
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen-doped micro-nano carbon spheres with multi-scale pore structure obtained from interpenetrating polymer networks for electrochemical capacitors†
Bing Hua,
Wei-Bin Zhanga,
Kun Yana,
Tong Zhanga,
Kai Lia,
Xi-Wen Chena,
Long Kangb and
Ling-Bin Kong *ab
*ab
aState Key Laboratory of Advanced Processing and Recycling of Non-ferrous Metals, Lanzhou University of Technology, Lanzhou 730050, P. R. China. E-mail: konglb@lut.cn; Fax: +86-931-2976578; Tel: +86-931-2976579
bSchool of Materials Science and Engineering, Lanzhou University of Technology, Lanzhou 730050, P. R. China
First published on 12th October 2018
Abstract
A chemical process was developed to prepare N-doped micro-nano carbon spheres with multi-scale pore structures via carbonization of N-PF/PMMA interpenetrating polymer networks, which contain melamine resin as the nitrogen source, PF as the carbon source, and polymethylmethacrylate (PMMA) as the pore-former. The N-content of N-doped micro-nano carbon spheres was controlled by adjusting the mass ratio of melamine and phenol before polymerization. The N-doped micro-nano carbon spheres as electrode materials possess appropriate pore size distribution, higher specific surface area (559 m2 g−1) and consistently dispersed nitrogen atoms with adjustable doping content. These distinct characteristics endow the prospective electrode materials with excellent performance in electrochemical capacitors. In particular, N-CS-IPN-4 exhibits the highest specific capacitance of 364 F g−1 at 0.5 A g−1 in 6 M KOH aqueous electrolyte in a three-electrode system. It also possesses superior rate capability (57.7% retention at current densities ranging from 0.5 to 50 A g−1) and excellent cycling performance at 2 A g−1 (100% retention after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles). All these results confirm that the N-doped micro-nano carbon spheres are promising electrochemical capacitor materials, which possesses the advantages of simple preparation procedure, multi-scale pore structures, higher specific surface areas, easy adjustment of N-content and excellent electrochemical properties.
000 cycles). All these results confirm that the N-doped micro-nano carbon spheres are promising electrochemical capacitor materials, which possesses the advantages of simple preparation procedure, multi-scale pore structures, higher specific surface areas, easy adjustment of N-content and excellent electrochemical properties.
1. Introduction
Electrochemical double-layer capacitors (EDLCs) have been widely used in portable equipments, emergency power supplies and electric vehicles1,2 due to their unique advantages, including rapid charge–discharge, good cycling performance, high rate capability, large energy density and low maintenance cost.3–6 Electrode materials have been normally considered to be important components impacting the performance of EDLCs. Among these electrode materials, carbon materials play an important role in development of EDLCs because of their large specific surface areas and superior conductivity.7–10In order to improve the performance of an electrochemical capacitor, an effective method is to introduce pseudocapacitance via a transition metal oxide or by doping heteroatoms (N and O).11 Transition metal oxides associated with carbon-based electrode materials generally suffer from low rate capacity as well as short cycling life due to their poor conductivity and instability. In contrast, introducing heteroatoms can enhance the performance of carbon-based electrode materials and maintain the superior intrinsic features of carbon-based materials.12–14 On the one hand, the primary method for the introduction of heteroatoms into carbon-based materials is the post-treatment approach. In this approach, N-containing chemicals (e.g., ammonia and urea) can be treated with preformed carbon-based materials such as graphenes, activated carbon, and templated carbon.15,16 Li et al. fabricated N-doped graphene by employing annealing treatment of graphene oxide in ammonia, which can enhance the conductivity and specific capacitance of N-doped graphene.17 Nevertheless, it is difficult to fabricate stable N-doped graphene with uniform dispersion and controlled N-doping using this method.18 On the other hand, for fabricating more uniform dispersions of heteroatoms in carbon-based materials, in situ synthesis approach has been developed. In this method, N-containing polymer were used, such as poly(acrylonitrile), melamine, and polyaniline.19–21 Li et al. fabricated N-doped mesoporous carbon with excellent electrochemical properties.12 This N-doped mesoporous carbon, synthesized from the melamine resins, possesses specific capacitance of 289 F g−1 at 0.2 A g−1. Furthermore, Lee et al. fabricated monodispersed N-doped carbon nanospheres by pyrolysis of polymer spheres made of polystyrene and melamine.22 These monodispersed N-doped carbon nanospheres have a specific capacitance of 191.9 F g−1 at 0.1 A g−1. However, these methods did not concretely adjust the N doping content, and the specific capacitance of these carbon-based materials was not quite high. Recently, investigators also fabricated a nitrogen-doped porous carbon by surface activation with HNO3 solution.19 However, this method is time-consuming and harmful for our environment.
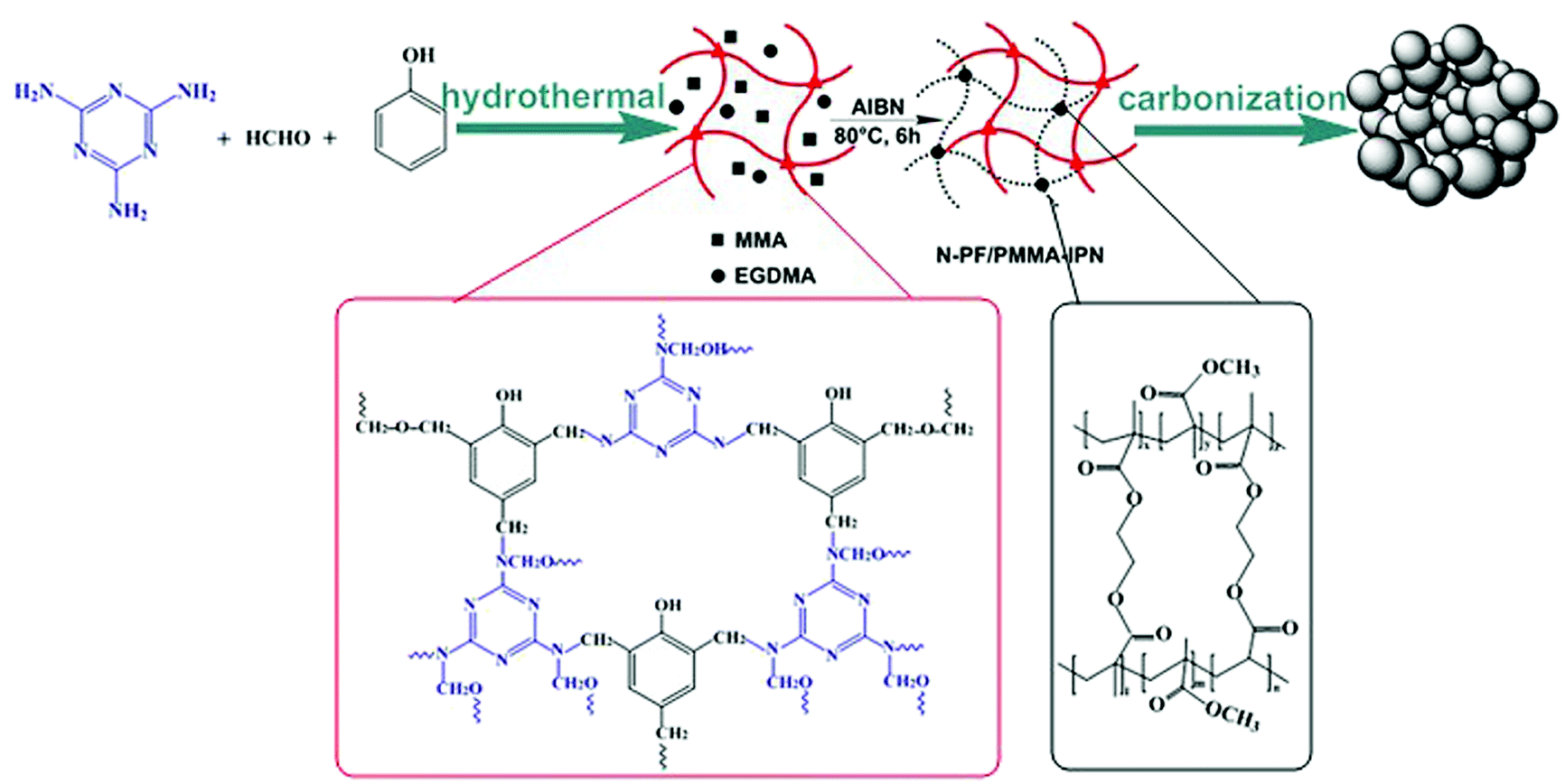
Herein, we present a facile and effective method that involved the preparation of nitrogen-doped micro-nano carbon spheres with multi-scale pore structure by employing melamine resin as the N source. The N-doped micro-nano carbon spheres (N-CS-IPNs) were successfully fabricated via straightforward carbonization of N-PF/PMMA interpenetrating polymer networks with different N-content (Scheme 1). Melamine resin had been selected as the N source due to its high nitrogen content and amino reactivity. In addition, N doping content was easily controlled by adjusting the weight ratio of melamine and phenol prior to polymerization. Moreover, the low cross-linking density-PMMA network was pyrolyzed, releasing gaseous products and leaving pores on carbon spheres. In this study, we also investigated the morphology of N-doped micro-nano carbon spheres, configurations of doped nitrogen and electrochemical performance of the carbon spheres. Excellent specific capacitance of 364 F g−1 at 0.5 A g−1 was obtained by the 6.44% N-doped micro-nano carbon spheres with multi-scale pore structure. In summary, the as-prepared N-CS-IPNs possess potential as promising electrode materials for electrochemical capacitors with higher specific capacitance as well as superior rate performance.
 | ||
| Scheme 1 Schematic of the preparation procedure for micro-nano carbon spheres with multi-scale pore structures obtained from interpenetrating polymer networks. | ||
2. Experimental section
2.1 Synthesis of N-doped micro-nano carbon sphere
In a typical process (Scheme S1†), 2 g melamine and 0.9 g phenol were added to into a three-neck flask and melted at 45 °C; then, 22.5 mL of 0.1 M NaOH was added gradually following continuous stirring. Subsequently, 10 mL of formalin solution (37 wt%) was added slowly. The reaction mixture was stirred at 70 °C for 3 h. Subsequently, 150 mL of deionized water was added into reaction mixture, and the reaction was continued for 17 h under 70 °C without N2 safeguard. The obtained dark-pink solution had been transferred into an autoclave after fixing the pH to 9; then the solution was heated to and maintained at 150 °C for 9 h. The resulting material was obtained using centrifugation, washed with ethanol and dried in a freeze-drying facility. The final product was used as the N-PF polymer (I) network for subsequent preparation of IPN.For the preparation of N-PF/PMMA-IPNs (Scheme S2†), the N-PF polymer (I) was treated with the monomer methyl methacrylate (MMA) (II) in the presence of a crosslinking agent ethylene glycol dimethacrylate (EGDMA, 3% by weight with respect to MMA) and an initiator 2,2-azobisisobutyronitrile (AIBN, 0.5% by weight with respect to MMA). The second monomer (MMA) mixture was allowed to polymerize and crosslink in situ in 30 g of N,N-dimethylformamide (DMF) in a 100 mL three-necked flask. Then, this compound was stirred, purged with nitrogen for 1 h and polymerized at 80 °C for 6 h. The as-obtained sample was transferred to a ceramic plate, followed by evaporation of the solvent at 120 °C for 6 h in a drying oven. The final product was an interpenetrating polymer network of N-PF/PMMA (N-PF/PMMA-IPNs).
The N-PF/PMMA-IPNs were ground into a fine powder. N-CS-IPNs were prepared by carbonizing the N-PF/PMMA-IPNs powder at 700 °C under nitrogen atmosphere for 2 h at a heating rate of 2 °C min−1. The as-obtained carbon was named as N-CS-IPN-2. For the preparation of N-CS-IPN-3 and N-CS-IPN-4, the amounts of melamine used were 3 g and 4 g, respectively. The other parameters were the same as those for the preparation of N-CS-IPN-2.
2.2 Characterization
Fourier transform infrared (FTIR) spectra were measured using a Bruker Equinox 55 FTIR spectrophotometer. The specimens were prepared using casting pellets from sample/KBr compound. TGA and DSC were measured under N2 flow with a heating rate of 10 °C min−1. Morphologies and structures of the samples were obtained using scanning electron microscopy (SEM) and transmission electron microscopy (TEM, 200 kV). Raman spectra were obtained by a Renishaw inVia spectrometer with a 520 nm Ar-ion laser. Powder X-ray Diffraction (XRD) patterns were measured on a Bruker Focus D8 diffractometer with Cu Kα radiation (40 kV, λ = 0.15418 nm) between 10° and 90°. Pore structure was determined using a Micromeritics ASAP 2020 surface area and porosity analyzer. Specific surface areas were analyzed by Brunauer–Emmett–Teller (BET) theory. Pore size distribution (PSD) was calculated according to the nonlocal density functional theory (NLDFT). Total pore volumes were calculated from the amount adsorbed at a relative pressure (P/P0) of 0.99. X-ray photoelectron spectroscopy (XPS) data were collected using a Sigma probe (Thermo VG Scientific) for analyzing the chemical structures of N-CS-IPNs.2.3 Electrochemical measurements
N-CS-IPN electrodes were prepared by mixing electroactive material powder (80 wt%), conductive graphite (7.5 wt%), acetylene black (7.5 wt%) and polytetrafluorethylene (PTFE, 5 wt% dispersion in water) in ethanol. Then, the compound was pressed onto a nickel foam current collector (surface area: 1 cm × 1 cm). The mass of electroactive material powder in each electrode is about 4 mg. The treated nickel foam was pressed under 10 MPa and dried at 110 °C for 12 h in a drying oven.In a normative three-electrode system, the EDLC properties were measured in 6 M KOH electrolyte using an electrochemical working station (CHI660E, Shanghai, China) with a platinum foil counter electrode, saturated calomel reference electrode (SCE), and the as-prepared N-CS-IPN working electrode. Cyclic voltammetry (CV) was performed under different scan rates, and galvanostatic charge–discharge (GCD) profiles were obtained under various current densities. Specific capacitances (Cm) were calculated according to the equation Cm = C/m = I × Δt/(m × ΔV), where Cm (F g−1), m (g), I (A), Δt (s), and ΔV (V) signify the specific capacitance, the mass of carbon-based working electrodes, the constant current density, discharging time and discharge potential, respectively. Electrochemical impedance spectroscopy (EIS) measurement was performed in frequencies ranging from 105 to 10−2 Hz under open circuit potential with an AC perturbation of 5 mV.
For a two-electrode system, two N-CS-IPN-4 electrodes containing 8 mg electroactive material were constructed into a symmetric capacitor. Specific capacitance (Cm, F g−1) of the electrode material, specific power (P, kW kg−1) and specific energy (E, W h kg−1) of the capacitor were calculated from the discharge curves using the following relationships: E = Cm × Δ2V/2 × 1000/3600; P = E/Δt × 3600, where Cm (F g−1), ΔV (V), Δt (s), and m (g) represent specific capacitance, change in discharge potential, discharge time and mass of carbon-based material used in the two working electrode, respectively.
3. Results and discussion
The fabrication of nitrogen-doped micro-nano carbon spheres is illustrated in Scheme 1. First, N-PF, which was the (I) polymer network, was prepared, as shown in Scheme S1.†11 Subsequently, the N-PF polymer (I) was treated with MMA (II) monomers using a crosslinking agent EGDMA and an initiator AIBN, and the second monomer (MMA) mixture was allowed to polymerize and crosslink in situ (Scheme S2†).51As shown in Fig. 1, the thermal stability of the as-prepared materials was monitored by TGA. The microphase-separation of polymers is usually studied by DSC.23,24 The weight loss stages of N-PF/PMMA interpenetrating polymer networks are clearly observed during the heating course, indicating the existence of three distinct stages in the TGA curves.25,26 The first mass loss stage is observed because the nanonetwork structure of N-PF undergoes sintering and partial dehydrogenation, which agrees with the exothermic peak at 150–280 °C. The second stage occurs in the temperature range of 300–430 °C due to the decomposition of PMMA networks. In fact, the PMMA networks are completely decomposed at 410 °C due to the low cross-linking density (Fig. S1†). The third mass loss stage is observed at 440–700 °C, which is associated with the complete carbonization of N-PF for transformation into a local graphitic structure.27,28 Furthermore, the char yields of N-PF/PMMA-IPN-2, N-PF/PMMA-IPN-3, and N-PF/PMMA-IPN-4 are 10.83 wt%, 17.56 wt%, and 17.30 wt% at 700 °C, respectively. Two melting transitions are obtained from the DSC curves, which indicate two glass transition temperatures (Tg) for the three samples. The two Tg of N-PF/PMMA-IPN-2, N-PF/PMMA-IPN-3, and N-PF/PMMA-IPN-4 are 292.88 °C and 393.91 °C, 291.81 °C and 394.80 °C, and 291.44 °C and 390.30 °C, respectively. The existence of two melting transitions indeed implies the microphase-separation in the N-PF/PMMA interpenetrating polymer networks.27,29
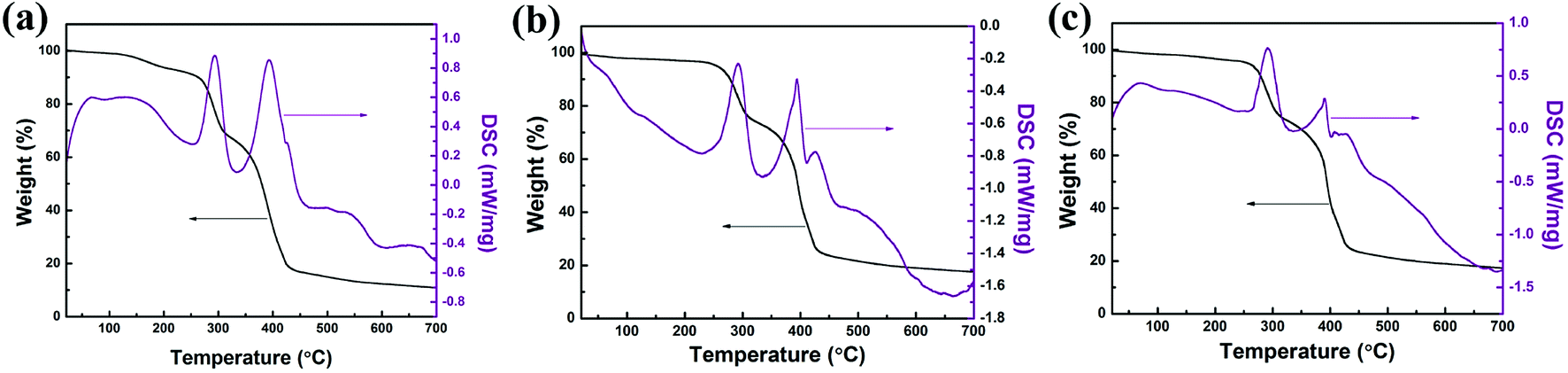 | ||
| Fig. 1 TGA and DSC curves of (a) N-PF/PMMA-IPN-2, (b) N-PF/PMMA-IPN-3, and (c) N-PF/PMMA-IPN-4. | ||
Nanostructure morphologies of N-CS-IPN-2, N-CS-IPN-3, and N-CS-IPN-4 were observed by SEM and TEM (Fig. 2). The SEM photographs (Fig. 2a–c) indicate that all the carbon materials are composed of micro-nano carbon spheres. The spherical shapes of N-CS-IPN-3 and N-CS-IPN-4 are better than those of N-CS-IPN-2. We find that the size of carbon spheres increased following the increase in mass of melamine (nitrogen source). The TEM images (Fig. 2e and f) of N-CS-IPNs also demonstrate the carbon spheres with increasing order of diameter. Each unit of the N-doped carbon spheres has micropores, and the decomposition of PMMA leaves pores in carbon spheres in the form of mesopores. In addition, the high resolution transmission electronic microscopy (HRTEM) image also shows partial graphitic structure, which results in an enhanced electrical conductivity (Fig. S2†). Electron diffraction diagrams of the three samples, shown as insets in Fig. S2†, indicate that the carbon samples are approximately crystalline.
 | ||
| Fig. 2 SEM images of (a) N-CS-IPN-2, (b) N-CS-IPN-3, and (c) N-CS-IPN-4. TEM images of (d) N-CS-IPN-2, (e) N-CS-IPN-3, and (f) N-CS-IPN-4. | ||
As can be seen in Fig. 3a, the chemical composition of the as-prepared samples was characterized using FTIR. In the FTIR spectrum of the carbon sphere before calcination (N-PF/PMMA-IPN), several carbon functional groups, such as C–OH (1270 cm−1) and C–H (1060 cm−1), are observed, while nitrogen-based functional groups of N–H (1342 cm−1 and 1469 cm−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (1566 cm−1) are also found. In addition, the functional groups of carbonyl (1729 cm−1), ester (1151 cm−1 and 1191 cm−1), and carbon–carbon band of the benzene ring (1660 cm−1) can be observed, which indicates that PMMA has been successfully synthesized by monomer MMA polymerization in the polymer blend systems. The N-doped carbon spheres (N-CS-IPN-4) display similar FTIR spectra and absorption peaks. Hence, the presence of N functional groups in the final product could be confirmed by FTIR analysis.
N (1566 cm−1) are also found. In addition, the functional groups of carbonyl (1729 cm−1), ester (1151 cm−1 and 1191 cm−1), and carbon–carbon band of the benzene ring (1660 cm−1) can be observed, which indicates that PMMA has been successfully synthesized by monomer MMA polymerization in the polymer blend systems. The N-doped carbon spheres (N-CS-IPN-4) display similar FTIR spectra and absorption peaks. Hence, the presence of N functional groups in the final product could be confirmed by FTIR analysis.
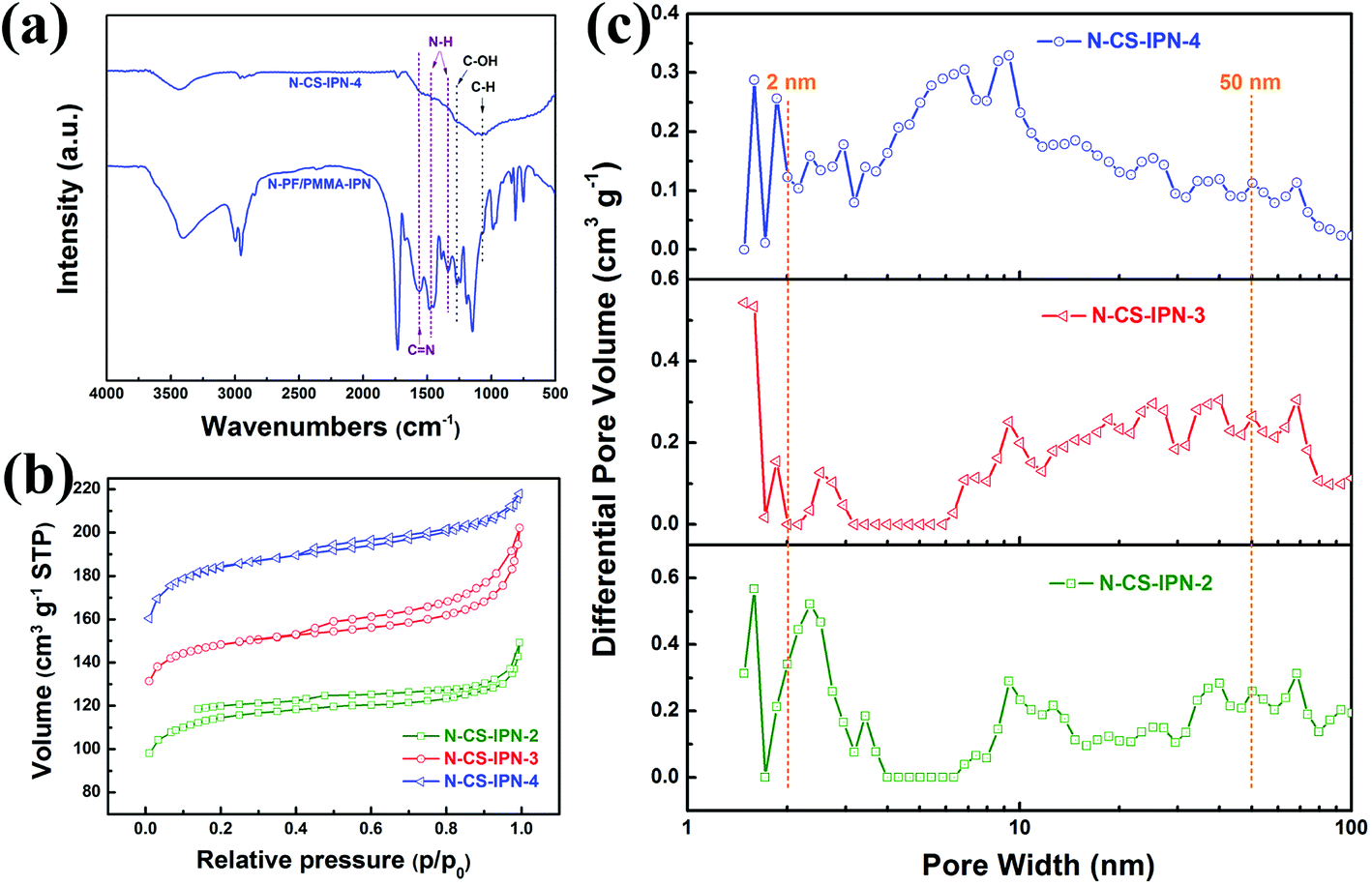 | ||
| Fig. 3 (a) FTIR spectra of N-PF/PMMA-IPN and N-CS-IPN-4. (b) Nitrogen absorption–desorption isotherms of N-CS-IPN-2, N-CS-IPN-3 and N-CS-IPN-4. (c) Pore size distributions of N-CS-IPN-2, N-CS-IPN-3 and N-CS-IPN-4. | ||
The specific surface areas of N-CS-IPNs were determined by employing N2 adsorption–desorption technique. As clearly shown in Fig. 3b, all N-CS-IPNs possess similar type I/IV adsorption–desorption isotherms with hysteresis loops between the relative pressures (P/P0) of 0.4–1.0. This indicates the existence of mesopore structures because of capillary condensation of the N2 molecules into mesopores.30 A high nitrogen uptake confirms the presence of abundant micropores within the micro-nano carbon spheres at low relative pressure. As shown in Fig. 3c, pore size distributions were calculated by employing nonlocal density functional theory (NLDFT).31 N-CS-IPNs show hierarchical porous structures as well as increased in mesopore size (such as 6.69 and 9.30 nm) following the increase in N-content, which was attributed to the pyrolysis of PMMA network and the removal of heteroatoms (N and H) from the mesoporous surface during the carbonization process.19 Moreover, the existence of micropores of varying sizes was confirmed from the PSD curves of these carbon materials due to the removal of individual atoms (C, H, and N); e.g., micropores with pore size in the range of 1.58–1.84 nm. The specific surface areas (SBET) of N-CS-IPN-2, N-CS-IPN-3, and N-CS-IPN-4 are 349, 449, and 559 m2 g−1, respectively. Moreover, the total pore volumes (Vtotal) of N-CS-IPN-2, N-CS-IPN-3, and N-CS-IPN-4 are 0.22, 0.30, and 0.33 cm3 g−1, respectively. The abovementioned results confirm that the interpenetrating polymer networks can successfully form nitrogen-doped micro-nano carbon spheres with multi-scale pore structures when pyrolyzed at 700 °C. Table 1 illustrates the structure parameters of N-CS-IPN-2, N-CS-IPN-3, and N-CS-IPN-4, displaying the increase in SBET, Smic, and Vtotal with the increase in nitrogen content from 3.98% to 6.44%. The abovementioned results eventually lead to more effective employment of the SBET of carbon samples.
| Sample | N-PF/PMMA (mg mg−1) | SBETa (m2 g−1) | Smicb (m2 g−1) | Smesoc (m2 g−1) | Vtotald (cm3 g−1) | Vmice (cm3 g−1) | Daf (nm) | N (%) | Cgh (Fg−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Specific surface areas were estimated by BET approach.b Micropores surface area.c Mesopores surface area.d Total pore volume measured at P/P0 = 0.99.e Micropore volume.f The average pore size was estimated from adsorption isotherms according to DFT model.h Galvanostatic capacitances estimated from discharge curves at 0.5 A g−1 in 6 M KOH solution. | |||||||||
| N-CS-IPN-2 | 20/80 | 349 | 280 | 69 | 0.22 | 0.13 | 2.3 | 3.98 | 189 |
| N-CS-IPN-3 | 20/80 | 449 | 364 | 85 | 0.30 | 0.19 | 2.5 | 5.02 | 243 |
| N-CS-IPN-4 | 20/80 | 559 | 445 | 114 | 0.33 | 0.23 | 2.6 | 6.44 | 364 |
Structural properties of N-CS-IPN-2, N-CS-IPN-3, and N-CS-IPN-4 were further investigated by XRD. As shown in Fig. 4a, the (002) characteristic peak located at 23° and the (100) characteristic peak located at 43° were obtained in the XRD patterns of the carbon samples. The characteristic (002) peaks indicate the existence of a graphite-like microcrystalline structure generally observed for pyrolytic carbon materials. Furthermore, interlayer distances (0.38 nm) estimated using Bragg equation are larger than those of graphite (0.34 nm),31 which indicates a random combination of graphitic and turbostratic stacking.32 The (100) characteristic peaks are attributed to the planes of amorphous carbon, indicating the formation of interlayer condensation and a mild degree of graphitic structures of the carbon materials. Structural characteristics of the as-obtained carbon samples were further researched by recording Raman spectra. As shown in Fig. 4b, two peaks at around 1353 cm−1 and 1589 cm−1, denoted as D (disordered) peak and G (graphitic) peak, respectively, are observed for all the three samples.27,33 The D band, which is related to crystal defects, evolves due to the existence of dopant atoms and absence of graphitic carbons in N-CS-IPN materials.34 The G band, which is attributed to atomic vibrations of large graphite crystallites,35 corresponds to the presence of graphitic carbon structures in N-CS-IPNs. It is well-known that the ID/IG ratio indicates the degree of graphitization or defect density in carbon materials, and the higher value of ID/IG indicates more disordered carbon structures or enhanced graphitization degree of amorphous carbon.36–38 In case of N-CS-IPNs materials, the value of ID/IG increases from 1.55 to 1.69 (Table S1†) with an increase in N content from 2 to 4, which indicates an increase in graphitization degree of the amorphous carbon.36 It is generally known that graphitization of carbon cloud improve electronic conductivity, whereas the type and content of doped heteroatoms are important in creating active catalytic sites in the carbon materials.39,40 The crystallite size (La) of N-CS-IPNs can be calculated by employing the empirical formula La = 4.35 IG/ID (nm). La of N-CS-IPN-2, N-CS-IPN-3, and N-CS-IPN-4 are 2.79, 2.71, and 2.56 nm (Table S1†), respectively, indicating that the carbon materials possess a graphite-like microcrystalline structure.
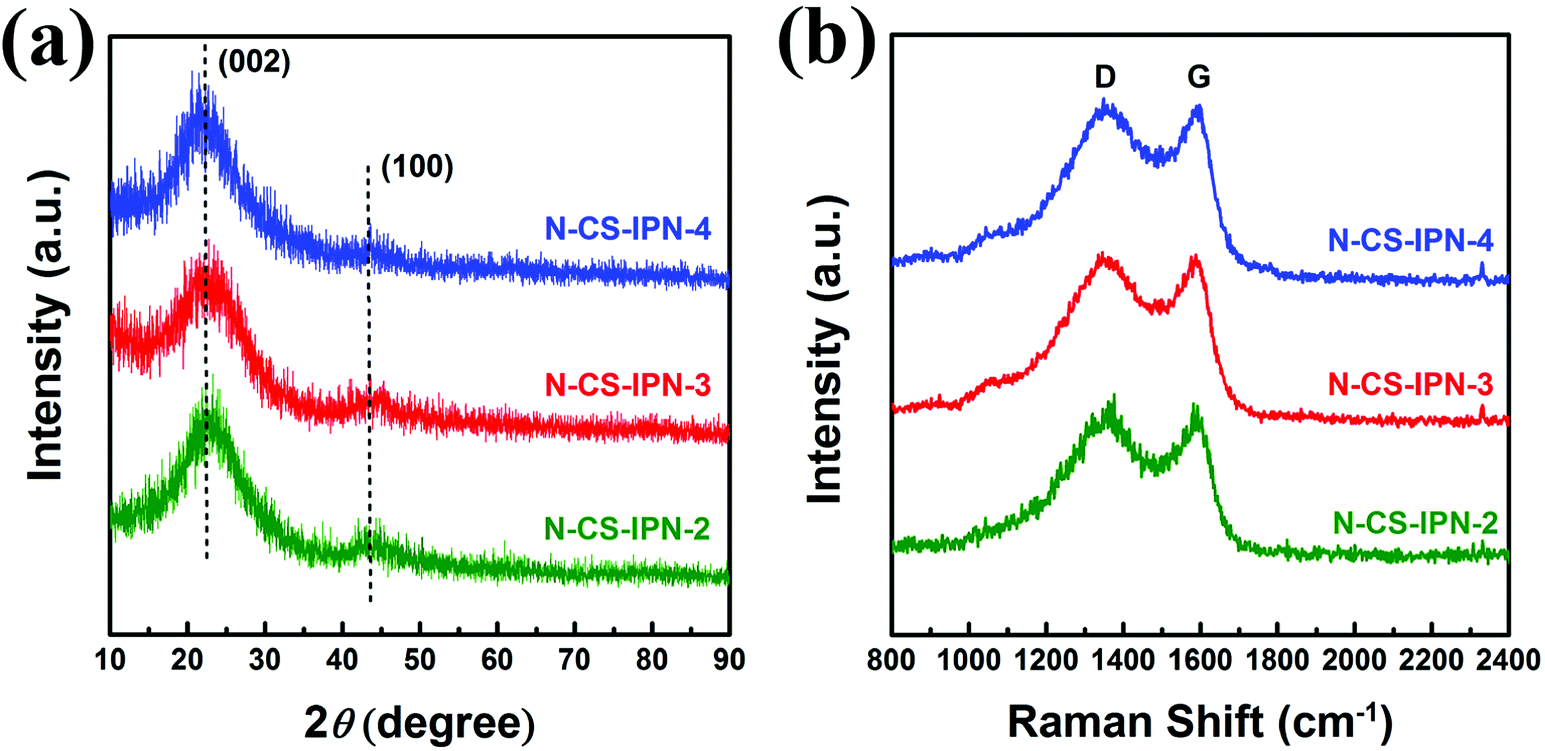 | ||
| Fig. 4 (a) XRD patterns of N-CS-IPN-2, N-CS-IPN-3 and N-CS-IPN-4. (b) Raman spectra of N-CS-IPN-2, N-CS-IPN-3 and N-CS-IPN-4. | ||
To investigate N bonding configurations and N doping degree, XPS measurements were performed for the N-doped carbon materials. Fig. 5a clearly displays the presence of carbon, nitrogen, and oxygen elements, indicating the incorporation of nitrogen atom within the precursor. Moreover, the N atomic percent of N-CS-IPN-2, N-CS-IPN-3, and N-CS-IPN-4 is 3.98%, 5.02%, and 6.44%, respectively (Table 1). Fig. 5b–d display N 1s spectra of N-CS-IPN-2, N-CS-IPN-3, and N-CS-IPN-4, which are fitted by three peaks: pyrrolic/pyridine N (N-5, 401 eV), pyridinic N (N-6, 398 eV) bonded to two C atoms of a six-membered ring located at the edge of a graphene plane, and a quaternary nitrogen (N-Q, 404 eV) bonded to three C atoms located at the center or valley of a graphene plane.39–42 Table 2 compares the relative amounts of N bonding configurations for N-CS-IPN-2, N-CS-IPN-3, and N-CS-IPN-4. N-CS-IPNs exhibit a relatively large content of N-6 and N-5 as well as a low portion of N-Q. Moreover, the N-Q and N-6 configurations are decreased, while N-5 configuration is increased. As reported previously, higher annealing temperature or longer treatment time for nitrogen doping could be beneficial to achieve N-Q configuration over the other configurations.20,41,43 In addition, the formation N-Q configuration requires higher energy according to a theoretical analysis.44 Therefore, it is possible to find that other configurations besides N-Q are generated with greater degree, while the doping amount of nitrogen is enhanced. According to the literature, N atoms located at the edge of a graphite layer (N-5, N-6) were more positive than those located at the center of a graphite layer (N-Q).45–47 Prominently, the percentage of nitrogen located at the edge in our samples of N-CS-IPN-2 (87.73%), N-CS-IPN-3 (88.42%), and N-CS-IPN-4 (89.11%) are very high. It is also reported that the N-5 and N-6 configurations enhance pseudocapacitance and capacitance performance, while the N-Q configuration improves conductivity of the carbon samples.44,48
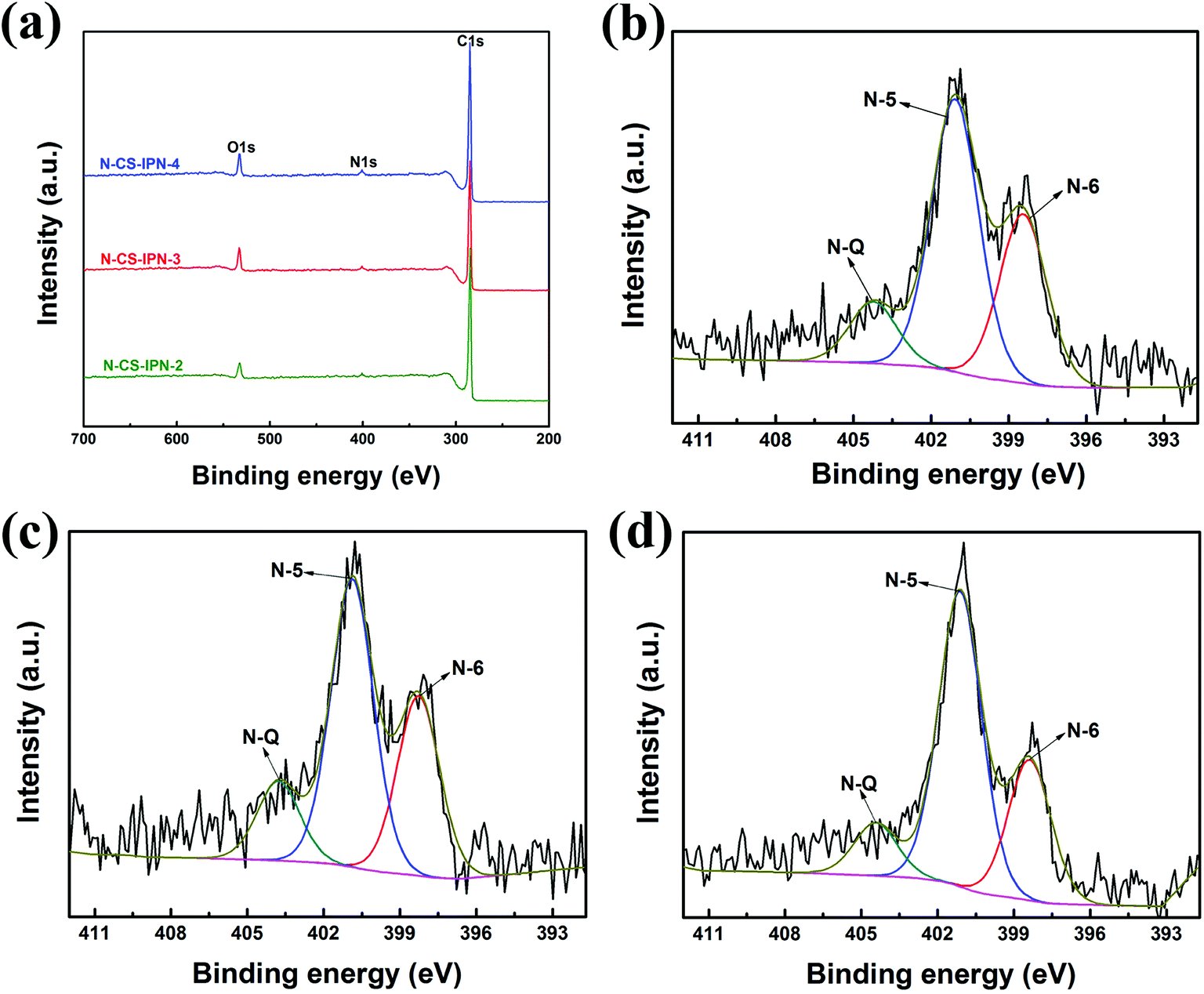 | ||
| Fig. 5 (a) XPS survey spectra of N-CS-IPN-2, N-CS-IPN-3 and N-CS-IPN-4. (b) N 1s spectrum of N-CS-IPN-2, (c) N 1s spectrum of N-CS-IPN-3, (d) N 1s spectrum of N-CS-IPN-4. | ||
| Functional group | N-6/N (%) | N-5/(%) | N-Q/(%) |
|---|---|---|---|
| B.E. (eV) | 398 | 401 | 404 |
| N-CS-IPN-2 | 33.57 | 54.16 | 12.27 |
| N-CS-IPN-3 | 32.64 | 55.78 | 11.58 |
| N-CS-IPN-4 | 28.72 | 60.39 | 10.89 |
Fig. 6a displays CV curves of N-CS-IPN-2, N-CS-IPN-3, and N-CS-IPN-4 in 6 M KOH electrolyte at a scan rate of 5 mV s−1. CV curves of the three samples exhibit quasi-rectangular shapes, indicating superior double-layer capacitance behavior with good charge transport due to their reasonable PSD and large SBET. Clearly, N-CS-IPN-4 exhibits the largest quasi-rectangular area with excellent shapes, which implies the highest specific capacitance. Fig. 6b displays galvanostatic charge–discharge curves of N-CS-IPN-2, N-CS-IPN-3, and N-CS-IPN-4 at 0.5 A g−1. The GCD curves are almost symmetric aside from partial deviations due to the presence of nitrogen in these carbon materials, which enhances their pseudocapacitance and capacitance performance as well as improves the conductivity. Specific capacitances of these carbon materials were calculated based on the discharging time of GCD curves. Distinctly, N-CS-IPN-4 possesses the longest discharging time as well as the highest specific capacitance at 0.5 A g−1, which corroborates the results obtained from CV measurements. The highest specific capacitance of N-CS-IPN-4 might be connected with the reasonable pore size distribution and the highest surface area, which are in favour of the development of an electric double layer. Moreover, the nitrogen can undergo reversible redox reactions, which could contribute to the pseudocapacitance and improve the surface wettability. However, the absence of distinct redox peak in CV curves implies that the capacitance is mainly derived from the electric double layer. EIS measurements were conducted for N-CS-IPN-2, N-CS-IPN-3, and N-CS-IPN-4, as shown in Fig. 6c. The three impedance spectra are similar, exhibiting a small semicircle at high frequencies as well as an almost vertical line at low frequencies. Nevertheless, distinct deviations exist among the three EIS curves. The internal resistance (Rb) comprises intrinsic resistances of carbon electrodes and total electrolyte ionic resistance. The internal resistances of N-CS-IPN-2, N-CS-IPN-3, and N-CS-IPN-4 electrodes were 0.93, 0.62, and 0.56 Ω, respectively. Clearly, the smaller Rb of N-CS-IPN-4 electrode than that of the others indicates that ion diffusion and electrolyte transfer into the pore channels easily occur, thus promoting the formation of the electric double layer. EIS curves of all the N-CS-IPNs electrodes possess angles larger than 45°, which reveal smaller Warburg impedances because of the enhanced electrolyte ions diffusion due to the hierarchical porous structures. For example, the diffusion resistance of N-CS-IPN-4 was smaller than that of N-CS-IPN-2 and N-CS-IPN-3 electrodes because of introduction of hydrophilic N and O functional groups during the N doping process. Moreover, relatively larger resistances of N-CS-IPN-2 and N-CS-IPN-3 than that of N-CS-IPN-4 may be caused by relatively smaller pore sizes and specific surface areas that hinder ion transport and electrolyte penetration. Fig. 6d exhibits the specific capacitances of N-CS-IPN-2, N-CS-IPN-3, and N-CS-IPN-4 at different current densities. It is distinctly seen that the specific capacitance has been largely increased with the increase in N content. The N-CS-IPN-4 electrode provides larger specific capacitance (364 F g−1) than N-CS-IPN-2 (189 F g−1) and N-CS-IPN-3 (243 F g−1) at 0.5 A g−1. Rate capabilities of N-CS-IPN-2, N-CS-IPN-3, and N-CS-IPN-4 were 42.3%, 49.4%, and 57.7%, respectively, when current densities increased from 0.5 to 50 A g−1. N-CS-IPN-4 possesses 76.9%, 68.7%, 63.5%, and 57.7% capacitance retention at 10, 20, 30, and 50 A g−1, respectively. This implies low resistance and superior retention rate capability, indicating that the hierarchical porous structure offers effective electrolyte transfer into the carbon material. The abovementioned results for N-CS-IPN-4 relate with better electrical conductivity and formation of porous structures. Moreover, the nitrogen-containing functional groups (N-5 and N-6) could also provide pseudocapacitance. Fig. 6e displays the CV curves of N-CS-IPN-4 at different scan rates. The CV curves of N-CS-IPN-4 possess good rectangular shapes from 5 to 100 mV s−1, indicating that N-CS-IPN-4 exhibits optimal double-layer capacitance behavior. Notably, the CV curves if N-CS-IPN-4 maintain the quasi-rectangular shape at a scan rate as high as 500 mV s−1, which reveals rapid electrolyte ions diffusion and electron transfer. Fig. 6f shows the GCD curves of N-CS-IPN-4 at different current densities. It is noteworthy that N-CS-IPN-4 still retains typical symmetric triangle shapes at current densities as large as 50 A g−1, indicating that N-CS-IPN-4 as an electrode material has excellent coulombic efficiency and perfect electric capacitive behavior. Specific capacitances were calculated to be 364, 334, 312, 285, 270, 250, 231, and 210 F g−1 at different current densities of 0.5, 1, 2, 5, 10, 20, 30, and 50 A g−1, respectively.
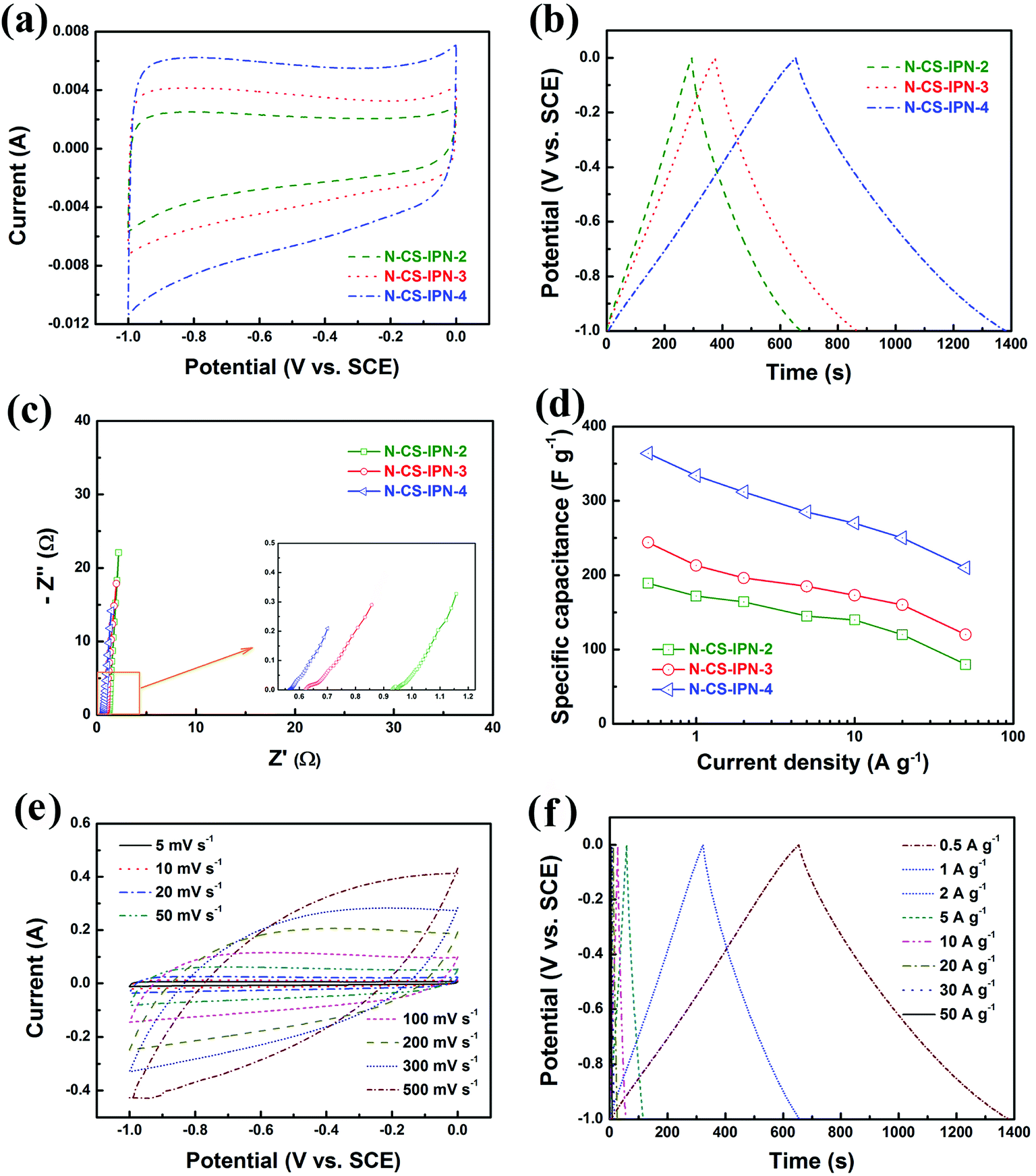 | ||
| Fig. 6 (a) Cyclic voltammograms of N-CS-IPN-2, N-CS-IPN-3 and N-CS-IPN-4 at a scan rate of 5 mV s−1. (b) Galvanostatic charge–discharge curves of N-CS-IPN-2, N-CS-IPN-3 and N-CS-IPN-4 at a current density of 0.5 A g−1. (c) Electrochemical impedance spectra of N-CS-IPN-2, N-CS-IPN-3 and N-CS-IPN-4. (d) Specific capacitances of N-CS-IPN-2, N-CS-IPN-3 and N-CS-IPN-4 at different current densities. (e) Cyclic voltammograms of N-CS-IPN-4 at different scan rates. (f) Galvanostatic charge–discharge curves of N-CS-IPN-4 at different current densities. | ||
For further research on the electrochemical performance of N-CS-IPN-4, the long-term cycling stability (10000 cycles) of this electrode was evaluated by repeating the charge–discharge test at a current density of 2 A g−1 in 6 M KOH electrolyte. As shown as Fig. 7, N-CS-IPN-4 electrode possesses excellent cycling property with 100% specific capacitance retained after 10000 cycles, which demonstrates its good reversibility and superior cycle stability in continuous charge–discharge process. The inset is the SEM image of N-CS-IPN-4 after 10000 cycles, indicating that the carbon spheres still maintain good shape.
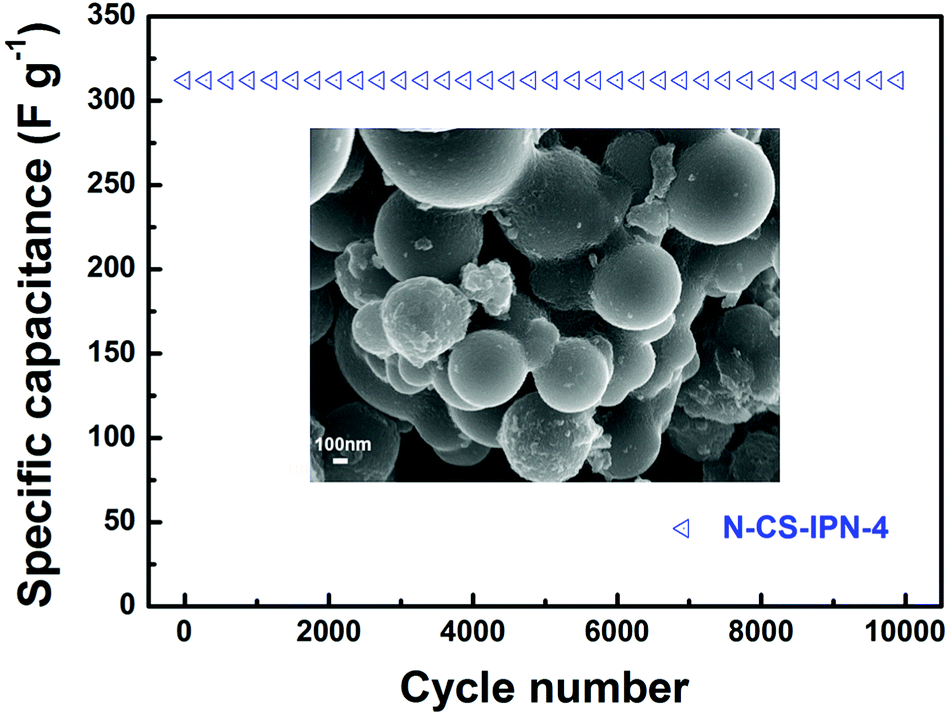 | ||
| Fig. 7 Cycling performance of N-CS-IPN-4 measured at a constant current density of 2 A g−1 (inset: SEM image after 10000 cycles). | ||
A symmetric two-electrode system was employed for further confirming the electrochemical properties of N-CS-IPN-4 in 6 M KOH electrolyte (Fig. 8). Fig. 8a displays the CV curves of N-CS-IPN-4//N-CS-IPN-4-based electrochemical capacitor at different scan rates, revealing a good rectangular shape at scan rates as high as 50 mV s−1 and suggesting that this capacitor possesses good rate capability. Fig. 8b shows triangle-like galvanostatic charge–discharge curves of N-CS-IPN-4//N-CS-IPN-4, which indicates its superior reversible capacitive behavior. Moreover, this capacitor shows superior capacitance retention of 75.6% at different current densities from 0.5 to 10 A g−1. For comparison, the electrochemical properties of N-CS-IPN-4 and AC were investigated in symmetric two-electrode systems. The CV curves of N-CS-IPN-4//N-CS-IPN-4 and AC//AC are shown in Fig. 8c, where the rectangular shape of the symmetric N-CS-IPN-4 electrode was larger than that for the symmetric AC electrode, indicating that the specific capacitance of symmetric N-CS-IPN-4 electrode was higher than that for AC. Fig. 8d displays the triangle-like galvanostatic charge–discharge curves of the two symmetric electrode systems. The N-CS-IPN-4 and AC capacitors exhibit specific capacitances of 90 F g−1 and 36 F g−1, respectively, at a current density of 0.5 A g−1, which implies that the symmetric N-CS-IPN-4 electrode possesses better electrochemical properties than AC. The EIS curve of N-CS-IPN-4//N-CS-IPN-4 is shown in Fig. 8e. A small semicircle could be observed for the N-CS-IPN-4 capacitor (Fig. 8e, inset) in the high frequency region, indicating its correspondingly smaller charge transfer resistance and better conductivity.49,50 The specific energy and specific power are key parameters to determine the electrochemical properties of electrode materials, which are estimated from the galvanostatic charge–discharge curves. The Ragone plots of N-CS-IPN-4//N-CS-IPN-4 and AC//AC are displayed in Fig. 8f. The specific energy is much larger (12.5 W h kg−1) for N-CS-IPN-4//N-CS-IPN-4 than for the AC//AC symmetric capacitor (5 W h kg−1) at 0.5 A g−1. In addition, the specific energy of N-CS-IPN-4//N-CS-IPN-4 remained at 9.4 W h kg−1 with a high specific power of 4.98 kW kg−1 at a current density as high as 10 A g−1, which was excellent compared with that of the AC//AC symmetric capacitor. Hence, the as-prepared N-CS-IPN-4 would be a promising electrode material for highly efficient electrochemical capacitors.
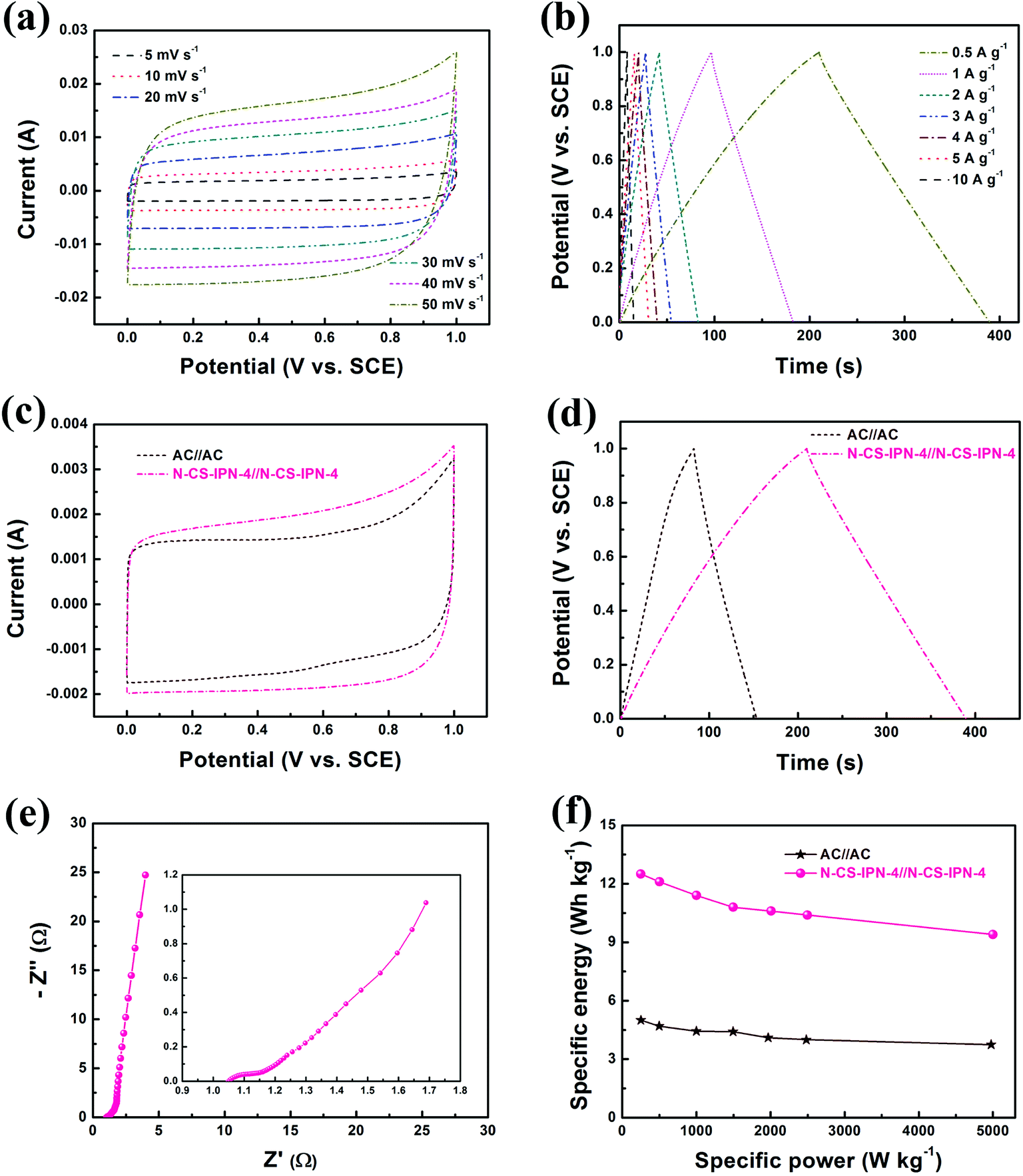 | ||
| Fig. 8 (a) Cyclic voltammograms of N-CS-IPN-4//N-CS-IPN-4 at various scan rates. (b) Galvanostatic charge–discharge curves of N-CS-IPN-4//N-CS-IPN-4 at various current densities. (c) CVs of AC//AC and N-CS-IPN-4//N-CS-IPN-4 at a scan rate of 5 mV s−1. (d) Charge–discharge curves of N-CS-IPN-4//N-CS-IPN-4 and AC//AC at a current density of 0.5 A g−1. (e) EIS curve of N-CS-IPN-4//N-CS-IPN-4. (f) Ragone plots of N-CS-IPN-4//N-CS-IPN-4 and AC//AC devices. | ||
4. Conclusion
In summary, we successfully designed a synthesis protocol for preparing nitrogen-doped micro-nano carbon spheres with multi-scale pore structure by employing melamine resin as the N source. The N-doped micro-nano carbon spheres were successfully fabricated via straightforward carbonization of N-PF/PMMA interpenetrating polymer networks with different N content. The as-prepared N-CS-IPNs electrode materials possess applicable pore size distribution, higher surface area, and consistently dispersed nitrogen functional groups with controlled doping content. Their particular electrochemical performances endow them as promising electrode materials for highly efficient electrochemical capacitors with excellent properties. On the one hand, particularly, the N-CS-IPN-4 electrode possesses higher specific surface area of 559 m2 g−1 and better specific capacitance of 364 F g−1 at 0.5 A g−1 compared with other N-CS-IPNs in a three-electrode system. Moreover, N-CS-IPN-4 reveals excellent rate capability (57.7% retention at current densities ranging from 0.5 to 50 A g−1) and excellent cycling performance at 2 A g−1 (100% retention after 10000 cycles). On the other hand, the electrochemical performance of the obtained carbon electrode materials was also researched using symmetrical two-electrode systems. The N-CS-IPN-4//N-CS-IPN-4 device possesses much higher specific energy density (12.5 W h kg−1) than the AC//AC symmetric capacitor (5 W h kg−1) at 0.5 A g−1. In conclusion, these abovementioned results confirm that N-doped micro-nano carbon spheres with multi-scale porous structures obtained from interpenetrating polymer networks will be promising electrode materials for electrochemical capacitors.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (No. 51762031) and the Foundation for Innovation Groups of Basic Research in Gansu Province (No. 1606RJIA322).References
- X. Sun, G.-P. Hao, X. Lu, L. Xi, B. Liu and W. Si, J. Mater. Chem. A, 2016, 4, 10166–10173 RSC.
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef PubMed.
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS PubMed.
- H. Jiang, L. Yang, C. Li, C. Yan, P. S. Lee and J. Ma, Energy Environ. Sci., 2011, 4, 1813–1819 RSC.
- M. Huang, Y. X. Zhang, F. Li, L. L. Zhang, Z. Y. Wen and Q. Liu, J. Power Sources, 2014, 252, 98–106 CrossRef CAS.
- F. X Ma, L. Yu, C. Y. Xu and X. W. Lou, Energy Environ. Sci., 2016, 9, 862–866 RSC.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- K. Xie, X. Qin, Y. Wang, H. Tao, Q. Wu, L. Yang and Z. Hu, Adv. Mater., 2012, 24, 347–352 CrossRef CAS PubMed.
- J. Gamby, P. L. Taberna, P. Simon, J. F. Fauvarque and M. Chesneau, J. Power Sources, 2001, 101, 109–116 CrossRef CAS.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 RSC.
- W. Li, D. Chen, Z. Li, Y. Shi, Y. Wan, J. Huang, J. Yang, D. Zhao and Z. Jiang, Electrochem. Commun., 2007, 9, 569–573 CrossRef CAS.
- O. M. Li and J. M. Xue, J. Phys. Chem. C, 2014, 118, 2507–2517 CrossRef.
- G. Lota, B. Grzyb, H. Machnikowska, J. Machnikowski and E. Frackowiak, Chem. Phys. Lett., 2005, 404, 53–58 CrossRef CAS.
- R. J. White, M. Antonietti and M. M. Titirici, J. Mater. Chem., 2009, 19, 8645–8650 RSC.
- D. Hulicova-Jurcakova, M. Kodama, S. Shiraishi, H. Hatori, Z. H. Zhu and G. Q. Lu, Adv. Funct. Mater., 2009, 19, 1800–1809 CrossRef CAS.
- H. M. Jeong, J. W. Lee, W. H. Shin, Y. J. Choi, H. J. Shin, J. K. Kang and J. W. Choi, Nano Lett., 2011, 11, 2472–2477 CrossRef CAS PubMed.
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS PubMed.
- Z. Wen, X. Wang, S. Mao, Z. Bo, H. Kim, S. Cui, G. Lu, X. Feng and J. Chen, Adv. Mater., 2012, 24, 5610–5616 CrossRef CAS PubMed.
- K. Yan, L. B. Kong, K. W. Shen, Y. H. Dai, M. Shi, B. Hu, Y. C. Luo and L. Kang, Appl. Surf. Sci., 2016, 364, 850–861 CrossRef CAS.
- L. F. Chen, X. D. Zhang, H. W. Liang, M. Kong, Q. F. Guan, P. Chen, Z. Y. Wu and S. H. Yu, ACS Nano, 2012, 6, 7092–7102 CrossRef CAS PubMed.
- L. Li, E. Liu, J. Li, Y. Yang, H. Shen, Z. Huang, X. Xiang and W. Li, J. Power Sources, 2010, 195, 1516–1521 CrossRef CAS.
- W. H. Lee and J. H. Moon, ACS Appl. Mater. Interfaces, 2014, 6, 13968–13976 CrossRef CAS PubMed.
- R. Dattani, J. H. Bannock, Z. Fei, R. C. I. MacKenzie, A. A. Y. Guilbert, M. S. Vezie, J. Nelson, J. C. de Mello, M. Heeney, J. T. Cabral and A. J. Nedoma, J. Mater. Chem. A, 2014, 2, 14711–14719 RSC.
- P. Willot, J. Teyssandier, W. Dujardin, J. Adisoejoso, S. DeFeyter, D. Moerman, P. Leclere, R. Lazzaroni and G. Koeckelberghs, RSC Adv., 2015, 5, 8721–8726 RSC.
- K. A. Smith, B. Stewart, K. G. Yager, J. Strzalka and R. Verduzco, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 900–906 CrossRef CAS.
- Y. Chen, H. Cui, L. Li, Z. Tian and Z. Tang, Polym. Chem., 2014, 5, 4441–4445 Search PubMed.
- M. J. Zhong, E. K. Kim, J. P. McGann, S. E. Chun, J. F. Whitacre, M. Jaroniec, K. Matyjaszewski and T. Kowalewski, J. Am. Chem. Soc., 2012, 134, 14846–14857 CrossRef CAS PubMed.
- C. T. Nguyen and D. P. Kim, J. Mater. Chem., 2011, 21, 14226–14230 RSC.
- K. T. Cho, S. B. Lee and J. W. Lee, J. Phys. Chem. C, 2014, 118, 9357–9367 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- K. T. Cho, S. B. Lee and J. W. Lee, J. Phys. Chem. C, 2014, 118, 9357–9367 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Carbon, 2013, 56, 155–166 CrossRef CAS.
- D. C. Wu, H. C. Dong, J. Pietrasik, E. K. Kim, C. M. Hui, M. J. Zhong, M. Jaroniec, T. Kowalewski and K. Matyjaszewski, Chem. Mater., 2011, 23, 2024–2026 CrossRef CAS.
- Y. Meng, D. Voiry, A. Goswami, X. Zou, X. Huang, M. Chhowalla, Z. Liu and T. Asefa, J. Am. Chem. Soc., 2014, 136, 13554–13557 CrossRef CAS PubMed.
- F. Xu, Z. W. Tang, S. Q. Huang, L. Y. Chen, Y. R. Liang, W. C. Mai, H. Zhong, R. W. Fu and D. C. Wu, Nat. Commun., 2015, 6, 7221–7231 CrossRef PubMed.
- G. Panomsuwan, N. Saito and T. Ishizaki, J. Mater. Chem. A, 2015, 3, 9972–9981 RSC.
- G. Nanda, S. Goswami, K. Watanabe, T. Taniguchi and P. F. A. Alkemade, Nano Lett., 2015, 15, 4006–4012 CrossRef CAS PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- S. Shrestha and W. E. Mustain, J. Electrochem. Soc., 2010, 157, B1665–B1672 CrossRef CAS.
- F. Kapteijn, J. A. Moulijn, S. Matzner and H. P. Boehm, Carbon, 1999, 37, 1143–1150 CrossRef CAS.
- F. Su, C. K. Poh, J. S. Chen, G. Xu, D. Wang, Q. Li, J. Lin and X. W. Lou, Energy Environ. Sci., 2011, 4, 717–724 RSC.
- D. Hulicova-Jurcakova, M. Kodama, S. Shiraishi, H. Hatori, Z. H. Zhu and G. Q. Lu, Adv. Funct. Mater., 2009, 19, 1800–1809 CrossRef CAS.
- T. Van Khai, H. G. Na, D. S. Kwak, Y. J. Kwon, H. Ham, K. B. Shim and H. W. Kim, J. Mater. Chem., 2012, 22, 17992–18003 RSC.
- D. W. Wang, F. Li, L. C. Yin, X. Lu, Z. G. Chen, I. R. Gentle, G. Q. Lu and H. M. Cheng, Chem.–Eur. J., 2012, 18, 5345–5351 CrossRef CAS PubMed.
- C. O. Ania, V. Khomenko, E. Raymundo-Piñero, J. B. Parra and F. Beguin, Adv. Funct. Mater., 2007, 17, 1828–1836 CrossRef CAS.
- E. J. Ra, E. Raymundo-Piñero, Y. H. Lee and F. Beguin, Carbon, 2009, 47, 2984–2992 CrossRef CAS.
- Z. Li, L. Zhang, B. S. Amirkhiz, X. Tan, Z. Xu, H. Wang, B. C. Olsen, C. M. B. Holt and D. Mitlin, Adv. Energy Mater., 2012, 2, 431–437 CrossRef CAS.
- X. Yang, D. Wu, X. Chen and R. Fu, J. Phys. Chem. C, 2010, 114, 8581–8586 CrossRef CAS.
- L. Yang, S. Cheng, Y. Ding, X. Zhu, Z. L. Wang and M. Liu, Nano Lett., 2011, 12, 321–325 CrossRef PubMed.
- S. Biswas and L. T. Drzal, Chem. Mater., 2010, 22, 5667–5671 CrossRef CAS.
- J. Zhang, X. F. Zhong, H. B. Chen, Y. Gao and H. M. Li, Electrochim. Acta, 2014, 148, 203–210 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05851b |
| This journal is © The Royal Society of Chemistry 2018 |