
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic ordering in a vanadium-organic coordination polymer using a pyrrolo[2,3-d:5,4-d′]bis(thiazole)-based ligand†
Yulia A. Getmanenko‡
 *a,
Christopher S. Mullinsb,
Vladimir N. Nesterovc,
Stephanie Lakea,
Chad Riskob and
Ezekiel Johnston-Halperina
*a,
Christopher S. Mullinsb,
Vladimir N. Nesterovc,
Stephanie Lakea,
Chad Riskob and
Ezekiel Johnston-Halperina
aDepartment of Physics, The Ohio State University, Columbus, Ohio 43210-1173, USA. E-mail: ygetmanenko@scripps.edu
bDepartment of Chemistry and Center for Applied Energy Research, University of Kentucky, Lexington, Kentucky 40506-0055, USA
cDepartment of Chemistry, University of North Texas, Denton, Texas 76201, USA
First published on 25th October 2018
Abstract
Here we present the synthesis and characterization of a hybrid vanadium-organic coordination polymer with robust magnetic order, a Curie temperature TC of ∼110 K, a coercive field of ∼5 Oe at 5 K, and a maximum mass magnetization of about half that of the benchmark ferrimagnetic vanadium(tetracyanoethylene)∼2 (V·(TCNE)∼2). This material was prepared using a new tetracyano-substituted quinoidal organic small molecule 7 based on a tricyclic heterocycle 4-hexyl-4H-pyrrolo[2,3-d:5,4-d′]bis(thiazole) (C6-PBTz). Single crystal X-ray diffraction of the 2,6-diiodo derivative of the parent C6-PBTz, showed a disordered hexyl chain and a nearly linear arrangement of the substituents in positions 2 and 6 of the tricyclic core. Density functional theory (DFT) calculations indicate that C6-PBTz-based ligand 7 is a strong acceptor with an electron affinity larger than that of TCNE and several other ligands previously used in molecular magnets. This effect is due in part to the electron-deficient thiazole rings and extended delocalization of the frontier molecular orbitals. The ligand detailed in this study, a representative example of fused heterocycle aromatic cores with extended π conjugation, introduces new opportunities for structure–magnetic-property correlation studies where the chemistry of the tricyclic heterocycles can modulate the electronic properties and the substituent at the central N-position can vary the spatial characteristics of the magnetic polymer.
1 Introduction
Metal–organic hybrid magnetic materials have the potential to contribute to the realization of spintronics,1 a fast-developing technology where the spin of the electron, not its charge, carries information. Vanadium tetracyanoethylene (V·(TCNE)x; V = vanadium, TCNE = tetracyanoethylene, x ∼1.5–2), a room temperature organometallic semiconducting ferrimagnet, is the most widely studied material in the class of the V·(ligand)x family of molecular magnets.2 The application of V·(TCNE)x as a spin injection/detection layer in hybrid devices3 and demonstration of material suitability for high-quality magnonic circuits4,5 indicate that metal–organic hybrid materials are competitive with traditional inorganic magnetic materials. Further, the air-sensitivity common to metal–ligand materials can be addressed by encapsulation,6 directly enabling their use in devices that operate under ambient conditions.The molecular designs for the organic ligands previously investigated in the preparation of hybrid vanadium-based magnetic materials can be divided into three major classes7 (Fig. 1): (1) acyclic polycyanoalkenes I, e.g. TCNE,2 hexacyanobutadiene,8 methyl tricyanoethylenecarboxylate9,10 and ethyl tricyanoethylenecarboxylate11,12 and several larger sized derivatives Ie–h containing substituted benzenes;13,14 (2) quinoidal systems, e.g. tetracyanoquinodimethane (TCNQ) IIa and its substituted analogues IIb–f,15 and N,7,7-tricyanoquinomethanimine III;16 (3) polycyano-substituted aromatic one-ring systems IV, e.g. tetracyanobenzene (TCNB),17 tetracyanopyridine,18 tetracyanopyrazine.19 The discovery of magnetically ordered vanadium–organic hybrid materials using α,α′-dicyanoperfluorostilbene Id13 provided evidence that relatively large ligands can be used for construction of new hybrid materials; however, the majority of the previously examined ligands are small in size. The choice of the material preparation method of vanadium-organic coordination polymers has impact on the composition, quality, and magnetic properties of the materials. Physical vapor deposition (PVD), chemical vapor deposition (CVD), and solution synthesis are typically used to prepare V-based hybrid materials. Optimized PVD and CVD allow for the preparation of reproducible, high-quality thin-films of the solvent-free material.20 Solution synthesis often results in solvent inclusion, which impacts composition and magnetic properties of the hybrid material,13,21 though it is a fairly straightforward method that allows for fast preparation of the bulk material using standard techniques and serves as an effective tool for preliminary investigation of magnetic properties of novel hybrid materials.
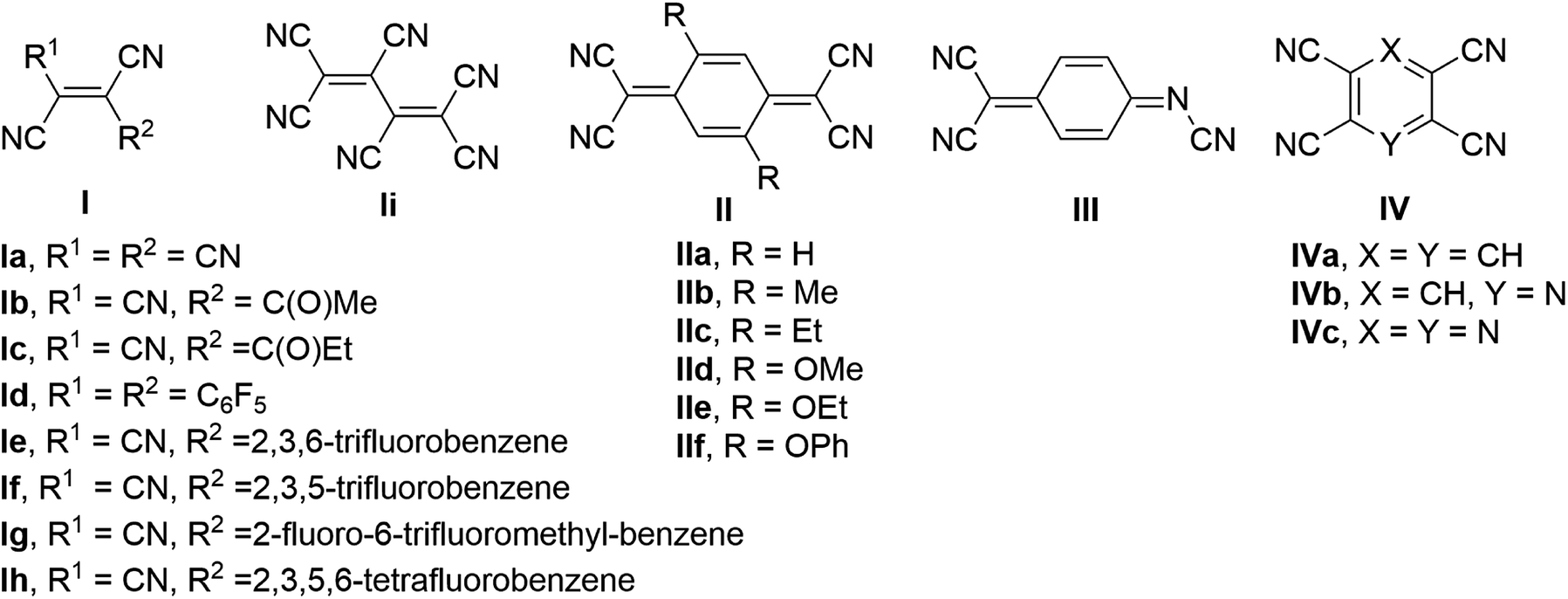 | ||
| Fig. 1 Organic ligands previously explored in vanadium-based hybrid magnetic materials. | ||
In order to expand the chemical library of ligands used in hybrid magnetic materials, here we synthesize a new tetracyano-quinoidal ligand, 7, based on the tricyclic core 4-hexyl-4H-pyrrolo[2,3-d:5,4-d′]bis(thiazole) (C6-PBTz).22 Single crystal structures of two intermediates and ligand 7 were studied by X-ray diffraction analysis. To gain insight into the electronic characteristics of 7, density functional theory calculations (DFT) calculations were carried out and the results compared with those for several ligands that were previously explored in a preparation of hybrid magnets, as well as closely related quinoidal systems23,24 based on the tricyclic cores dithieno[3,2-b:2′,3′-d]pyrrole (DTP) and diselenopheno[3,2-b:2′,3′-d]pyrrole (DSP). We then examine the reaction of 7 with V(CO)6 as a vanadium precursor in dichloromethane, and investigation of the magnetic properties of the V·(ligand 7)x hybrid material 8 indicates a presence of robust magnetic ordering.
2 Results and discussion
2.1 Preparation and characterization of the ligand (7) and X-ray analysis
A six-step synthesis of the target ligand 7 begins with commercially available 2-bromothiazole 1 (Scheme 1). Dibromo-2,2′-bis(triisopropylsilyl)-5,5′-bithiazole (3), a key intermediate for the cyclization reaction, was prepared as described previously.25 4-n-Hexyl-4H-pyrrolo[2,3-d:5,4-d′]bis(thiazole) (5) was prepared from dibromide 3 in two steps22 using optimized reaction conditions. Buchwald–Hartwig amination of dibromide 3 with hexylamine using anhydrous toluene as a solvent at 170–175 °C in a pressure vessel26 improved the yield of 4 up to 45% in comparison with 13–31% yield previously observed for an open vessel reaction using boiling mesitylene as a solvent.22 The introduction of iodine functional groups was achieved in good yield of 63–78% by a reaction of C6-PBTz (5) with N-iodosuccinimide (NIS) in anhydrous N,N-dimethylformamide (DMF) at room temperature. The Pd-catalysed reaction of diiodo-C6-PBTz (6) with deprotonated malononitrile followed by oxidative work up with bromine water proved to be a challenging step on a gram scale, and the best result was obtained on a small scale when ∼350 mg of diiodide 6 with 50% catalyst load were used. The target ligand 7 was isolated as a dark coloured solid (its solution in dichloromethane is red-wine coloured, Fig. S7†) and was characterized by nuclear magnetic resonance (1H, 13C{1H} and DEPT-135 NMR) and high-resolution mass spectrometry.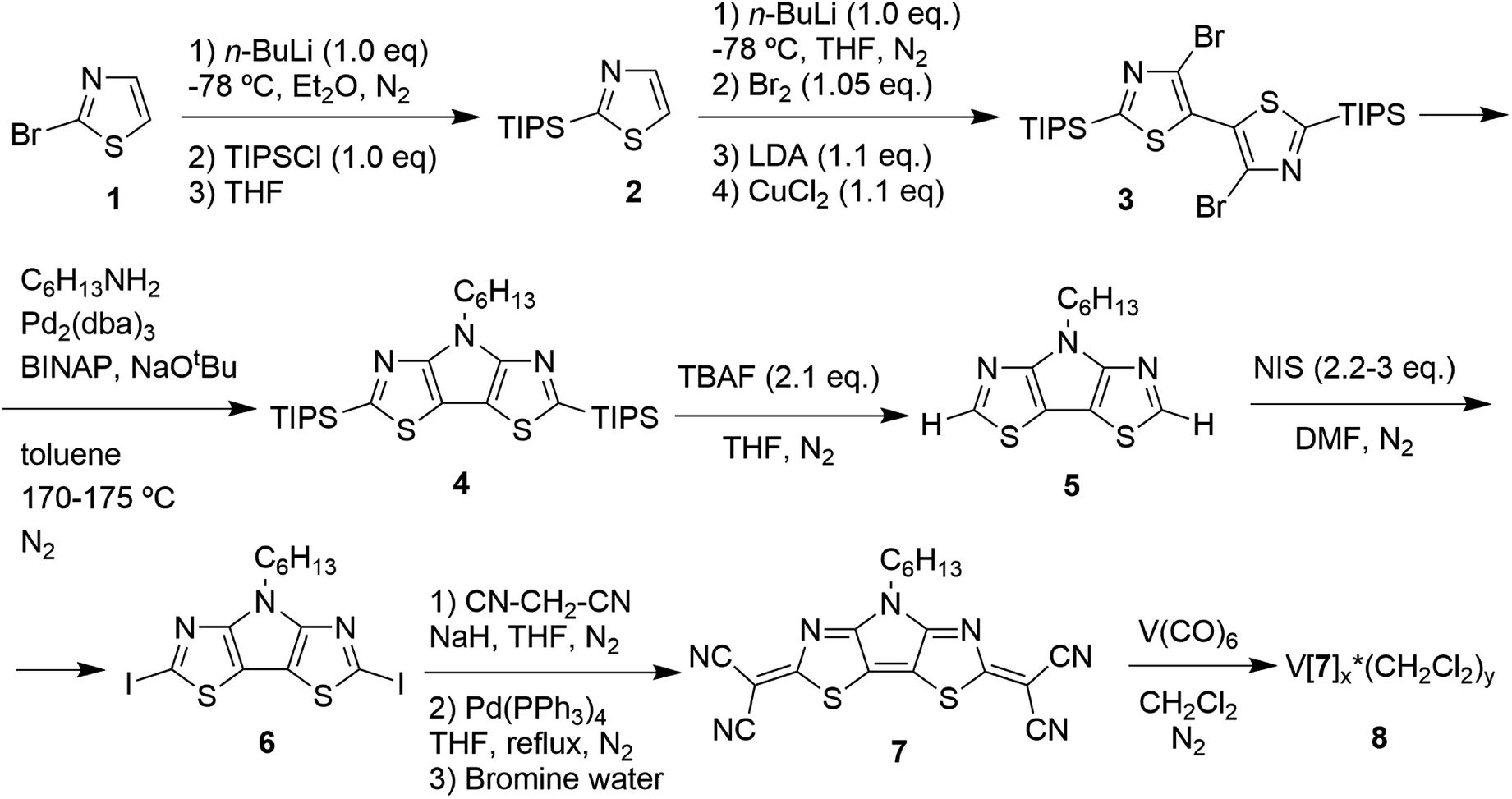 | ||
| Scheme 1 Synthesis of the ligand 7 and its vanadium-based hybrid material 8. | ||
Single crystals of intermediate C6-PBTz (5) (Fig. S1†) were obtained from a purified material, which was initially isolated as an oil after column chromatography purification and then solidified on standing. Single crystals of the diiodo-derivative 6 (Fig. S2†) were obtained by slow solvent evaporation from ethanol–dichloromethane mixture. Ligand 7 produced single crystals (Fig. S3†) by slow removal of dichloromethane or chloroform. The refinement details and structural parameters from the single crystal X-ray analyses of intermediates 5 and 6 and ligand 7 are summarized in Table S1.†
Compound C6-PBTz (5) crystallizes in the monoclinic space group Cc with one molecule per asymmetric unit (Fig. 2). The hexyl chain in this molecule is perpendicular to the tricyclic core with dihedral angle of 89.83(6)°. The dihedral angle between the two thiazole rings is 1.7(1)° indicating a nearly planar tricyclic core. This analysis is consistent with recently published results.27 Analysis of the crystal packing in 5 reveals the existence of two short intermolecular contacts, N(1)⋯S(2) (3.109(5) Å) and N(3)⋯S(2) (3.083(5) Å), which link molecules into layers parallel to the ab plane (Fig. 2b).
 | ||
| Fig. 2 (a) General view of molecule 5; (b) fragment of the crystal packing diagram along the c axis. | ||
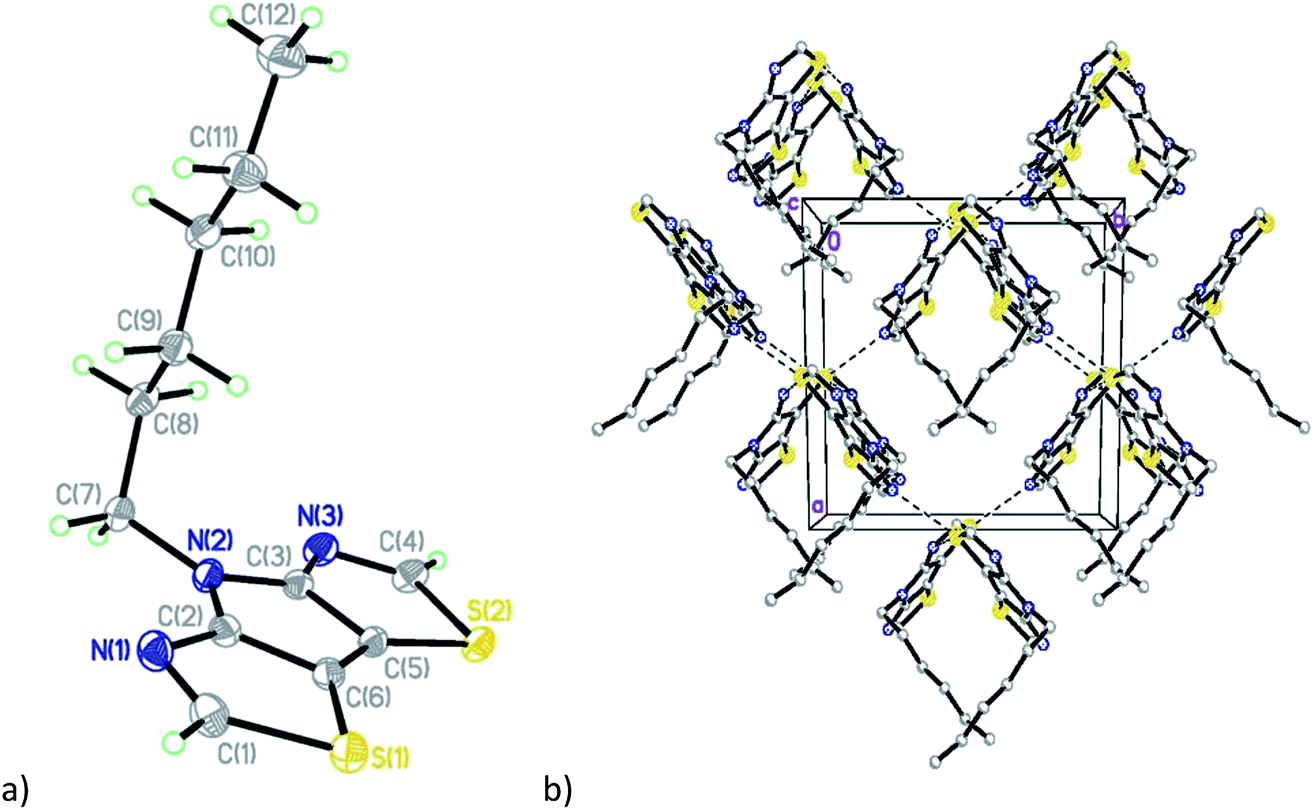
The diiodo-derivative 6 crystallizes in the monoclinic space group Cm with two independent molecules per asymmetric unit with a practically flat tricyclic core (Fig. 3). In a crystal of the compound 6 one of the independent molecules occupies a special position in the unit cell and forms short intermolecular contacts with two other molecules: I(1)/I(1B)⋯I(1A)/I(1AA) with distance of 4.008(5) Å and I(1)/I(1B)⋯N(1A)/N(1AA) with distance of 3.007(5) Å (Fig. 3b).
 | ||
| Fig. 3 (a) General view of one independent molecule 6 (the disordered part was removed for clarity); (b) fragment of the crystal packing diagram with short intermolecular contacts shown as dotted lines. | ||
The mutual orientation of the molecules in the crystal of 6 leads to the formation of another set of intermolecular contacts: I(1)/I(1B)⋯I(2A) equal to 4.144(5) Å, I(1)/I(1B)⋯I(2A) equal to 4.170(5) Å, I(1A)⋯I(2A) equal to 4.034(5) Å, N(1)/N(1B)⋯I(2A) 3.040(5) Å, I(1A)⋯N(3A) = 2.973(5) Å, and I(2A)⋯S(2A) equal to 3.867(5) Å. These close contacts link molecules into a three-dimensional framework (Fig. 4).
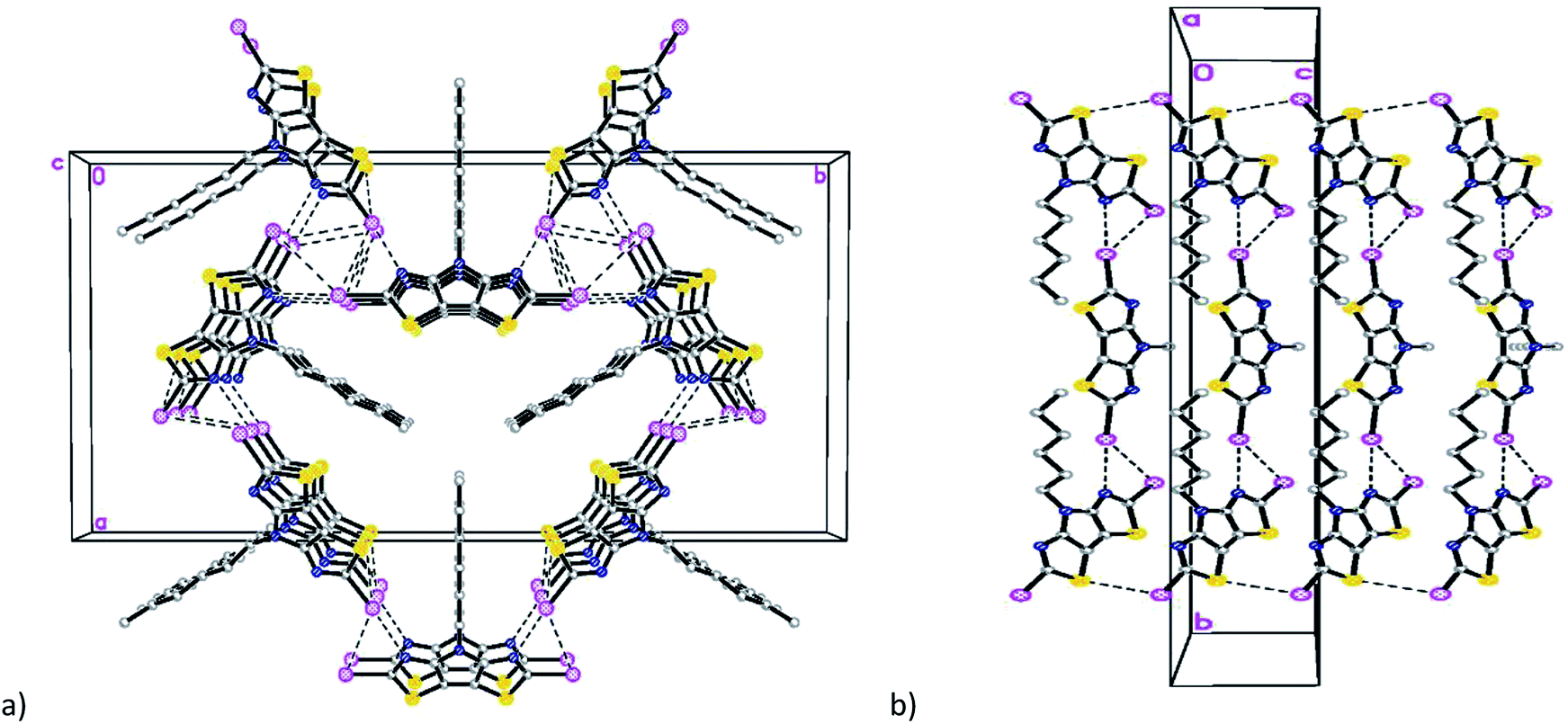 | ||
| Fig. 4 Fragment of the crystal packing diagrams of 6 along (a) c axis and (b) a axis. | ||
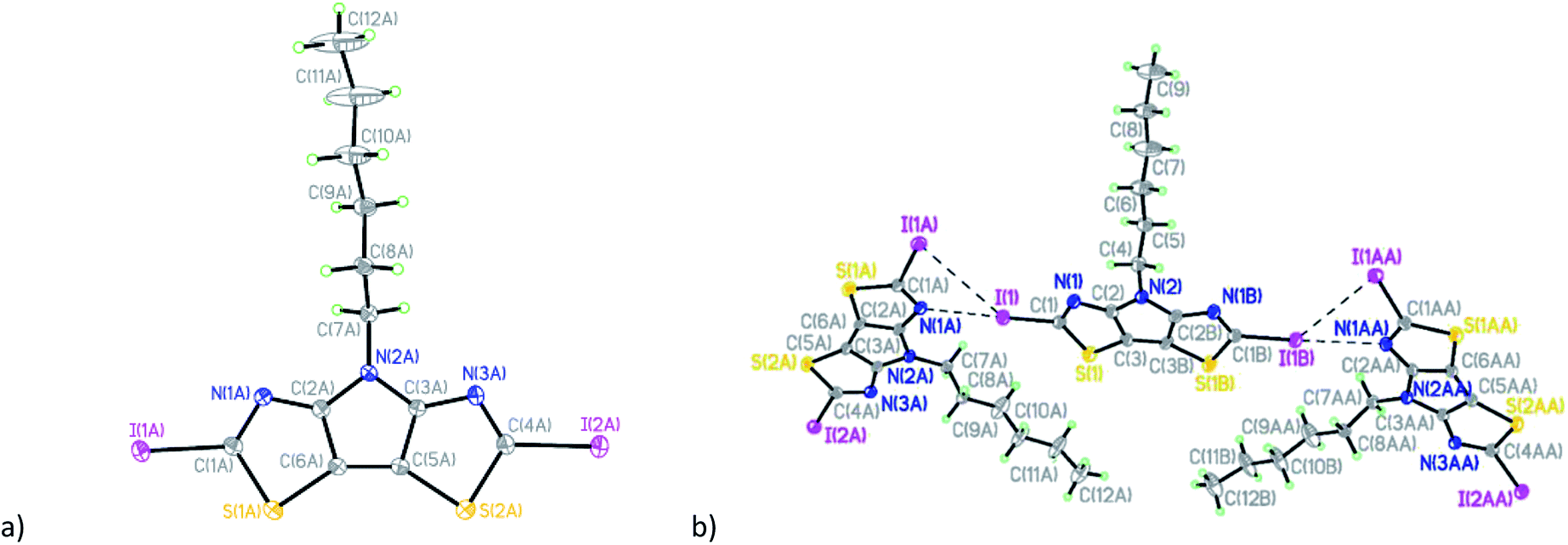
Compound 7 (Fig. 5a) crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with four crystallographically-independent molecules that have different geometry of the hexyl side chain per asymmetric unit (Table S1 and Fig. S5†).
with four crystallographically-independent molecules that have different geometry of the hexyl side chain per asymmetric unit (Table S1 and Fig. S5†).
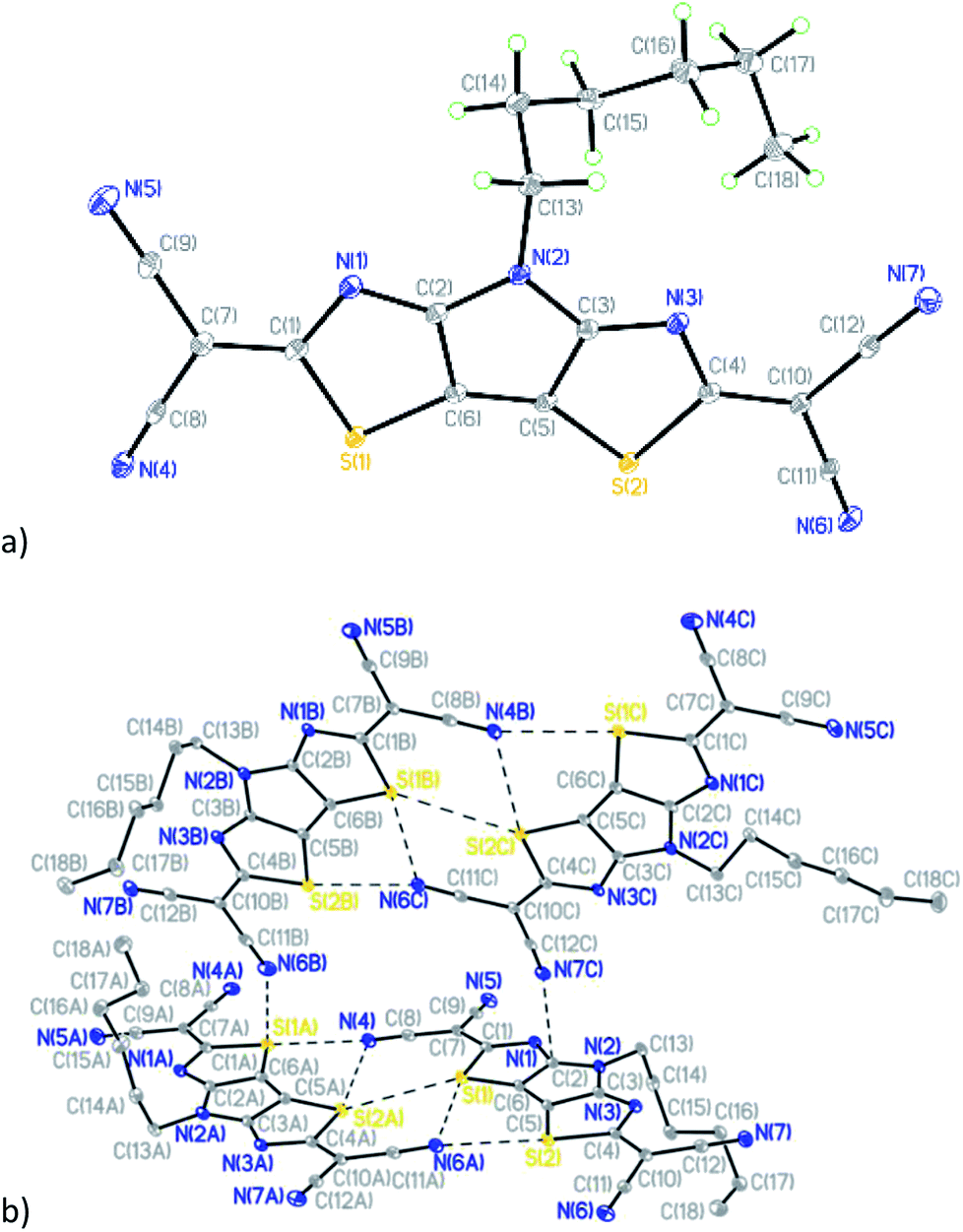 | ||
| Fig. 5 (a) General view of one independent molecule of 7; (b) mutual orientation and short intermolecular contacts between molecules in 7. | ||
Similar to compounds 5 and 6, the tricyclic core is nearly planar in all four molecules of 7 with values of dihedral angles between the two thiazole rings ranging from 1.34(1) to 2.98(1)°. The deviations of dicyanomethylene groups from the nearly planar cores of the molecules are in the range between 3.0(2) and 11.7(2)°, which does not preclude a possibility of conjugation through these molecules. Analysis of the crystal packing revealed the existence of short intermolecular contacts linking molecules into pairs (Fig. 5b and Table S2†). The other contacts C(2)⋯N(7C) 3.006(2) Å and S(1A)⋯N(6B) 3.325(2) Å link such pairs of molecules together in the crystal (Fig. 5b).
Density functional theory (DFT) calculations were carried out by means of the optimally tuned (OT) long-range corrected (LC) ωPBE (OT-LC-ωPBE)28,29 functional and the cc-PVDZ basis set,30,31 as implemented in the Gaussian16 software suite,32 to examine the electronic, redox, and optical properties of 7 (here, we investigate L7-methyl where the hexyl chain of 7 is truncated to a methyl group to reduce the computational cost). Normal modes of all optimized geometries were evaluated to assess whether the geometries represented minima on the potential energy surface. The range separation parameter, ω, was optimized through the non-empirical ionization potential (IP)-tuning procedure;33,34 the ω for each system investigated is provided in Table 1. We note that the assessment of the electron affinity (EA) of TCNQ via DFT methods has been a topic of interest in the literature, and it has recently been shown that non-empirically tuned LC functionals provide reasonable EA assessments for this and related molecules.35,36
| Molecule | ω | HOMO | LUMO | Δ(HOMO − LUMO) | IP | EA | Δ(IP − EA) |
|---|---|---|---|---|---|---|---|
| a At the OT-LC-ωPBE/cc-PVDZ level of theory, the optimized geometry of the TCNB cation resulted in one negative frequency at −292 cm−1. Multiple attempts to overcome this negative frequency were unsuccessful. Tests with different functionals and at the HF/cc-PVDZ level of theory suggest that it is the amount of Hartree–Fock (HF) exchange in the functional that leads to the non-minimum structure. | |||||||
| L7-methyl | 0.201 | −9.31 | −3.03 | 6.27 | 8.52 | −3.80 | 4.72 |
| TCNQ | 0.236 | −9.73 | −3.07 | 6.67 | 9.11 | −3.63 | 5.49 |
| TCNE | 0.324 | −11.99 | −2.82 | 9.17 | 11.63 | −3.28 | 8.35 |
| TCNB | 0.278 | −11.32 | −1.91 | 9.41 | 10.86a | −2.40 | 8.46a |
| DTP (9) | 0.182 | −8.55 | −2.60 | 5.95 | 7.69 | −3.42 | 4.27 |
| DSP (10) | 0.180 | −8.50 | −2.53 | 5.98 | 7.64 | −3.37 | 4.27 |
In general, the geometric parameters determined for neutral L7-methyl are in good agreement with the crystallographic data for 7 (Table S3†), providing confidence in the level of theory to describe the characteristics of this molecule. The OT-LC-ωPBE/cc-PVDZ calculations reveal that 7 has an EA of −3.80 eV, which is larger than that of TCNQ (−3.63 eV), TCNE (−3.28 eV) and TCNB (−2.40 eV) (Table 1). The combination of the tetracyano groups and extended π-conjugation in L7-methyl, when compared to the commonly used TCNE ligand in molecular magnets results in a larger EA, a smaller IP, and smaller fundamental (IP-EA) gap. The S0 → S1 optical and HOMO–LUMO gaps are also smallest for L7-methyl (Table S4†). It is expected that the extended π conjugation will play an important role in the implementation of L7-methyl in molecular magnetic materials.
Ligand 7 is structurally similar to previously published quinoidal DTP and DSP systems,23,24 which have first half-wave reduction potentials in dichloromethane at −0.23 V (vs. Ag/AgCl)23 and at −0.66 V (vs. Cp2Fe0/+)24 for DTP-based derivatives and −0.61 V (vs. Cp2Fe0/+) for DSP.24 Incorporation of electron-deficient thiazole rings in lieu of thiophene in DTP or selenophene in DSP is expected to positively shift the reduction potential for ligand 7 by ∼200 mV, based on observations for like tricyclic systems.37 OT-LC-ωPBE/cc-PVDZ calculations for DTP (9) and DSP (10) derivatives (chemical structures are shown in Fig. S4†), each with known electrochemical and UV-vis absorption properties, were carried out for direct comparison. The EA of L7-methyl is 0.38 eV larger than the DTP derivative 9, confirming that the thiazole-based ligand 7 is expected to be a strong acceptor. Both the HOMO and the LUMO in the tricyclic systems L7-methyl, 8, and 9 are delocalized across the π-conjugated backbones (Fig. S6†), with only marginal impact on the frontier molecular orbital energies and the band gaps upon substituting the sulphur atoms in 9 with selenium atoms in 10, a result consistent with experimental electrochemical and UV-vis absorption properties.24
2.2 Synthesis and characterization of hybrid material 8
With the expected reduction potential of 7 at around −0.2 to −0.4 V vs. Cp2Fe0/+, which is comparable to that of TCNE (half-wave reduction potential of −0.28 V vs. Cp2Fe0/+ in acetonitrile38), a facile reaction of 7 with vanadium hexacarbonyl V(CO)6 was expected. Indeed, a red-wine coloured solution of ligand 7 quickly changed its colour to dirty-brown during a dropwise addition of a freshly prepared bright yellow solution of V(CO)6 in dichloromethane, and a dark-coloured precipitate of 8 formed after several minutes (Fig. S7†). Combustion elemental analysis results for two independently prepared vanadium-hybrid materials 8 are close to composition of V·(ligand 7)2·(CH2Cl2)0.5 (calculated 50.03; H, 3.11; N, 22.38; found: C, 49.58; H, 3.73; N, 20.66; found: C, 50.47; H, 3.64; N, 20.93). The magnetic properties of the hybrid material 8 were investigated by exploring the field- and temperature-dependent magnetization from 5 to 300 K. In the zero field cooled (ZFC) experiment, the magnetization increased on warming from 5 K and reached a broad maximum at 45–60 K, followed by rapid decrease. In the field-cooled (FC) experiment (10 Oe field), the magnetization only slightly decreased from 5 K to 60 K with a maximum magnetization observed at 5 K (Fig. 6). Qualitatively the observed behaviour of 8 is similar to that of the vanadium-tetracyanobenzene hybrid material.17 The fitting parameter β for temperature-dependent magnetization using the equation M(T) = M(0)(1 − βT3/2) for two samples of 8 was extracted from the data (Fig. S8†), and the best fit was found for β = 0.0115 and 0.0105 K−3/2. This parameter yields TC of 110 K and 115 K, respectively (magnetization vanishes at around 130 K). Field-dependent magnetization measurements were carried out at 5 K (Fig. 7), and 50 K and 100 K (Fig. S9†) (for a second independently prepared sample M(T) curve is shown in Fig. S10† and M(H) curves at 5 K, 50 K and 95 K are shown in Fig. S11 and S12†). From the saturation magnetization measurements at 5 K (7.8 emu g−1) (Fig. 7), at 50 K (6.0 emu g−1) and at 100 K (3.1 emu g−1) (Fig. S9†), the fitting parameter β of 0.01065 appears to give the best fit and is comparable to β estimated from the M(T) curves (Fig. 6 and S8†). This run to run variation is well within the variability typically observed in this class of materials prepared using solution synthesis, and the agreement to within less than 5% of TC further supports the presence of a magnetic moment arising from ferrimagnetic order.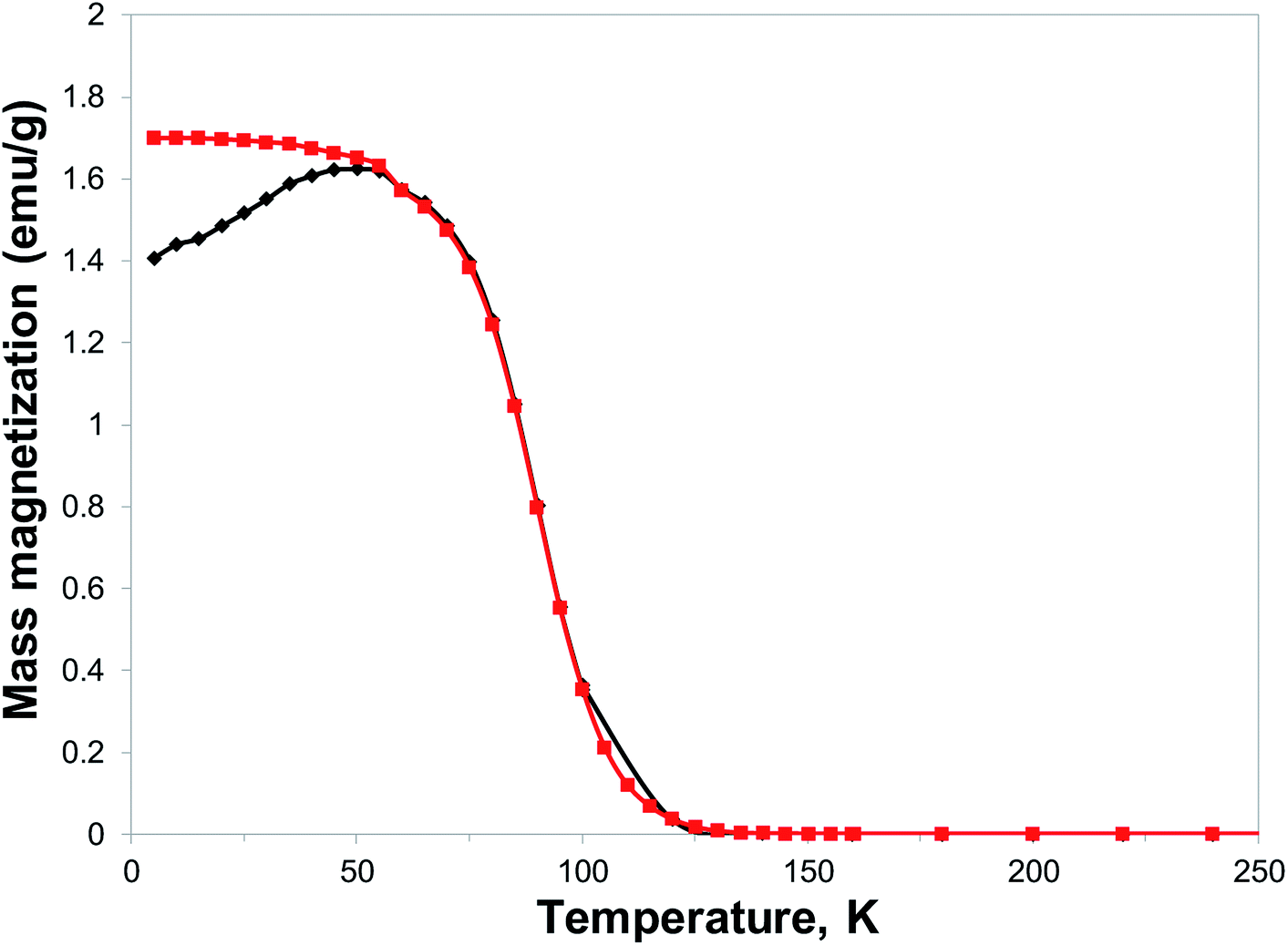 | ||
| Fig. 6 Temperature dependence of magnetization of material 8: (black diamond) ZFC in 10 Oe applied field, (red squares) FC in 10 Oe applied field. | ||
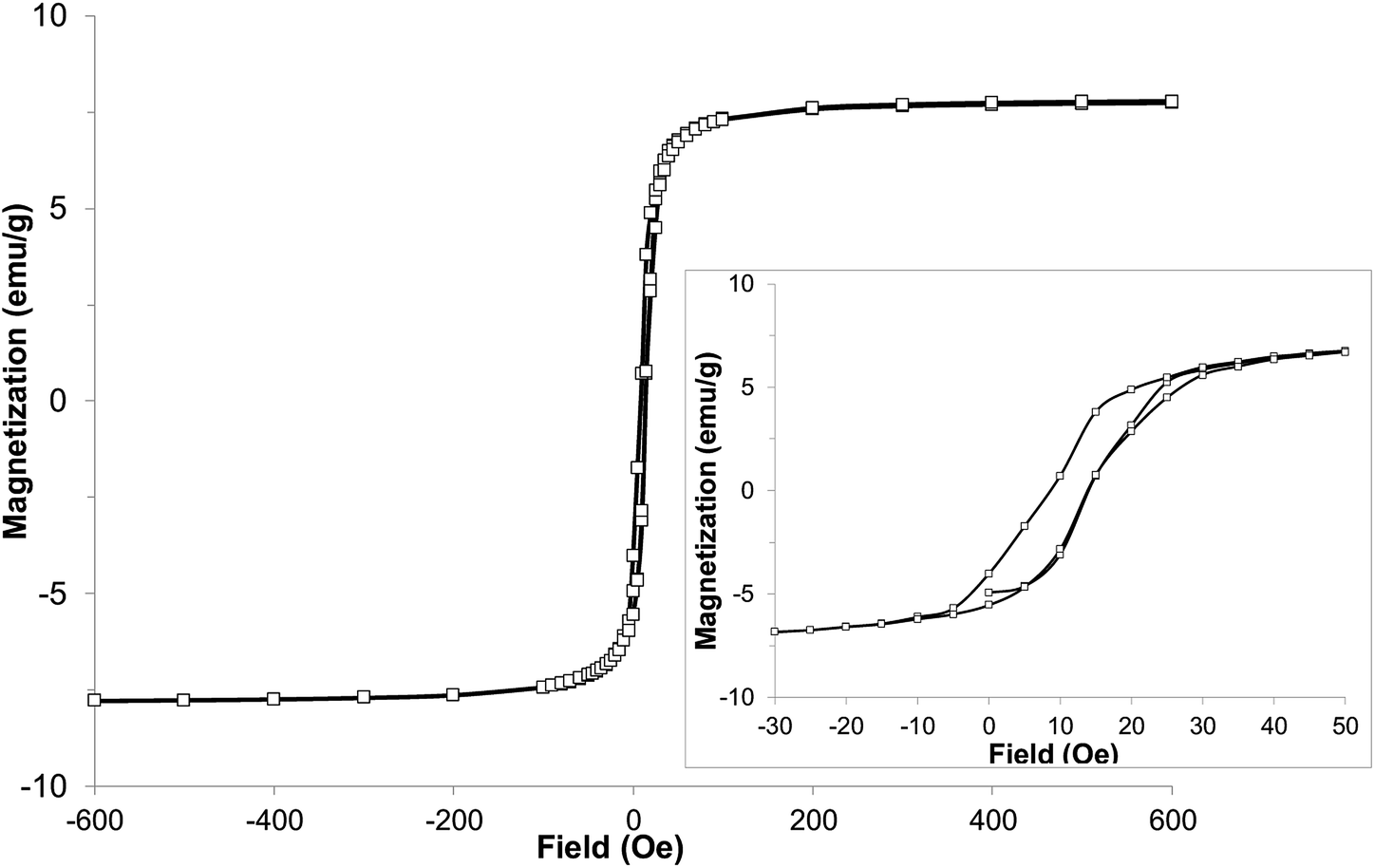 | ||
| Fig. 7 Hysteresis loop of hybrid material 8 at 5 K (inset with enlarged region of the same data shows small coercive field). | ||
At 5 K a rapid increase of magnetization was observed with increasing field to 100 Oe. Further increase of the field up to 600 Oe resulted in a very slight almost linear increase of magnetization up to 7.8 emu g−1 (for comparison, V(TCNE)∼2·(CH2Cl2)∼0.5 has mass magnetization of 17.1 emu g−1).39 The saturation mass magnetization of material 8 at 5 K is in the range of 7.8–8.4 emu g−1. This value is about 45–49% of that of the benchmark V(TCNE)∼2 ferrimagnet (17.1 emu g−1), while the molar magnetization exceeds that of V(TCNE)∼2 considering that the molecular weight of the ligand 7 is 3.3 times larger than that of TCNE. The small coercive field of about 5 Oe at 5 K indicates that material 8 is a soft ferrimagnet.
3 Experimental details
V(CO)6 was prepared from Et4N[V(CO)6] and anhydrous ortho-phosphoric acid.41 Compounds 2,25 3,25 4,22 and 522 were synthesized according to previously published procedures. Yield of the compound 4 was improved in comparison with the published procedure25 using a pressure vessel26 (see below for details).Anhydrous solvents (tetrahydrofuran, dichloromethane, and N,N-dimethylformamide) were used as received. 1H, DEPT-135 and 13C{1H} NMR spectra were acquired using 300 and 400 MHz Bruker instruments; signals were referenced to a residual CHCl3 peak (7.27 ppm for 1H NMR and 77.0 for 13C{1H} NMR) or tetramethylsilane (0 ppm) as internal standard. HRMS was obtained using a Bruker Solarix 15T ICR with laser desorption ionization (LDI) with no matrix added. The sample was diluted into chloroform and spotted directly on to a stainless-steel plate before MS analysis.
Preparation of the hybrid material was performed in the nitrogen glovebox (<5 ppm O2). Superconducting quantum interference device (SQUID) was used for determination of the field and temperature dependence of magnetization of the bulk samples.
X-ray data for compound 5 was collected at 220(2) K on a Bruker SMART APEX2 CCD-based X-ray diffractometer equipped with a low-temperature cryostat (Oxford Instruments) and a Mo X-ray tube (λ = 0.71073 Å). Data collection, indexing, and initial cell refinements were carried out using APEX2,42 with the frame integrations and final cell refinements carried out using SAINT.43 An absorption correction was applied using the program SADABS.44 Non-hydrogen atoms were refined anisotropically and all hydrogen atoms were placed in idealized positions and were refined using a riding model using ShelXL program.45 The structure was examined using the Addsym subroutine of PLATON, in order to ensure that no additional symmetry could be applied to the finalized model.46
Single crystal X-ray data for compounds 6 and 7 were collected using a Rigaku XtaLAB Synergy-S diffractometer equipped with a HyPix-6000HE Hybrid Photon Counting (HPC) detector and dual Mo and Cu microfocus sealed X-ray source as well as a low-temperature Oxford Cryostream 800 at 100(2) K.
Data collection strategy was calculated within CrysAlisPro47 to ensure desired data redundancy and percent completeness. Unit cell determination, initial indexing, data collection, frame integration, Lorentz-polarization corrections and final cell parameter calculations were carried out using CrysAlisPro. An absorption correction was performed using the SCALE3 ABSPACK scaling algorithm embedded within CrysAlisPro. The structures were solved using ShelXT,48 all non-hydrogen atoms were refined anisotropically using ShelXL45 and their space groups were unambiguously verified by PLATON.46 All hydrogen atoms were attached via the riding model at calculated positions. Olex2 (ref. 49) was used for the preparation of the publication materials. Crystal data and refinement details are summarized in Table S1.†
3.1 Et4N[V(CO)6]
In an oven-dried flask anthracene (56.36 mmol, 10.05 g) was mixed with anhydrous THF (120 mL) at room temperature under an atmosphere of argon. Sodium (53.62 mmol, 1.23 g, cut in pieces) was added, and colourless suspension became dark blue in colour after several minutes of stirring. This mixture was stirred for 15.5 hours and then cooled in an ice-water bath. A red solution of VCl3 in THF (prepared in a separate flask by mixing VCl3·(THF)3 (12.47 mmol, 4.60 g) with anhydrous THF (120 mL) at room temperature and stirring for 15 hours under argon atmosphere) was added in portions to a flask with sodium anthracenide using a glass syringe and a stainless-steel needle. The resulting very dark-coloured mixture (brown-grey with a hint of purple) was stirred for ∼11 hours and then cooled in ice-water bath. Argon atmosphere was replaced with CO, and the mixture quickly became yellow-brown in colour. The reaction mixture was slowly warmed up to room temperature overnight without removal of the cooling bath, and after ∼13 hours of stirring the atmosphere of argon was introduced, and adapter for vacuum distillation was fitted. About 200 mL of THF was distilled out under vacuum, and water (∼200 mL, degassed by bubbling nitrogen for 1–2 hours) was added to the dark-coloured residue. The flask (under reduced pressure) was transferred to nitrogen glovebox, the dark-coloured mixture was filtered twice, the solid was washed with additional amount of water (∼150 mL), and the yellow filtrate was added to a colourless solution of Et4NBr (61.85 mmol, 13 g) in ∼30 mL of degassed water. Yellow precipitate was separated by vacuum filtration, and dried (2.0 g, 46% yield). Lower yields of the product were observed for two other runs on similar scale without any obvious reason.3.2 4-Hexyl-2,6-bis(triisopropylsilyl)-4H-pyrrolo[2,3-d:5,4-d′]bis(thiazole) (4)
In a pressure vessel 4,4′-dibromo-2,2′-bis(triisopropylsilyl)-5,5′-bithiazole (3) (8.0 mmol, 5.10 g) was mixed with Pd2(dba)3 (5 mol%, 0.4 mmol, 0.37 g), BINAP (20 mol%, 1.6 mmol, 0.37 g) and NaOtBu (7.5 eq., 60.0 mmol, 5.77 g) under nitrogen atmosphere. Anhydrous toluene (20 mL) and n-hexylamine (8.0 mmol, 0.80 g) were added, the pressure vessel was sealed, and resulting mixture was stirred at 170 °C overnight. The mixture was cooled to room temperature, treated with water, and organic matter was extracted with hexanes several times. The solvents were removed from combined organic phases, and the crude product was purified by column chromatography (silica gel, hexanes, then hexanes![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dichloromethane (10:1) as eluant). Product 4 was obtained as yellowish solid in 45% yield (2.0 g). 1H NMR and 13C{1H} NMR analyses were consistent with the previously published data.22
:dichloromethane (10:1) as eluant). Product 4 was obtained as yellowish solid in 45% yield (2.0 g). 1H NMR and 13C{1H} NMR analyses were consistent with the previously published data.22
3.3 4-Hexyl-2,6-diiodo-4H-pyrrolo[2,3-d:5,4-d′]bis(thiazole) (6)
4-Hexyl-2,6-bis(triisopropylsilyl)-4H-pyrrolo[2,3-d:5,4-d′]bis(thi-azole) (5) (1.09 mmol, 0.60 g) was dissolved in anhydrous THF (30 mL) under nitrogen atmosphere, and tetrabutylammonium fluoride (TBAF) (2.3 mmol, 0.73 g) was added to a yellowish solution. The yellow reaction mixture was stirred for ∼1 h, treated with water, and organic matter was extracted with hexanes several times. Combined organic phases were dried over MgSO4, the drying agent was filtered off, and the solvents were removed by rotary evaporation. Crude 4-hexyl-4H-pyrrolo[2,3-d:5,4-d′]bis(thiazole) (5) was mixed with N-iodosuccinimide (2.1 eq., 2.29 mmol, 0.52 g) and anhydrous DMF (10 mL), and the mixture was stirred at room temperature under nitrogen atmosphere overnight. The mixture was treated with water, and organic matter was extracted with dichloromethane. Combined organic phases were subjected to rotary evaporation, and crude product was purified by silica gel chromatography (100 mL of silica gel, chloroform as eluant). Solvent was removed from combined fractions by rotary evaporation, and the residue was heated with ethanol (∼100 mL) and dichloromethane (∼20 mL) until all solid was dissolved. The mixture was cooled to room temperature, then in the freezer, and the off-white solid was separated by vacuum filtration (0.35 g, 62.5% yield for two steps; 78.2% yield was obtained for a larger scale reaction using 3.65 mmol of 4). 1H NMR (400 MHz, CDCl3, δ): 4.47 (t, J = 7.3 Hz, 2H), 1.97 (m, 2H), 1.32 (m, 6H), 0.97 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3, δ): 152.87, 109.13, 95.52, 45.68 (CH2), 31.27 (CH2), 29.94 (CH2), 26.30 (CH2), 22.51 (CH2), 13.98 (CH3). Anal. calcd for C12H13I2N3S2: C 27.87, H 2.53, N 8.12; found: C 28.00, H 2.51, N 7.83.3.4 2,2′-(4-Hexyl-2H-pyrrolo[2,3-d:5,4-d′]bis(thiazole)-2,6(4H)-diylidene)dimalononitrile (7)
Malononitrile (4.0 eq., 2.70 mmol, 0.18 g) was dissolved in anhydrous THF (30 mL) under nitrogen atmosphere, and sodium hydride (∼60% in mineral oil, 8 eq., 0.22 g) was added at room temperature. The mixture was stirred for ∼0.5 h, and 2,6-diiodo-hexyl-4H-pyrrolo[2,3-d:5,4-d′]bis(thiazole) (6) (1.0 eq., 0.68 mmol, 350 mg) and Pd(PPh3)4 (1.0 eq., 0.68 mmol, 0.79 g) were added. The mixture was heated to reflux for ∼0.5 h, cooled to room temperature, and added to bromine water (∼200–250 mL). The organic phase was extracted with dichloromethane several times, and the solvent was removed from combined organic phases. Crude product was purified by column chromatography (∼100 mL of silica gel, dichloromethane as eluant). Dark red-wine coloured fractions were combined, the solvent was removed and the purified product 7 was obtained as a dark solid (∼100 mg, ∼38% yield). This material was purified by column chromatography four times prior to use in the preparation of hybrid material 8. 1H NMR (300 MHz, CDCl3, δ): 4.16 (t, J = 7.3 Hz, 2H), 1.93 (m, 2H), 1.37 (m, 6H), 0.92 (t, J = 7.0 Hz, 3H); DEPT-135 (CDCl3, δ): 46.29 (CH2), 30.95 (CH2), 28.00 (CH2), 26.06 (CH2), 22.41 (CH2), 13.93 (CH3). Due to low solubility in CDCl3 13C{1H} spectrum of 7 was not recorded. 13C{1H} NMR (CD2Cl2): 182.60, 179.66, 127.07, 112.60, 111.30, 46.75, 31.57, 28.50, 26.61, 23.00, 14.27. HRMS (LDI) calculated for (C18H13N7S2 + H) 392.0752, found 392.0744. Anal. calcd for C18H13N7S2: C, 55.23, H, 3.35, N, 25.05; found C, 55.67, H, 3.63, N 24.11.3.5 Synthesis of hybrid material 8
Solution synthesis of the hybrid material 8 was accomplished by addition of bright yellow solution of V(CO)6 (∼0.038 mmol) in dichloromethane (∼1 mL) to a solution of the ligand 7 (30.0 mg, 0.077 mmol) in dichloromethane (3 mL) at room temperature in a nitrogen glovebox. The red-wine coloured solution of ligand 7 quickly changed its colour to dirty-brown, and then precipitate formed. After about 0.5 h a very dark-coloured solid was separated by vacuum filtration (21 mg, 66% yield). Part of this sample (14 mg) was transferred to a 5 mm quartz tube, dried under vacuum using O-ring adapter in the nitrogen glovebox, and then sealed under vacuum for the SQUID measurements. The rest of the sample was transferred to the vial and was stored in the freezer (about −35 °C) in a glovebox.Two more solution syntheses were carried out on a smaller scale using V(CO)6 (0.0191 mmol, 4.3 mg) in 0.5 mL of dichloromethane and ligand 7 (0.0383 mmol, 15 mg) in 1.5 mL of dichloromethane. Anal. calcd for V·(C18H13N7S2)2·(CH2Cl2)0.5: 50.03; H, 3.11; N, 22.38. Found: C, 49.58; H, 3.73; N, 20.66; found: C, 50.47; H, 3.64; N, 20.93.
4 Conclusions
A new organic ligand, 2,2′-(4-hexyl-2H-pyrrolo[2,3-d:5,4-d′]bis(thiazole)-2,6(4H)-diylidene)dimalononitrile 7, which represents a class of tetracyano-substituted tricyclic aromatic cores with quinoidal structure, forms a magnetically ordered vanadium-organic hybrid material 8 in the reaction with V(CO)6. 8 is a soft ferrimagnet with coercive field of about 5 Oe, a Curie temperature of ∼110–115 K, and saturation mass magnetization of 7.8–8.4 emu g−1 at 5 K, which is 45–49% of that of the benchmark V·(TCNE)x ferrimagnet (17.1 emu g−1). Combustion elemental analysis suggests V·(ligand 7)∼2·(CH2Cl2)∼0.5 as the composition of the hybrid material 8. Some discrepancy in combustion elemental analysis between two materials prepared in almost identical fashion indicates that every solution synthesis produces a slightly different material. However, the magnetic properties (Curie temperature and saturation magnetization) reproduce well for materials prepared independently using various batches of both ligand 7 and V(CO)6. This is consistent with the solution synthesis of V(TCNE)x, and as a consequence it is expected that moving to a thin-film synthesis (i.e. CVD or PLD) would yield similar gains in uniformity and improvement in magnetic properties.40 As observed with other polycyanoorganic-based vanadium coordination polymers, 8 is air-sensitive and should be stored in a sealed tube under inert atmosphere to minimize the degradation of magnetic properties, or encapsulated if synthesized in thin-film form.6Single crystal X-ray analysis of the parent 4-n-hexyl-4H-pyrrolo[2,3-d:5,4-d′]bis(thiazole), its 2,6-diiodo derivative and ligand 7 indicate presence of the disorder in hexyl chains and a linear arrangement between the substituents in positions 2 and 6. The discovery of magnetic ordering in compound 8, despite the flexible alkyl groups and the larger size of the ligand in comparison with majority of the previously studied organic polycyano compounds, demonstrates the suitability of the new class of tricyclic organic ligands, and potentially a large variety of other tetracyano quinoidal systems with extended cores, for the preparation of novel hybrid magnets. In addition, modification of the end of the alkyl chain with a functional group might be used for the attachment of the ligand to the metal or metal oxide surface and formation of a V·(ligand)-(mono)layer. The ability to fine-tune the electronic properties by modification of the fused tricyclic core, e.g. use of selenophene24 and thiophene-based ligands,23,24 and packing motifs, by choosing various alkyl chains attached to the nitrogen atom in a central pyrrole unit, opens up opportunities for better understanding of the molecular requirements for design of novel metal–organic hybrid materials with magnetic functionality.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Center for Emergent Materials, an NSF MRSEC under award number DMR-1420451, NSF DMR-1523611, and Infrastructure Seed Award (NSF Grant #IIA-1301346). We acknowledge the NSF MRI Program (CHE-1726652) and the UNT for supporting the acquisition of the Rigaku XtaLAB Synergy-S X-ray diffractometer. Y. A. G. thanks Professor Yang Qin (University of New Mexico) for providing synthetic facilities for the preparation of the ligand and for helpful discussions, Dr Camelia Selcu (The Ohio State University) for assistance with the SQUID measurements, Dr Tanya Whitmer for assistance with NMR experiment (The Ohio State University), and Mr Michael Chilcote and Mr Andrew Franson for helpful discussions. The computational work at the University of Kentucky (UK) was supported in part by start-up funds received from the UK Vice President for Research and the UK Center for Applied Energy Research (CAER). Supercomputing resources on the Lipscomb High Performance Computing Cluster were provided by the University of Kentucky Information Technology Department and Center for Computational Sciences (CCS).Notes and references
- S. A. Wolf, D. D. Awchalom, R. A. Buhrman, J. M. Daughton, S. von Molnár, M. L. Roukes, A. Y. Chtchelkanova and D. M. Treger, Science, 2001, 294(5546), 1488 CrossRef CAS PubMed.
- J. M. Manriquez, G. T. Yee, R. S McLean, A. J. Epstein and J. S. Miller, Science, 1991, 252(5011), 1415 CrossRef CAS PubMed.
- J.-W. Yoo, C.-Y. Chen, H. W. Jang, C. W. Bark, V. N. Prigodin, C. B. Eom and A. J. Epstein, Nat. Mater., 2010, 9, 638 CrossRef CAS PubMed.
- N. Zhu, X. Zhang, I. H. Froning, M. E. Flatté, E. Johnston-Halperin and H. X. Tang, Appl. Phys. Lett., 2016, 109(8), 82402 CrossRef.
- H. Liu, C. Zhang, H. Malissa, M. Groesbeck, M. Kavand, R. McLaughlin, S. Jamali, J. Hao, D. Sun, R. A. Davidson, L. Wojcik, J. S. Miller, C. Boehme and Z. V. Vardeny, Nat. Mater., 2018, 17, 308 CrossRef CAS PubMed.
- I. H. Froning, M. Harberts, Y. Lu, H. Yu, A. J. Epstein and E. Johnston-Halperin, Appl. Phys. Lett., 2015, 106, 122403 CrossRef.
- M. Chilcote, Y. Lu and E. Johnston-Halperin, World Scientific Reference on Spin in Organics, 2018, vol. 4, p. 125 Search PubMed.
- J. Hao, R. A. Davidson, M. Kavand, K. J. van Schooten, C. Boehme and J. S. Miller, Inorg. Chem., 2016, 55, 9393 CrossRef CAS PubMed.
- K. I. Pokhodnya, B. Lefler and J. S. Miller, Adv. Mater., 2007, 19, 3281 CrossRef CAS.
- Y. Lu, M. Harberts, C.-Y. Kao, H. Yu, E. Johnston-Halperin and A. J. Epstein, Adv. Mater., 2014, 26, 7632 CrossRef CAS PubMed.
- Y. Lu, H. Yu, M. Harberts, A. J. Epstein and E. Johnston-Halperin, RSC Adv., 2015, 5, 82271 RSC.
- Y. Lu, H. Yu, M. Harberts, A. J. Epstein and E. Johnston-Halperin, J. Mater. Chem. C, 2015, 3, 7363 RSC.
- J. P. Fitzgerald, B. B. Kaul and G. T. Yee, Chem. Commun., 2000, 49 RSC.
- M. K. Amshumalia, M. D. Harveyb and G. T. Yeeb, Synth. Met., 2014, 188, 53 CrossRef.
- E. B. Vickers, T. D. Selby, M. S. Thorum, M. L. Taliaferro and J. S. Miller, Inorg. Chem., 2004, 43(20), 6414 CrossRef CAS PubMed.
- J. L. Arthur, S. H. Lapidus, C. E. Moore, A. L. Rheingold, P. W. Stephens and J. S. Miller, Adv. Funct. Mater., 2012, 22, 1802 CrossRef CAS.
- M. L. Taliaferro, M. S. Thorum and J. S. Miller, Angew. Chem., Int. Ed., 2006, 45, 5326 CrossRef CAS PubMed.
- J. Hao, R. A. Davidson, C. Kareis, M. Kavand, K. J. van Schooten, C. Boehme, E. Wöβ, G. Knör and J. S. Miller, Chem.–Eur. J., 2016, 22, 14273 CrossRef CAS PubMed.
- E. B. Vickers, T. D. Selby and J. S. Miller, J. Am. Chem. Soc., 2004, 126, 3716 CrossRef CAS PubMed.
- K. I. Pokhodnya, A. J. Epstein and J. S. Miller, Adv. Mater., 2000, 12(6), 410 CrossRef CAS.
- B. G. Morin, P. Zhou, C. Hahm, A. J. Epstein and J. S. Miller, J. Appl. Phys., 1993, 73, 5648 CrossRef CAS.
- Y. A. Getmanenko, S. Singh, B. Sandhu, C. Y. Wang, T. Timofeeva, B. Kippelen and S. R. Marder, J. Mater. Chem. C, 2014, 2(1), 124 RSC.
- T. M. Pappenfus, B. J. Hermanson, T. J. Helland, G. W. Lee, S. M. Drew, K. R. Mann, K. A. McGee and S. C. Rasmussen, Org. Lett., 2008, 10(8), 1553 CrossRef CAS PubMed.
- Y. A. Getmanenko, T. A. Purcell, D. K. Hwang, B. Kippelen and S. R. Marder, J. Org. Chem., 2012, 77, 10931 CrossRef CAS PubMed.
- Y. A. Getmanenko, P. Tongwa, T. V. Timofeeva and S. R. Marder, Org. Lett., 2010, 12(9), 2136 CrossRef CAS PubMed.
- M. Al-Hashimi, J. G. Labram, S. Watkins, M. Motevalli, T. D. Anthopoulos and M. Heeney, Org. Lett., 2010, 12(23), 5478 CrossRef CAS PubMed.
- E. J. Uzelac, C. B. McCausland and S. C. Rasmussen, J. Org. Chem., 2018, 83, 664 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- O. A. Vydrov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 234109 CrossRef PubMed.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007 CrossRef.
- D. E. Woon and T. H. Dunning Jr, J. Chem. Phys., 1993, 98, 1358 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Lian, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi; J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc, 2016 Search PubMed.
- T. Stein, L. Kronik and R. Baer, J. Am. Chem. Soc., 2009, 131, 2818 CrossRef CAS PubMed.
- T. Stein, H. Eisenberg, L. Kronik and R. Baer, Phys. Rev. Lett., 2010, 105, 266802 CrossRef PubMed.
- B. Milian, R. Pou-Amerigo, R. Viruela and E. Ortı, Chem. Phys. Lett., 2004, 391, 148 CrossRef CAS.
- L. Gallandi, N. Marom, P. Rinke and T. Körzdörfer, J. Chem. Theory Comput., 2016, 12(2), 605 CrossRef CAS PubMed.
- Y. A. Getmanenko, C. Risko, P. Tongwa, E. G. Kim, H. Li, B. Sandhu, T. Timofeeva, J.-L. Bredas and S. R. Marder, J. Org. Chem., 2011, 76(8), 2660 CrossRef CAS PubMed.
- M. Moscherosch, E. Waldhdr, H. Binder, W. Kaim and J. Fiedlers, Inorg. Chem., 1995, 34, 4326 CrossRef CAS.
- J. S. Miller and A. J. Epstein, Angew. Chem., Int. Ed., 1994, 33, 385 CrossRef.
- M. Harberts, Y. Lu, H. Yu, A. J. Epsein and E. Johnston-Halperin, J. Visualized Exp., 2015, 101, e52891 Search PubMed.
- X. Liu, J. E. Ellis, T. D. Miller, P. Ghalasi and J. S. Miller, Inorg. Synth., 2004, 34, 96 CrossRef CAS.
- Bruker APEX2, Bruker Advanced Analytical X-ray Systems, Madison, WI, 2007 Search PubMed.
- Bruker Saint, Bruker Advanced Analytical X-ray Systems, Madison, WI, 2007 Search PubMed.
- Bruker SADABS, Bruker Advanced Analytical X-ray Systems, Madison, WI, 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., 2009, D65, 148 CrossRef PubMed.
- Version 1.171.40.12b, Rigaku Oxford Diffraction, 2018 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, A71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1819816, 1819817 and 1868490 for compounds 5, 6 and 7, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra05697h |
| ‡ Current affiliation: The Scripps Research Institute, 130 Scripps Way, Jupiter, Florida 33458, USA; Email: E-mail: ygetmanenko@scripps.edu |
| This journal is © The Royal Society of Chemistry 2018 |