inside the channels created by the Fe octahedral.
The material is described as “analogous in structure to basic Fe sulfate, but with a –Fe–O– cage, as shown in Fig. 1B. The Sch possessed a channel-like structure in the c axis orientation with a diameter of ca. 0.50 nm.5 The SO42− ions in the channel pores can be easily substituted by other ions that have a similar radius, such as PO43− (r = 0.238 nm), CrO42− (Cr: chromium) (0.240 nm), or AsO43− (0.248 nm).29 Bigham et al.30 reported that SO42− ions have a mass percentage of between 10–15%. A third of them are adsorbed on the mineral surface and two-thirds are found in the channels.
2. Synthetic strategy – structural characteristics
2.1 Synthetic strategies
The most common formation process for natural Sch (formula (1)) usually goes through two main steps, which are the oxidation of Fe2+ and co-precipitation of Fe3+. However, after nearly 20 years of development and improvement, Sch can be synthesized using various techniques that involve the chemical oxidation of ferrous species, abiotic precipitation of ferrous species (fast hydrolysis and slow dialysis), and/or biological ferrooxidation of ferrous species by Fe2+-oxidizing bacteria, such as Acidithiobacillus ferrooxidans, as Table 2 listed. Essentially, Sch is an Fe mineral salt, which means there can be various Fe chemical sources, such as Fe2+ salts and Fe3+ salts, but only if the SO42− concentration is high enough. Usually, if the Fe source materials have a low valence, another oxidation step is necessary, whereas, when they are in a high valence state, then only phase transformation is needed.| 8Fe(SO4) + 2O2 + (4 + x)H2O → Fe8O8(OH)x(SO4)y↓ + (8 − y)SO42− + 2(8 − y)H+ | (1) |
| Synthetic methods | Fe2+ oxidation | Fe3+ hydrolysis | ||
|---|---|---|---|---|
| Chemical oxidation | Biological ferrooxidation | Fast hydrolysis | Slow dialysis | |
| Fe source | Fe2+ salts (sulfate) | Fe3+ salts (sulfate, nitrate, chloride) | ||
| Temperature | 24–75 | 25–40 | 60–85 | 60 |
| pH value | 2.4–3.0 | 2.5–3.2 | 1.66–4.0 | — |
| Oxidant | H2O2 | — | — | — |
| Time | 24 h | 5–7 days | 1 h–25 days | 30 d |
| References | 20,32,34,35,38,43,78,89 | 24,38,40,41,44,51,53,83,84,90 | 1,6,52,61,62,89,92 | 20,26,35,38,45,46,53,102,103 |
| 2Fe2+ + H2O2 + 2H+ → 2Fe3+ + 2H2O | (2) |
| Fe3+ + 6H2O → [Fe(H2O)6]3+8[Fe(H2O)6]3+ + (24 − 2x)OH− + xSO42− → Sch + 56H2O | (3) |
In comparison to the chemical oxidation method, the abiotic precipitation of Fe3+ species is carried out at higher temperatures, which are usually higher than 60 °C and lead to fast precipitation. Furthermore, the nucleation rate for abiotic precipitation is relatively higher than for chemical oxidation. Different abiotic precipitation techniques may lead to differences in crystal-growing time, but not the nucleation rate (fast completed in short time).
It needs to clear that all the three methods we mentioned above involves chemical phase-transformation of Fe3+, without interferes of other composites. Such processes need precise control of condition factors to make sure the purity and structure of aimed product. Yet, the process droved by biological force may more effective and eco-environmental.
In summary, the chemical oxidation methods were the earliest Sch production methods, whereas slow dialysis and the biogenic method can be more efficiently upscaled. However, chemical oxidation is more suitable for theoretical studies, especially when investigating crystal structure and surface morphology, because of its stricter requirement for heterogeneous synthesis and separation of the product.
In nature, the final properties of Sch are not only dependent on the fabrication method, but are also closely tied with the post-processing procedure, which includes the separation, drying, and smashing processes. Usually, drying methods in the laboratory involve natural drying, heat drying, freeze drying, and air drying. Freeze drying is more suitable for stable repeated preparation when the diffusion and transfer of water molecules in the samples is taken into consideration. After the drying step, physical steps, such as smashing and grinding, are used to obtain the powdered sample. These processes change the surface colour. After the post-processing steps and the separation process mentioned above, a whole preparation procedure that uses slow fabrication and freeze drying may result in Sch products with better crystallinity and larger particle sizes.
2.2 Characteristics
Previously reported studies on Sch materials have mainly focused on the structure–properties relationship. However, it is necessary to discuss the structure-related reports on synthetic methods because the different structures/morphologies are closely dependent on the synthetic strategies used and their tunable parameters.Fig. 2 shows that most of the Sch samples obtained by chemical oxidation contain spherical particles that are larger than 0.5 μm and at the μm scale. Models ii and iii produce particles that have a hedge-hog-like or a sea urchin-like morphology, which is possibly caused by a higher synthetic temperature or a slow addition rate for H2O2.
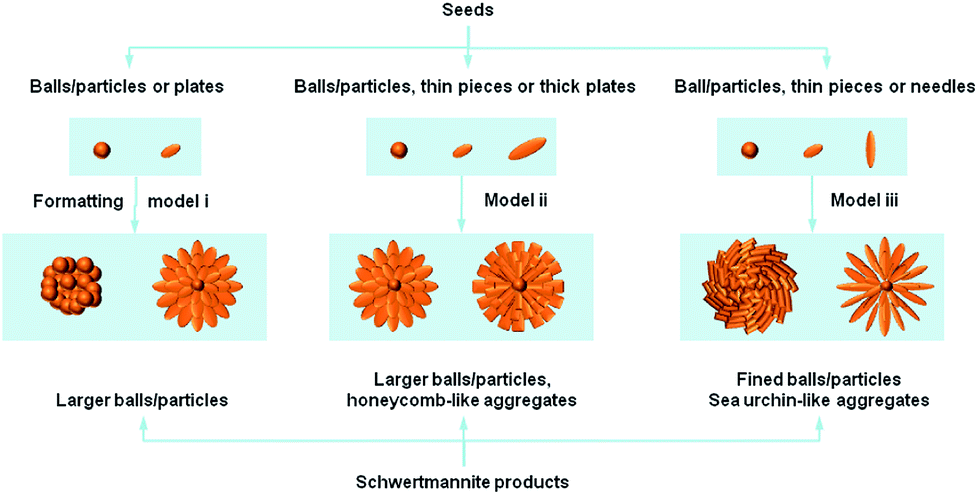 | ||
| Fig. 2 Schematic illustration of the formation process from seeds to schwertmannite products. | ||
The reaction temperature is important in the oxidation process. French et al.43 stated that the synthetic Sch produced via chemical oxidation should not be described as a single phase with a repeating unit cell, but as a polyphasic nano-mineral with crystalline areas spanning less than a few nano-meters in diameter within a characteristic “pin-cushion”-like amorphous matrix. The difference in synthetic temperature affects the density of the needles on the Sch surface. The needles on Sch produced at higher temperatures had a dendritic morphology, whereas the needles on Sch produced at room temperature were more closely packed.
The H2O2 addition rate is intensively optimized for stable mineralization. Liu et al.31 reported that a slow H2O2 supply rate led to a low ferrous ions oxidation efficiency and low total Fe precipitation efficiency in Sch synthesis systems, which increased the SSA value by changing the morphology from a spherical to a “hedge-hog” structure. This improved the heavy metal ion removal efficiency.
There are too many examples of fast hydrolysis for an in-depth discussion. Model ii in Fig. 2 shows that two kinds of remarkably different morphologies can be produced. The first has a hedge-hog like structure with a diameter of ca. 100 nm, whereas the second type aggregates with each other and produces particles that cover a board range of sizes. After studying a large numbers of literatures, it is discovered that, the hedge-like morphology of Sch minerals are mainly formed at higher temperature (ca. 85 °C) without control of pH value, whereas the aggregated one of Sch minerals are mainly formed at lower temperature (<60 °C) and lower pH values (2.4–3.0). Considering the pH value are mainly decided by the detail strategies, such as lower pH values for oxidation reaction while relatively higher pH values for hydrolysis, it was firstly settled as an environmental factor. Different from that, the temperature factor may influence the nucleation and aging process of crystallization, which in great degree decides the final morphology of Sch products. Thus, once the pH condition has been set, the temperature factor becomes the main cause of morphology formation.
