
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural modification of oridonin via DAST induced rearrangement†
Dong-Dong Luo‡
a,
Kai Peng‡a,
Jia-Yu Yanga,
Pawinee Piyachaturawatb,
Witchuda Saengsawangb,
Lei Aoc,
Wan-Zhou Zhaoc,
Yu Tang *a and
Sheng-Biao Wan*a
*a and
Sheng-Biao Wan*a
aLaboratory for Marine Drugs and Bioproducts of Qingdao National Laboratory for Marine Science and Technology, Key Laboratory of Marine Drugs, Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, Yushan Road 5, Qingdao 266003, China. E-mail: biaowan@ouc.edu.cn; tangyu@ouc.edu.cn; Tel: +86-532-82031087
bDepartment of Physiology, Faculty of Science, Mahidol University, Bangkok 10400, Thailand
cThe Nanjing Han & Zaenker Cancer Institute (NHZCI), Nanjing OGpharma Co. Ltd., Nanjing 210036, China
First published on 20th August 2018
Abstract
A simple and efficient protocol was developed for the syntheses of oridonin analogues, i.e. 6,20-epoxy ent-kaurane diterpenoid analogues from oridonin via diethylaminosulfur trifluoride (DAST) promoted rearrangement, most of which exhibited superior anticancer activities compared with their precursor.
Introduction
Oridonin, an ent-kaurane diterpenoid extracted from Isodon rubescens (Chinese name “Donglingcao”), has received particular interest from the pharmaceutical community1,2 due to its pharmacological utilities, e.g. unique and prominent anticancer activity3 as well as its safety. It has been demonstrated that oridonin could significantly suppress cancer cell migration via regulation of non-muscle myosin IIA.4 The anticancer mechanism of oridonin in vitro may involve multiple pathways,5–13 for instance, oridonin can not only inhibit the proliferation of the breast cancer cell McF-7, but also induce apoptosis through the pathways of hampering the cell cycle and activation of mitochondria.9,14–18 Targeting AML1-ETO (AE) fusion protein which plays a critical role in leukemogenesis shows potent antitumor activity with low adverse effects on t(8;21) leukemia in vitro and in vivo.19 However, the clinical application of oridonin has been significantly impeded by its poor aqueous solubility, moderate potency, low bioavailability20 and metabolic instability.21 Thus it's highly desirable to synthesize the oridonin analogues via rational chemical modification of its structure for better pharmacological properties such as anticancer activity.Oridonin belongs to the class of 7,20-epoxy ent-kaurane diterpenoid structures, which features 1,6,14-trihydroxy groups, 7-hemiacetal moiety in the B-ring and the α-methylene cyclopentanone in the D-ring. The strong hydrogen bonding interaction exists between 6β-OH and carbonyl group at C-15 (Fig. 1). It's difficult for 6β-OH and 7β-OH to participate in nucleophilic reaction due to the adjacent steric hindrance and the intramolecular hydrogen bonding interaction. The previous studies of oridonin on the structure–activity relationship (SAR) have proved that D-ring is crucial to anti-cancer activity,22,23 and any modification of enone moiety24 would lead to loss of anti-cancer activity.25 As shown in Fig. 1, previous efforts on structural modification of oridonin were mainly focused on A ring,2,26–28 6-O positions29 and 14-O positions17,30,31 as well as B-ring opening via oxidative cleavage of C–C bond between C-6 and C-7.31
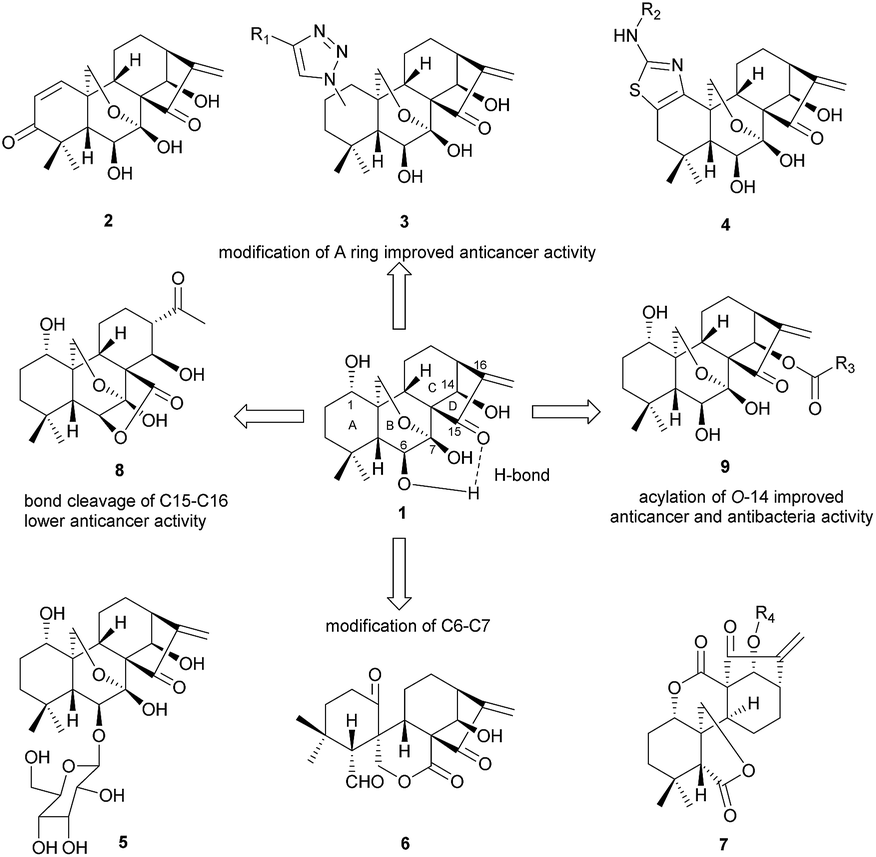 | ||
| Fig. 1 Structures of oridonin and biologically active analogues. | ||
Diethylaminosulfur trifluoride (DAST) has been employed as a versatile fluorinating agent for various fluorination reactions,32–34 e.g. fluorination of carbohydrate in which structural rearrangements could be observed.34–37 Therefore, it can be envisioned that the similar structural rearrangements of oridonin could occur in the presence of DAST, yielding new oridonin analogues. As a continuation of our research works on pharmaceutical molecules,38,39 herein, we disclosed a simple and efficient method for preparation of novel 6,20-epoxy ent-kaurane diterpenoid analogues from oridonin, in which 6,20-endo ring was formed via DAST promoted rearrangement.
Results and discussion
Our currently ongoing research works indicated that new potent antitumor agents could be produced via installing protected amino acid residues or cinnamyl group at C-14 position of oridonin analogues. Thus, oridonin analogues 11a–11r were prepared in good yields through esterification of oridonin, 1-acetyl-oridonin and 1-oxo-oridonin derivatives with 4-methoxy cinnamylic acid, 3,4-dimethoxy cinnamic acid, 3,4,5-trimethoxy cinnamic acid, Boc-Gly, Boc-L-Val, Boc-L-Leu, Boc-L-Ile, Boc-L-Pro, Boc-L-Phe, N-Fmoc-L-Asp-1-OtBu, Fmoc-L-Phe (Scheme 1), which would be exploited as substrates for rearrangement.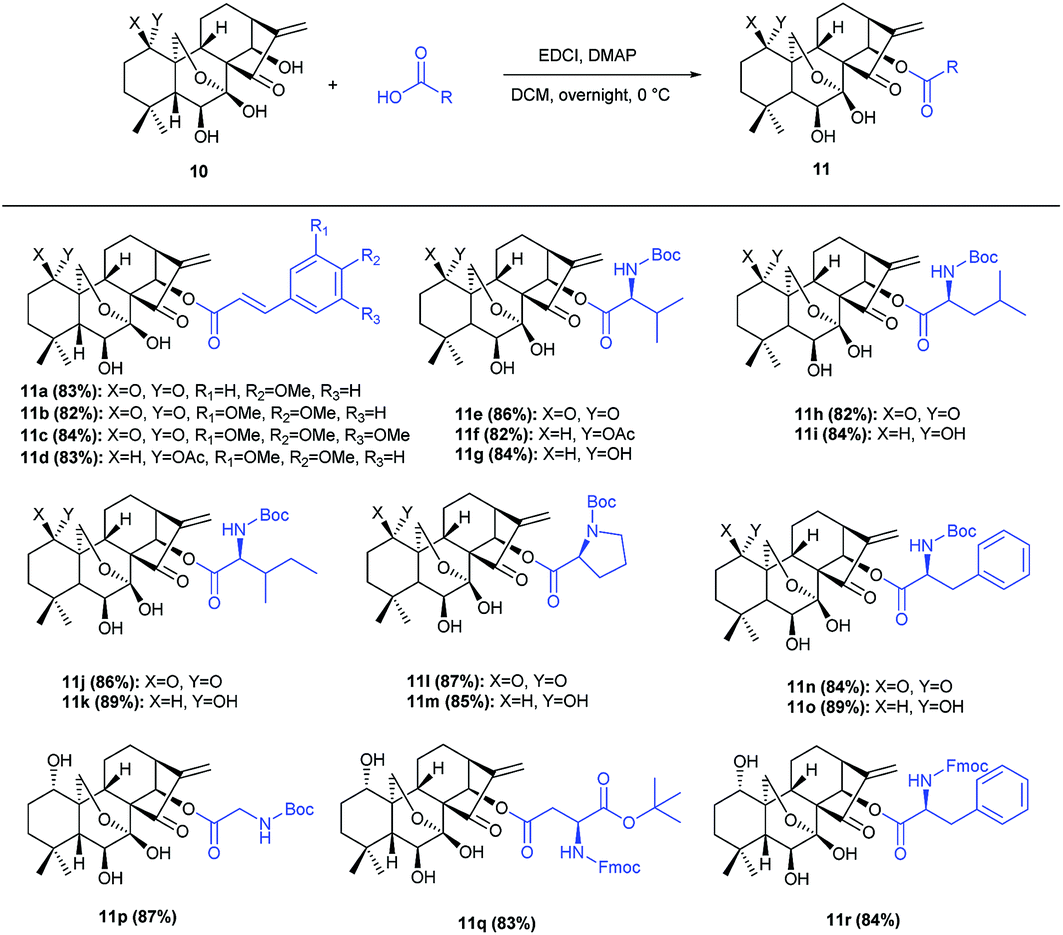 | ||
| Scheme 1 Synthesis of 14-acyl oridonin analogues 11. | ||
Initially, the feasibility of rearrangement was investigated using readily accessed 1-oxo-14-acyl oridonin analogue 11b as the model substrate in the presence of DAST (1 equiv.) with dichloromethane (DCM) as solvent, and gratifyingly the desired product 12b was furnished in 31% yield, the structure of which was determined unambiguously by X-ray crystallography (CCDC 1590323) (Table 1, entry 1). Afterwards, the impact of DAST amount on the reaction was examined, and the yields of 12b could be increased to 40%, 57% and 80%, respectively with 3 equivalents, 5 equivalents and 10 equivalents of DAST employed (Table 1, entries 2–4). However, further increasing the amount of DAST led to inferior yield (Table 1, entry 5). The reaction temperature was also evaluated, and the reaction could be significantly accelerated at higher temperature, which, however, had a detrimental influence on the yield (Table 1, entry 6). Subsequently, various solvents were screened, and DCM was identified as the optimal one which furnished 12b in highest yield (Table 1, entry 4). The employment of other solvents such as THF, MeCN, DMF and DMSO resulted in inferior yields (Table 1, entry 7–11), and notably, only a trace amount of 12b was observed using acetone as solvent (Table 1, entry 11).
|
|||
|---|---|---|---|
| Entry | Reagent (equiv.) | Solvent | Yieldb (%) |
| a Reaction condition: 11b (0.09 mmol), solvent (5.0 mL), −78 °C for 10 min, then warmed up to room temperature for 8 h.b Yield of isolated products.c −78 °C for 10 min, then warmed up to rt for 2 h.d −78 °C for 10 min, then warmed up to 40 °C for 1 h.e −78 °C for 10 min, then warmed up to rt for 3 h.f Room temperature for 3 h. | |||
| 1 | DAST (1) | DCM | 31 |
| 2 | DAST (3) | DCM | 40 |
| 3 | DAST (5) | DCM | 57 |
| 4c | DAST (10) | DCM | 80 |
| 5c | DAST (12) | DCM | 77 |
| 6d | DAST (10) | DCM | 68 |
| 7e | DAST (10) | THF | 61 |
| 8e | DAST (10) | MeCN | 54 |
| 9e | DAST (10) | DMF | 61 |
| 10f | DAST (10) | DMSO | 36 |
| 11e | DAST (10) | Acetone | Trace |
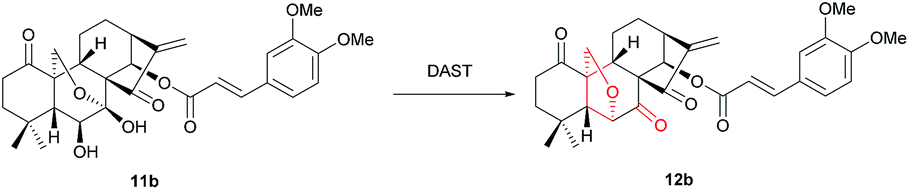
Other strategies for this rearrangement were also investigated for more efficient transformation and more environmentally benign conditions. Intriguingly, a trace amount of 12b could be observed under Swern oxidation condition using DMSO, (COCl)2 and i-Pr2NEt (for details, see the ESI† control experiments). Afterwards, 11b was subjected to Mitsunobu condition using 3,4,5-trimethoxybenzoic acid, PPh3 and DIAD in anhydrous THF, whereas no 12b was found. Finally, various Brønsted and Lewis acids, e.g. p-TsOH, CF3SO3H, HCl, AlCl3 (for details see the ESI Table S-1†), were evaluated as catalyst for dehydroxylation, however, no 12b could be detected, which suggested that the acidic additive could thoroughly block the transformation.
Under the optimal conditions, the generality of this protocol was investigated with sterically and electronically diverse substrates subjected to this rearrangement reaction. Gratifyingly, most of the substrates 11a–11r, were well tolerated, furnishing a series of 6,20-epoxy-14-acyl ent-kaurane diterpenoids 12a–12j in 79–84% yields (Scheme 2). Notably, the incorporated functionalities had a trivial impact on the transformation. Subsequently, aiming to prepare oridonin analogues carrying unprotected hydroxyl at C-14, the substrate 13 was synthesized via protection of 7,14-dihydroxyl of oridonin with 2,2-dimethoxypropane followed by acetylation with Ac2O, and satisfyingly the correspondingly 6,20-epoxy-14-OH ent-kaurane diterpenoid 14 was delivered in 89% yield with 13 treated with DAST (Scheme 3).
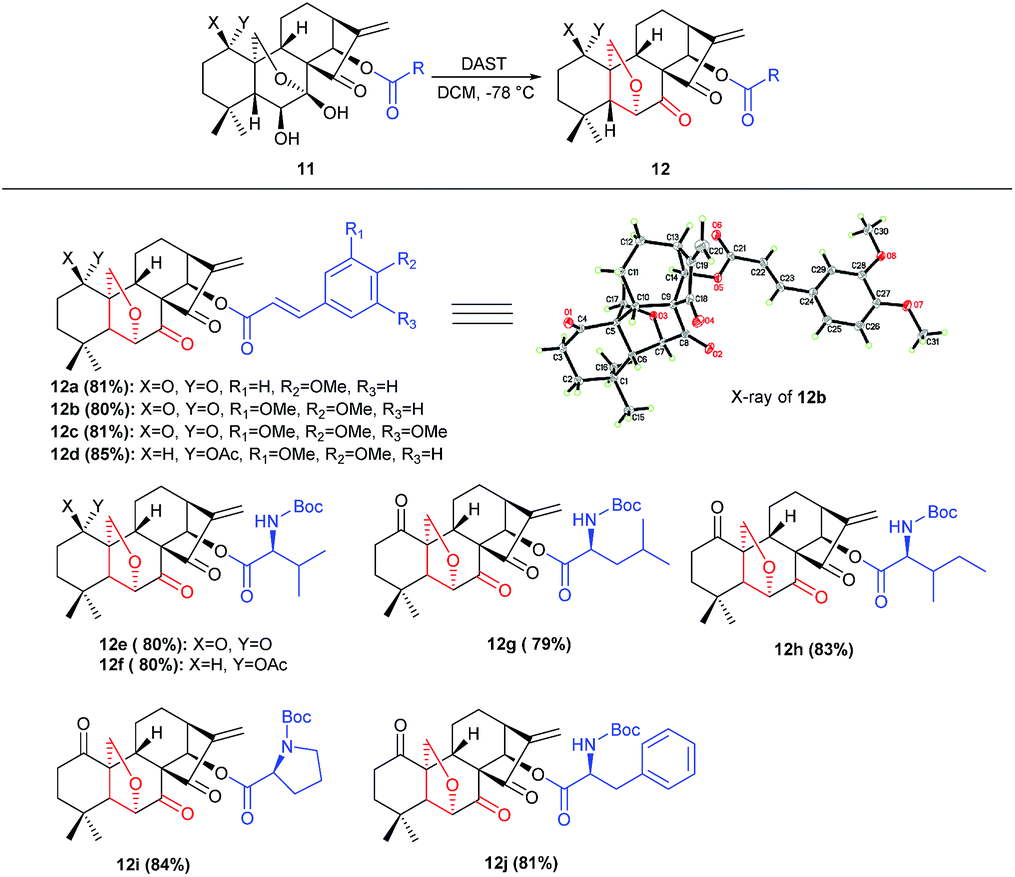 | ||
| Scheme 2 Synthesis of novel 6,20-epoxy ent-kaurane diterpenoid 12. | ||
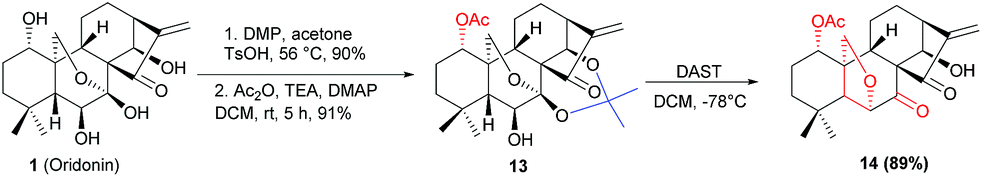 | ||
| Scheme 3 Synthesis of 6,20-epoxy-14-OH ent-kaurane diterpenoid 14. | ||
Interestingly, when 11d was treated with DAST in the presence of (E)-3-(3,4-dimethoxyphenyl)acrylic acid, the 1-acetyl-6,14-diacyl oridonin analogue 15 was isolated as the main product instead in 83% yield and only trace of 12d was obtained (Scheme 4).
 | ||
| Scheme 4 Synthesis of 1-acetyl-6,14-diyl oridonin analogue 15. | ||
Based on these experimental results, a plausible mechanism was proposed (Scheme 5). Initially, 11d reacts with DAST to produce the intermediate 16, which undergoes an intramolecular nucleophilic substitution to furnish the bicyclic oxiranium ion intermediate 17. Due to the basic nature of fluorine anion and comparative strong acidity of 7-OH, the hydroxyl group is readily deprotonated, followed by the opening of oxiranium ion moiety to yield 12d (route a). In contrast, an alternative reaction pathway (route b) might operate in the presence of 3,4-dimethoxycinnamic acid, the oxiranium ion moiety can be preferentially attacked by the more nucleophilic carboxylate anion of 3,4-dimethoxycinnamic acid to give the product 15.
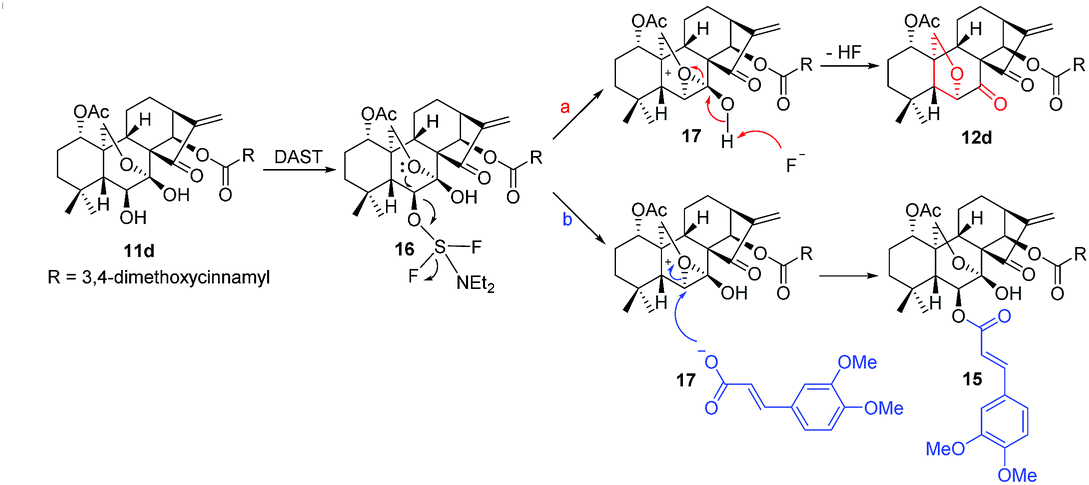 | ||
| Scheme 5 A possible reaction mechanism. | ||
The in vitro cytotoxicity of some products were determined by the methylthiazol tetrazolium (MTT) assay on the human hepatic carcinoma cell (HepG2), human multiple myeloma cell (RPMI-8226), human lung carcinoma (A549) cell lines with the commercial anticancer drug, paclitaxel (PTX), as the positive control. The results were summarized in Table 2. Notably, 11i (IC50 = 0.98 μM) and 14 (IC50 = 2.07 μM) exhibited potent inhibitory activities against HepG2 cell line. Compound 11e (IC50 = 7.60 μM) was found to be a potent cytotoxic agent against A549 cell lines (Fig. 2). In addition, to obtain the cytotoxicity of these new compounds on normal human cells, the effect of compounds 11e, 11i, 14 and oridonin was evaluated in human liver cancer cell line HepG2 and normal liver cell line L-O2. Compared with LO2 cells, oridonin was approximately 2-fold more selective in inhibiting the growth of HepG2 cells. The tested analogues 11i (8.84-fold) and 14 (3.43-fold) exhibited higher selectivity than oridonin (see ESI Table S-2†). Particularly, compound 11i seemed to be more selective than oridonin, with an SI (selectivity index, IC50 of normal cells/IC50 of tumor cells) value of 8.84. These results suggest that these 14-acyl oridonin analogues and novel 6,20-epoxy ent-kaurane diterpenoid analogues may serve as promising antitumor agents.
| Compounds | Cytotoxicity (IC50, μM) | ||
|---|---|---|---|
| HepG2 | RPMI-8226 | A549 | |
| a IC50 values were presented as the mean ± SD (standard error of the mean) from three separated experiments. | |||
| Oridonin | 7.93 ± 1.25 | 9.84 ± 0.41 | 22.64 ± 1.28 |
| PTX | 0.19 ± 0.03 | 1.40 ± 0.50 | 0.44 ± 0.26 |
| 11e | 13.81 ± 2.27 | 19.55 ± 2.07 | 7.60 ± 0.74 |
| 11g | 10.75 ± 1.21 | >100 | 21.14 ± 1.43 |
| 11h | 16.07 ± 0.53 | 10.96 ± 1.06 | 19.35 ± 1.05 |
| 11i | 0.98 ± 0.10 | 11.53 ± 1.33 | 19.82 ± 1.14 |
| 11j | 15.28 ± 1.90 | >100 | 18.13 ± 3.20 |
| 11k | 15.28 ± 1.90 | >100 | 18.13 ± 3.20 |
| 11l | 8.71 ± 1.25 | 10.23 ± 0.40 | 15.08 ± 1.46 |
| 11m | 13.53 ± 3.16 | 9.87 ± 0.97 | 14.01 ± 1.61 |
| 11o | 14.62 ± 1.68 | 14.52 ± 0.90 | 14.81 ± 1.99 |
| 11p | 45.00 ± 3.59 | 17.98 ± 1.38 | 72.22 ± 4.38 |
| 11q | 11.37 ± 0.77 | 16.61 ± 2.04 | 21.08 ± 3.53 |
| 11r | 23.68 ± 2.25 | 9.57 ± 0.92 | 23.80 ± 2.05 |
| 12a | 13.96 ± 0.68 | 9.66 ± 1.50 | 23.56 ± 2.76 |
| 12b | 16.07 ± 1.26 | 7.33 ± 1.42 | 16.73 ± 1.73 |
| 12d | 18.13 ± 1.20 | 19.66 ± 1.8 | 11.03 ± 1.70 |
| 12e | 9.42 ± 1.03 | 33.21 ± 3.87 | 18.06 ± 2.29 |
| 12g | 11.00 ± 2.68 | 10.05 ± 1.31 | 13.34 ± 1.95 |
| 12h | 9.37 ± 0.65 | 10.62 ± 0.95 | 14.13 ± 2.01 |
| 12i | 16.58 ± 2.93 | 14.64 ± 1.75 | 12.85 ± 2.20 |
| 12j | 21.60 ± 3.17 | 11.95 ± 1.34 | 17.58 ± 2.32 |
| 14 | 2.07 ± 0.29 | 9.54 ± 0.57 | 14.95 ± 3.57 |
| 15 | 9.02 ± 0.80 | 6.94 ± 0.41 | 10.31 ± 1.89 |
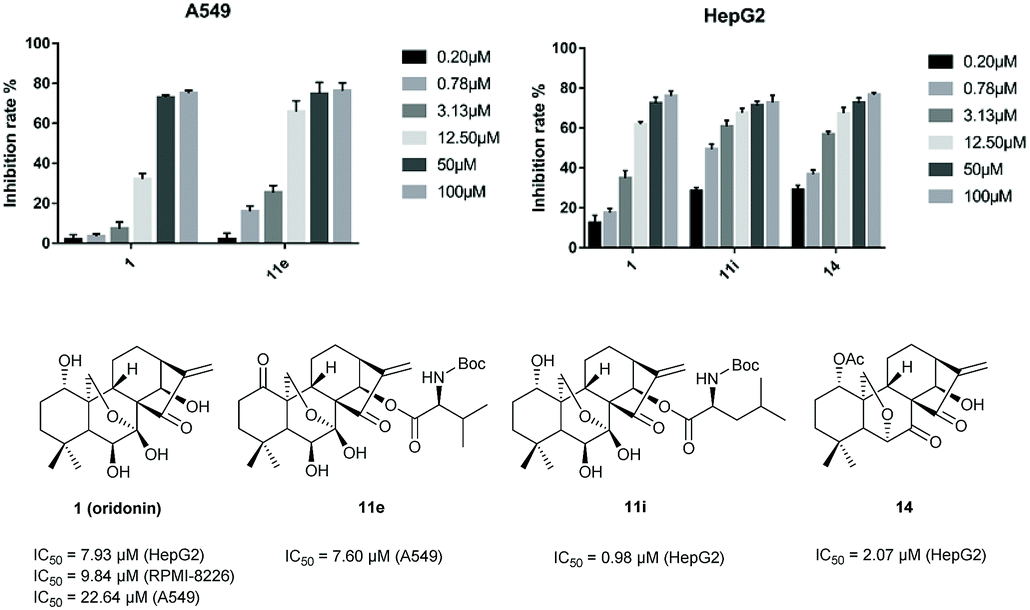 | ||
| Fig. 2 Comparison of the activity and IC50 of 1, 11e in inhibiting A549 and 1, 11i, 14 in inhibiting HepG2. | ||
Conclusions
In summary, we report a novel and concise protocol for syntheses of 6,20-epoxy ent-kaurane diterpenoid analogues from oridonin with α-methylene cyclopentanone core intact. The reaction features mild conditions and good yield. Remarkably, the prepared 14-acyl oridonin analogues 11e, 11i and 6,20-epoxy ent-kaurane diterpenoid analogue 14 exhibited superior anticancer activities compared to oridonin in vitro. Oridonin was endowed with more potent bioactivities via chemical modification, which will open a new avenue for development of novel anticancer agents. The further anticancer activity investigation of oridonin analogues is under the way.Experimental
Chemistry section
Unless otherwise stated, all commercial reagents were used without additional purification and solvents were distilled prior to use. All reactions were carried out under nitrogen atmosphere. 1H NMR and 13C NMR spectra were recorded in CDCl3 at 500 MHz, using CDCl3 as a reference standard (δ = 7.26 ppm) for 1H NMR and (δ = 77.00 ppm) for 13C NMR or DMSO-d6 as a reference standard (δ = 2.50 ppm) for 1H NMR and (δ = 39.52 ppm) for 13C NMR. Melting points were measured with a Laboratory Device MEL-TEMP and were uncorrected. TLC was performed using commercially prepared silica gel plates (GF254), and visualization was effected at 254 nm and 365 nm. High resolution mass spectra (HRMS) were recorded on the Exact Mass Spectrometer equipped with ESI ionization source.General procedure for the synthesis of compounds 11a–11r
To a stirring solution of oridonin (500 mg, 1.37 mmol) in acetone (40 mL) was added Jones reagent (0.6 mL) dropwise at ice-water bath. The resulting mixture was stirred at 0 °C for 20 min, and isopropyl alcohol was added to quench excess Jones reagent. Then the mixture was diluted with water and extracted with dichloromethane (3 × 10 mL). The extract was then washed with brine, dried over anhydrous MgSO4, filtered, and evaporated to give a solid crude product. The crude residue was recrystallized from acetone–hexane to give compound 10a as a white solid, 440 mg, 88% yield.Treatment of oridonin (500 mg, 1.37 mmol) with 2,2-dimethoxypropane in the presence of p-TsOH in acetone afforded 7,14-(1-methylethylene)-dioxy-oridonin derivative (498 mg, 1.23 mmol) in 90% yield. Subsequently, this derivative (200 mg, 0.50 mmol) was treated with Ac2O (0.05 mL, 0.50 mmol), Et3N (1 mL) and DMAP (183 mg, 1.50 mmol) in 15 mL dichloromethane to yield the corresponding compound 13 (221 mg, 91% yield). Deprotection of compound 13 (200 mg, 0.44 mmol) with 2% HCl solution in 10 mL tetrahydrofuran gave the corresponding compound 10b (155 mg, 87% yield).
Compound 10a (100 mg, 0.25 mmol) was mixed with 4-methoxycinnamic acid (50 mg, 0.25 mmol), EDCI (143 mg, 0.75 mmol) and DMAP (92 mg, 0.75 mmol) in 10 mL anhydrous dichloromethane, and the resulting mixture was stirred under nitrogen atmosphere at room temperature overnight. The reaction was poured into 1 M HCl solution, and the mixture was extracted with dichloromethane (3 × 5 mL). The organic layers were combined, washed with water and saturated NaCl solution, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (SiO2, DCM/MeOH) to give the compound 11a. White solid, mp 136–137 °C. 121 mg, 83% yield. Rf = 0.35 (1: 25 MeOH in CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.60 (d, J = 15.9 Hz, 1H), 7.44 (d, J = 8.7 Hz, 2H), 6.88 (t, J = 5.8 Hz, 2H), 6.27 (s, 1H), 6.20 (d, J = 15.9 Hz, 1H), 5.92 (s, 1H), 5.62 (d, J = 0.9 Hz, 1H), 5.47 (d, J = 8.8 Hz, 1H), 4.32 (dd, J = 10.6, 1.0 Hz, 1H), 4.05 (dd, J = 10.6, 1.5 Hz, 1H), 3.83 (d, J = 6.1 Hz, 3H), 3.25 (d, J = 9.6 Hz, 1H), 2.62 (dt, J = 14.0, 8.8 Hz, 1H), 2.47 (ddd, J = 15.4, 10.9, 6.6 Hz, 1H), 2.38–2.24 (m, 3H), 2.08–2.01 (m, 1H), 1.99 (d, J = 8.6 Hz, 1H), 1.95–1.87 (m, 1H), 1.74 (ddd, J = 13.8, 8.8, 6.7 Hz, 2H), 1.68–1.60 (m, 1H), 1.37–1.30 (m, 1H), 1.21 (s, 3H), 1.01 (s, 3H). 13C NMR (125 MHz, CDCl3) d 211.78, 204.97, 165.43, 161.79, 149.32, 146.32, 130.08, 126.56, 122.20, 114.37, 114.06, 97.13, 75.54, 73.03, 64.93, 61.18, 60.35, 55.39, 50.77, 48.56, 41.32, 38.54, 35.83, 32.89, 30.53, 30.00, 29.69, 23.30, 19.05. HRMS (m/z) (ESI): calcd for C30H35O8 523.2326 [M + H]+ found 523.2318.
All of the products 11b–11r were synthesized according to above described procedure.
General procedure for the synthesis of compounds 12a–12k
Compound 11a (94 mg, 0.18 mmol) was mixed with 0.24 mL DAST (diethylaminosulfur trifluoride) in anhydrous dichloromethane (10 mL), and the resulting mixture was stirred under nitrogen atmosphere at −78 °C for 10 min, then warmed up to room temperature and stirred for 2 h. The mixture was poured into water, and extracted with dichloromethane (3 × 5 mL). The organic layers were combined, washed with water and saturated NaCl solution, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (petroleum ether/ethyl acetate) to give the compound 12a. White solid, mp 119–120 °C. 73 mg, 81% yield. Rf = 0.46 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 petroleum ether/ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.58 (d, J = 15.9 Hz, 1H), 7.44 (d, J = 8.7 Hz, 2H), 6.86 (d, J = 8.7 Hz, 2H), 6.20 (d, J = 15.9 Hz, 1H), 6.16 (s, 1H), 5.72 (s, 1H), 5.50 (s, 1H), 4.38 (d, J = 9.7 Hz, 1H), 4.31–4.26 (m, 2H), 3.82 (s, 3H), 3.23 (d, J = 7.9 Hz, 1H), 2.74 (ddd, J = 14.6, 11.7, 6.3 Hz, 1H), 2.56–2.45 (m, 2H), 2.35 (dt, J = 14.6, 4.6 Hz, 1H), 2.18–2.10 (m, 2H), 1.96–1.81 (m, 3H), 1.77–1.69 (m, 1H), 1.66 (s, 1H), 1.19 (d, J = 15.1 Hz, 3H), 1.05 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 209.87, 202.56, 199.65, 166.04, 161.48, 147.69, 145.74, 129.98, 126.97, 119.22, 114.68, 114.21, 83.31, 77.27, 77.01, 76.76, 75.61, 70.10, 64.24, 61.58, 61.31, 55.34, 47.17, 42.05, 41.18, 36.37, 31.96, 31.33, 30.17, 29.68, 23.23, 19.17. HRMS (m/z) (ESI): calcd for C30H33O7 505.2221 [M + H]+ found 505.2218.
:1 petroleum ether/ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.58 (d, J = 15.9 Hz, 1H), 7.44 (d, J = 8.7 Hz, 2H), 6.86 (d, J = 8.7 Hz, 2H), 6.20 (d, J = 15.9 Hz, 1H), 6.16 (s, 1H), 5.72 (s, 1H), 5.50 (s, 1H), 4.38 (d, J = 9.7 Hz, 1H), 4.31–4.26 (m, 2H), 3.82 (s, 3H), 3.23 (d, J = 7.9 Hz, 1H), 2.74 (ddd, J = 14.6, 11.7, 6.3 Hz, 1H), 2.56–2.45 (m, 2H), 2.35 (dt, J = 14.6, 4.6 Hz, 1H), 2.18–2.10 (m, 2H), 1.96–1.81 (m, 3H), 1.77–1.69 (m, 1H), 1.66 (s, 1H), 1.19 (d, J = 15.1 Hz, 3H), 1.05 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 209.87, 202.56, 199.65, 166.04, 161.48, 147.69, 145.74, 129.98, 126.97, 119.22, 114.68, 114.21, 83.31, 77.27, 77.01, 76.76, 75.61, 70.10, 64.24, 61.58, 61.31, 55.34, 47.17, 42.05, 41.18, 36.37, 31.96, 31.33, 30.17, 29.68, 23.23, 19.17. HRMS (m/z) (ESI): calcd for C30H33O7 505.2221 [M + H]+ found 505.2218.
All of the products 12b–12j were synthesized according to above described procedure.
General procedure for the synthesis of compound 14
Compound 13 (100 mg, 0.22 mmol) was mixed with 0.30 mL DAST (diethylaminosulfur trifluoride) in anhydrous dichloromethane (10 mL) and the resulting mixture was stirred under nitrogen atmosphere at −78 °C for 10 min, then warmed up to room temperature and stirred for 2 h. The mixture was poured into water, and the mixture was extracted with dichloromethane thrice (3 × 5 mL). The organic layers were combined, washed with brine, dried over anhydrous MgSO4, filtered and concentrated in vacuo. The crude product was purified by column chromatography (6:1 petroleum ether/ethyl acetate) to give the compound 14. Yellow solid, mp 115–116 °C. 77 mg, 89% yield. Rf = 0.27 (1:1 petroleum ether/ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 6.13 (s, 1H), 5.53 (s, 1H), 4.88 (dd, J = 10.9, 6.0 Hz, 1H), 4.88 (dd, J = 10.9, 6.0 Hz, 1H), 4.74 (s, 2H), 4.58 (s, 1H), 4.17 (s, 1H), 4.12 (d, J = 10.0 Hz, 1H), 4.10 (q, J = 10.0 Hz, 2H), 4.09 (d, J = 10.0 Hz, 1H), 3.83 (dd, J = 43.1, 15.7 Hz, 1H), 3.11 (d, J = 8.7 Hz, 1H), 3.11 (d, J = 8.7 Hz, 1H), 2.44–2.35 (m, 1H), 2.45–2.33 (m, 1H), 2.19 (dd, J = 12.2, 6.1 Hz, 1H), 2.19 (dd, J = 12.2, 6.1 Hz, 1H), 1.99 (s, 3H), 1.89 (s, 1H), 1.64–1.57 (m, 2H), 1.49 (s, 1H), 1.41 (d, J = 8.7 Hz, 1H), 1.21 (s, 1H), 1.04 (s, 3H), 1.01 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 205.36, 203.22, 169.94, 149.04, 120.20, 82.53, 76.74, 73.59, 68.21, 64.94, 55.04, 52.97, 51.04, 43.79, 36.97, 32.26, 30.83, 29.68, 29.37, 24.60, 22.45, 21.40, 18.95. HRMS (m/z) (ESI): calcd for C22H29O6 389.1959 [M + H]+ found 389.1959.
General procedure for the synthesis of compound 15
Compound 11d (50 mg, 0.08 mmol) was mixed with (E)-3-(3,4-dimethoxyphenyl)acrylic acid (33 mg, 0.16 mmol), 0.11 mL DAST (diethylaminosulfur trifluoride) in anhydrous dichloromethane (10 mL) and the resulting mixture was stirred under nitrogen atmosphere at −78 °C for 10 min, then warmed up to room temperature and stirred overnight. The mixture was poured into water, and the mixture was extracted with dichloromethane thrice (3 × 5 mL). The organic layers were combined, washed with brine, dried over anhydrous MgSO4, filtered and concentrated in vacuo. The crude product was purified by column chromatography (6:1 petroleum ether/ethyl acetate) to give the compound 15. White solid, mp 125–126 °C. 52 mg, 83% yield. Rf = 0.63 (1:1 petroleum ether/ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.88 (d, J = 15.9 Hz, 1H), 7.59 (d, J = 15.9 Hz, 1H), 7.18 (d, J = 8.3 Hz, 1H), 7.15 (s, 1H), 7.04 (d, J = 8.3 Hz, 1H), 6.99 (s, 1H), 6.88 (d, J = 8.2 Hz, 1H), 6.81 (t, J = 9.1 Hz, 1H), 6.56 (d, J = 15.9 Hz, 1H), 6.20 (dd, J = 15.8, 6.4 Hz, 1H), 6.01 (d, J = 13.7 Hz, 2H), 5.36–5.31 (m, 2H), 5.02 (s, 1H), 4.73 (dd, J = 11.2, 5.5 Hz, 1H), 4.41 (d, J = 10.6 Hz, 1H), 4.28 (t, J = 8.9 Hz, 1H), 3.93 (d, J = 6.4 Hz, 6H), 3.89 (d, J = 5.4 Hz, 6H), 3.17 (d, J = 9.9 Hz, 1H), 2.61–2.53 (m, 1H), 2.36–2.31 (m, 1H), 2.04 (d, J = 7.4 Hz, 3H), 1.90–1.78 (m, 2H), 1.71 (d, J = 6.1 Hz, 1H), 1.56 (d, J = 12.9 Hz, 1H), 1.46 (t, J = 12.7 Hz, 1H), 1.40–1.30 (m, 3H), 1.21 (d, J = 8.9 Hz, 3H), 0.90 (d, J = 8.9 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 200.70, 169.93, 168.85, 165.90, 151.30, 151.20, 150.25, 149.16, 146.21, 146.05, 127.58, 127.10, 123.20, 117.17, 115.70, 114.89, 110.93, 110.86, 109.77, 109.31, 96.81, 75.94, 75.16, 74.05, 63.40, 60.14, 56.04, 52.18, 41.97, 40.46, 40.23, 37.70, 33.68, 31.65, 30.37, 29.68, 27.00, 25.17, 21.58, 21.38, 17.82. HRMS (m/z) (ESI): calcd for C44H51NO13 787.3324 [M + H]+ found 787.3342.
In vitro cytotoxicity
The HepG2, RPMI-8226, A549, L-O2 cell lines used in this study were all purchased from EnoGene company. RPMI-8226 and A549 cells were cultured in RPMI 1640 media containing 10% heat inactivated FBS and HepG2 and L-O2 cells were cultured in DMEM media containing 10% heat inactivated FBS at 37 °C with 5% CO2. In order to investigate the antitumor activity of some compounds, a commercial Paclitaxel (PTX) was used as a positive control drug.10000 cells of HepG2, RPMI-8226, A549 or L-O2 were prepared into 200 μL cell suspension in each well of 96-well plates and the plates were incubated for 24 h at 37 °C with 5% CO2. 100 μL medium with compounds was mixed into each well of 96-well plates, respectively. And the negative control group, the solvent control group, the positive control group were established, respectively. The plates were incubated for 72 h at 37 °C with 5% CO2. Then 10 μL CCK-8 solution was mixed into each well of 96-well plates and the plates were incubated for 4 h. Optical absorbance at 450 nm was determined with microplate absorbance reader (Bio-Rad). IC50 values were calculated from the dose–response curves of the assay (Prism 7.0).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the financial support from National Natural Science Foundation of China (81561148012, 21572154, 21772181), NSFC-Shandong Joint Fund for Marine Science Research Centers (U1606403) and the Fundamental Research Funds for the Central Universities [201612013].Notes and references
- H. D. Sun, S. X. Huang and Q. B. Han, Nat. Prod. Rep., 2006, 23, 673–698 RSC.
- C. Y. Ding, Y. S. Zhang, H. J. Chen, Z. D. Yang, C. Wild, L. L. Chu, H. L. Liu, Q. Shen and J. Zhou, J. Med. Chem., 2013, 56, 5048–5058 CrossRef PubMed.
- P. H. Abelson, Science, 1990, 247, 513 CrossRef PubMed.
- Y. C. Li, M. R. Sun, Y. H. Zhao, X. Z. Fu, H. W. Xu and J. F. Liu, Cytotechnology, 2016, 68, 389–397 CrossRef PubMed.
- T. Ikezoe, Y. Yang, K. Bandobashi, T. Saito, S. Takemoto, H. Machida, K. Togitani, H. P. Koeffler and H. Taguchi, Mol. Cancer Ther., 2005, 4, 578–586 CrossRef PubMed.
- Q. Cui, S. Tashiro, S. Onodera, M. Minami and T. Ikejima, Biol. Pharm. Bull., 2007, 30, 859–864 CrossRef.
- C. Y. Li, E. Q. Wang, Y. Cheng and J. K. Bao, Int. J. Biochem. Cell Biol., 2011, 43, 701–704 CrossRef PubMed.
- S. Wang, Z. Zhong, J. Wan, W. Tan, G. Wu, M. Chen and Y. Wang, Am. J. Chin. Med., 2013, 41, 177–196 CrossRef PubMed.
- R. F. Bao, Y. J. Shu, X. S. Wu, H. Weng, Q. Ding, Y. Cao, M. L. Li, J. S. Mu, W. G. Wu, Q. C. Ding, Z. J. Tan, T. Y. Liu, L. Jiang, Y. P. Hu, J. F. Gu and Y. B. Liu, BMC Cancer, 2014, 14, 217 CrossRef PubMed.
- Y. Dong, T. Zhang, J. J. Li, H. Y. Deng, Y. J. Song, D. Zhai, Y. Peng, X. L. Lu, M. Y. Liu, Y. X. Zhao and Z. F. Yi, PLoS One, 2014, 9, e113830 CrossRef PubMed.
- Y. Li, Y. Wang, S. Wang, Y. Gao, X. Zhang and C. Lu, Med. Oncol., 2015, 32, 365 CrossRef PubMed.
- M. Zheng, Z. Zhu, Y. Zhao, D. Yao, M. Wu and G. Sun, Mol. Med. Rep., 2017, 15, 375–379 CrossRef PubMed.
- J. Yang, X. Ren, L. Zhang, Y. Li, B. Cheng and J. Xia, Biomed. Pharmacother., 2018, 100, 226–232 CrossRef PubMed.
- T. Zhang, Y. Tan, R. Zhao and Z. Y. Liu, Wspolczesna Onkol., 2013, 17, 38–44 CrossRef PubMed.
- D. L. Liu, H. Q. Bu, H. M. Jin, J. F. Zhao, Y. Li and H. Huang, Mol. Med. Rep., 2014, 10, 3027–3034 CrossRef PubMed.
- S. Y. Gao, H. X. Tan, N. Zhu, H. Y. Gao, C. Y. Lv, J. Gang and Y. B. Ji, Internet J. Oncol., 2016, 48, 2453–2460 CrossRef PubMed.
- S. T. Xu, S. S. Luo, H. Yao, H. Cai, X. M. Miao, F. Wu, D. H. Yang, X. M. Wu, W. J. Xie, H. Q. Yao, Z. S. Chen and J. Y. Xu, J. Med. Chem., 2016, 59, 5022–5034 CrossRef PubMed.
- S. T. Xu, H. Yao, S. S. Luo, Y. K. Zhang, D. H. Yang, D. H. Li, G. Y. Wang, M. Hu, Y. Y. Qiu, X. M. Wu, H. Q. Yao, W. J. Xie, Z. S. Chen and J. Y. Xu, J. Med. Chem., 2017, 60, 1449–1468 CrossRef PubMed.
- G. B. Zhou, H. Kang, L. Wang, L. Gao, P. Liu, J. Xie, F. X. Zhang, X. Q. Weng, Z. X. Shen, J. Chen, L. J. Gu, M. Yan, D. E. Zhang, S. J. Chen, Z. Y. Wang and Z. Chen, Blood, 2007, 109, 3441–3450 CrossRef PubMed.
- W. Xu, J. Sun, T. T. Zhang, B. Ma, S. M. Cui, D. W. Chen and Z. G. He, Acta Pharmacol. Sin., 2006, 27, 1642–1646 CrossRef PubMed.
- Y. H. Ma, W. W. Xie, T. T. Tian, Y. R. Jin, H. J. Xu, K. R. Zhang and Y. F. Du, Anal. Biochem., 2016, 511, 61–73 CrossRef PubMed.
- E. Fujita, Y. Nagao, K. Kaneko, S. Nakazawa and H. Kuroda, Chem. Pharm. Bull., 1976, 24, 2118–2127 CrossRef PubMed.
- E. Fujita, Y. Nagao, T. Kohno, M. Matsuda and M. Ozaki, Chem. Pharm. Bull., 1981, 29, 3208–3213 CrossRef PubMed.
- M. Zhang, Y. M. Zhang, W. Lu and F. J. Nan, Org. Biomol. Chem., 2011, 9, 4436–4439 RSC.
- S. X. Huang, Y. Zhou, J. X. Pu, R. T. Li, M. Li, W. L. Xiao, L. G. Lou, Q. B. Han, L. S. Ding, S. L. Peng and H. D. Sun, Tetrahedron, 2006, 62, 4941–4947 CrossRef.
- C. Ding, Y. Zhang, H. Chen, Z. Yang, C. Wild, N. Ye, C. D. Ester, A. Xiong, M. A. White, Q. Shen and J. Zhou, J. Med. Chem., 2013, 56, 8814–8825 CrossRef PubMed.
- C. Y. Ding, Y. S. Zhang, H. J. Chen, C. Wild, T. Z. Wang, M. A. White, Q. Shen and J. Zhou, Org. Lett., 2013, 15, 3718–3721 CrossRef PubMed.
- C. Ding, L. Wang, H. Chen, C. Wild, N. Ye, Y. Ding, T. Wang, M. A. White, Q. Shen and J. Zhou, Org. Biomol. Chem., 2014, 12, 8442–8452 RSC.
- X. B. Yan, M. Lei, Y. J. Zhang and H. M. Liu, Chin. J. Org. Chem., 2005, 25, 222–224 Search PubMed.
- J. Y. Xu, J. Y. Yang, Q. Ran, L. Wang, J. Liu, Z. X. Wang, X. M. Wu, W. Y. Hua, S. T. Yuan, L. Y. Zhang, M. Q. Shen and Y. F. Ding, Bioorg. Med. Chem. Lett., 2008, 18, 4741–4744 CrossRef PubMed.
- D. H. Li, L. Wang, H. Cai, Y. H. Zhang and J. Y. Xu, Molecules, 2012, 17, 7556–7568 CrossRef PubMed.
- S. E. Boiadjiev and D. A. Lightner, J. Org. Chem., 1997, 62, 399–404 CrossRef PubMed.
- T. Mase, I. N. Houpis, A. Akao, I. Dorziotis, K. Emerson, T. Hoang, T. Iida, T. Itoh, K. Kamei, S. Kato, Y. Kato, M. Kawasaki, F. Lang, J. Lee, J. Lynch, P. Maligres, A. Molina, T. Nemoto, S. Okada, R. Reamer, J. Z. Song, D. Tschaen, T. Wada, D. Zewge, R. P. Volante, P. J. Reider and K. Tomimoto, J. Org. Chem., 2001, 66, 6775–6786 CrossRef PubMed.
- K. Suzuki, Y. Ito and O. Kanie, Carbohydr. Res., 2012, 359, 81–91 CrossRef PubMed.
- P. BorracheroMoya, F. CabreraEscribano, M. GomezGuillen and F. MadridDiaz, Tetrahedron Lett., 1997, 38, 1231–1234 CrossRef.
- P. Borrachero, F. Cabrera-Escribano, A. T. Carmona and M. Gomez-Guillen, Tetrahedron: Asymmetry, 2000, 11, 2927–2946 CrossRef.
- T. S. Lin, W. T. Tsai and P. H. Liang, Tetrahedron, 2016, 72, 5571–5577 CrossRef.
- C. Yang, I. L. Wong, K. Peng, Z. Liu, P. Wang, T. Jiang, T. Jiang, L. M. Chow and S. B. Wan, Eur. J. Med. Chem., 2017, 125, 795–806 CrossRef PubMed.
- I. L. Wong, B. C. Wang, J. Yuan, L. X. Duan, Z. Liu, T. Liu, X. M. Li, X. Hu, X. Y. Zhang, T. Jiang, S. B. Wan and L. M. Chow, J. Med. Chem., 2015, 58, 4529–4549 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: CCDC 1590323. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra05728a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |