
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn ‘‘off–on–off’’ sensor for sequential detection of Cu2+ and hydrogen sulfide based on a naphthalimide–rhodamine B derivative and its application in dual-channel cell imaging†
Shuai
Wang
,
Haichang
Ding
,
Yuesong
Wang
,
Congbin
Fan
 ,
Yayi
Tu
,
Gang
Liu
* and
Shouzhi
Pu
*
,
Yayi
Tu
,
Gang
Liu
* and
Shouzhi
Pu
*
Jiangxi Key Laboratory of Organic Chemistry, Jiangxi Science and Technology Normal University, Nanchang, Jiangxi 330013, P. R. China. E-mail: liugang0926@163.com; pushouzhi@tsinghua.org.cn; Fax: +86-791-83831996; Tel: +86-791-83831996
First published on 25th September 2018
Abstract
A novel colorimetric and fluorometric sensor with unique dual-channel emission to sequentially detect Cu2+ and hydrogen sulfide (H2S) was synthesized from naphthalimide–rhodamine B through the PET and FRET mechanism. The sensor showed a selective “off–on” fluorescence response with a 120-fold increase toward Cu2+, and its limits of detection were 0.26 μM and 0.17 μM for UV-vis and fluorescence measurements, respectively. In addition, 1–Cu2+ was an efficient “on–off” sensor to detect H2S with detection limits of 0.40 μM (UV-vis measurement) and 0.23 μM (fluorescence measurement), respectively. Furthermore, the sensor can also be used for biological imaging of intracellular staining in living cells. Therefore, the sensor should be highly promising for the detection of low level Cu2+ and H2S with great potential in many practical applications.
Introduction
As many metal ions/anions play a significant role in various biological and environmental processes,1 Cu2+ is an essential transition metal ion in some physiological and pathological processes and as a significant catalytic cofactor for the synthesis of hemoglobin, elastin and collagen.2,3 However, an abnormal amount of Cu2+ has significant negative effects on human health since excessive Cu2+ can cause oxidative stress and neurological disorders, such as Alzheimer's, Parkinson's and Wilson's diseases.4–7 Therefore, the development of sensitive and selective methods to detect Cu2+ in biological and environment samples is very important. In the past decade, several techniques have been developed to detect Cu2+, such as high-performance liquid chromatography, inductively coupled plasma atomic emission spectrometry, mass spectrometry and atomic absorption spectrometry.8–10 However, these techniques are laborious and time-consuming, and require expensive instruments. On the contrary, fluorescent chemosensors are better and preferable approach for the recognition of Cu2+ due to their simplicity, high selectivity and sensitivity, and rapid response. Therefore, sensitive fluorescent chemosensors to trace and visualize Cu2+ are highly demanded.Over the past few years, selective recognition of anions has also gained much attention due to their vital role in biological systems,11 but detection of anion under aqueous condition is still very challenging because water molecules compete with anions for binding sites.12 Recently, many metal complex-based receptors have been employed, such as Cu2+/CN−,13–16 Cu2+/H2S,17 Hg2+/I−,18 Zn2+/H2S,19 Al3+/F−,20,21 Cr3+/F−,22 Ni2+/CN−,23 and Zn2+/H2PO4−/CN−.24 In the metal replacement process, the binding capacity of anions and metal ions is greater than that of metal ions and sensors, and metal ions escape from the complex and restore to its original state, which leads to a change in the intensity or wavelength of absorption or emission spectra.25 Compared with the traditional detection approaches, such as colorimetric and electrochemical assays, gas chromatography, the required equipments are complicated, time-consuming and costly, the utilization of known metal-anion affinities that displays the metal ion from the sensor-ions complex releasing the ligand into the solution is an important method for sensing anions, due to its simple operation and high detection sensitivity and simplicity.
Additionally, hydrogen sulfide (H2S) is closely associated with these physiological processes, such as mediation of neurotransmission, relaxation of vascular smooth muscle and inhibition of insulin signaling.26–28 However, excessive H2S can have a negative impact on human health.29 Therefore, it is also significant to develop powerful recognition and detection systems to monitor H2S. So far, utilization of copper-sulfide affinity for sensing H2S has been reported (ESI Table 1†), most of them exhibit changes only in fluorescence intensity at one emission wavelength. By contrast, a dual-channel fluorescence-enhanced/quenched sensor displays fluorescence enhancement/quenched at two emission channels simultaneously upon binding with an analyte. Therefore, the development of dual-channel fluorescent sensors is exceedingly sought because of their particularly favorable features such as high sensitivity, selectivity, and convenient visible emission assays due to improved signal-to-noise ratio.30 However, to the best of our best knowledge, no sensor with fluorescence-enhanced for sensing Cu2+ and fluorescence-quenched in dual-channel emissions for detecting H2S has been previously reported.
In this study, a new sensor for the sequential detection of Cu2+ and H2S was synthesized based on naphthalimide–rhodamine B derivative. The framework of rhodamine B was used to detect Cu2+, and the strategy of metal displacement was employed to detect H2S. In addition, the sensor was successfully applied to the biological imaging application in living cells. The whole detecting process for Cu2+ and H2S not only provides a vivid sight by “naked eyes” with significant changes in both color (light yellow-pink-light yellow), but also displays a selective fluorescence “off–on–off” switching.
Experimental
Reagents
All reagents were of analytical grade and purchased from commercial suppliers without further purification before use. The stock solutions of metal ions were prepared through dissolving HgCl2, KCl, MnCl2·4H2O, Al(NO3)3·9H2O, Cd(NO3)2·4H2O, Ba(NO3)2, Cu(NO3)2·3H2O, Ni(NO3)2·6H2O, Pb(NO3)2, Zn(NO3)2·6H2O, Fe(NO3)3·9H2O, Cr(NO3)3·9H2O, Sr(NO3)2, Co(NO3)2·6H2O, Ca(NO3)2·4H2O and Mg(NO3)2·6H2O in distilled water, respectively. To get an aqueous solution (0.1 mol L−1) of each inorganic salt, these inorganic salts (1 mmol) dissolved in distilled water (10 mL), respectively. The solutions of anion (0.1 mol L−1 of NO3−, NO2−, SO42−, SO32−, HSO4−, HSO3−, S2O32−, PO43−, SCN−, H2S, CN−, CO32−, HCO3−, CH3COO−, F−, Cl−, Br− or I−) were prepared in deionized water. Aqueous sulfide (Na2S) served as the H2S donor,31–33 and was used to produce H2S. The obtained stock solutions, before use, were diluted to required concentrations when needed.Instrumentation
NMR spectra were recorded on a Bruker AV400 (1H NMR 400 MHz, 13C NMR 100 MHz) instrument for sample solution in CD2Cl2 with tetramethylsilane (TMS) as an internal reference. Mass spectra were collected on an Agilent 1100 ion trap MSD spectrometer. Melting point was measured with a WRS-1B melting point apparatus. Elemental analysis was carried out with a PE CHN 2400 analyzer. UV-vis spectra were measured on an Agilent 8453 UV/vis spectrophotometer. Fluorescence spectra were recorded on a Hitachi F-4600 fluorescence spectrophotometer. Infrared spectra (IR) were collected on a Bruker Vertex-70 spectrometer. Fluorescence imaging of cells were performed with an OLYMPUS FV1000 confocal laser scanning microscope.Methods
A stock solution of 1 (1.0 × 10−3 mol L−1) was prepared in CH3CN. The fluorescence and absorption studies on 1 were performed with 2 mL CH3CN–H2O (9/1, v/v, 2.0 × 10−5 mol L−1) solution and appropriate amounts of metal ions at room temperature. In order to make the metal ions chelate with the sensors sufficiently, solutions were kept for about 2 h before the absorption and fluorescent intensity was measured. In the FT-IR experiments, samples (1, 1–Cu2+ and 1–Cu2+ + H2S) were prepared in advance and dried in a drying oven and were mixed with KBr prior to spectral acquisition. A pure KBr background was collected and subtracted from all sample spectra prior to statistical. All spectra were recorded in the range 600–4000 cm−1 with an accumulation of 16 scans and 4 cm−1 spectral resolution. In the anion detection experiments, 1–Cu2+ complex solution was gained after 2.0 eq. Cu2+ was added into 2 mL CH3CN–H2O (7/3, v/v) solution of 1 (2.0 × 10−5 mol L−1), and the absorbance and fluorescence spectral data were recorded after 5 min since the addition of the various anions. Fluorescence measurements were carried out with excitation and emission slit of 5 nm and 5 nm (λex = 325 nm).Calculation of detection limits
The emission intensity and absorbance of sensor 1 were measured 10 times and the standard deviation of blank measurements was determined. To gain the slope, the fluorescence intensity of 1 (2.0 × 10−5 mol L−1) at 610 nm or the ratio change of absorbance (A564/A425) was plotted as a concentration of Cu2+.The detection limit was calculated with the following equation:34,35
| Detection limit = 3σ/k |
Cell culture and imaging
HeLa cells (human cervical cancer cell) were cultured in DMEM (Dulbecco modified Eagle's medium) supplemented with 10% FBS (fetal bovine serum) in an atmosphere of 5% CO2 and 95% air at 37 °C. The cells were plated in 20 mM cell culture dishes and allowed to adhere overnight before experiments. After the HeLa cells were washed with phosphate-buffered saline (PBS) buffer, the cells were incubated with sensor 1 (20 μM) in the culture medium for 30 min. After the HeLa cells were rinsed with PBS three times, the cells were imaged with an OLYMPUS FV1000 confocal laser scanning microscope and the fluorescence emissions were collected at 460–560 nm for channel 1 and 570–670 nm for channel 2 (excited at 405 nm).Synthesis of the sensor 1
The synthetic route for 1 was shown in Scheme 1. Compound 2, 3, 4 and 5 were synthesized by the reported methods.36–39 To a stirring mixture of compound 3 (0.15 g, 0.40 mmol) and compound 5 (0.24 g, 0.45 mmol) in CH3CN (40 mL), and K2CO3 (0.069 g, 0.50 mmol) was added. After refluxing for 12 h, the mixture was cooled to room temperature. Then the solvent was removed and the crude product purified by column chromatography on silica gel using petroleum ether/ethyl acetate (v/v, 2/1) as the eluent to obtain target compound 1 (0.23 g, 0.27 mmol) in 67.5% yield. Mp 432–433 K; 1H NMR (400 MHz, CD2Cl2), δ (ppm): 8.56 (d, J = 8.0 Hz, 1H), 8.47 (d, J = 8.0 Hz, 1H), 8.36 (d, J = 4 Hz, 2H), 7.92 (d, J = 8.0 Hz, 1H), 7.70 (t, J = 8.0 Hz, 1H), 7.57–7.50 (m, 2H), 7.12–7.07 (m, 2H), 6.62 (d, J = 8.0 Hz, 2H), 6.33 (d, J = 8.0 Hz, 2H), 6.28 (s, 2H), 4.39 (t, J = 4.0 Hz, 2H), 3.81 (t, J = 4.0 Hz, 2H), 3.62 (s, 4H), 3.32–3.22 (m, 8H), 3.11 (s, 2H), 3.02 (s, 4H), 2.64 (s, 4H), 1.05 (t, J = 8.0 Hz, 12H) (Fig. S1†); 13C NMR (100 MHz, CD2Cl2), δ (ppm): 166.30, 164.11, 163.56, 163.09, 154.96, 152.64, 150.57, 148.05, 132.15, 131.29, 130.06, 129.37, 128.88, 128.33, 127.42, 125.18, 124.76, 122.89, 122.19, 122.13, 115.68, 113.85, 107.03, 103.11, 96.36, 71.36, 67.36, 64.98, 60.68, 59.36, 43.30, 38.41, 11.30 (Fig. S2†); ESI-MS (m/z): 866.42 [1 + H+]+ (Fig. S3†). Anal. calcd for C50H55N7O7: C, 69.34; H, 6.41; N, 11.32; O, 12.93%. Found: C, 69.27; H, 6.45; N, 11.35; O, 12.90%.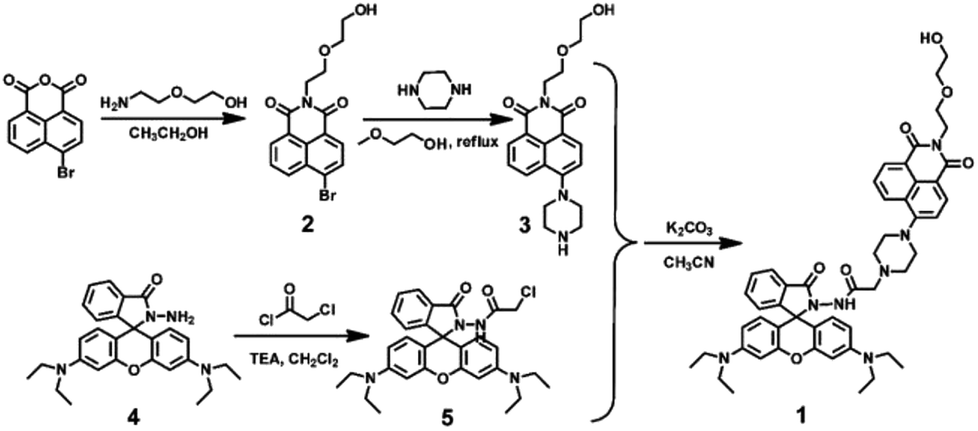 | ||
| Scheme 1 Structure and synthesis of 1. | ||
Results and discussion
Absorption and fluorescence spectral responses of sensor 1 to metal ions
Firstly, selectivity studies were performed with various metal ions (Fe3+, Al3+, Ca2+, Cd2+, Co2+, Cu2+, Ba2+, Cr3+, Hg2+, K+, Sr2+, Mg2+, Mn2+, Ni+, Pb2+ and Zn2+). As shown in Fig. 1A and D, only Cu2+ caused significant absorption change of the light yellow solution of 1 with a distinct absorption peak at 564 nm and a color change from light yellow to pink, which corresponded to the characteristic absorption peak of the ring-opened form of the rhodamine B moiety. Meanwhile, no significant response could be observed upon the addition of other metal ions. Similarly, as shown in Fig. 1B, the selectivity of the sensor 1 to various cations was observed in the fluorescence spectra. The sensor 1 showed non-fluorescence upon its excitation at 325 nm in the absence of metal ions in CH3CN–H2O (v/v, 9/1) solution. However, two fluorescence emission bands were generated with the maximum at 528 nm and 610 nm upon the addition of Cu2+. Simultaneously, the fluorescent color changed from green to light yellow due to formation of complex 1–Cu2+ (Fig. 1E). Although other competing metal ions caused slight fluorescent intensity to enhance, such as Fe3+, Al3+ and Cr3+, the enhanced fluorescent intensity was far less than that of Cu2+ under the same conditions. These results indicated that sensor 1 could be a “turn-on” chemosensor and serve as a “naked-eyes” chemosensor to selectively detect Cu2+ over the other metal ions.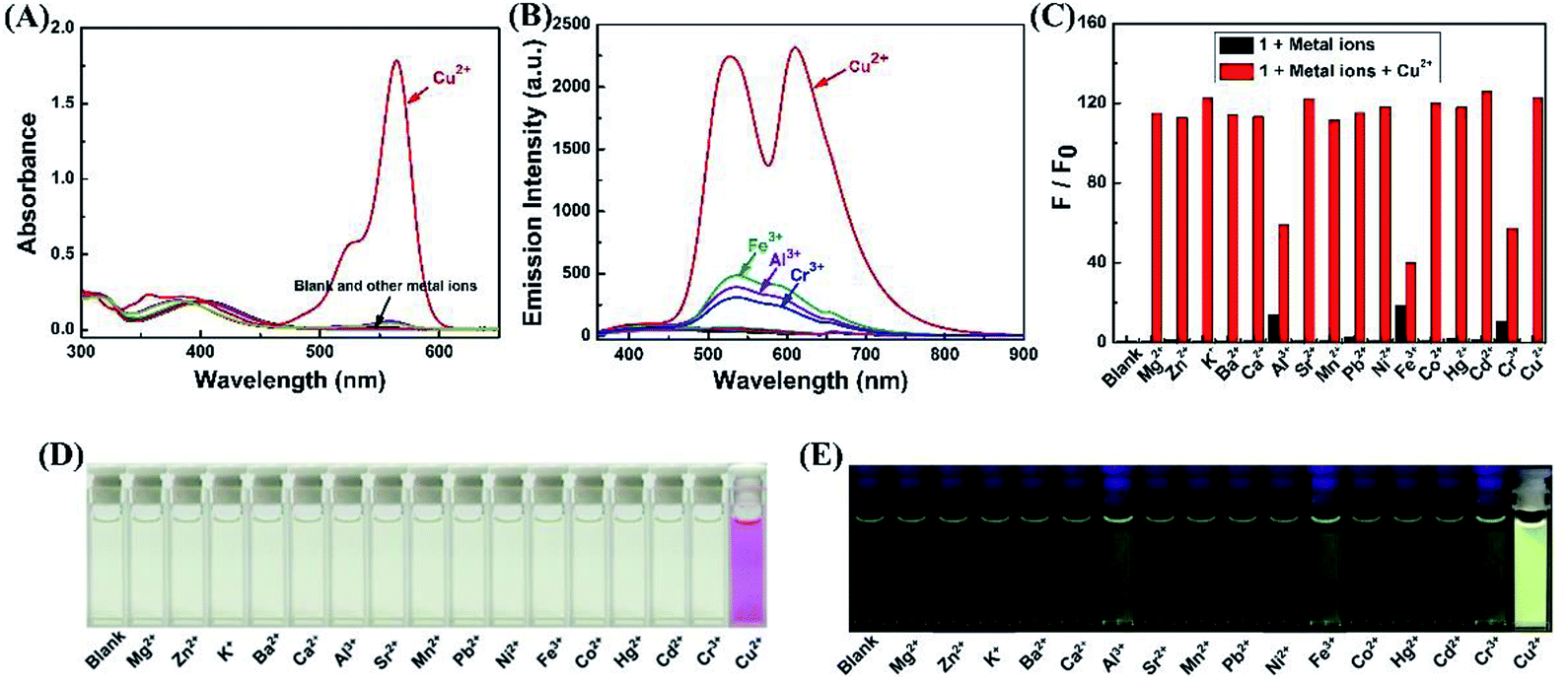 | ||
| Fig. 1 Spectroscopic changes of 1 (20 μM) with the addition of various metal ions in CH3CN–H2O (9/1, v/v) solution. (A) Absorption spectral changes. (B) Fluorescence emission intensity changes. (C) The fluorescence intensity changes at 610 nm (black bars: 1 with other ions, red bars: 1 with Cu2+ and other metal ions). (D) The color image with the addition of various metal ions. (E) The fluorescence image with the addition of various metal ions. | ||
Subsequently, as shown in Fig. 1C, competition experiments were carried out through the monitoring of the change in the fluorescence intensity at 610 nm upon the addition of 2.0 eq. of Cu2+ to a solution of the sensor 1 in the presence of other metal ions (5.0 eq.). The result suggested that the Cu2+-induced fluorescence responses were not affected by the competitive metal ions, expect for Fe3+, Al3+ and Cr3+, the fluorescent intensity was much weaker than that of Cu2+. Thus, the Cu2+-selective binding and “turn-on” response could take place in the coexistence of the competitive metal ions, indicating the designed sensor could recognize Cu2+ with high anti-interference ability even in the presence of other metal ions.
To acquire a better understanding of the reaction of sensor 1 with Cu2+, time dependent modulations in the fluorescence spectra were monitored. The fluorescence intensity at 610 nm was plotted as a function of time. As shown in Fig. 2A and B, upon the addition of Cu2+ (2.0 eq.) to the CH3CN–H2O (9/1, v/v) solution of sensor 1. With the extended response time, the fluorescence intensity at 610 nm increased dramatically within 10 min. The fluorescence intensity at 610 nm slowly increased, when the reaction time was more than 10 min. Finally, the fluorescence intensity no longer changes and remains at its maximum after 1 h.
 | ||
| Fig. 2 (A) Time-dependent fluorescence intensity changes of sensor 1 to Cu2+ in CH3CN–H2O (9/1, v/v) solution. (B) Relative fluorescence intensity at 610 nm as a function of time in CH3CN–H2O (9/1, v/v) solution. | ||
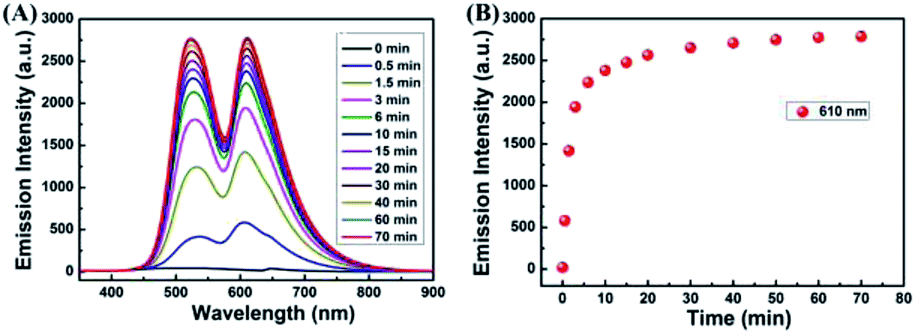
In order to investigate the optical response of the sensor to Cu2+, absorption and fluorescence titration studies were performed. Different concentrations of Cu2+ from 0–2.0 eq. were added into in CH3CN–H2O (v/v, 9/1) solution of the sensor 1 (20 μM). As shown in Fig. 3A, upon the addition of Cu2+ to the solution of 1, three new absorption bands centered at 357 nm, 425 nm and 564 nm changed with significant color change from light yellow to pink. Upon the addition of Cu2+, the absorption band at 425 nm was gradually decreased, the bands at 357 nm and 564 nm were gradually increased, and two isosbestic points were shown at 403 nm and 465 nm, respectively. These results indicated that several components existed in the equilibrium.40,41 A linear dependence of the ratio change of absorbance (A564/A425, red dots line) and the ratio change of absorbance (A357/A425, black dots line) as a function of Cu2+ concentration were observed. When the concentration of Cu2+ was increased up to 2.0 eq., the ratio of absorbance (A564/A425, A357/A425) all stayed unchanged. For example, within a certain concentration range (0–35 μM), the ratio change of absorbance (A564/A425) exhibited a good linear relationship (Y = 0.52887X − 0.44648, R = 0.9957) to the concentration of Cu2+ (Fig. S4†), and the detection limit was determined to be 0.26 μM based on 3σ/k (Fig. S5†) (σ is the standard deviation of the blank signal and k is the slope of the linear calibration plot).42
 | ||
| Fig. 3 Absorbance spectra (A) and fluorescence spectra (B) of 1 (20 μM) in the presence of different amounts of Cu2+ in CH3CN–H2O (9/1, v/v) solution. Inset of (A): the ratio of absorbance at 564 nm and absorbance at 425 nm (red dots line) and the ratio of absorbance at 357 nm and absorbance at 425 nm (black dots line) as a function of Cu2+ concentration. Inset of (B): the fluorescence intensity at 528 nm and 610 nm as a function of Cu2+ concentration. | ||
As shown in Fig. 3B, without Cu2+, the sensor exhibited non-fluorescence, upon the addition of Cu2+ (0–2.0 eq.) to the CH3CN–H2O (9/1, v/v) solution of 1, two fluorescence emission bands appeared with the maximum at 528 nm and 610 nm, which is the characteristic of the naphthalimide moiety and rhodamine B group, respectively. However, beyond a certain concentration (2.0 eq.) until reached 5.0 eq., further addition of Cu2+ did not cause any change in the fluorescence intensity. For example, the fluorescence titration curve showed a steady increase with the increase of Cu2+ concentration at 610 nm, which demonstrated an efficient fluorescence response. A good linearity was found between the concentration of Cu2+ and the fluorescence intensity was displayed in the range 0–40 μM (Y = 60.5712X + 109.64832 (R = 0.9876)) (Fig. S6†). The fluorescence detection limit was 0.17 μM based on 3σ/k (Fig. S7†), which was much lower than the maximum level of Cu2+ (20 μM) in drinking water (Environmental Protection Agency defined limit).41,43 These results indicated that the sensor 1 is a potential fluorescent sensor for the detection of low level Cu2+ in practical applications.
Absorption and fluorescence spectral responses of 1–Cu2+ complex to anions
Due to the high stability of CuS, the Cu2+ should bind H2S stronger over sensor 1, and the 1–Cu2+ complex should act as a detecting ensemble for H2S recognition.As shown in Fig. 4A, when the same amount of the various anions (8 eq., NO3−, NO2−, SO42−, SO32−, HSO4−, HSO3−, S2O32−, PO43−, SCN−, H2S, CN−, CO32−, HCO3−, CH3COO−, F−, Cl−, Br− and I−) was added to the 1–Cu2+ complex, only H2S caused significant change in the absorption spectrum, the absorbance at 564 nm was decreased remarkably with visible color change from pink to light yellow (Fig. 4D). Similarly, as shown in Fig. 4B and E, only H2S caused two fluorescence emission peaks at 528 nm and 610 nm to quench dramatically and the fluorescence color was changed from bright yellow to green. With the addition of other anions to the solution of 1–Cu2+, none of those other anions caused significant change in the fluorescent spectrum, except that CO32− caused negligible change in the fluorescence intensity compared to H2S. The anti-interference performance of the 1–Cu2+ to H2S was also further confirmed in competitive tests with other anions. As shown in Fig. 4C, the fluorescence emission spectra at 610 nm displayed a similar pattern to that with H2S alone, and coexistence of equal amounts of other anions did not have any significant interference on the H2S recognition, suggesting that all the tested anions did not interfere the sensing of H2S. Therefore, the 1–Cu2+ complex had remarkable selectivity and excellent anti-interference toward H2S.
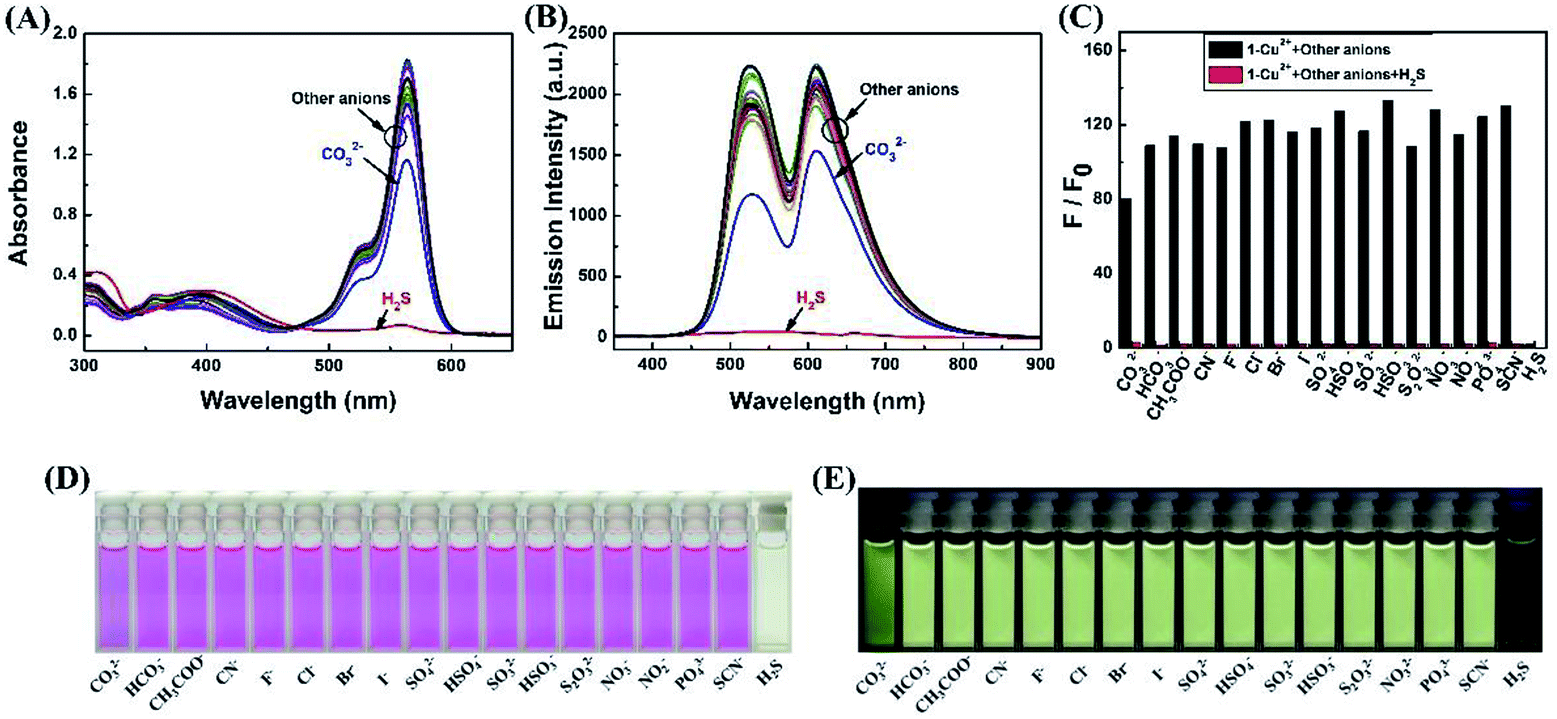 | ||
| Fig. 4 (A) Spectroscopic changes of 1–Cu2+ complex (20 μM) with the addition of various anions in CH3CN–H2O (7/3, v/v) solution. (A) Absorption spectral changes. (B) Fluorescence emission intensity changes. (C) The fluorescence intensity changes at 610 nm (black bars: 1–Cu2+ complex with other anions, red bars: 1–Cu2+ complex with H2S and other anions). (D) The color image with the addition of various anions. (E) The fluorescence image with the addition of various anions. | ||
To research the time-dependence of 1–Cu2+ complex on H2S detection, the fluorescence emission intensity of 1–Cu2+ complex solution was studied. As shown in Fig. 5A and B, when H2S was present in the CH3CN–H2O (7/3, v/v) solution of 1–Cu2+ complex, the fluorescence intensity of 1–Cu2+ complex completely quenched and reached a steady state within 30 s. These results showed that the sensor 1 would provide method for the detection of Cu2+, and the H2S detection by 1–Cu2+ complex could be potentially used for real-time detection.
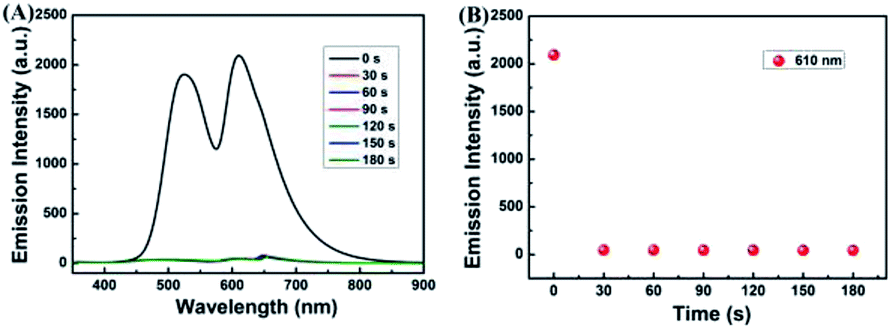 | ||
| Fig. 5 (A) Time-dependent fluorescence intensity changes of sensor 1–Cu2+ to H2S in CH3CN–H2O (7/3, v/v) solution. (B) Relative fluorescence intensity at 610 nm as a function of time in CH3CN–H2O (7/3, v/v) solution. | ||
To further investigate the interaction between H2S and 1–Cu2+ complex, the absorption and fluorescence titration experiments were performed in CH3CN–H2O (7/3, v/v) solution. As shown in Fig. 6A, the absorption titration of 1–Cu2+ complex with an increasing amount of H2S in the range of 0–8.0 eq. in CH3CN–H2O (7/3, v/v) solution was investigated. With the increase of H2S concentration, the absorbance was gradually decreased at 564 nm and the solution of color was changed from pink to light yellow, which indicated that the coordination of 1–Cu2+ system was broken and spirolactam ring of the rhodamine turned to the closing state. An excellent linear correlation (Y = −0.1638X + 16.31734, R2 = 0.9772) between the ratio change of absorbance (A564/A425) and the H2S concentrations from 0 to 80 μM was observed (Fig. S8†), and the detection limit was estimated to 0.40 μM based on 3σ/k (Fig. S9†). Similarly, as shown in Fig. 6B, when the 1–Cu2+ complex was excited at 325 nm, the fluorescence intensity around 528 nm and 615 nm was decreased with the continuous addition of H2S, and the fluorescence intensity of 1–Cu2+ complex was completely quenched when 4.0 eq. H2S were added. As shown in Fig. S10,† there is a linear response relationship between the fluorescence intensity of 1–Cu2+ complex at 610 nm and H2S concentration within the range of 0–40 μM, and the linear equation was found to be Y = −39.5825X + 1920.65909 (R2 = 0.9985). The fluorescence detection limit was measured to be 0.23 μM based on 3σ/k (Fig. S11†).
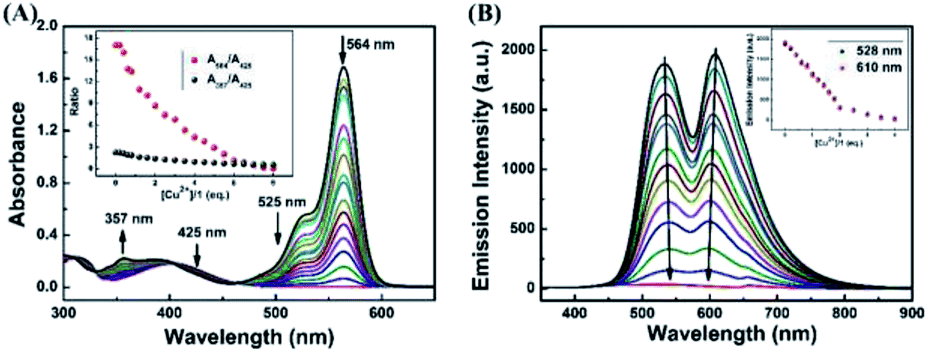 | ||
| Fig. 6 (A) Absorbance and (B) fluorescence spectra of 1–Cu2+ complex in the presence of different amounts of H2S in CH3CN–H2O (7/3, v/v) solution. Inset of (A): the ratio of absorbance at 564 nm and absorbance at 425 nm (red dots line) and the ratio of absorbance at 357 nm and absorbance at 425 nm (black dots line) as a function of Cu2+ concentration. Inset of (B): the fluorescence intensity at 528 nm and 610 nm as a function of Cu2+ concentration. | ||
Detection mechanism for Cu2+ and H2S
The possible mechanism to detect Cu2+ was studied through emission spectra and ESI-MS spectra. A Job plot experiments was carried out to study the binding stoichiometry between Cu2+ and sensor 1 (Fig. 7A). In this experiment, the total concentration of 1 and Cu2+ was kept constant (40 μM). The maximum fluorescence emission intensity at 610 nm was reached when the molar fraction was 0.5, which implied that the binding stoichiometry between Cu2+ and 1 is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Further confirmation of 1:1 stoichiometry was done with ESI-MS analysis, in which the peak at m/z 888.4084, 928.3478, 963.3177 and 1027.3954 in the mass spectrum could be assigned to the mass of [1+ Na+]+, [1+ Cu2+ − H+]+, [1+ Cu2+ + 2H2O − H+]+ and [1+ Cu2+ + 2H2O + CH3CN + Na+ − 2H+]+, respectively. These results confirmed the 1:1 stoichiometry between Cu2+ and sensor 1 in the 1–Cu2+ complex (Fig. 7B). Meanwhile, rhodamine B group structure changed from spirolactam (non-fluorescent) to ring-opening form in response to Cu2+ could be explained with 13C NMR spectrum, in which the 13C NMR signal of the tertiary carbon (64.98 ppm) in the spirolactam ring of sensor 1 disappeared in the presence of Cu2+ (Fig. 7C).
:1. Further confirmation of 1:1 stoichiometry was done with ESI-MS analysis, in which the peak at m/z 888.4084, 928.3478, 963.3177 and 1027.3954 in the mass spectrum could be assigned to the mass of [1+ Na+]+, [1+ Cu2+ − H+]+, [1+ Cu2+ + 2H2O − H+]+ and [1+ Cu2+ + 2H2O + CH3CN + Na+ − 2H+]+, respectively. These results confirmed the 1:1 stoichiometry between Cu2+ and sensor 1 in the 1–Cu2+ complex (Fig. 7B). Meanwhile, rhodamine B group structure changed from spirolactam (non-fluorescent) to ring-opening form in response to Cu2+ could be explained with 13C NMR spectrum, in which the 13C NMR signal of the tertiary carbon (64.98 ppm) in the spirolactam ring of sensor 1 disappeared in the presence of Cu2+ (Fig. 7C).
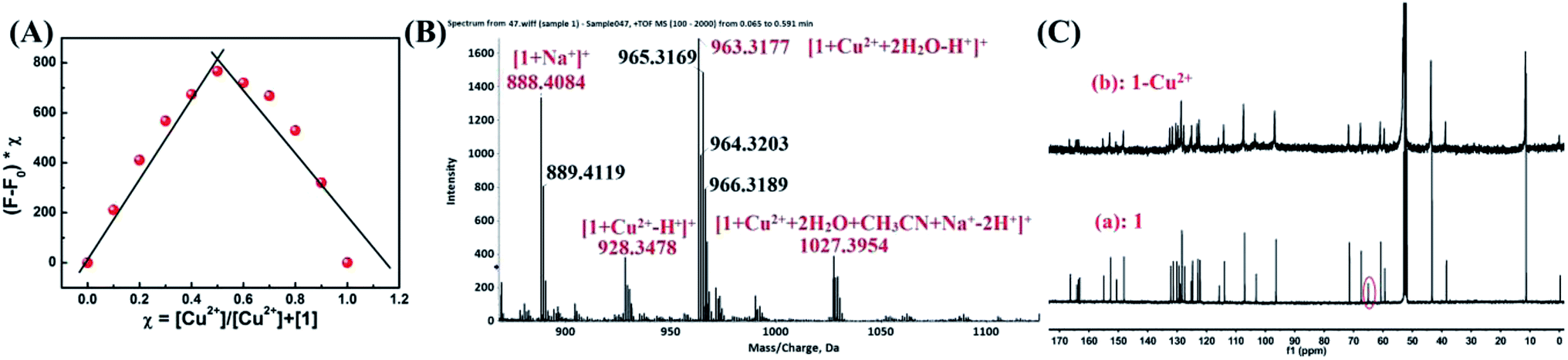 | ||
| Fig. 7 (A) Job's plot for the complexation of 1 with Cu2+, indicating the formation of a 1:1 complex. The total [1] + [Cu2+] = 40 μM. (B) ESI-MS spectrum of 1–Cu2+. (C) 13C NMR spectra of 1 upon addition of Cu2+ in CD2Cl2. | ||
FT-IR measurements were performed to investigate the mechanism of interactions between 1–Cu2+ complex and H2S. The FT-IR spectra of 1, 1–Cu2+ and 1–Cu2+ + H2S were shown in Fig. 8. As illustrated in Fig. 8A, the two absorption peaks at 1724 cm−1 and 1616 cm−1 could be attributed to the antisymmetric and symmetric stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (the rhodamine B unit).44 By comparing the FT-IR spectra of 1 (Fig. 8A) and 1–Cu2+ (Fig. 8B), the absorption band of 1–Cu2+ at 1724 cm−1 disappeared and the absorption intensity at 1590 cm−1 was significantly enhanced, which could be caused by the absorption peak shifting from 1610 cm−1 to 1590 cm−1 after the addition of Cu2+, indicating that the oxygen atom of spirolactam of 1 in the carbonyl group did participate in the coordination. The strong absorption peak at 1384 cm−1 was attributed to the excess Cu(NO3)2. As shown in Fig. 8C, the υ(CO) (the rhodamine B unit) at 1724 cm−1 and 1616 cm−1 were restored, which indicated the re-formation of spirolactam from ring-opened amide. Therefore, it can be concluded that the rhodamine spirolactam of sensor 1 was transformed to ring-opened amide through the forming 1–Cu2+ complex, then re-formed spirolactam after the addition of H2S, which led the escape of Cu2+ from 1–Cu2+ complex and the Cu2+ form a very stable species with the targeting H2S because CuS (Ksp of CuS = 1.27 × 10−36)45 was more stable than 1–Cu2+ complex.
O (the rhodamine B unit).44 By comparing the FT-IR spectra of 1 (Fig. 8A) and 1–Cu2+ (Fig. 8B), the absorption band of 1–Cu2+ at 1724 cm−1 disappeared and the absorption intensity at 1590 cm−1 was significantly enhanced, which could be caused by the absorption peak shifting from 1610 cm−1 to 1590 cm−1 after the addition of Cu2+, indicating that the oxygen atom of spirolactam of 1 in the carbonyl group did participate in the coordination. The strong absorption peak at 1384 cm−1 was attributed to the excess Cu(NO3)2. As shown in Fig. 8C, the υ(CO) (the rhodamine B unit) at 1724 cm−1 and 1616 cm−1 were restored, which indicated the re-formation of spirolactam from ring-opened amide. Therefore, it can be concluded that the rhodamine spirolactam of sensor 1 was transformed to ring-opened amide through the forming 1–Cu2+ complex, then re-formed spirolactam after the addition of H2S, which led the escape of Cu2+ from 1–Cu2+ complex and the Cu2+ form a very stable species with the targeting H2S because CuS (Ksp of CuS = 1.27 × 10−36)45 was more stable than 1–Cu2+ complex.
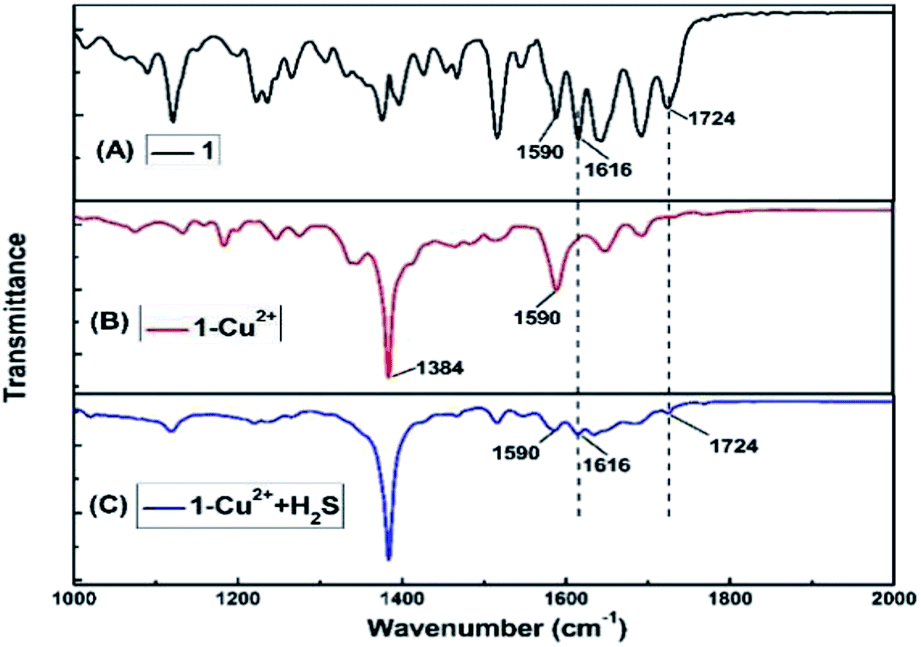 | ||
| Fig. 8 Partial FT-IR spectra of 1 (A), 1–Cu2+ (B) and 1–Cu2+ + H2S (C). | ||
As shown in Fig. 9, without Cu2+, the sensor 1 showed non-fluorescence upon its excitation at 325 nm because the fluorescence of amino-naphthalimide fluorophore was quenched by the electron lone pair in the amino group of piperazine moiety through photo-induced electron transfer (PET) process.46,47 When Cu2+ was added, Cu2+ formed 1–Cu2+ complex with the amino group on the piperazine of sensor 1, which interrupted the PET process and caused fluorescence emission peaks of naphthalimide moiety at 528 nm, which indicated that the position of N atom in sensor 1 participate in the coordination with Cu2+ complexes.48 Meanwhile, rhodamine B group changed from spirocyclic (non-fluorescent) form to ring-opening one in response to Cu2+, resulting in fluorescence emission at 610 nm. In this process, the absorption spectrum of the open spirocycle form of the rhodamine B developed a significant overlap with the emission spectrum of naphthalimide moiety and clearly offered the possibility of a FRET process (Fig. S12†).
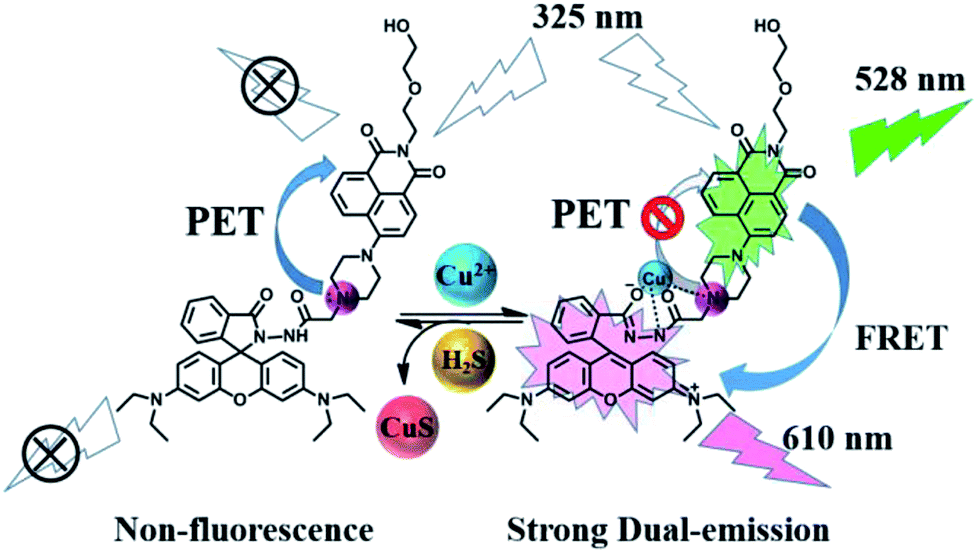 | ||
| Fig. 9 The proposed mechanism of 1 to sense Cu2+ and 1–Cu2+ to sense H2S. | ||
Bioimaging applications
To demonstrate the potential application in the biological systems, HeLa cells were chosen to investigate the preliminarily application viability of the sensor 1 in intracellular imaging. The cells treated were imaged with confocal microscopy (OLYMPUS FV1000) upon the laser irradiation at 405 nm. The HeLa cells were incubated with sensor 1 (20 μM) for 30 min, and then HeLa cells were washed three times with PBS. As shown in Fig. 10B and C, the cells showed fluorescence from the intracellular region at channel 1 (460–560 nm) and channel 2 (570–670 nm). These preliminary results suggested that the sensor is permeable to cell membrane and can be used to the biological imaging application for intracellular staining with dual-channel fluorescence emission in living cells, which may have great potentials in bio-medical applications. | ||
| Fig. 10 Confocal fluorescence images of HeLa cells: (A) bright field image (B) dark field image of channel 1 (emission collected at 460–560 nm, pseudo-color: green); (C) dark field image of channel 2 (emission collected at 560–670 nm, pseudo-color: red); (D) merged images of A, B and C. Fluorescence emissions were collected at an excitation wavelength of 405 nm with a [40×] objective. | ||
Conclusions
In conclusion, a fluorescent “off–on–off” sensor for the sequential detection of Cu2+ and H2S based on naphthalimide–rhodamine B was designed and synthesized. The sensor displayed high selectivity and sensitivity to Cu2+ over other cations. The Cu2+ detection of sensor 1 with a 1:1 binding stoichiometry was adequately supported by ESI-MS data and Job's plot analysis. Additionally, 1 could be used to the biological imaging application in living cells. More importantly, the 1–Cu2+ complex could be successfully applied to selectively detect H2S over other anions through a de-complexation reaction. Therefore, the sensor 1 could be used to detect trace amount of Cu2+ and H2S with remarkable changes in both color and fluorescence, which was greatly desirable for biological detection and imaging.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (21662015, 21363009, 41867052), the “5511” science and technology innovation talent project of Jiangxi, the key project of Natural Science Foundation of Jiangxi Province (20171ACB20025), the Science Funds of Natural Science Foundation of Jiangxi Province (20171BAB203014, 20171BAB203011), the Masters' Innovative Foundation of Jiangxi Science and Technology Normal University (YC2017-X30).Notes and references
- D. T. Quang and J. S. Kim, Chem. Rev., 2010, 110, 6280–6301 CrossRef CAS PubMed.
- Y. Wang, C. Zhang, X. Chen, B. Yang, L. Yang, C. Jiang and Z. Zhang, Nanoscale, 2016, 8, 5977–5984 RSC.
- C. Xie, L. Xiao, S. Peng and X. Shi, New J. Chem., 2014, 38, 6095–6102 RSC.
- Z. Hu, J. Hu, Y. Cui, G. Wang, X. Zhang, K. Uvdal and H. W. Gao, J. Mater. Chem. A, 2014, 2, 4467–4472 CAS.
- G. Multhaup, A. Schlicksupp, L. Hesse, D. Beher, T. Ruppert, C. L. Masters and K. Beyreuther, Science, 1996, 271, 1406–1409 CrossRef CAS.
- A. Pal, M. Siotto, R. Prasad and R. Squitti, J. Alzheimer's Dis., 2015, 44, 343–354 CAS.
- E. Carboni and P. Lingor, Metallomics, 2015, 7, 395–404 RSC.
- Z. Xiao, F. Loughlin, G. N. George, G. J. Howlett and A. G. Wedd, J. Am. Chem. Soc., 2004, 126, 3081–3090 CrossRef CAS.
- S. Wustoni, S. Hideshima, S. Kuroiwa, T. Nakanishi, Y. Mori and T. Osaka, Analyst, 2015, 140, 6485–6488 RSC.
- D. S. Volkov, M. A. Proskurnin and M. V. Korobov, Carbon, 2014, 74, 1–13 CrossRef CAS.
- P. A. Gale, S. E. García-Garrido and J. Garric, Chem. Soc. Rev., 2008, 37, 151–190 RSC.
- H. Sharma, V. K. Bhardwaj, N. Kaur, N. Singh and D. O. Jang, Tetrahedron Lett., 2013, 54, 5967–5970 CrossRef CAS.
- G. J. Park, I. H. Hwang, E. J. Song, H. Kim and C. Kim, Tetrahedron, 2014, 70, 2822–2828 CrossRef CAS.
- A. Helal, S. B. Kim and H. S. Kim, Bull. Korean Chem. Soc., 2011, 32, 3123–3126 CrossRef CAS.
- L. Tang and M. Cai, Sens. Actuators, B, 2012, 173, 862–867 CrossRef CAS.
- L. Tang, N. Wang, Q. Zhang, J. Guo and R. Nandhakumar, Tetrahedron Lett., 2013, 54, 536–540 CrossRef CAS.
- D. Zhang and W. Jin, Spectrochim. Acta, Part A, 2012, 90, 35–39 CrossRef CAS.
- F. Ma, W. Shi, H. Mi, J. Luo, Y. Lei and Y. Tian, Sens. Actuators, B, 2013, 182, 782–788 CrossRef CAS.
- Z. Dong, X. Le, P. Zhou, C. Dong and J. Ma, New J. Chem., 2014, 38, 1802–1808 RSC.
- Y. Mi, Z. Cao, Y. Chen, S. Long, Q. Xie, D. Liang, W. Zhu and J. Xiang, Sens. Actuators, B, 2014, 192, 164–172 CrossRef CAS.
- Y. S. Mi, D. M. Liang, Y. T. Chen, X. B. Luo and J. N. Xiang, RSC Adv., 2014, 4, 42337–42345 RSC.
- H. Liu, X. Wan, T. Liu, Y. Li and Y. Yao, Sens. Actuators, B, 2014, 200, 191–197 CrossRef CAS.
- J. H. Kang, S. Y. Lee, H. M. Ahn and C. Kim, Sens. Actuators, B, 2017, 242, 25–34 CrossRef CAS.
- A. Pandith, N. Uddin, C. H. Choi and H. S. Kim, Sens. Actuators, B, 2017, 247, 840–849 CrossRef CAS.
- H. Sharma, N. Kaur, A. Singh, A. Kuwar and N. Singh, J. Mater. Chem. C, 2016, 4, 5154–5194 RSC.
- K. Abe and H. Kimura, J. Neurosci., 1996, 16, 1066–1071 CrossRef CAS.
- G. Yang, L. Wu, B. Jiang, W. Yang, J. Qi, K. Cao, Q. Meng, A. K. Mustafa, W. Mu, S. Zhang, S. H. Snyder and R. Wang, Science, 2008, 322, 587–590 CrossRef CAS.
- Y. Kaneko, Y. Kimura, H. Kimura and I. Niki, Diabetes, 2006, 55, 1391–1397 CrossRef CAS.
- W. J. Cai, M. J. Wang, L. H. Ju, C. Wang and Y. C. Zhu, Cell Biol. Int., 2014, 34, 565–572 CrossRef.
- M. Ren, B. Deng, J. Y. Wang and Z. R. Liu, J. Mater. Chem. B, 2015, 3, 6746–6752 RSC.
- Y. Qian, J. Karpus, O. Kabil, S. Y. Zhang, H. L. Zhu, R. Banerjee, J. Zhao and C. He, Nat. Commun., 2011, 2, 495 CrossRef.
- C. Zhang, G. Zhang, L. Feng and J. Li, Sens. Actuators, B, 2015, 216, 412–417 CrossRef CAS.
- M. L. Aulsebrook, S. Biswas, F. M. Leaver, M. R. Grace, B. Graham, A. M. Barrios and K. L. Tuck, Chem. Commun., 2017, 53, 4911–4914 RSC.
- D. Guo, Z. P. Dong, C. Luo, W. Y. Zan, S. Q. Yan and X. J. Yao, RSC Adv., 2014, 4, 5718–5725 RSC.
- C. Kar, M. D. Adhikari, A. Ramesh and G. Das, Inorg. Chem., 2013, 52, 743–752 CrossRef CAS.
- M. V. Ramakrishnam Raju, E. Chandra Prakash, H. C. Chang and H. C. Lin, Dyes Pigm., 2014, 103, 9–20 CrossRef CAS.
- S. Liu, H. Bai, Q. Sun, W. Zhang and J. Qian, RSC Adv., 2015, 5, 2837–2843 RSC.
- S. Goswami, S. Maity, A. C. Maity, A. K. Maity, A. K. Das and P. Saha, RSC Adv., 2014, 4, 6300–6305 RSC.
- H. C. Ding, B. Q. Li, S. Z. Pu, G. Liu, D. C. Jia and Y. Zhou, Sens. Actuators, B, 2017, 247, 26–35 CrossRef CAS.
- W. N. Wu, P. D. Mao, L. Jia, Y. Wang and Z. Q. Xu, Spectrochim. Acta, Part A, 2016, 166, 44–48 CrossRef CAS.
- Z. Q. Xu, X. J. Mao, Y. Wang, W. N. Wu, P. D. Mao, X. L. Zhao, Y. C. Fan and H. J. Li, RSC Adv., 2017, 7, 42312–42319 RSC.
- L. He, X. Yang, Y. Liu and L. Weiying, Anal. Methods, 2016, 8, 8022–8027 RSC.
- Y. Ding, S. Z. Shen, H. Sun, K. Sun and F. Liu, Sens. Actuators, B, 2014, 203, 35–43 CrossRef CAS.
- R. Tabit, O. Amadine, Y. Essamlali, K. Danoun, A. Rhihil and M. Zahouily, RSC Adv., 2018, 8, 1351–1360 RSC.
- Y. F. Zhu, D. H. Fan and W. Z. Shen, J. Phys. Chem. C, 2008, 112, 10402–10406 CrossRef CAS.
- X. Zhou, F. Su, H. Lu, P. Senechal-Willis, Y. Tian, R. H. Johnson and D. R. Meldrum, Biomaterials, 2012, 33, 171–180 CrossRef CAS.
- J. Fan, M. Hu, P. Zhan and X. Peng, Chem. Soc. Rev., 2013, 42, 29–43 RSC.
- W. H. Zhu, L. J. Shen and H. Tian, Angew. Chem., 2007, 119, 5645–5649 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05963b |
| This journal is © The Royal Society of Chemistry 2018 |