Another method involving slow dialysis is also based on the hydrolysis of Fe3+ species and is quite similar to fast hydrolysis. However, the precipitation time for the dialysis is much longer than the fast hydrolysis pathway. This process mainly utilizes an extra cellulose membrane, which requires consistent treatment of the precipitate. Fig. 2 shows that the samples produced by model ii all possess a typical hedge-hog like structure, which may be due to the use of 60 °C as the synthetic temperature. A higher temperature promotes dialysis, which thermally stabilizes the 3D structure.
In contrast, the biological ferrooxidation method can be easily classified and evaluated. This micro-organism mediated process is also an oxidation method. As model iii in Fig. 2 shows, this method produces many more defined particles than the other methods. However, these biogenic Sch particles have a limited SSA value of <50 m2 g−1, which may be due to the ultrafine Sch particles.44
The XRD data shows that Sch consists of 8-line diffraction profiles of between 0.486 and 0.146 nm (mainly at ca. 26.3, 35.2, 55.3, and 61.3°) as Fig. 3 shows.45 In all cases, the broad but consistent character of the diffraction maxima indicates a material that is poorly crystallized, but certainly not X-ray amorphous. However, the XRD diffraction patterns of the Sch samples are slightly different depending on the synthetic pathway used.
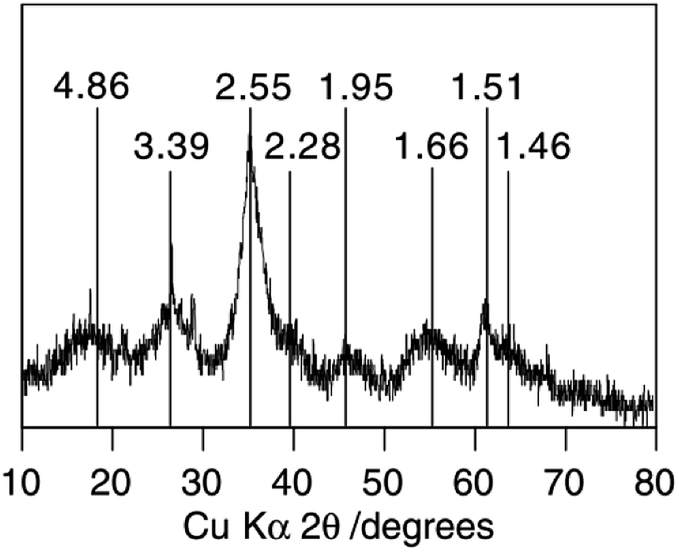 | ||
| Fig. 3 The classic XRD pattern of schwertmannite. (Reproduced from ref. 45 with permissions from the Elsevier). | ||
Three dominant SO42− groups are present in synthetic Sch according to the FI-IR spectra (Fig. 4) produced by Boily et al.46 which are tentatively assigned to H-bonded (I and III) and Fe-bonded (I) sulfate ions that are attached to Sch; and protonated (II) sulfate species with the following features: (I) bands at 1120, 1070, 1033, and 990 cm−1, which is similar to sulfate complexes adsorbed to hematite, goethite, and Sch in the presence of water; (II) bands at 1210, 1180, 1130, 1033, 972, and 966 cm−1, which is comparable to the sulfate spectra for hematite and has the greatest degree of ν3 splitting; and (III) bands at 1108 and 972 cm−1.
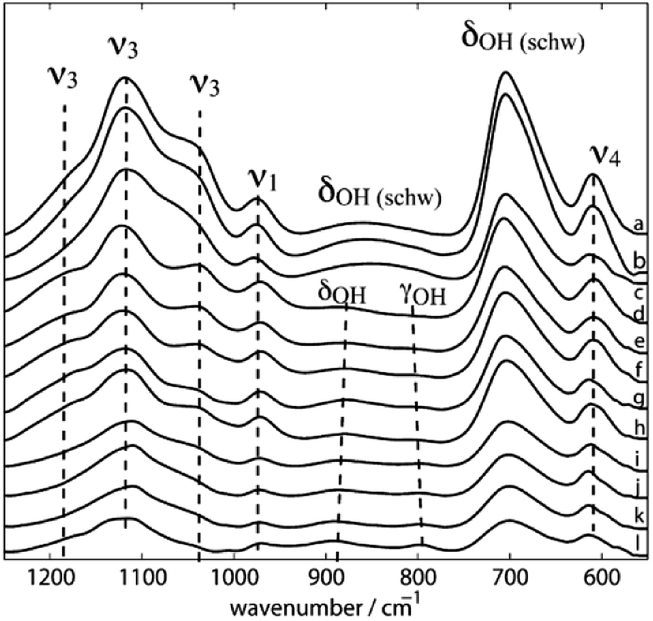 | ||
| Fig. 4 ATR FT-IR spectra of synthetic schwertmannite at different pH values at 298.2 K (a–l: 3.14, 3.44, 4.00, 4.40, 5.05, 5.50, 6.13, 6.52, 6.65, 8.04, 8.44, and 9.04). (Reproduced from ref. 46 with permissions from the American Chemical Society). | ||
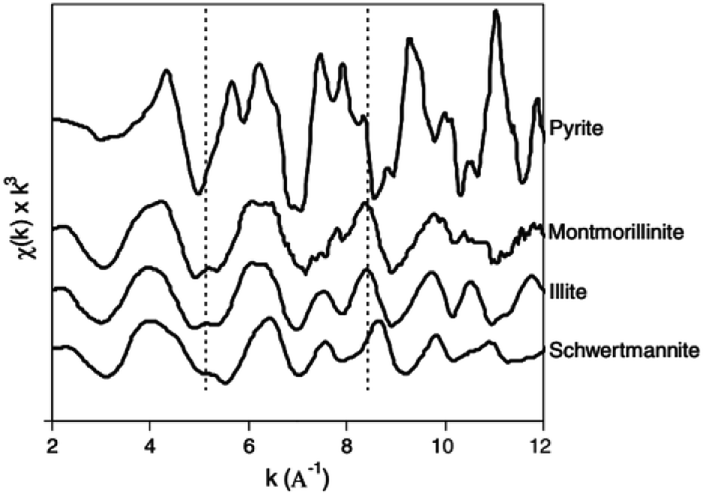 | ||
| Fig. 5 Iron K-edge EXAFS spectra of schwertmannite and reference minerals. (Reproduced from ref. 47 with permissions from the Elsevier). | ||
Besides direct characterization, these technologies are now used to monitor As and Cr ions' ads/desorption on the Sch surface. In 1996, synthetic Sch, where the sulfate had been substituted by As and Se, was examined by X-ray absorption structure to characterize the location of anionic complexes. The results showed the direct substitution of SO42− by Se within tunnels and the sorption of Se outside the tunnels. However, As was mainly found outside the structure, which destabilized it and reduced growth.49 The X-ray absorption fine structure (XAFS) results for As adsorption on Sch50 showed that As was bound to Fe octahedral, and individual Fe octahedral in the Sch became more regular after adsorption.
The relationship between pore/channel structure and the SSA and pore size distribution parameters for the Sch samples is not quite consistent with the relationship between fabrication method and structure/morphology. Pore size distribution is mainly decided by the primary formation of mineral nanoparticles, whereas SSA is preferentially confirmed by the secondary stacking of primary nanoparticles during nucleation and aging. This may explain some of the structure/morphology aspects of Sch, but more research needs to be undertaken in this area. Zhang et al.53 introduced fractal geometry (Df), a novel mathematical tool, to describe the irregular and complex geometries of Sch that involves morphological characteristics and pore structures. The surface (shown by denoised SEM images) and inner core (shown by low-pressure N2 adsorption analysis) Df values are considered to be the operative measurements of surface roughness and pore complexity, respectively. However, this structural evaluation model is based on microstructure and the entire morphology, which means that it needs further research and improvement.
Due to the structural similarities between Sch and ferrihydrite, it is easy to deduce that the most common functional group on the surface of Sch is Fe–OH. The Fe–OH bands exist widely in Fe-based clay or minerals.54 The surface charge density of a Sch powder sample can be calculated via the experimental method reported by Yang et al.55 or simulated by a theoretical model.56 The surface electric properties are key factors and are based on the Zeta potential.57 Two of them are mentioned frequently. These are the pH value at zero point of charge (pHzpc) and the pI value, which are the pH values of materials or systems58 when the surface charge becomes zero and the surface charge is not affected by a solution (not necessarily zero). The pHzpc values of Sch samples are reasonable and varies from 3.0–7.2 depending on the fabrication and test methods/conditions. The pI value order (called pHzpc in most reported references) is as follows: 7.2 (AMD)45 > 5.4 (bio-ferrooxidation)59 > 5.1 (chem. oxidation)34> 4.2 (fast hydrolysis)60 > 3.7 (fast hydrolysis)61 > 3.05 (fast hydrolysis).62 According to the above order, natural Sch may have a higher pHzpc value, and Sch obtained using fast hydrolysis may have the lowest pHzpc value.
The estimated log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K values for Sch are 2.01 ± 0.30, which seems to vary depending on the sulfate content. In comparison, the value for 2-line ferrihydrite is 8.46 ± 1.40 and 10.12 ± 0.74 for 6-line ferrihydrite.1
K values for Sch are 2.01 ± 0.30, which seems to vary depending on the sulfate content. In comparison, the value for 2-line ferrihydrite is 8.46 ± 1.40 and 10.12 ± 0.74 for 6-line ferrihydrite.1
2.3 Stability
Although Sch has a tunable chemical composition and a unique pore/channel structure, its practical application is still somewhat limited. There are two possible reasons: unstable fabrication and storage, both of which originate from the low crystallinity of the Sch phase. For example, Sch can reductively transform into two 2nd Fe2+ minerals: siderite and mackinawite, and also relatively rapidly into goethite in ca. 25–65% anoxic reducing sediments,63,64 depending on the SO42− concentration.37,65 However, many factors can influence this transformation process, such as pH, Fe3+ concentration, thermal treatment, existence of organic matter, and coexisting ions, etc.French et al.43 used high resolution transmission electron microscopy to show that the synthetic temperature in the chemical oxidation process used to produce Sch with “pin-cushion”-like morphology affects the density of needles on the surface. The needles on Sch produced at higher temperatures have a dendritic morphology, whereas those on Sch produced at room temperature are more closely packed. The low temperature treatment may cause different structural changes compared to the high temperature treatment. This factor does not change the inherent structure but does affect the acquired properties. Qiao et al.44 reported that the temperatures used during the bio-Sch process do not greatly change Sch up to 250 °C. Above this, there is a broad peak at 2θ ≈ 30–35°, which indicates a change from Sch to 2-line ferrihydrite (especially above 350 °C). The SSA and As removal increased when the sample was heated at 250 °C. However, removal then decreased at 550 °C, but this was dependent on the loss of –OH from the Sch surface. Johnston et al.68 used higher thermal temperatures (200–800 °C) to transform As5+–Sch. Their results suggested that Sch was transformed into a nanocrystalline hematite with a greater SSA and a smaller particle size when heated to temperatures above 400 °C.
Fulvic acid (FA) and humic acid (HA) are also often present when Sch is applied.76,77 Fulvic acid is well-known to interact strongly with both Sch and As. Furthermore, As concentration has a strong influence on the liberation of acidity from pure Sch and As–Sch.78 However, knowledge about the fate of Fe and Cr after the reduction and complexation of CrO42−-substituted Sch by FA is poorly understood. The interaction between FA and CrO42−–Sch was investigated by Xie et al.,79 who showed that the Fe and Cr concentrations in solution increased at first, but then decreased when FA was at 10 mg L−1 and the pH was 6.5. This was possibly due to synergistic effects, such as interactions between ligand-promotion and reduction. The effect of FA on the liberation of acidity and the mobilization of As results in a greater liberation of acidity by As–Sch at pH 4.5 and 6.5 compared to pure-Sch.78 The FA enhanced As mobilization at both pH values, but it was greater at pH 6.5. Humic acid has a skeleton of alkyl and aromatic units with carboxylic acid, phenolic hydroxyl, and quinine functional groups.80 These large molecules can strongly adsorb to the outer particle surfaces (not the interior surfaces) of Sch powders. Therefore, these organic molecules can effectively modify species mobilization and acidity liberation. Furthermore, FA seems to affect Sch transformation more than HA.
The factors mentioned above not only affect the formation of “fresh” Sch, but also influence the long-term storage of Sch products. When Sch is formed in AMD, its transformation to goethite occurs within 2 years in meteoric waters.85 Regenspurg et al.20 suggested that Sch occurrence is transient and restricted to certain environments, such as acidic mining lakes, where Fe3+ coordination is controlled by competition between SO42− and OH. In contrast, synthetic Sch samples are more chemically stable and can remain unchanged after years. This is due to their higher purity and better storage conditions compared to natural Sch forms, as previously mentioned. Furthermore, pH is an important factor that affects the long-term chemical stability of Sch. The effect of pH on the release of As from Sch–As is more obvious at higher pH values. This indicates that As release from Sch–As is mainly controlled by outside environmental factors, such as pH, rather than time.86
3. Environmental remediation application
3.1 Water treatment
Over recent decades, there have been many systematic studies on Sch and its potential use in environmental remediation.87 They have mainly focused on the removal of contaminants in the water phase, such as AsO43−, CrO42−, SbO43−, and F− etc. These reports provide a solid foundation for theoretical research. The details are listed in Table 4.| Source | Composition | Structural/E-K parameters | Conditions | Adsorptions | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fe/S | SO42− % | Morphology | SBET [m2 g−1] | pHzpc | Pollutanta | C0 [mM] | S/L [g L−1] | pHb | T [°C] | T [h] | Qe [mmol g−1] or Qm [mg g−1] | ||
| a The relative atomic masses of As (74.92 ≈ 75), Cr (51.996 ≈ 52), Cu (63.546 ≈ 64), Sb (121.75 ≈ 122), and Se (78.96 ≈ 79), respectively.b The optional pH for adsorption is chosen as a defined value rather than a value range, if provided. | |||||||||||||
| AMD or AMLs | 1.3–16 | 32–19.4 | Large spheroids | 72 (ml 77) | — | — | — | — | — | — | — | — | 20 |
| 5.37 | 16 | — | 173 | 2 | — | — | — | — | — | — | — | 45 | |
| 4.35 | 15.0 | Sphericity | 80 ± 8 | — | As | Trace | — | — | — | — | — | 81 | |
| 4.60 | — | Spherical/elliptical | — | — | — | — | — | — | — | — | — | 15 | |
| 5.30 | — | Spheroidal | 14.7 | — | AsIII | 0.067–1.33 | 5–100 | — | 2–25 | 2 | Qe: 0.0126 | 104 | |
| Chemical oxidation | 3.81 | — | — | — | — | — | — | — | — | — | — | — | 20 |
| 5 | — | — | 4–14 | — | AsIII | 0–60 | 68.0 | 9.0 | — | 60 | Qm: 280.0 | 32 | |
| AsV | 3.0 | Qm: 246.0 | |||||||||||
| 4.71 | 16.3 | Smooth rounded | 5.3 | — | AsIII | 0.13–1.33 | 10 | 2.6 | — | 120 | Qm: 14.16 | 38 | |
| — | — | 40 nm | — | — | — | — | — | — | — | — | — | 35 | |
| 4.32 | 16.2 | Spheroids | — | — | — | — | — | — | — | — | — | 78 | |
| — | — | Closely packed | — | — | — | — | — | — | — | — | — | 43 | |
| — | — | Dendritic | — | — | — | — | — | — | — | — | — | 43 | |
| — | — | Aggregates | 130.9 | 7.1 | SbV | 0–0.49 | 0.3 | 7.0 ± 0.2 | 5 ± 1 | 24 | Qm: ca. 90 | 34 | |
| 4.75 | — | Spheroidal | — | — | — | — | — | — | — | — | — | 63 | |
| 4.71 | — | Spheroidal | 10 | — | — | — | — | — | — | — | — | 64 | |
| 4.17 | — | Sphere shape | 48.18 | — | — | — | — | — | — | — | — | 89 | |
| 4.67–5.04 | — | Spheroids, hedge-hog | 2.06–16.30 | — | AsIII | (3.33–13.3) × 10−4 | 0.25 | 7.0 | 30 | 24 | — | 31 | |
| 5.30 | — | Spheroidal | 50.4 | — | Nitro-Ph | — | — | — | — | — | — | 33 | |
| Fast hydrolysis | 4.04–4.82 | 15.1–16.1 | — | — | — | — | — | — | — | — | — | — | 1 |
| — | — | — | 165 | 4.2 | CrVI | — | — | 4.2 | — | 5 | Qm: 178.6 mg g−1 | 60 | |
| 4.57 | — | Hedge-hog like | 325.52 | 3.05 | — | — | — | — | — | — | — | 62 | |
| — | — | — | 206.1 | — | AsV | (10–35) | 0.3 | 5.0 | 5 ± 1 | 24 | Qm: (104.2) | 82 | |
| 5.30 | 5.31 | — | — | — | AsV | — | 1.0 | 3–4 | 5 | 24 | Qm: 114 | 6 | |
| — | — | Hedge like | — | 3.7 | AsIII | (0.2–0.6) | 0.25–2 | 3–11 | 25 | 4 | Qm: 1.011 | 61 | |
| 4.57 | — | Hedgehog-like | 325.52 | — | AsV | 1.33 | 1 | 3 | 5 | 5 | Qm: 182.86 | 89 | |
| AsIII | 7 | 25 | Qm: 217.85 | ||||||||||
| — | 24 ± 1 | Agglomeration | 5.6 ± 1.2 | — | — | — | — | — | — | — | — | 52 | |
| — | — | — | 199.43 | — | SbII | 0.181 | 0.5/1/2/4 | 7.0 ± 0.2 | 25 ± 1 | 48 | Qm: 84.03 | 92 | |
| SbV | 0.189 | Qm: 78.60 | |||||||||||
| Slow dialysis | — | 13.5 | — | 154 | — | AsV | 0–100.5 | 1 | 3.95 ± 0.04 | 5 | 24 | Qm: 82.4 | 26 |
| 6.15 | — | Hedge-hog | — | — | — | — | — | — | — | — | — | 20 | |
| — | 12.3 | — | — | — | — | — | — | — | — | — | — | 45 | |
| — | — | — | — | — | — | — | — | — | — | — | — | 46 | |
| 5.30 | 15.4 | Uneven surfaces | 210.0 | — | AsIII | 0.13–1.33 | 10 | 2.6 | — | 120 | Qm: 20.08 | 38 | |
| 7.84 | 9.79 | — | 171 | — | As | 0.5–4 | 1 | 4.5–4.6 | 25 ± 1 | 72 | Qm: 196 mmol/molFe | 102 | |
| Cr | 0.15–2 | 4.5 | Qm: 115 mmol/molFe | ||||||||||
| Mo | 0.2–2.5 | 4.5 | Qm: 88 mmol/molFe | ||||||||||
| — | — | Aggregate | — | — | — | — | — | — | — | — | — | 35 | |
| — | — | Irregular-shaped | 199.43 | — | AsV | 0.02–0.27 | 0.5 | 7.0 | 25 ± 1 | 24 | Qm: 31.7 | 82 | |
| Cylindrical-shaped | 189.27 | Qm: 22.3 | |||||||||||
| Spherical-shaped | 32.52 | Qm: 3.6 | |||||||||||
| 5.13 | — | — | — | — | HAsO42- | 0–20 | 1 | 7.00 ± 0.28 | 25 | 24 | Qm: 76.6 | 103 | |
| CrO42- | Qm: 37.6 | ||||||||||||
| SeO42- | Qm: 24.7 | ||||||||||||
| — | — | Macropore/network | 14.81 | — | HA | — | — | — | — | — | Qe: 15.1 | 53 | |
| CrVI | Qe: 4.2 | ||||||||||||
| Biological ferrooxidation | 6.4 | 12.5 | Fibrous aggregates | 14.7 | — | AsIII | 0.13–1.33 | 10 | 3.1 | — | 120 | Qm: 103.39 | 38 |
| 4.7 | 14.9 | Spheroidal | 14.7 | — | AsIII | 2.0 | 10 | 3.0 | 25 | 132 | Qe: 3.746 | 90 | |
| — | — | — | — | — | CrIV | 10 | 0.1 | 2–8 | — | — | Qm: 35.30 | 83 | |
| — | — | Spherical, net like | — | — | CuII | 0.31–3.15 | — | 8.0 | — | 24 | Qm: 10.35–50.14 | 84 | |
| CrVI | 0.39–3.85 | 5.0 | Qm: 30.46–38.82 | ||||||||||
| — | — | Needle, whiskers | — | — | — | — | — | — | — | — | — | 40 | |
| 5.16 | — | Net-work like | 33.52 | — | As | — | — | — | — | — | — | 41 | |
| — | — | — | — | — | As | — | — | — | — | — | — | 42 | |
| — | — | Spherical | 5.14 | — | AsIII | 0.013 | 0.25 | 7.50 | 28 | 4 | Qe: 1.004 | 44 | |
| — | — | 110.06–78.82 | — | Qe: 3.72–2.928 | 44 | ||||||||
| — | — | Sea urchin, flower | 34.55 | — | HA | — | — | — | — | — | Qe: 12.3 | 53 | |
| CrVI | Qe: 5.5 | ||||||||||||
| 6.3 | — | — | 14.7 | — | — | — | — | — | — | — | 24 | ||
| 4.65 | — | Spheric | 92.92 | — | BPA | — | — | — | — | — | — | 51 | |
AsO43− and AsO33−. The presence of As in water and soil systems (dominantly originating from parent rock and human activities) is a serious environmental problem for humans and other living organisms due to its high toxicity and wide distribution in natural environment.88 Even at low concentrations, long-term exposure to inorganic As such as AsO43− or AsO33− can lead to several diseases. Usually, the valence state of As needs to be considered, and as the concentration and pH should be taken into account when predicting and managing As mobility in Sch rich systems.
In general, sorption of AsO43− is greatest at low pH, whereas high pH favours the sorption of AsO33−. Burton32 showed that the actual pH of equivalent AsO43− and AsO33− sorption was strongly load dependent when the pH decreased from ∼8.0 to pH ∼4.6. AsO43− is strongly partitioned to the Sch solid phase at low loadings while AsO33− is weakly adsorbed at low loadings, but has a greater affinity at high loadings. The sorption of AsO43− and AsO33− causes a significant release in SO42− from within the Sch solid-phase, which can be interpreted as As sorption via incorporation into the Sch structure rather than merely surface complexation at the mineral–water interface. Similar results were also reported by Song et al.,89 who found that more AsO43− was adsorbed on Sch at lower pH values, whereas AsO33− sorption increased as the pH rose.
The As loading may also have different influences on its removal. Sorption is governed by multilayer processes, as indicated by highly nonlinear Freundlich adsorption isotherms.38 A relationship between AsO33− uptake and sulfate release is only observed at high initial AsO33− concentrations, which suggests that AsO33− retention through ligand exchange is of minor relevance. Higher AsO33− loadings cause morphological degradation, which lead to angular particles with porous centres, and the extent of the degradation is affected by AsO33− partitioning. Biogenic Sch with an SSA of 14.7 m2 g−1 was reported by Paikaray et al.90 who showed that there was a rapid AsO33− uptake followed by slow retention, possibly into the internal absorbing sites through multilayer and heterogeneous sorption processes. The ionic exchange between SO42− and AsO33−, and surface precipitation governs the total AsO33− uptake, where lower dissolved SO42− and a higher sorbent mass enhance AsO33− retention. The AsO33− ratio affects surface oxidation to AsO43−, whereas AsO33− is the predominant redox species at high AsO33−:Fe3+ ratios. Only 0.83% of adsorbed AsO33− was released, which was subsequently re-adsorbed into Sch during a 4 months stabilization period without mineralogical transformation.
Schwertmannite can also be produced in other forms or shapes, such as irregular, cylindrical, and spherical granules.82 The irregular and cylindrical granules have larger Sch loadings, higher porosities, a more abundant pore structure and larger Vmicro values than the spherical forms. The AsO43− adsorption kinetics follows a pseudo second order rate equation and there is a two-stage of intraparticle diffusion process. The diffusion rate parameter order is irregular granules > cylindrical granules > spherical granules, which is in accordance with their Vp and interparticle porosity. Furthermore, Qe values of 34, 21, and 5 mg g−1, for irregular, cylindrical, and spherical granules, respectively, are achieved at an initial AsO43− concentration of 20 mg L−1 and 0.5 g L−1. Irregular and cylindrical granules perform much better over a wider pH range than spherical granules and can be used for four cycles without reducing the Qm value. Furthermore, the toxicity characteristic leaching procedure (TCLP) results show that the spent irregular and cylindrical granules are inert and could safely be disposed of in landfills.
CrO42−. Chromium is a heavy metal contaminant that is frequently detected in the natural environment. It poses a threat to fauna when present as the CrO42− species due to its higher solubility, mobility, and toxicity, when compared with Cr3+.91 Previous studies have shown that Sch used for CrO42− removal is usually prepared by biological ferrooxidation of the Fe2+ species. Many batches are prepared using slow hydrolysis.
Biogenic Sch has been used to absorb CrO42− in a continuous flow column (1 mL min−1, pH = 6) with a Qm value of 35.30 mg g−1. A possible adsorption mechanism for CrO42− is the anion exchange process between various Cr6+ species and SO42−, a constituent of Sch.83 Gan et al.84 modified biosynthetic Sch with AlPO4. The results showed that the bio-Sch changed from villous spherical aggregates to smooth globules. Furthermore, crystallinity decreased as the AlPO4 content increased. The optimum pH for CrO42− adsorption was at 5.0. The Qm for CrO42− reached 38.82 mg g−1. The modification enhanced CrO42− selective adsorption in a binary metals system. Furthermore, the materials could be effectively regenerated after they were washed in pH 2.0 water. Overall, the biogenic Sch samples had limited adsorption CrO42− capacities, even under acidic conditions, which favour CrO42− remediation. Therefore, the CrO42− absorption properties of Sch produced by alternative methods need to be discussed. Previous studies have suggested that Sch with relatively ordered channel structures may be more effective at CrO42− removal. Therefore, the preparation methods and conditions could possibly be a key issue that needs to be addressed by future research.
SbO43− and SbO33−. Compared to As and Cr, the references about Sb removal by Sch are easily summarized and much fewer in number. In general, the studies on Sb removal by Sch are quite similar to those about As, but are still limited to water phase removal. Previous research suggests that Sb is not easy to remove from water, even though it is in the same family as P and As, Dong et al.34 combined Sch in its main active phase (confirmed by X-ray photoelectron spectroscopy) with graphite oxide (GO) as a composite adsorbent for SbO43− removal so that there could be a synergistic effect between Sch and GO. The Qm value for SbO43− was 158.6 mg g−1 (8.0 mg L−1 SbO43−, pH = 7.0), which was superior to either GO or Sch alone. This was due to the effective incorporation of Sch among GO platelets and its high dispersion on the GO surface. They also used Sch granules to overcome the drawbacks of using small particle-sized adsorbents for SbO33− and SbO43− removal from water.92 Sch granules were able to remove 32.9 mg g−1 SbO33− and 23.2 mg g−1 SbO43, which was superior to most reported granular and powdered absorbents. The SbO33− was effectively removed over a wide pH range, while the removal of SbO43− was pH dependent and could be enhanced by lowering the solution pH. The SbO33−-loaded Sch was regenerated with 91.2% re-adsorption capacity, and this could be repeated five times. However, the Sch adsorption capacity for Sb still needs further research because it is toxic even at low concentrations in water.
Fluorions. Fluorions have a small volume, which means that they easily enter into channel pores to form stable complex compounds. Related studies have shown that many Fe oxides or hydroxides have good adsorption capacities for fluorions, especially, when the pH value is lower than the pHzpc value for Sch. In addition, Sch also possesses many –OH and SO42− groups on the surface, which also exchange with fluorions. Methods involving Sch in the water phase and within the soil system have great potential compared to traditional methods for fluorion removal.
Research by Eskandarpour et al.93 into the adsorption of fluorions onto Sch in a batch system suggests that the adsorption of F by Sch is high, but it is insensitive to changes in temperature and equilibrium pH. The inner-sphere complex-forming species had negative effects on fluorion adsorption, whereas outer-sphere complex-forming species slightly improved fluorion removal efficiency. A nano magnetic Sch prepared by introducing nano-magnetite into Sch94 achieved the permissible limit defined by World Health Organization for removing F from water with a Qe of 17.24 mg g−1 according to the isotherm data. Interference by PO43−, CO32−, and HCO3− was more than by Cl− and SO42−. The spent adsorbents were regenerated with basic solutions and they retained good adsorption efficiencies.
Cd2+, Cu2+, etc. Schwertmannite can also remove cationic contaminants. For example, Cd is a typical toxic contaminant and needs to be removed from wastewater. Fan et al.95 investigated the feasibility of using slow synthetic Sch to remove Cd2+ from aqueous solutions. They used batch experiments that tested different Cd2+ concentrations, temperatures, and pH values. The Langmuir isotherm successfully described the adsorption of Cd2+. A thermodynamic study suggested that absorption was spontaneous and endothermic. The Sch had a high Cd2+ adsorption capacity of 110 mg g−1 at a dosage of 1 g L−1 and an initial pH of 8.0 at 25 °C. At pH > 6.0, Cd adsorption dramatically increased and nearly 100% of the Cd was adsorbed at pH 8.0. In the natural pH range for Sch, the Sch that had adsorbed Cd2+ had good regeneration ability. The Cd2+ desorption proportion of the total sorbed quantity was 50–80% and the desorption rate increased as the pH decreased. Therefore, Sch can be employed as an efficient adsorbent for the removal of Cd from contaminated water. However, there are many other heavy metals, such as copper,84 that could be removed by Sch or Sch-based materials, but these are not discussed in this review.
As we concluded in Table 4, among all the related pollutants, Sch presents better adsorption properties for As and Cr-based pollutants than the other types. This is mainly caused by the unique channel-like structure whose pore diameter is quite close to the size of As or Cr-based ions like AsO43−, AsO33−, or CrO42−, promoting the easy ion-exchange process. As for As-based pollutants, the Fe element in Sch phase can be also used to firmly bond with As-based ions to form more stable phase. This point can also be confirmed by the amounts of literatures, as a proof.
These processes are not time/energy-consuming, which means they may become common Sch regeneration techniques. After regeneration, the adsorption capacity of Sch decreases to a certain degree, but it can still reach the adsorption capacity of fresh batches if the adsorption conditions are adjusted. There are numbers of reasons why Sch can be easily regenerated. It is possible that the basic aqueous solution destroys the surface bonds on the outer surface of Sch or the inner surface of the Sch channels. Fast washing and quick separation without complex processes and special reagents do not influence the Sch structure, which ensures that the adsorption capacity of Sch remains high in subsequent cycles. This easy separation of solid Sch and the regeneration process gives this material wide applications as adsorbent or catalyst in heterogeneous systems. Sch could also be reobtained by using some other functional composites, such as magnetic nanoparticles or hydrophilic/hydrophobic molecules. However, these possibilities require further research.
3.2 Soil remediation
Considering the huge amount of researches about Sch for water treatment, the potential of Sch for treatment on inorganic and organic pollutants could also be technically applied in soil remediation. The first research on the use of Sch for soil remediation appeared in 2016. However, there have not been many studies to date. Biogenetic Sch pre-treated by acidic and alkaline activation (A-Sch)41 achieved an immobilization efficiency of > 99.5% for water-soluble As with an A-Sch dosage of 5% and an Sch dosage of 10%, respectively, in As-contaminated soil. The immobilization percentages for NaHCO3-extractable As increased from 31.5% to 90.4% and from 40.2% to 93.8% when the dosage increase from 0.5 to 10 wt% for Sch and A-Sch, respectively. In general, both Sch and A-Sch effectively immobilize As in contaminated soil, and the immobilization performance of A-Sch is better than Sch, especially at lower dosages. This finding also has important implications for in situ immobilization of As in contaminated soils, especially in soils that have acidic Fe and sulfate-rich environments. This study was finished in 2016 and used an indirect application of Sch. The combination of microbial oxidation (AsO33−-oxidizing bacterium strain YZ-1) (CGMCC No. 8329) and bio-Sch immobilization by Yang et al.42 successfully treated highly As-contaminated soil with immobilization efficiencies of 99.3% and 82.6% for water-soluble and NaHCO3-extractable total As, respectively. This study suggested that the combination could potentially remediate highly As-contaminated soils.Studies about farmlands or other sites are needed so that the Sch remediation of heavy metal contaminated soils can be further evaluated. Sch is quite an effective remediator of As-contaminated soil and perhaps Sb and Cr contaminated soil, although there have been no published reports on these latter two metals. Furthermore, potential problems about production technology, quality control, and cost control have still to be resolved, and the sustainability of contaminated soil remediation by Sch also needs to be examined.
As comparisons, Schs especially the synthetic ones own a comprehensive advantage for water and soil remediation. For example, Sch shows much higher SSA value than the reported organo-bentonites,97 as well as wide application for more types of heavy metals. This mineral also needs facile fabrication, compared to the reported magnesium silicate-hydrothermal carbon composite98 and graphene oxide (or its composites).99,100 As for the adsorption for ionic typed pollutant like AsIII, Sch shows a significant advantage with much higher adsorption than the reported CaMgFe-LDH.101 Technically, the Sch mineral is an easily prepared, more effective, and more potential candidate for types of environmental process, even as a catalyst or carrier.
4. Remediation mechanisms
4.1 Inorganics
It is unrealistic to propose only one Sch method for the removal of heavy metals. Antelo et al.102 proposed two mechanisms: complexation with Fe hydroxyl surface groups and anion exchange with sulfate complexes. Arsenic oxyanion adsorption mainly occurs through the 1st mechanism, whereas CrO42− oxyanions are absorbed by the 2nd mechanism. The adsorption and post adsorption behaviour of Sch towards various oxyanions were investigated by Khamphila et al.103 The results implied that the AsO43−, PO43−, and CrO42− Sch adsorption mechanism was different from SeO42− and SO42−. The AsO43−, PO43−, and CrO42− form inner-sphere complexes, whereas SeO42− and SO42− ions form outer-sphere complexes with the Sch surface. An illustration about the main mechanism path of Sch for water treatment is presented as Fig. 6 shows.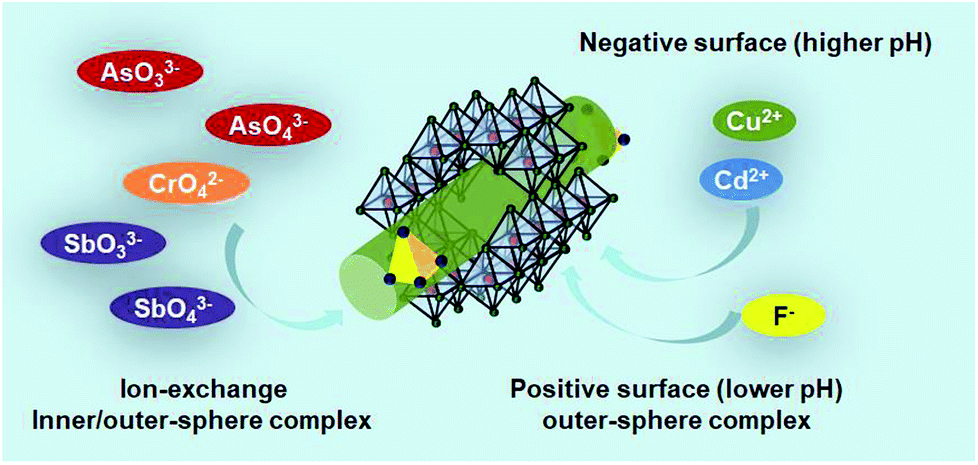 | ||
| Fig. 6 The treatment mechanism of As, Sb, Cr, Cd, Cu, and F-based pollutants by schwertmannite in water. | ||
For AsO33− species, the best fitting models are the Freundlich model38,90 and the Langmuir model.82 The former one shows that AsO33− is retained by multilayer sorption sites with heterogeneous sorption energies105 and the latter model indicates monolayer sorption.
| 0.61(Fe1)2SO4 + 0.39(Fe3)2SO4 + 1.61H2AsO4−→ 1.22Fe1H2AsO4 + 0.39(Fe3)2HAsO4 + 0.39H+ + SO42− | (4) |
The best fitting models for CrO42− are the Freundlich60 and Langmuir models,84 which have negligible differences of R2 constant.
4.2 Organics
The treatment of organic contaminants in water or soil is best undertaken using the Fenton reaction. Although the reactants may differ, the reaction mechanism is quite similar. A possible mechanism for the oxidation of phenol with H2O2 in the presence of Sch62 is shown in Fig. 7. In the oxidation process, intermediates, such as quinines, catechol, oxalic acid, and acetic acid are produced. These organics play an important role in reducing Fe3+ to Fe2+ and lead to the dissolution of Fe oxides. Most of the intermediates are further oxidized and transformed into H2O and CO2. | ||
| Fig. 7 The Fenton reaction when catalysed by schwertmannite. | ||
In this mechanism, the identification of ·OH is required. Methanol was chosen as an effective ·OH scavenger to identify whether the ·OH type reaction has degraded the nitrobenzene,33 and Fe leaching and H2O2 decomposition confirmed the success of the process. The effect of other kinds of ·OH scavengers was also investigated during the degradation of bisphenol A.51 In the degradation reaction, free ·OH was generated in the bulk solution along with some intermediates, including p-isopropenyl phenol, p-benzoquinone, and methyl propionate, which proved the occurrence of a Fenton-like process on the Sch surface.
5. Conclusions and perspectives
Schwertmannite is a potential material that can be used for environmental remediation. This mineral has a unique channel structure and ion-exchange properties, which gives it an excellent adsorption capacity for inorganic contaminants in the water phase and catalytic degradation ability for organic contaminants, resulting in potential application in environmental remediation of water and soil. In the remediation process, the stability of Sch is an important factor that significantly influences its application, owing to the easy transformation of metastable Sch into other minerals. So far, Sch is not just a mineral or clay, but also an adsorbent or catalyst. Novel functional Sch materials in further investigation are always concerned. However, there are still lots of questions about the synthesis and application of schwertmannite as follows:(1) The schwertmannite samples fabricated via various methods and conditions present quite remarkable differences in material's characteristics and the adsorption properties of pollutants. The chemical oxidation, fast hydrolysis, slow dialysis, and biological ferrooxidation process are closely connected with distinct microstructures, pore-structure, and most importantly the adsorption properties. This phenomenon can be observed even at similar conditions. The relationship between the synthetic strategies or methods and materials' properties should be discussed from different views to provide more clarity about the controllable optimization aiming at the practical application.
(2) The environmental application of schwertmannite mainly focuses on water treatment while those about soil remediation are still in starting stage. Technically, the researches about the former aspect mainly revolve industrial waste water and acid mine drainage. In 2016, the study in schwertmannite for soil remediation was firstly reported with lots of attentions. Afterwards, more discussions are eagerly necessary for further application.
(3) Schwertmannite is a metastable amorphous Fe-based mineral, which is quite easy to transform into goethite phase. Such phase-transforming process has become an important and necessary issue. Schwertmannite can exist in long-term during the remediation process of soil and underground water, requiring necessity about the study for its phase-transformation and stability. Several researches about these factors of natural/synthetic or As/Cr-doped Schwertmanites at different pH, temperature, co-existing ions, and organic matter have been reported in succession. Nevertheless, the issues about how the phase transformation occurs, how the pollutants release, and how long the release will last, are still lack of related simulation and tracking study, especially in the system of underground water and soil.
(4) As a potential catalytic material, schwertmannite can be used for the degradation of organic pollutants basically as efficient Fenton-like catalysts. Until now, kinds of aromatic compounds (such as phenolic and BPA) have already been examined as candidates, and excellent catalytic property in short time has been achieved. However, its recycling ability and chemical stability during the process of chemical reactions still need more deep study.
(5) Many paths can be used for the promotion of adsorption properties of Sch to various pollutants, such as thermal modification, acidic modification, organic modification, and even shape granulations. Other strategies and technologies for structure optimization and functionalization are particularly concerned for more systematic researches.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Engineering Laboratory for Site Remediation Technologies (NEL-SRT201705) and the National Natural Science Foundation of China (Grant No. 41071165) for their financial support.Notes and references
- J. Y. Yu, M. Park and J. Kim, Geochem. J., 2002, 36, 119–132 CrossRef CAS.
- D. Richard and G. W. Luther III, Chem. Rev., 2007, 107, 514–562 CrossRef PubMed.
- R. W. Fitzpatrick, L. M. Mosley, M. D. Raven and P. Shand, Geoderma, 2017, 308, 235–251 CrossRef CAS.
- J. M. Bigham, U. Schwertmann, S. J. Traina, R. L. Winland and M. Wolf, Geochim. Cosmochim. Acta, 1996, 60, 2111–2121 CrossRef CAS.
- R. J. J. Barham, Mater. Res., 1997, 12, 2751–2758 CrossRef CAS.
- S. HoungAloune, T. Kawaai, N. Hiroyoshi and M. Ito, Hydrometallurgy, 2014, 147–148, 30–40 CrossRef CAS.
- K. S. Brady, J. M. Bigham, W. F. Jaynes and T. J. Logan, Clays Clay Miner., 1986, 34, 226–274 CrossRef.
- E. J. Murad, J. Mag. Mag. Mater., 1988, 74, 153–157 CrossRef CAS.
- J. M. Bigham, U. Schwertmann, I. Carlson and E. Murad, Geochim. Cosmochim. Acta, 1990, 54, 2743–2758 CrossRef CAS.
- J. M. Bigham, L. Carlson and E. Murad, Mineral. Mag., 1994, 58, 641–648 CrossRef CAS.
- U. Schwertmann, J. M. Bigham and E. Murad, Eur. J. Mineral., 1995, 7, 547–552 CrossRef CAS.
- K. Fukushi, M. Sasaki, T. Sato, N. Yanase, H. Amano and H. Ikeda, Appl. Geochem., 2003, 18, 1267–1278 CrossRef CAS.
- G. S. Zhou, L. X. Zhou and F. X. Chen, Spectrosc. Spectral Anal., 2007, 27, 367–370 Search PubMed.
- Y. H. Huang, J. H. Cai and Y. W. Cao, Acta Petrol. Mineral., 1999, 18, 50–64 Search PubMed.
- H. F. Sun, F. H. Zhao, Z. Y. Cong, M. Yue and D. Y. Ren, Acta Mineral. Sin., 2006, 26, 38–42 CAS.
- M. Q. Chen, G. N. Lu, C. L. Guo, C. F. Yang, J. X. Wu, W. L. Huang, N. Yee and Z. Dang, Chemosphere, 2015, 119, 734–743 CrossRef CAS PubMed.
- C. F. Yang, G. N. Lu, M. Q. Chen, Y. Y. Xie, C. L. Guo, J. Reinfelder, X. Y. Yi, H. Wang and Z. Dang, Geoderma, 2016, 281, 21–29 CrossRef CAS.
- X. H. Shu, Q. Zhang, G. N. Lu, X. Y. Yi and Z. Dang, Int. Biodeterior. Biodegrad., 2016, 128, 122–128 CrossRef.
- Y. P. Bao, C. L. Guo, H. Wang, G. N. Lu, C. F. Yang, M. Q. Chen and Z. Dang, Geomicrobiol. J., 2017, 34, 692–705 CrossRef.
- S. Regenspurg, A. Brand and S. Peiffer, Geochim. Cosmochim. Acta, 2004, 68, 1185–1197 CrossRef CAS.
- E. Murad, J. M. Bigham, L. H. Bowen and U. Schwertmann, Hyperfine Interact., 1990, 58, 2373–2376 CrossRef CAS.
- J. L. Bishop and E. Murad, Mineral Spectroscopy: A Tribute to Roger G. Burns, ed. M. D.Dyar, C.McCammon and M. W.Schaefer, DLR, 1996, pp. 337–358 Search PubMed.
- E. Murad and P. Rojik, Super soil 3rd Australian New Zealand Soils Conference, 2004 Search PubMed.
- S. Paikaray, C. Schröder and S. Peiffer, Geochim. Cosmochim. Acta, 2017, 217, 292–305 CrossRef CAS.
- E. Murad and P. Rojík, Clay Miner., 2005, 40, 427–440 CrossRef CAS.
- M. Loan, J. M. Cowley, R. Hart and C. M. Parkinson, Am. Mineral., 2004, 89, 1735–1742 CrossRef CAS.
- K. Fukushi, T. Sato, N. Yanase, J. Minato and H. Yamada, Am. Mineral., 2004, 89, 1728–1734 CrossRef CAS.
- A. Fernandez-Martinez, V. Timon, G. Roman-Ross, G. J. Cuello, J. E. Daniels and C. Ayora, Am. Mineral., 2010, 95, 1312–1322 CrossRef CAS.
- S. Regenspurg and S. Peiffer, Appl. Geochem., 2005, 20, 1226–1239 CrossRef CAS.
- J. M. Bigham, U. Schwertmann and G. Pfab, Appl. Geochem., 1996, 11, 845–849 CrossRef.
- F. W. Liu, J. Zhou, S. S. Zhang, L. L. Liu, L. X. Zhou and W. H. Fan, PLoS One, 2015, 10, e0138891 CrossRef PubMed.
- E. D. Burton, R. T. Bush, S. G. Johnston, K. M. Watling, R. K. Hocking, L. A. Sulliwan and G. K. Parker, Environ. Sci. Technol., 2009, 43, 9202–9207 CrossRef CAS PubMed.
- H. Y. Duan, Y. Liu, X. H. Yin, J. F. Bai and J. Qi, Chem. Eng. J., 2016, 283, 873–879 CrossRef CAS.
- S. X. Dong, X. M. Dou, D. Mohan, C. U. Pittman Jr and J. M. Luo, Chem. Eng. J., 2015, 270, 205–214 CrossRef CAS.
- H. F. Sun, F. H. Zhao and S. Wu, Acta Petrol. Mineral., 2013, 32, 1006–1012 CAS.
- A. Stefansson, Environ. Sci. Technol., 2007, 41, 6117–6123 CrossRef CAS PubMed.
- L. Reichelt and M. Bertau, Z. Anorg. Allg. Chem., 2015, 641, 1696–1700 CrossRef CAS.
- S. Paikaray, J. Göttlicher and S. Peiffer, Chem. Geol., 2011, 283, 134–142 CrossRef CAS.
- H. X. Xiong, Y. H. Liao and L. X. Zhou, Environ. Sci. Technol., 2008, 42, 8681–8686 CrossRef CAS PubMed.
- J. F. Mori, S. P. Lu, M. Händel, K. U. TotSche, T. R. Neu, V. V. Iancu, N. Tarcea, J. Popp and K. Küsel, Microbiology, 2016, 162, 62–71 CrossRef CAS PubMed.
- L. Y. Chai, J. W. Tang, Y. P. Liao, Z. H. Yang, L. F. Liang, Q. Z. Li, H. W. Wang and W. C. Yang, J. Soils Sediments, 2016, 16, 2430–2438 CrossRef CAS.
- Z. H. Yang, Z. J. Wu, Y. P. Liao, Q. Liao, W. C. Yang and L. Y. Chai, Chemosphere, 2017, 181, 1–8 CrossRef CAS PubMed.
- R. A. French, N. Monsegue, M. Murayama and M. F. Hochella Jr, Phys. Chem. Miner., 2014, 41, 237–246 CrossRef CAS.
- X. X. Qiao, L. L. Liu, J. Shi, L. X. Zhou, Y. H. Guo, Y. Y. Ge, W. H. Fan and F. W. Liu, Minerals, 2017, 7, 1–14 CrossRef CAS.
- J. Jönsson, P. Persson, S. Sjöberg and L. Lövgren, Appl. Geochem., 2005, 20, 179–191 CrossRef.
- J. F. Boily, P. L. Gassman, T. Peretyahko, J. Szanyi and J. M. Zachara, Environ. Sci. Technol., 2010, 44, 1185–1190 CrossRef CAS PubMed.
- R. N. Collins, A. M. Jones and T. D. Waite, Geochim. Cosmochim. Acta, 2010, 74, 482–496 CrossRef CAS.
- F. Maillot, G. Morin, Y. Wang, D. Bonnin, P. Ildefonse, C. Chaneac and G. Calas, Geochim. Cosmochim. Acta, 2011, 75, 2708–2720 CrossRef CAS.
- G. A. Waychunas, N. Xu, C. C. Fuller, J. A. Davis and J. M. Bigham, Phys. B, 1995, 208&209, 481–483 Search PubMed.
- M. Sakamaki, H. Takahashi, T. Konishi, A. Inoue and T. Fujikawa, AIP Conf. Proc., 2007, 882, 271–273 CrossRef CAS.
- X. Li, Y. K. Zhang, Y. Xie, Y. Zeng, P. Y. Li, T. H. Xie and Y. B. Wang, J. Hazard. Mater., 2018, 344, 689–697 CrossRef CAS PubMed.
- L. Reichelt and M. Bertau, Chem. Eng. Res. Des., 2015, 98, 70–80 CrossRef CAS.
- C. Y. Zhang, Z. F. Zhang, M. N. Chen and D. G. Fu, RSC Adv., 2017, 7, 27895–27899 RSC.
- R. M. Taylor, Clay Miner., 1980, 15, 369–382 CrossRef CAS.
- X. F. Yang, Z. X. Sun, D. S. Wang and W. Forsling, J. Colloid Interface Sci., 2007, 308, 395–404 CrossRef CAS PubMed.
- X. Bi, T. Fan and H. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 20498–20509 CrossRef CAS PubMed.
- H. Zhang and X. N. Zhang, Geoderma, 1992, 54, 173–188 CrossRef CAS.
- G. A. Parks, Chem. Rev., 1965, 65, 177–198 CrossRef CAS.
- Y. H. Liao, J. R. Liang and L. X. Zhou, Chemosphere, 2011, 83, 295–301 CrossRef CAS PubMed.
- A. Eskandarpour, M. S. Onyango, M. Tananhashi, A. Ochieng, Y. Bando, K. Iwai, M. Okado and S. Asai, ISIJ Int., 2008, 48, 240–244 CrossRef CAS.
- A. Dey, R. Singh and M. K. Purkait, J. Water Process Eng., 2014, 3, 1–9 CrossRef.
- W. M. Wang, J. Song and X. Han, J. Hazard. Mater., 2013, 262, 412–419 CrossRef CAS PubMed.
- C. L. Vithana, L. A. Sullivan, E. D. Burton and R. T. Bush, Geoderma, 2015, 239–240, 47–57 CrossRef CAS.
- C. L. Vithana, L. A. Sullivan, R. T. Bush and E. D. Burton, Geoderma, 2015, 249–250, 51–60 CrossRef CAS.
- E. D. Burton, S. G. Johnston, P. Kraal, R. T. Bush and S. Claff, Environ. Sci. Technol., 2013, 47, 2221–2229 CrossRef CAS PubMed.
- S. Kumpulainen, M. L. Räisänen, F. Von der Kammer and T. Hofmann, Clay Miner., 2008, 43, 437–448 CrossRef CAS.
- S. Paikaray and S. Peiffer, Mine Water Environ., 2015, 34, 213–222 CrossRef CAS.
- S. G. Johnston, E. D. Burton and E. M. Moon, Environ. Sci. Technol., 2016, 50, 8010–8019 CrossRef CAS PubMed.
- W. Stumm, Colloids Surf., A, 1997, 120, 143–166 CrossRef CAS.
- J. D. Kubicki, D. Tunega and S. Kraemer, Chem. Geol., 2017, 464, 14–22 CrossRef CAS.
- A. Amirbahman, L. Sigg and U. V. Gunten, J. Colloid Interface Sci., 1997, 194, 194–206 CrossRef CAS PubMed.
- H. Carbbe, N. Fernandez and F. Jones, J. Cryst. Growth, 2015, 416, 28–33 CrossRef.
- Y. Y. Xie, G. N. Lu, H. Ye, C. F. Yang, X. Y. Yi and Z. Dang, J. Environ. Qual., 2017, 46, 1088–1097 CrossRef CAS PubMed.
- S. Wang and C. Mulligan, Environ. Geochem. Health, 2006, 28, 197–214 CrossRef CAS PubMed.
- J. Chen, B. Gu, R. A. Royer and W. D. Burgos, Sci. Total Environ., 2003, 307, 167–178 CrossRef CAS PubMed.
- L. Weng, W. H. V. Riemsdijk, L. K. Koopal and T. Hiemstra, Environ. Sci. Technol., 2006, 40, 7494–7500 CrossRef CAS PubMed.
- L. Weng, W. H. V. Riemsdijk and T. Hiemstra, Environ. Sci. Technol., 2009, 43, 7198–7204 CrossRef CAS PubMed.
- C. L. Vithana, L. A. Sullivan, E. D. Burton and R. T. Bush, Chem. Geol., 2014, 372, 1–11 CrossRef CAS.
- Y. Y. Xie, G. N. Lu, H. Ye, C. F. Yang, D. Xia, X. Y. Yi, J. Reinfelder and Z. Dang, Chem. Geol., 2017, 475, 52–61 CrossRef CAS.
- J. F. Liu, Z. S. Zhao and G. B. Jiang, Environ. Sci. Technol., 2008, 42, 6949–6954 CrossRef CAS PubMed.
- P. Acero, C. Ayora, C. Torrento and J. M. Nieto, Geochim. Cosmochim. Acta, 2006, 70, 4130–4139 CrossRef CAS.
- X. M. Dou, D. Mohan and C. U. Pittman Jr, Water Res., 2013, 47, 2938–2948 CrossRef CAS PubMed.
- C. Y. Yu, J. Zhang, X. L. Wu, Y. Q. Lan and L. X. Zhou, Geochem. J., 2014, 48, 1–7 CrossRef CAS.
- M. Gan, S. J. Sun, Z. H. Zheng, H. J. Tang, J. R. Sheng, J. Y. Zhu and X. X. Liu, Appl. Surf. Sci., 2015, 356, 986–997 CrossRef CAS.
- N. J. Welehan, K. A. Malatt and S. Vukcevic, Miner. Eng., 2000, 13, 911–931 CrossRef.
- S. HoungAloune, N. Hiroyoshi and M. Ito, Miner. Eng., 2015, 74, 51–59 CrossRef CAS.
- R. P. Schwarzenbach, T. Egli, T. B. Hofstetter, U. V. Gunten and B. Wehrli, Annu. Rev. Environ. Resour., 2010, 35, 109–136 CrossRef.
- P. Miretzky and A. F. Cirelli, Crit. Rev. Environ. Sci. Technol., 2010, 40, 93–115 CrossRef CAS.
- J. Song, S. Y. Jia, H. Y. Ren, S. H. Wu and X. Han, Int. J. Environ. Sci. Technol., 2015, 12, 1559–1568 CrossRef CAS.
- S. Paikaray, J. Gottlicher and S. Peiffer, Chemosphere, 2012, 86, 557–564 CrossRef CAS PubMed.
- B. Dhal, H. N. Thatoi, N. N. Das and B. D. Pandey, J. Hazard. Mater., 2013, 250–251, 272–291 CrossRef CAS PubMed.
- Y. H. Li, D. Mohan, C. U. Pittman Jr, Y. S. Ok and X. M. Dou, Desalin. Water Treat., 2016, 57, 25639–25652 CrossRef CAS.
- A. Eskandarpour, M. S. Onyango, A. Ochieng and S. Asai, J. Hazard. Mater., 2008, 152, 571–579 CrossRef CAS PubMed.
- A. Goswami and M. K. Purkait, J. Water Process Eng., 2014, 1, 91–100 CrossRef.
- L. J. Fan and X. J. Zhang, Desalin. Water Treat., 2017, 79, 243–250 CrossRef CAS.
- G. C. C. Yang, S. C. Huang, C. L. Wang and Y. S. Jen, Chemospheres, 2016, 159, 282–292 CrossRef CAS PubMed.
- K. Yu, J. Xu, X. H. Jiang, C. Liu, W. McCall and J. L. Lu, Chemosphere, 2017, 184, 884–891 CrossRef CAS PubMed.
- T. Xiong, X. Z. Yuan, X. H. Chen, Z. B. Wu, H. Wang, L. J. Leng, H. Wang, L. B. Jiang and G. M. Zeng, Appl. Surf. Sci., 2018, 427, 1107–1117 CrossRef CAS.
- T. Xiong, X. Z. Yuan, H. Wang, L. J. Leng, H. Li, Z. B. Wu, L. B. Jiang, R. Xu and G. M. Zeng, J. Environ. Manage., 2018, 205, 99–106 CrossRef CAS PubMed.
- T. Wen, X. L. Wu, X. L. Tan, X. K. Wang and A. W. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 3304–3311 CrossRef CAS PubMed.
- J. Z. Zhou, W. K. Shu, Y. Gao, Z. B. Cao, J. Zhang, H. Hou, J. Zhao, X. P. Chen, Y. Pan and G. R. Qian, RSC Adv., 2017, 7, 20320–20326 RSC.
- J. Antelo, S. Fiol, D. Gondar, R. López and F. Arce, J. Colloid Interface Sci., 2012, 386, 338–343 CrossRef CAS PubMed.
- K. Khamphila, R. Kodama, T. Sato and T. Otake, J. Miner. Mater. Charact. Eng., 2017, 5, 90–106 CAS.
- S. Paikaray, J. Essilfie-Dughan, J. Göttlicher, K. Pollok and S. Peiffer, J. Hazard. Mater., 2014, 265, 208–216 CrossRef CAS PubMed.
- W. J. Weber and F. A. DiGiano, Process Dynamics in Environmental Systems, Wiley Interscience, New York, 1996 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |