
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the role of the hexagonal polymorph on SrAl2O4-based phosphors†
Rocío Estefanía Rojas-Hernandez *ab,
Fernando Rubio-Marcosa,
Aida Serranocd,
Aydar Rakhmatulline,
Catherine Bessadae and
José Francisco Fernandeza
*ab,
Fernando Rubio-Marcosa,
Aida Serranocd,
Aydar Rakhmatulline,
Catherine Bessadae and
José Francisco Fernandeza
aElectroceramic Department, Instituto de Cerámica y Vidrio, CSIC, Kelsen 5, 28049, Madrid, Spain. E-mail: rociorojas@icv.csic.es
bCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal
cSpLine, Spanish CRG Beamline at the ESRF, F-38043 Grenoble, Cedex 09, France
dInstituto de Ciencia de Materiales de Madrid, CSIC, Cantoblanco, 28049, Madrid, Spain
eCNRS, CEMHTI UPR3079, Univ. Orléans, F-45071 Orléans, France
First published on 14th August 2018
Abstract
In persistent luminescence materials, the SrO–Al2O3 system has been mainly studied due to its chemical stability, higher photoluminescence response and longest green-afterglow times. Specifically, the research has focused on SrAl2O4 doped with europium and dysprosium. SrAl2O4 has two polymorphs: monoclinic polymorph (space group P21) and hexagonal polymorph (space group P6322). Besides, the coexistence of these two phases, monoclinic and hexagonal, appears in almost all the results. However, it is not clear how this coexistence influences optical response. Some authors have reported that only the monoclinic structure exhibits luminescence properties, while another suggests that the hexagonal SrAl2O4 polymorph has a higher emission efficiency than the monoclinic polymorph. Here we report a systematic evaluation of the effects of the stabilization of the hexagonal SrAl2O4 polymorph. We show that an interrelationship between the hexagonal polymorph and phosphorescent properties is the linchpin for the development of good luminescence properties. Remarkably, the stabilization of the hexagonal SrAl2O4 polymorph on the monoclinic–hexagonal polymorphic coexistence appears to be related to the preservation of the nanometric nature of the SrAl2O4-based system. Our results will help to understand the role of the hexagonal polymorph in the polymorphic coexistence on SrAl2O4-based systems and may facilitate the development of luminescent nanometric particles for the design and preparation of new light emitting materials.
The SrAl2O4 host has gathered wide interest because of its potential luminescent applications when it is doped mainly with rare earths. A large number of works have centered on Eu, Dy1,2 doping to obtain a persistent material and to a lesser extent with other combinations such as Ce3+;3 Eu2+; Dy3+; Gd3+;4 Eu2+; Nd3+;5 Eu3+; Ho3+;6 and Dy3+; Yb3+.7 Rare earth-doped alkaline earth aluminates have been the most extensively studied.8
It is well known that strontium aluminate, SrAl2O4, has two crystallographic polymorphs. Monoclinic symmetry, Mc, with space group P21 (a = 8.447 Å, b = 8.816 Å, c = 5.163 Å, β = 93.42, JCPDS (no. 34-0379)) is stable at temperatures below 650 °C and hexagonal symmetry, H, (ideal undistorted structure) with space group P6322 (a = 5.140 Å, c = 8.462 Å, JCPDS (no. 31-1336)) that is stable above this temperature and metastable at room temperature (RT). It is not clear whether the transition is direct or if there is a second-order transition. Escribano et al.9 suggest a direct transition from monoclinic phase, P21, into the hexagonal phase, P6322, in a reversible process. While other authors establish a monoclinic ↔ hexagonal phase transition at 680 °C in the space group P63 and a second-order transition to P6322 at 860 °C.10
The lack of an accurate study about the hexagonal phase is due to the complexity of synthesis. The introduction of Ba to obtain the hexagonal phase requires a content above 0.43 (ref. 11) or 0.37 (ref. 12) mol, or the incorporation of Ca.13 On the other hand, the excess of Al when SrAl4O7 is synthesized at 1000 °C by solidification from the high-temperature liquid state13 provokes the appearance of SrAl2O4 hexagonal phase and stabilization of γ-Al2O3. Also, Singh et al.14 produce SrAl2O4 hexagonal phase with 20 wt% of ZnO by the combustion technique.
Taking into account these studies and previous results that suggest the coexistence of hexagonal and monoclinic phase at RT by molten salt synthesis,15 here we propose the combination of an excess of Al together the incorporation of Zn2+ to synthesize the hexagonal phase at RT and as processing synthesis molten salt route. This synthetic route (that is, molten salt process) has been used due to relevant advantages in particle control and phosphorescence efficiency over the previous methods than may use for the synthesis of strontium aluminates as microemulsion process.16 The control of particle size overcomes limiting aspect (particles with average particle size > ca. 3–50 μm) for current applications. In addition, the phosphorescence efficiency is attained by the synthesis of defect-free nanopowders with high crystallinity.
Referring to luminescent properties as a function of the strontium aluminate phase, majority of works establish that Mc polymorph has the best performance,17,18 and the presence of hexagonal phase in coexistence with Mc phase decreases the emission intensity.19 There are, however, divergent views in that regard since a higher emission efficiency for hexagonal phase has been already reported.20 Therefore, the stabilization of one polymorph to the detriment of the other polymorph (that is, the hexagonal polymorph versus the monoclinic, or vice versa) may meddle with the emission mechanism and, thus, directly influence the macroscopic luminescence response of SrAl2O4-based system.
Here, we explore the relationship between the polymorphic coexistence and its influence on the macroscopic luminescence response of SrAl2O4-based system. For that, we have stabilized the hexagonal polymorph (H) by means of the synergistic effect between Zn2+ incorporation and the addition of an Al3+ excess on SrAl2O4-based system, using molten salt synthesis. This approach was adopted taking in accounts the results related to hexagonal polymorph stabilization, obtained previously by combustion technique14 and by spray-drying process.21 Specifically, it is important to remark, that both the alumina precursor and the Al2O3/SrO ratio play an important role in the hexagonal polymorph stabilization. Ultimately, high resolution transmission electron microscope of the hexagonal phase suggests that nanometric particle size preserves the stabilization of the hexagonal polymorph at RT.
Results & discussion
1. Effect of Zn2+ incorporation on the SrAl2O4-based system
A ZnO incorporation study was carried out employing two alumina precursors: γ-Al2O3 and α-Al2O3 with average particle size ≈ 0.1 μm. Fig. 1 shows the XRD patterns corresponding to the powder synthesized by the molten salt method without (Fig. 1(a) and (c)) and with 20 wt% of ZnO (Fig. 1(b) and (d)), which were thermally treated at 1000 °C for 2 h in air atmosphere, employing a salt/SrAl2O4 molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and Al2O3/SrO ratio of 1, and two alumina precursors: γ-Al2O3 (Fig. 1(a) and (b)) and α-Al2O3 (Fig. 1(c) and (d)). As shown in Fig. 1, all samples present a polymorphic behavior, which is associated with the coexistence of the monoclinic (Mc, P21) and hexagonal (H, P6322) symmetries at RT. However, the polymorphic behavior does not show a clear dependency with the ZnO addition (see Fig. 1) in contrast to the result of Singh et al.,7 and therefore this strategy results not effective in achieving our objective (that is, the stabilization of hexagonal SrAl2O4 polymorph). This fact can be related to the limited solubility of Zn2+ on the SrAl2O4-based trydymite structure.
:1 and Al2O3/SrO ratio of 1, and two alumina precursors: γ-Al2O3 (Fig. 1(a) and (b)) and α-Al2O3 (Fig. 1(c) and (d)). As shown in Fig. 1, all samples present a polymorphic behavior, which is associated with the coexistence of the monoclinic (Mc, P21) and hexagonal (H, P6322) symmetries at RT. However, the polymorphic behavior does not show a clear dependency with the ZnO addition (see Fig. 1) in contrast to the result of Singh et al.,7 and therefore this strategy results not effective in achieving our objective (that is, the stabilization of hexagonal SrAl2O4 polymorph). This fact can be related to the limited solubility of Zn2+ on the SrAl2O4-based trydymite structure.
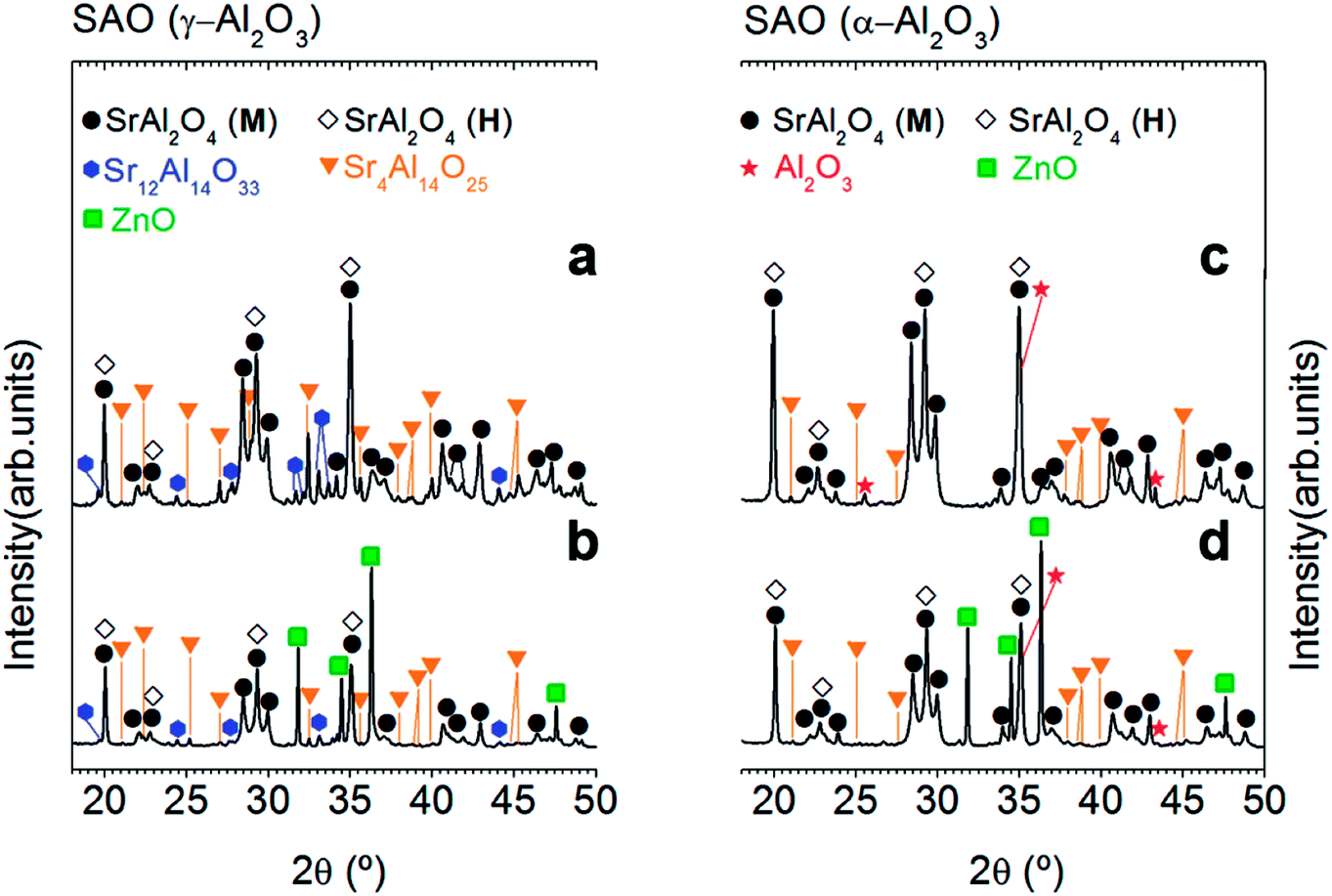 | ||
| Fig. 1 Effect of Zn incorporation on the structure of the SrAl2O4-based system: XRD patterns of SrAl2O4: Eu, Dy (SAO) phosphor employing two alumina precursors: γ-Al2O3 (a and b) and α-Al2O3 (c and d) as well as without (a and c) and with 20 wt% of ZnO (b and d), respectively. Additionally, the alumina precursor used in the synthesis process are shown at the top of the panels (a) and (c). The symbols highlight SrAl2O4 (monoclinic, black circle), SrAl2O4 (hexagonal, black-open diamonds), Sr12Al14O33 (blue hexagon), Sr4Al14O25 (orange triangle), ZnO (green square) and Al2O3 (red start). | ||
On the other hand, ZnO incorporation decreases rich strontium secondary phases, it can be attributed to the role of ZnO as a mineralizing agent.22 The absence of secondary phases in the doped powders should be a remarkable result since these phases are commonly present in SrAl2O4-based systems affecting the luminescence response. However, the presence of ZnO diffraction peaks demonstrates again the low interaction of ZnO with Al2O3, and consequently the low solubility of Zn2+ cations in the SrAl2O4 trydymite structure.
2. Effective stabilization of the hexagonal polymorph by the Al3+ cation excess addition on SrAl2O4-based system
As the stabilization of hexagonal phase with the ZnO incorporation by molten salt route was not satisfactorily achieved, an excess of Al3+ was evaluated. For this reason, our next step will be to use a relation 1 SrO/2 Al2O3 (Al/Sr: 2). Capron et al.21,23 were able to obtain the SrAl2O4 hexagonal symmetry as metastable solid solution when they were trying to synthesize SrAl4O7 phase by solidification from the high-temperature liquid state. Fig. 2 illustrates XRD patterns corresponding to the powder synthesized at 1000 °C for 2 h in air atmosphere, employing a salt/SrAl2O4 molar ratio of 3:1 and Al2O3/SrO ratio of 2 (that is, an Al3+ cation excess), and two alumina precursors: γ-Al2O3 (Fig. 2(a)) and α-Al2O3 (Fig. 2(b)). As observed, the mainly reflection of hexagonal polymorph located at 2θ = 29.06° is the most intense peak in comparison with the monoclinic reflections at 28.38° and 29.92° in 2θ. In this way, we can infer that the hexagonal phase contribution increases significantly with the Al3+ cation excess; demonstrating that effectively a major content of alumina stabilizes the hexagonal polymorph at RT. Note that the stabilization is more effective as γ-Al2O3 is used as a precursor. Such behavior must be related to the high reactivity of this alumina precursor. It is important to remark that hexagonal phase stabilization is modulated by both the alumina precursor reactivity and the Al2O3/SrO ratio. In the case of γ-Al2O3, the quick diffusivity of Sr2+ cations assisted by the molten salt starts in early reaction state, allowing the SrAl2O4 formation at low temperatures (1000 °C) in comparison to the high temperature required for conventional solid state route (>1400 °C). Fig. 2(c) shows agglomerates formed by primary particles, with average particle size ≤ 250 nm. This confirms that effectively, the stabilization at RT of the high-temperature hexagonal polymorph is in part related to nanometric crystalline domains. The field emission scanning electron microscopy (FE-SEM) micrograph, shown in Fig. 2(d), illustrates the morphology of the phosphor obtained using as precursor α-Al2O3, observing that the powders are composed of particles with sizes of 800 nm.
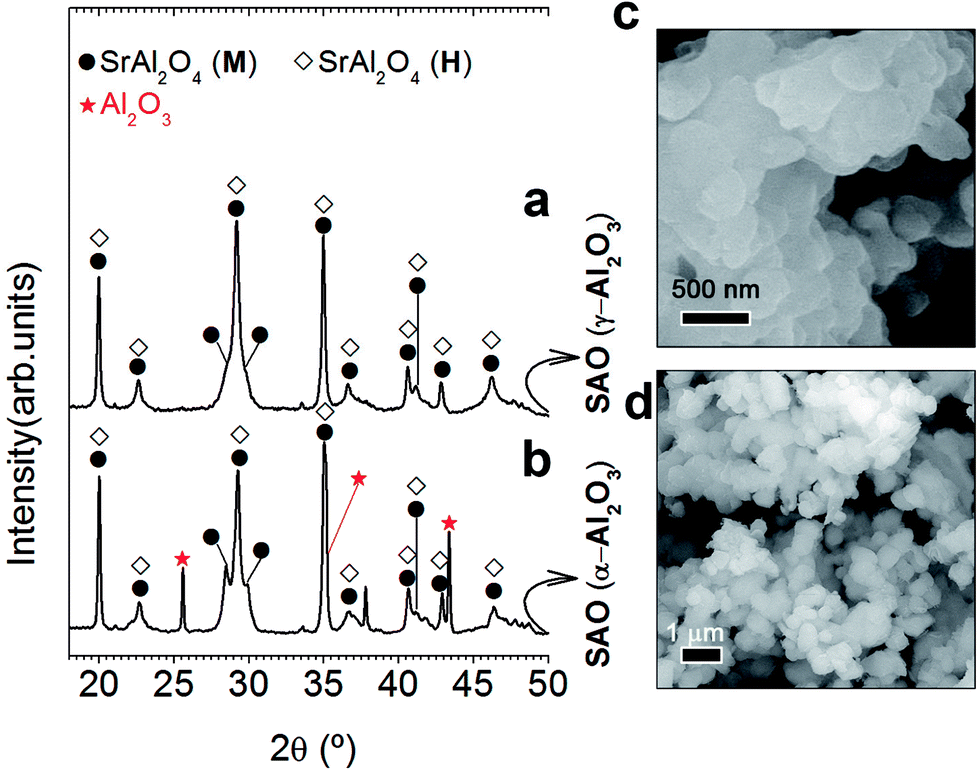 | ||
| Fig. 2 Role of the alumina precursor excess in structural and microstructural characterization: XRD patterns of SrAl2O4: Eu, Dy (SAO) phosphor employing two alumina precursors: γ-Al2O3 (a) and α-Al2O3 (b) and an Al2O3/SrO ratio of 2. The symbols highlight SrAl2O4 (monoclinic, black circle), SrAl2O4 (hexagonal, black-open diamonds), and Al2O3 (red start). Detail micrograph of SrAl2O4: Eu, Dy (SAO) phosphor using as precursors γ-Al2O3 (c) and α-Al2O3 (d). | ||
3. Two are better than one: taking advantage of the opportunity offered by ZnO incorporation
Our previous results (Fig. 1) establishes that ZnO incorporation do not promote hexagonal phase stabilization at RT when a stoichiometric ratio (1 SrO/1 Al2O3) is employed. However, the experimental proofs evidenced that ZnO acts as mineralizing agent. For this reason, we believe that it can be interesting to evaluate the synergy between ZnO (that is, mineralizing agent) and the Al3+ cation excess i.e., stabilizing agent of the hexagonal phase. For that, different ZnO contents are evaluated: 1.25, 2.5, 3.75 and 5 wt%, using the γ-Al2O3 as Al3+ precursor since at lower values of particle average size are expected and a γ-Al2O3/SrO ratio of 2.Fig. 3(a) shows XRD patterns of powder synthesized at 1000 °C for 2 h in 90N2–10H2 atmosphere, using a salt/SrAl2O4 molar ratio of 3:1 and γ-Al2O3/SrO ratio of 2, with different ZnO contents. In all the cases peaks related to hexagonal polymorph are visible, without the presence of peaks associated with ZnO. Bear in mind that any change in the phase formation is noticeable, lattice cell parameters and crystalline cell volume as a function of ZnO content are also calculated (ESI S1†). A slight reduction of lattice cells (a, b) with a higher content of ZnO is observed. Concerning the lattice cell parameter, c, there is not a clear evidence of its modification (see ESI Fig. S1†). The crystallite size is also calculated; being 38, 34, 31, 29 and 30 nm for the sample without and with 1.25, 2.5, 3.75 and 5 wt% of ZnO, respectively.
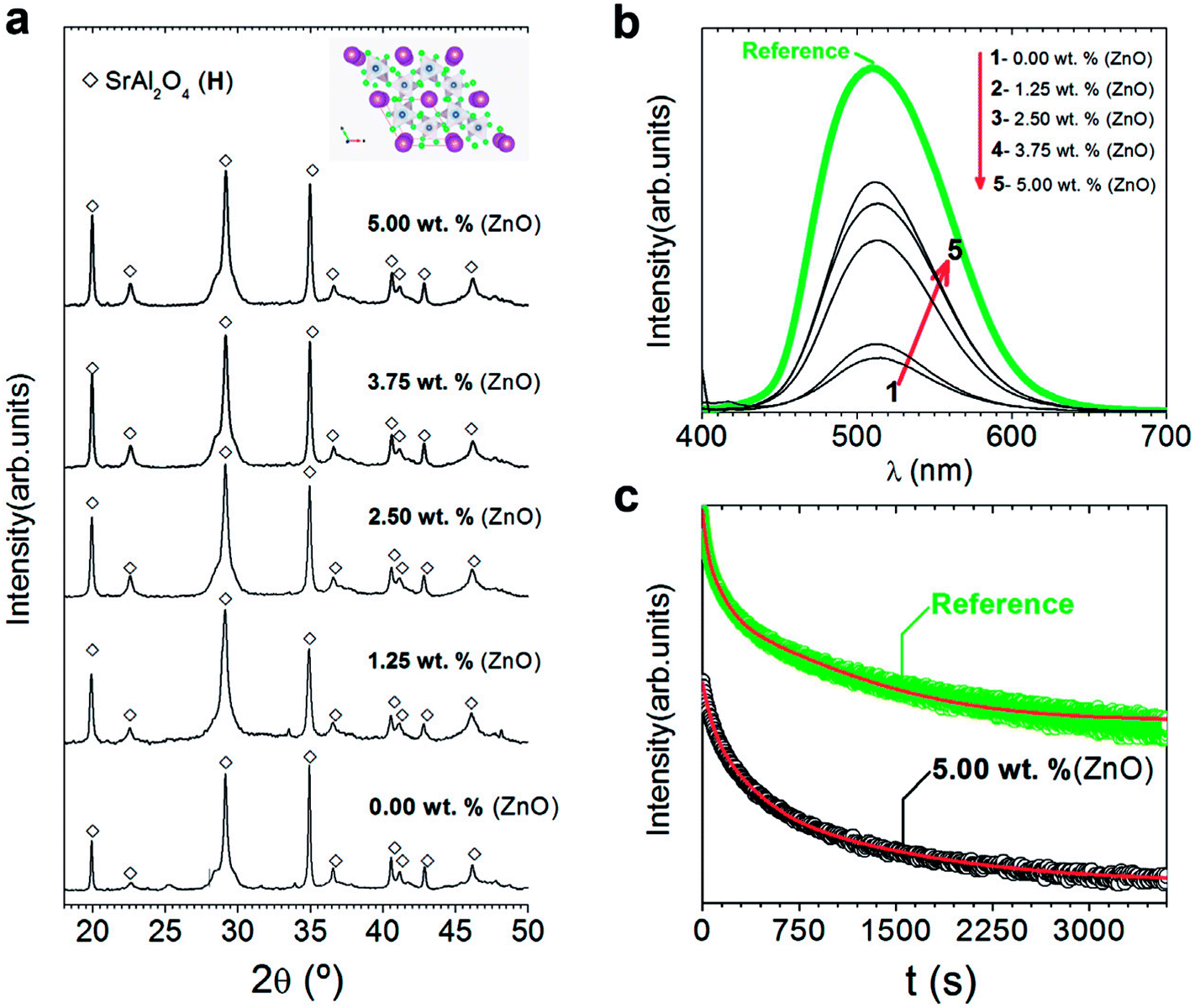 | ||
| Fig. 3 Structural characterization of the powder and their photoluminescent response: (a) XRD patterns of synthesized SrAl2O4: Eu, Dy (SAO) phosphor as a function of the ZnO content. The symbols highlight SrAl2O4 (hexagonal, black-open diamonds). Additionally, the scheme shown at the top of a represents the hexagonal SrAl2O4 polymorph determined using VESTA.24,25 (b) Emission spectra upon excitation at 380 nm of synthesized SrAl2O4: Eu, Dy compared with the commercial powder (from Jinan Chenghao Technology Co., Ltd) as received. The compositions represented as a function of the ZnO content in (b) are labelled as follows: (1) 0.00; (2) 1.25; (3) 2.50; (4) 3.75; and (5) 5.00 wt%. (c) Afterglow decay curve of SrAl2O4: Eu, Dy powder synthesized without and with 5 wt% of ZnO. | ||
Despite the fact that most studies describe that hexagonal phase is not luminescent, discrepancies in the literature encourage for evaluating the photoluminescence response of hexagonal phase. Fig. 3(b) shows the photoluminescence emission spectrum (λexc = 380 nm) of SrAl2O4: Eu, Dy powder with different ZnO contents compared to a commercial powder (from Jinan Chenghao Technology Co., Ltd) as received, which has an average particle size d50∼ 20 μm. The emission band centered at 515 nm is assigned to the transition 4f65d1→ 4f7 (8S7/2) of Eu2+ ions.26 Therefore, hexagonal phase shows green emission. The intensity of the emission is lower than the commercial reference due to larger particle size (20 μm) of the reference in comparison with particle size of powders synthesized with hexagonal phase in this work. As increasing ZnO content, photoluminescence intensity emission increases, so a high content of zinc oxide (10 wt%) is also evaluated (see ESI S2.†). By XRD, secondary phases or the presence of unreacted ZnO are not observed (see ESI S2.†). However, it is possible to detect the incipient formation of a new Sr3Al2O6 phase (JCPDS file 24-1187). This fact related to Zn2+ seems to facilitate the SrAl2O4 (H) formation only up to a certain Zn2+ concentration (∼5 wt% of ZnO), above which the hexagonal lattice is unable to accommodate the nominal Zn2+ content implying an excess in the A-site of the AB2O4 trydymite structure. Thus, the corresponding excess should be compensated by the eviction of some Sr2+ ions that together with the Al3+ excess from the alumina results in the incipient appearance of the new phase (Sr3Al2O6 phase). The photoluminescence intensity decreases with a high content of zinc oxide (10 wt%). This reduction can be related to the incipient the formation of secondary phases, such as Sr3Al2O6 (see ESI S2†).
Fig. 3(c) shows the afterglow decay curve of synthesized SrAl2O4: Eu, Dy powder with 5 wt% of ZnO compared to the commercial powder after the light source was cut off, irradiated for 10 min by a λexc = 380 nm. The initial intensity decreases significantly in comparison with the commercial counterpart. Decay phosphorescent measurement only for the sample with 5 wt% of ZnO is presented because the reduction is higher than 1 order of magnitude for samples with lower content of ZnO. These results confirm that the long-lasting luminescence drops meaningfully for nanometric particles. Therefore, hexagonal phase is luminescent, but the persistence of the emission is lower than that of monoclinic phase partly due to the nanometric particle size obtained when hexagonal phase is stabilized at RT.
4. The key to stabilizing the hexagonal polymorph is its nanometric nature
In order to study in depth the role of ZnO incorporation in the phase formation, a detailed morphological characterization is carried out by FE-SEM and high-resolution transmission electron microscopy (HRTEM). Fig. 4 illustrates the micrograph by FE-SEM of the synthesized powders (a–d) without and (e–h) with 5 wt% of ZnO. A general view of the micrographs reveals that the samples are composed by two different morphologies. Hereafter, it will be referred as type I and type II. Type I morphology involves discrete particles as shown in Fig. 4(a) with an average particle size ≤ 200 nm and clearly observed in the high magnification micrograph in Fig. 4(b). Whereas, type II morphology comprises particles with a high agglomeration grade as illustrated in Fig. 4(c). In both cases, there is a coexistence of these morphologies types (Fig. 4(d) and (f)), however, it is remarkable that mainly type I morphology is formed.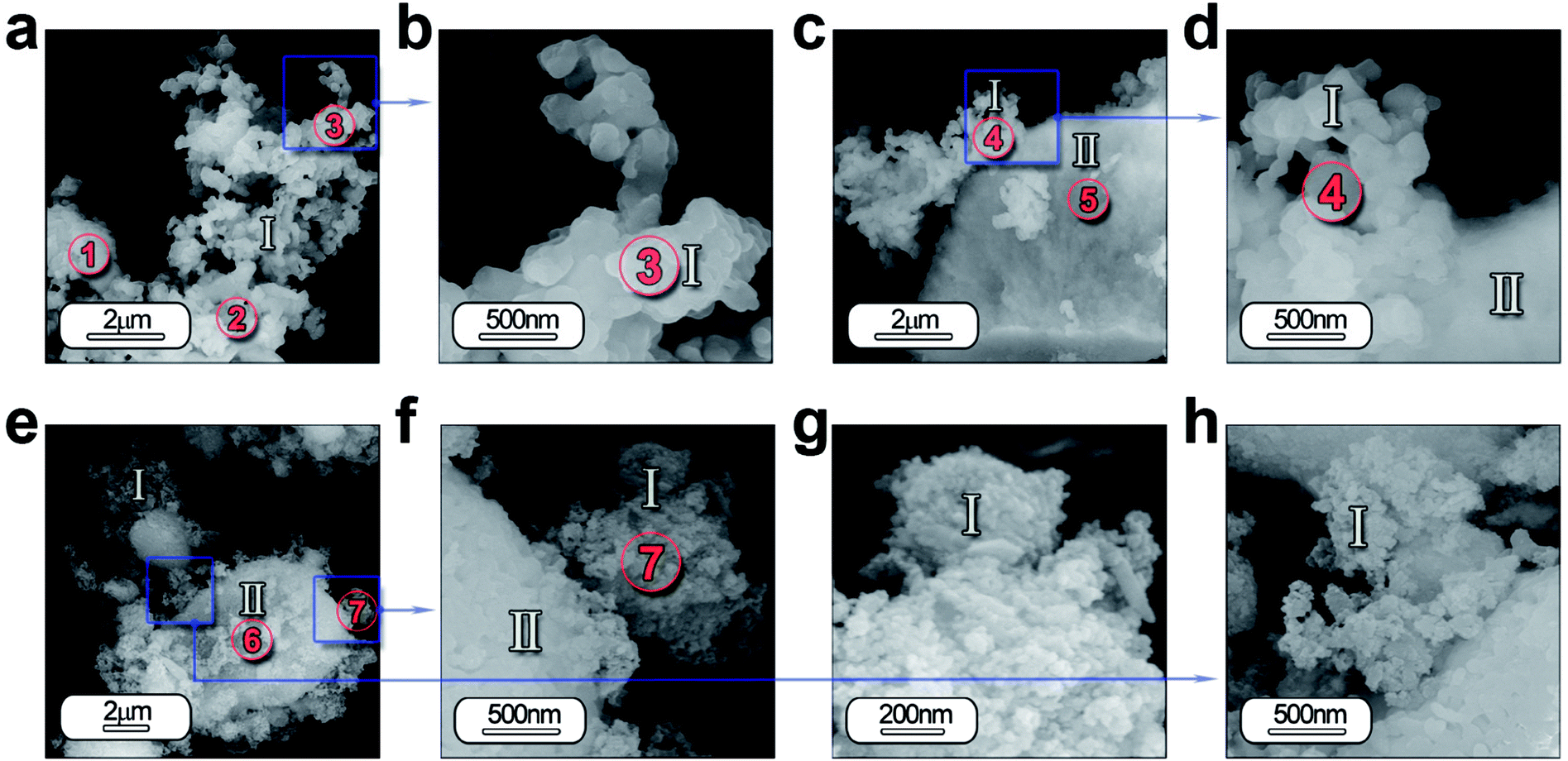 | ||
| Fig. 4 Morphological characterization of the powder synthesized by molten salt method by SEM: low-and high-magnification micrographs of the powder SrAl2O4: Eu, Dy synthesized without ZnO (a–d) and with 5 wt% of ZnO (e–h). In both cases it can be detected that samples are composed by two different morphologies, which are categorized as type I and type II depending mainly on its particle size and agglomeration state. Additionally, regions marked as circles and indicated as 1–7 show the positions where EDS analyses are accomplished. | ||
Furthermore, the EDS analysis is carried out on all the specimens in order to identify the composition of the both morphologies as well as its homogeneity. Chemical analysis by EDS from the areas of interest 1, 2 and 3 in Fig. 4(a) and 4 and 5 in Fig. 4(c) detects the following elements: Sr, Al, O, Eu and Dy. This analysis confirmed that the atomic% of the elements has a close agreement between the theoretical and calculated value considering the presence of SrAl2O4 and Al2O3, as shown in Table S1 (see ESI).†
Concerning the particles with ZnO, the two morphologies are also observed (Fig. 4(e) and (f)). However the morphology type I is formed by particles with lower particle size (see Fig. 4(g) and (h)). Chemical analyses by EDS from the areas of interest 6, 7 in Fig. 4(e) detects the following elements: Sr, Al, O, Eu, Dy and Zn. Area 7 reveals a higher content of Sr, indicating that these particles have a chemical composition near to stoichiometric composition, while in area 6, particles are more agglomerated and a higher presence of unreacted Al2O3 (see ESI Table S1†).
HRTEM is also performed to obtain a detailed phase and morphological characterization. TEM and HRTEM analyses of the powder without ZnO are illustrated in Fig. 5(a–f) and with ZnO in Fig. 6(a–d). The low magnification TEM image (Fig. 5(a)) shows the presence of discrete particles (that is, type I morphology) and agglomerates (that is, type II morphology) as shown in SEM characterization. The magnified micrograph is shown in Fig. 5(b) with particles of average size ≤ 50 nm. The interplanar spacing is calculated as 3.13 Å from the inverse FFT pattern, matching to the (103) (d103 Theoretical = 3.07 Å) reflection of the SrAl2O4 pattern (JCPDS no. 31-1336). The selected area electron diffraction (SAED) pattern recorded at the former location reveals ring patterns containing bright spots which confirm the presence of crystalline phase in the sample (Fig. 5(d)). The SAED pattern is indexed according to (101), (110), (104) reflections of the hexagonal phase of SrAl2O4, corresponding to 3.94, 2.57, and 1.91 Å calculated d-spacing, respectively. The dimension of the crystalline regions is lower than 20 nm. These results justify the hypothesis that a smaller particle size preserves the stability of hexagonal polymorph.
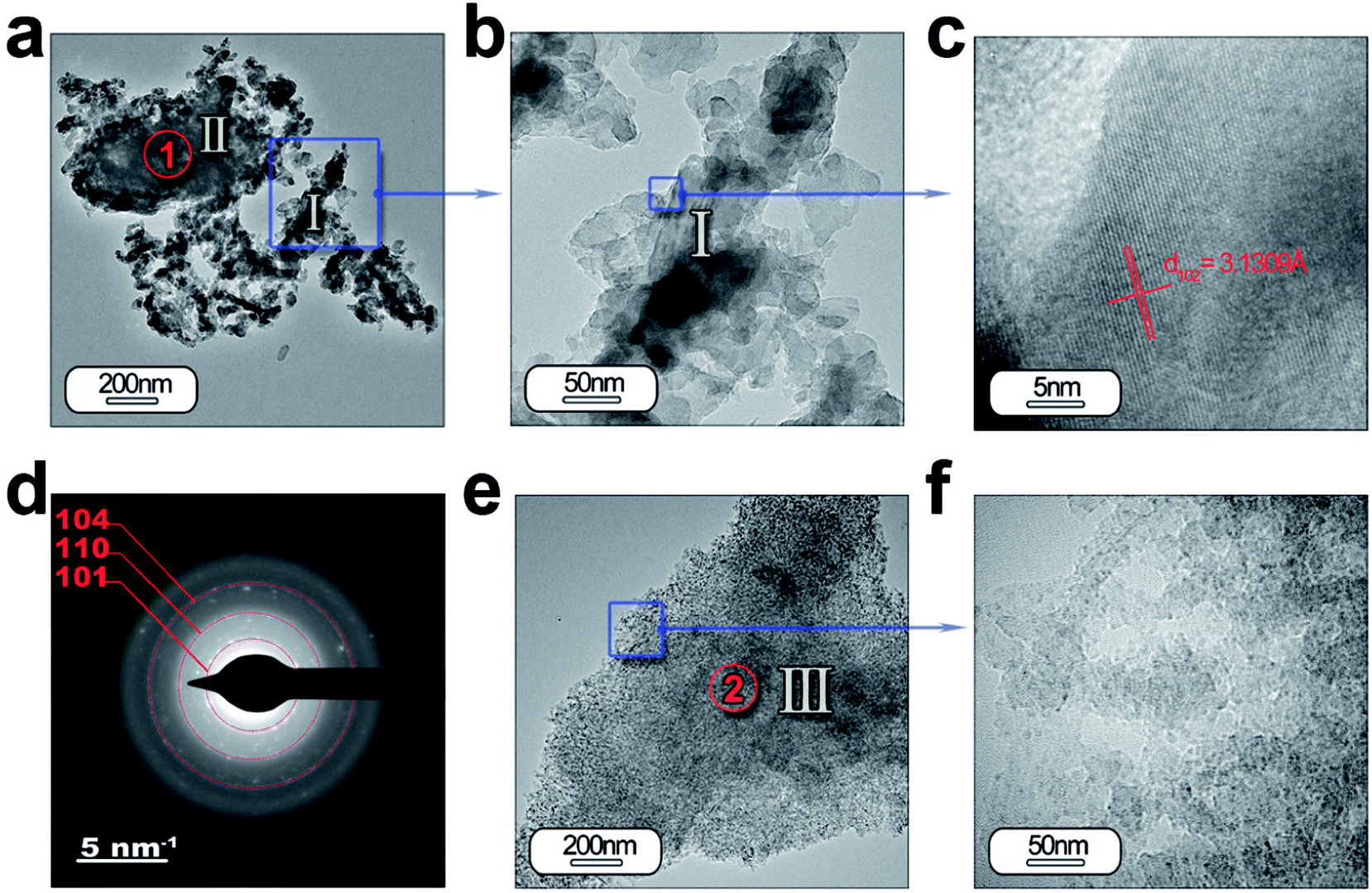 | ||
| Fig. 5 Detailed morphological characterization of the SrAl2O4: Eu, Dy phosphor by TEM: (a) low-magnification TEM micrographs of the powder synthesized without ZnO. From the panel (a) it can be observed the two different morphologies which were categorized as type I and type II in Fig. 4. The panel (b) and (c) show a detail of the type I particles, and its lattice fringe, the d-spacing at 3.13 Å corresponding to SrAl2O4 (H) polymorph. (d) The rings are indexed based on the SrAl2O4 (H) polymorph. (e and f) Low- and high-resolution micrographs showing a new type of particle morphology, which is categorized as type III, and is associated with Al2O3 unreacted. Additionally, the regions marked as red circles and indicated as 1 and 2 show the positions where EDS analyses are accomplished. | ||
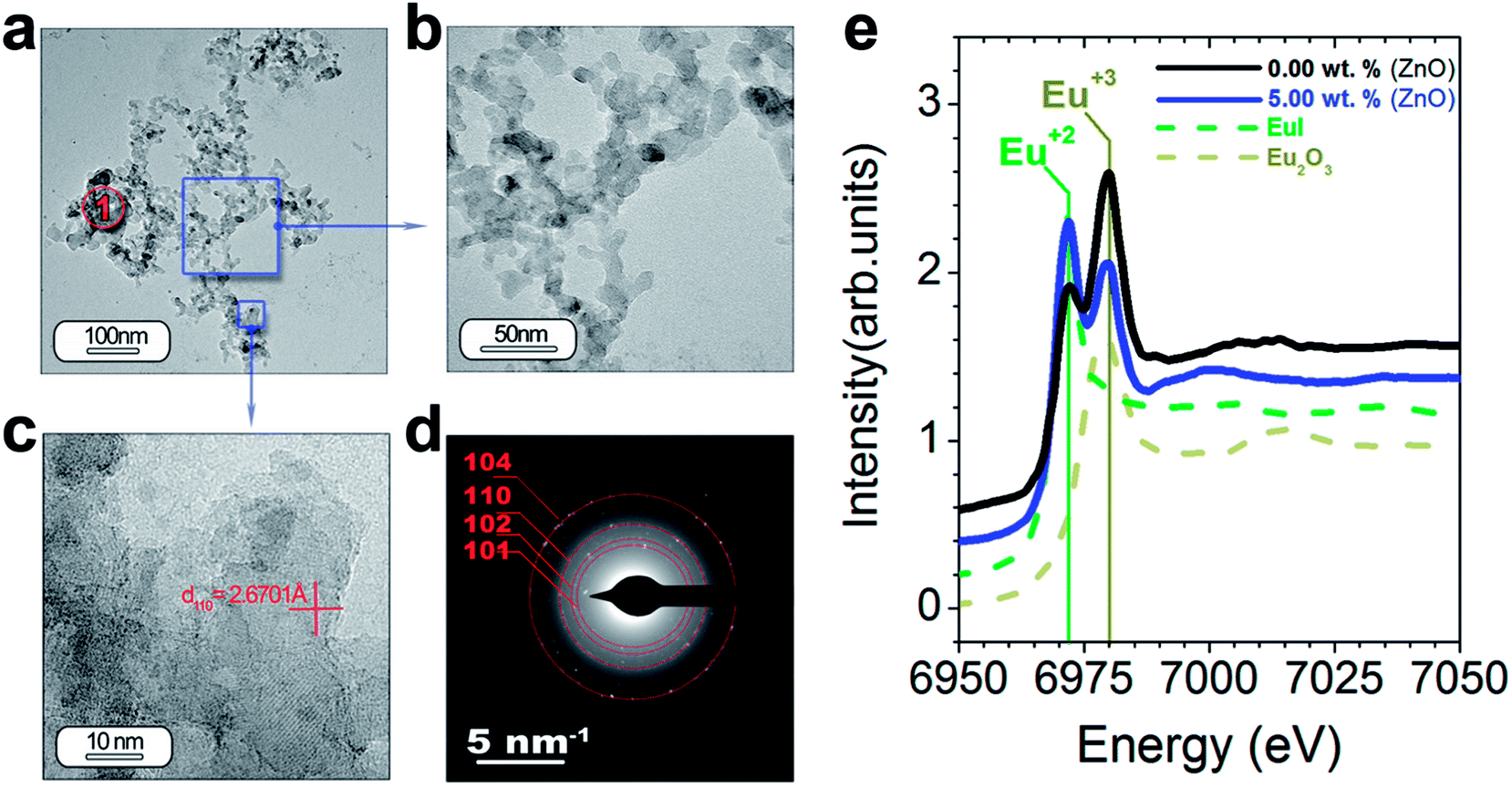 | ||
| Fig. 6 Study of the morphology and functional response of the SrAl2O4 (H) polymorph: (a–d) low and high magnification of TEM micrographs of the powder SrAl2O4: Eu, Dy synthesized with 5 wt% of ZnO. The lattice fringe observed in panel (c), the d-spacing at 2.67 Å, is indexed as the (110) plane of the SrAl2O4 (H) polymorph. The rings are indexed based on the SrAl2O4 (H) polymorph. (e) XANES spectra measured at the Eu-L3 edge of the powders and the Eu2O3 and EuI reference. The region marked as red circle and indicated as 1′ shows the positions where EDS analysis is accomplished. | ||
Fig. 5(e) and (f) illustrate another type of morphology particles (that is, type III morphology); the size of the particles is lower than the other morphology (that is, type I morphology). Chemical analyses reveals that particles located at point 1 (Fig. 5(a)) are formed by SrAl2O4 and unreacted Al2O3, while the particles located at point 2 (Fig. 5(e)) are associated with alumina (see ESI Table S2.†). The particles with type I and II morphology could be formed by shell-like SrAl2O4 formation and a core of Al2O3, as has been observed previously,27 when the dissolution–diffusion transport mechanism (“template mechanism”) is the dominant mechanism in molten salt route.
TEM and HRTEM of particles with ZnO (Fig. 6(a–f)) reveal that ZnO addition decreases significantly the particle size in good agreement with the SEM results (Fig. 5(e–h)). The interplanar spacing is calculated as 2.67 Å from the inverse FFT pattern, matching to the (110) (d110 Theoretical = 2.57 Å). The SAED pattern recorded at the former location reveals ring patterns containing bright spots which confirm the presence of crystalline phase in the sample (Fig. 6(d)). The SAED pattern is indexed according to (101), (102), (110) and (104) reflections of the hexagonal phase of SrAl2O4, corresponding to 3.99, 3.13, 2.54, and 1.81 Å calculated d-spacing, respectively. Chemical analyses do not reveal ZnO presence from particles located at point 1′ (Fig. 6(a)) (see ESI Table S2†).
As we just observed, the photoluminescence intensity is dependent on the extrinsic (i.e., morphology and particle size) and intrinsic features of the particles (i.e., polymorphic coexistence, crystallinity, etc.).28 Within these last features, the oxidation state for the Eu element has a great influence mainly on the macroscopic photoluminescence response of the SrAl2O4-based system.27 To quantify the relative amounts of the two oxidation states for Eu element, X-ray absorption near-edge structure spectroscopy (XANES) measurements at the Eu L3-edge were performed. The reference spectra of Eu2+ (EuI) and Eu3+ (Eu2O3) are shown in Fig. 6(e). The difference in the energy can serve as a fingerprint of the two oxidation states. The spectra of the powder synthesized without ZnO (black spectrum) and with 5 wt% ZnO (blue spectrum) by using Al2O3/SrO molar ratio of 2 are shown in Fig. 6(e). These spectra consist of two peaks coinciding with the reference spectra of Eu2+ and Eu3+, and definitively confirming the coexistence of both oxidation states. ZnO addition results in a notable reduction of the Eu3+ and an increase of Eu2+ peak intensity. It should be emphasized that the photoluminescence intensity increases accordingly with the Eu2+ fraction.27 The addition of a mineralizing agent as ZnO favors the diffusion of Eu cations during the thermal treatment under reduction atmosphere in the trydymite structure and their stabilization as Eu2+. For this reason the photoluminescence intensity increases when ZnO is added.
5. A combined experimental 27Al MAS NMR and 2D MQ-MAS 27Al NMR study: one more step to reveal the origin of the macroscopic photoluminescence response of the hexagonal polymorph
Solid-state NMR has specific and important advantages compare to XRD by studying the local structure around a selected nucleus, with no limitations due to disorder in the sample. NMR is a local probe that detects all the structures regardless of whether they are related to crystalline phase, amorphous phase.The 27Al quantitative one pulse MAS NMR spectra at 20 T of the powders synthesized with different ZnO content at 1000 °C are shown in Fig. 7. They are exhibiting two peak regions at around 12 and 80 ppm, which can be assigned to octahedral (AlO) and tetrahedral (AlT) coordination, respectively. Neither the peak positions nor the lineshapes of AlO and AlT sites change significantly as a function of ZnO loading. However, a slight lineshape change and the different peak intensity indicate modification in the distribution and coordination on the AlO and AlT site.
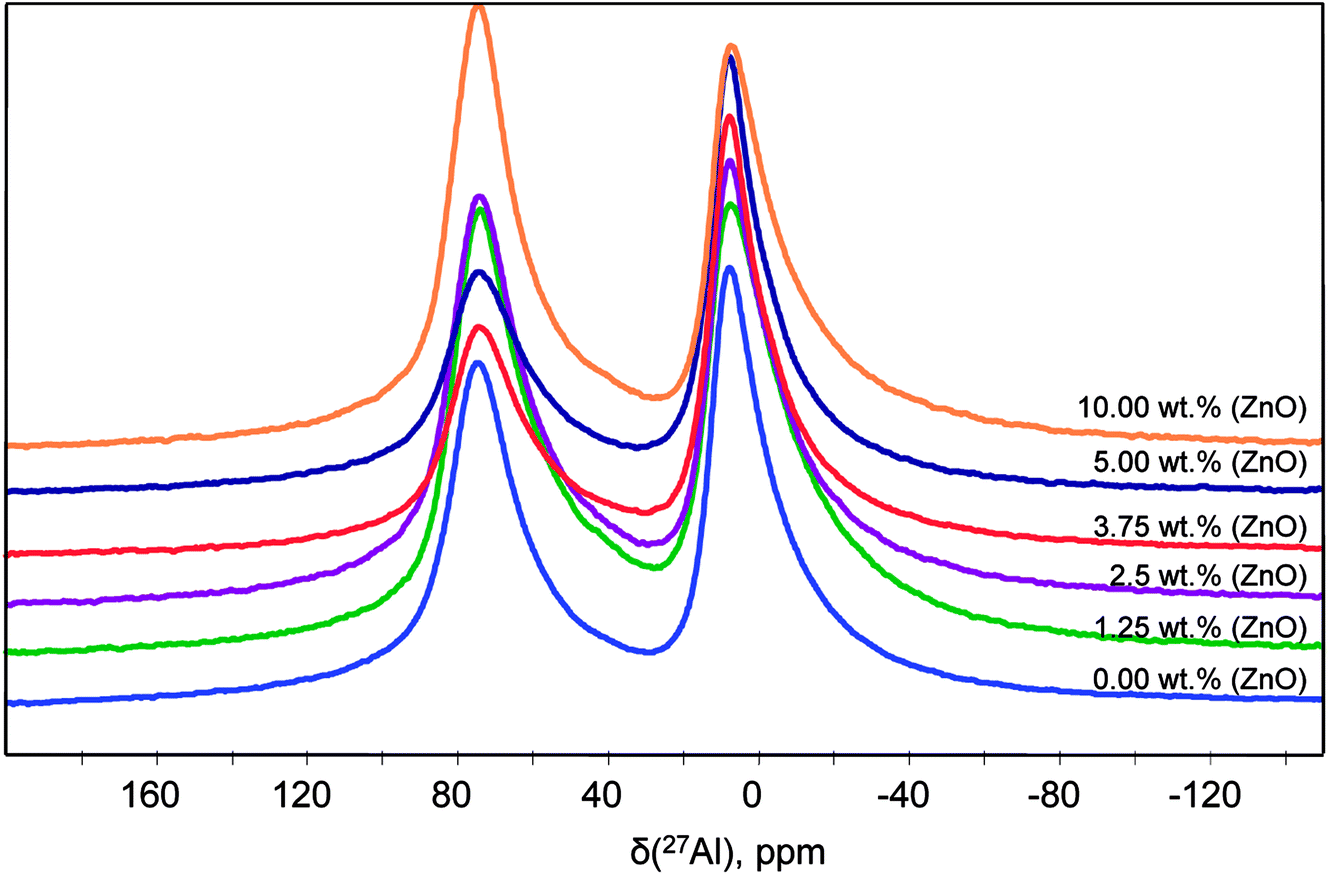 | ||
| Fig. 7 27Al NMR spectra of the powder SrAl2O4: Eu, Dy synthesized with different content of ZnO. The compositions represented as a function of the ZnO content in are labelled as follows: 0; 1.25; 2.5; 3.75; and 5 according to the ZnO wt%. | ||
To improve the resolution, we have used the two dimensional MQ-MAS technique. Fig. 8(a) illustrates the 27Al MAS (experimental (blue) and simulated (red)) and MQ-MAS spectra and with its simulation of the alumina precursor (γ-Al2O3 phase). The observed 27Al line shapes are characteristic of a distribution of 27Al chemical shift and quadrupolar coupling constant. 1D and 2D spectra are very satisfactorily simulated using a fully random distribution of quadrupolar and isotropic chemical shift interactions as expressed within the GIM model.29 The retrieved average isotropic chemical shift δiso, mean quadrupolar coupling constant CQ and population of each site are given in Table 1. The whole set of 1D and 2D spectra were modeled using a single consistent set of parameters.
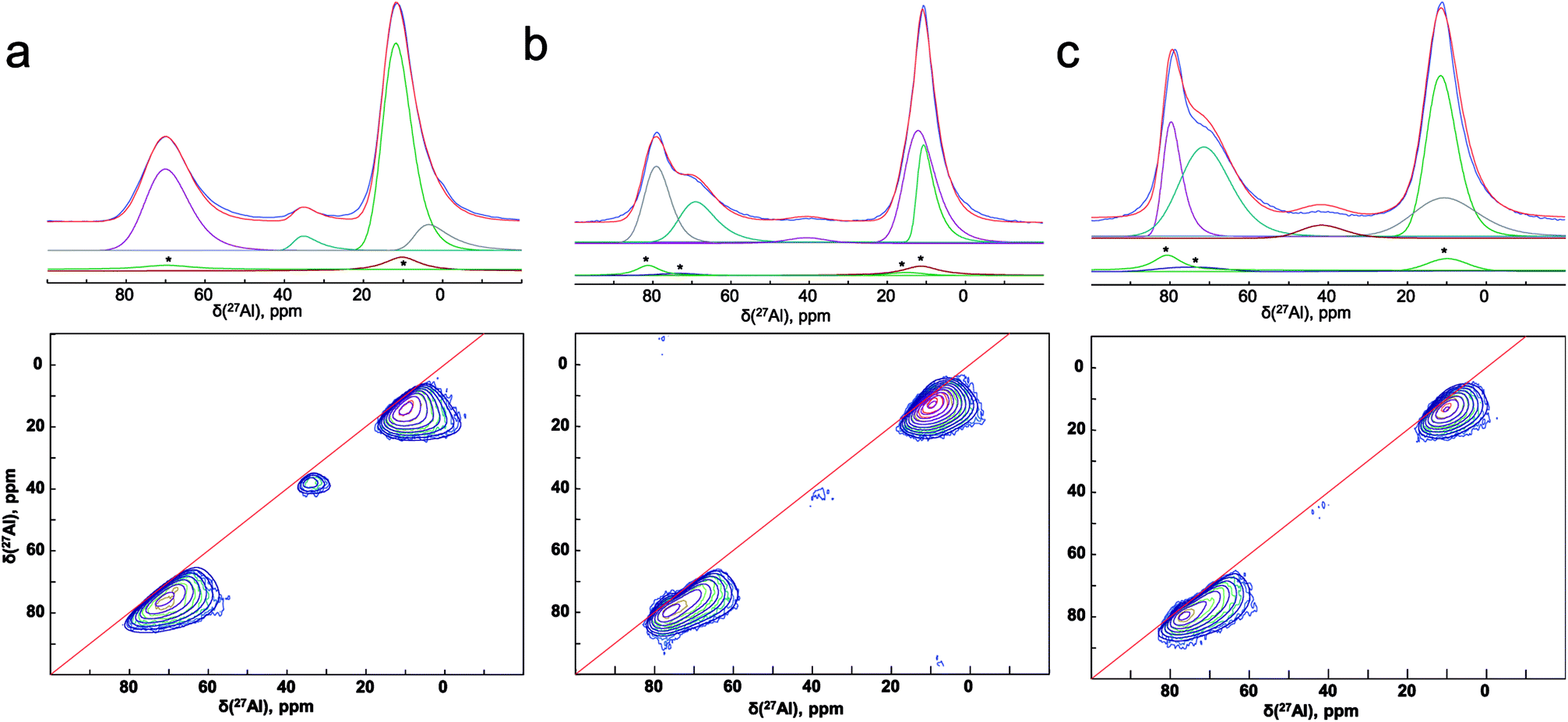 | ||
| Fig. 8 (Experimental (blue) and simulated (red)) 27Al 1D MAS (upper figures) and 2D MQ-MAS spectra (lower figures) spectra (color lines) and its simulation (black lines) with the parameters reported in Table 1 for the (a) alumina precursor, and the synthesized SrAl2O4: Eu, Dy without (b) ZnO and (c) with 5% of ZnO. 1D spectra are fitted along with their respective MAS-related sidebands (*) which correspond to the bands n = 0 of the external transitions. | ||
| 27Al site | δiso, ppm (±0.5 ppm) | MHz, (±0.2 MHz) | I, % (±2%) | |
|---|---|---|---|---|
| γ-Al2O3 | #1 | 7.3 | 5 | 9 |
| #2 | 14.6 | 6.7 | 55 | |
| #3 | 37.8 | 5.6 | 4 | |
| #4 | 74.1 | 6.0 | 32 | |
| SAO γ-Al2O3 Al/Sr: 2 | #1 | 12.5 | 5.6 | 18 |
| #2 | 15.3 | 4.8 | 37 | |
| #3 | 40.6 | 5.6 | 3 | |
| #4 | 72.8 | 6.0 | 17 | |
| #5 | 81.4 | 4.5 | 25 | |
| SAO γ-Al2O3 Al/Sr: 2 + 5 wt% ZnO | #1 | 14.4 | 4.9 | 34 |
| #2 | 15.9 | 6.5 | 16 | |
| #3 | 45.3 | 5.4 | 4 | |
| #4 | 75.9 | 6.0 | 31 | |
| #5 | 81.8 | 4.4 | 15 |
The spectra reveals one peak in the AlT region (85 to 60 ppm) with a maximum located at about 74 ppm. Likewise, two peaks can also be found in the AlO region (25 to 0 ppm) with located at about 14.6 and 7.3 ppm. It corresponds to the γ-Al2O3 phase with an isotropic chemical shift at about 12 ppm30 and unassigned alumina (Alun) phase that appears at the lowest isotropic chemical shift of about 7 ppm.31–33
In addition, the pentahedral (AlP) coordination is observed; around ca. 37.8 ppm. These broad peaks are characteristic of the highly disordered γ-Al2O3 phase.31 Two classes of quadrupolar lineshapes in the AlO spectral region, two in the AlT region and one in the AlP region can distinguish for γ-Al2O3 precursor. The simulated parameters are summarized in Table 1. The NMR spectra for the synthesized SrAl2O4: Eu, Dy powder without and with 5 wt% of ZnO are shown in Fig. 8(b) and (c), respectively. SrAl2O4 phase formation shows the appearance of a shoulder at the high chemical shift side of the tetrahedral Al3+ feature. In SrAl2O4 structure all the aluminum atoms are located into AlO4 tetrahedral (isotropic chemical shift at about 80 ppm). Also, the peak with chemical shift of about 7 ppm disappears and 27Al nucleation sites that eventually evolve into the θ-Al2O3 phase (near 13 ppm) that appears. Specifically the relatively broad and asymmetric peak of the AlO initially centered at around 14 ppm becomes symmetric, and encompasses at least two distinct components at 11 and 12 ppm chemical shifts.
Therefore, for the powders synthesized without and with ZnO, two unique quadrupolar lineshapes are observed in the AlT region that can be related to γ-Al2O3 phase (near 74 ppm), and to the SrAl2O4 structure. Furthermore, the isotropic chemical shift of AlP sites located at about 38 ppm is associated with γ-Al2O3 phase. Concomitantly, the AlT peak centered at 74 ppm loses intensity for the sample without ZnO, while the shoulder at 80 ppm gains in intensity, indicating the formation of hexagonal SrAl2O4 polymorph.
The presence of resonances associated with γ-Al2O3 reinforces the hypothesis of strontium aluminate formation on the surface of alumina agglomerates and the presence of the excess of unreacted alumina inside nanostructured strontium aluminate particles.
The relative intensities of each 27Al resonances (Alo, AlT, AlP and Alun) are calculated, allowing us to compare the Al sites quantitatively between the raw material and the samples with and without ZnO. Since the amount of each type of Al site is proportional to its corresponding integrated peak intensity, the results clearly show that the amount AlT (near 74 ppm) decreases with SrO incorporation, because of the SrAl2O4 formation. AlT site (near 74 ppm) keeps constant regarding alumina precursor with ZnO loading, and AlT site (near 81 ppm) appears; indicating that ZnO could act as an agent that inhibits alumina reaction.
Conclusions
In conclusion, we have shown that the stabilization in a single step of SrAl2O4: Eu2+, Dy3+ hexagonal polymorph at room temperature is possible. Through a combination of synthesis strategies such as the modulation by alumina precursor and optimization of the Al2O3/SrO ratio, we have demonstrated that the preservation of the nanometric nature of the SrAl2O4-based system is closely related to the stabilization of the hexagonal SrAl2O4 polymorph. In addition, Zn doping facilitates the diffusion of Eu and their stabilization as Eu2+ in hexagonal SrAl2O4 polymorph as is required for efficient emission intensity. Although the long-lasting luminescence of these materials is limited in comparison with monoclinic counterpart, the hexagonal SrAl2O4 polymorph offers a several remarkable advantage: the nanometric nature of the hexagonal SrAl2O4 polymorph that will allow designing and preparing of new light emitting materials, inks for security printing field, medicine, and military fields as well as in our daily life including cosmetic or dermatological compositions.Methods
1. Materials
α-Al2O3 (Almatis, CTSG10, specific surface area, BET: 13 m2 g−1, average particle size, d50 ≈ 0.1 μm), γ-Al2O3 (Sasol, Puralox SCCa, 98%), SrCO3 (Merck, 99.9%, d50 ≈ 1.1 μm), Eu2O3 (Metall Rare Earth Limited, 99.5%, d50 ≈ 3.8 μm), Dy2O3 (Metall Rare Earth Limited, 99.5%, d50 ≈ 3.1 μm) and ZnO, were used as precursors.2. Synthesis of sub-micron particles
SrAl2O4: Eu2+, Dy3+ particles were synthesized by molten salt synthesis. In all the experiments, the raw materials were first dried at 120 °C for 1 h due to their hygroscopic nature. Sr1−x−yEuxDyyAl2O4 compositions with x = 0.02 to 0.01 and y = 0.01 to 0.05 were prepared. The molten salt was composed of a mixture of NaCl (99.5% purity) and KCl (99.5% purity) using a 0.5/0.5 molar ratio (eutectic mixture hereafter abbreviated as (NaCl–KCl)e). It was dry homogenized by grinding in a 60 cm3 nylon container for 20 minutes by using a turbula-type mixer at 50 rpm with ZrO2 balls with a diameter of 0.5 mm. The above materials were mixed in the same dry conditions using the turbula-type mixer. The salt/SrAl2O4 molar ratio was kept at 3:1. The homogenized mixture was placed in an alumina crucible with a platinum foil to avoid reaction with the crucible. The powders were heated to a given temperature (1000 °C) in air or 90N2–10H2 atmosphere and held for 2 h in order to reduce the europium and obtain a phosphorescent material. A white powder was finally obtained.
3. Structural and microstructural characterization
The crystalline phases were characterized by X-ray diffraction (XRD, D8, Bruker) using a Lynx Eye detector and a Cu Kα1,2 radiation. The morphology of the nanostructured powders was evaluated using secondary electron images of field emission scanning electron microscopy (FE-SEM, Hitachi S-4700). The particle size and morphology at the nanoscale of the samples were also evaluated using a transmission electron microscope (TEM/HRTEM, JEOL 2100F) operating at 200 kV and equipped with a field emission electron gun providing a point resolution of 0.19 nm. The microscope is coupled with an INCA x-sight energy dispersive X-ray spectrometer (EDXS, Oxford Instruments) used for chemical analysis.X-ray absorption near-edge structure spectroscopy (XANES) measurements were carried out at Eu-L3 edge at the Spanish CRG beamline BM25A (SpLine) at the European Synchrotron (ESRF), France. X-ray absorption (XAS) spectra were collected in fluorescence mode at 45° incidence and at RT. Signal was measured using a 13 element Si (Li) solid state detector from 2 eV Scientific Instruments. Final spectra represent an average of three X-ray absorption (XAS) scans. The X-ray absorption data were analysed using ATHENA software.34
Solid-state NMR experiments were performed using Bruker Avance III spectrometer operating at magnetic field strength of 20.0 T, using a 2.5 mm resonance probe at a MAS frequency of 30 kHz. The 27Al chemical shifts are referenced to 1 M Al(NO3)3. Triple-quantum 27Al MQMAS 2D spectra were acquired using a three-pulse sequence with a zero-quantum filter,35 a repetition delay of 1.0 s, and rotor-synchronized sampling of the indirect dimension. Phase cycling involved the States method36 for acquisition of pure absorption line shapes. The optimized excitation and conversion pulse width was 2 and 0.9 μs, respectively. The NMR parameters (chemical shifts and quadrupolar parameters) were fitted by means of a DMfit program.37
4. Luminescent characterization
Optical properties of these materials were investigated by measuring emission and excitation spectra. The photoluminescence spectra of the phosphor particles were recorded with a spectrofluorometer (Fluorolog®-3, HORIBA Jobin Yvon) at RT. The emission spectrum was measured over the wavelength 400–650 nm, a xenon arc lamp was used as excitation source (λexc = 380 nm). The decay profiles were also recorded using the same instrument after the samples were exposed to monochromatic light, λexc = 380 nm, for about 10 minutes.Author contributions
The samples were prepared by R. E. R.-H. Characterization of the samples was performed by R. E. R.-H., F. R.-M., A. S., C. B., A. R. and J. F. F. F. R.-M cooperated carefully on TEM characterization and analysis. R. E. R.-H collaborated specifically on photoluminescence characterization, C. B and A. R. particularly on NMR analysis and A. S intensely on XANES analysis. Manuscript was written by Dr R. E. R.-H., edited by Dr F. R.-M., A. S., J. F. F. and C. B., A. R., and approved by all authors. All authors contributed to discussions and reviewed the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the Spanish Ministry of Economy and Competitiveness (MINECO) under the projects MAT2017-86450-C4-1-R, and the Spanish National Research Council (CSIC) under the project NANOMIND CSIC 201560E068. F. R.-M. is also indebted to MINECO for a ‘Ramon y Cajal’ contract (ref: RyC-2015-18626), which is co-financed by the European Social Fund. We acknowledge BM25-SpLine, the Spanish CRG, and the European Synchrotron (ESRF) for the use of their facilities, hospitality and financial help.References
- T. Matsuzawa, Y. Aoki, N. Takeuchi and Y. Murayama, J. Electrochem. Soc., 1996, 143, 2670–2673 CrossRef.
- W. Peng, H. Xia, Z. Zhang, T. Qi, S. Kong, W. Dai and Z. Huang, J. Alloys Compd., 2018, 753, 35–40 CrossRef.
- K. E. Foka, F. B. Dejene and H. C. Swart, Phys. B, 2013, 439, 1–4 Search PubMed.
- H. Song, D. Chen, W. Tang and Y. Peng, Displays, 2008, 29, 41–44 CrossRef.
- S. O. Eu, H. Ryu and K. S. Bartwal, Phys. B, 2009, 404, 1714–1718 CrossRef.
- Z. Gyri, V. Havasi, D. Madarász, D. Tátrai, T. Brigancz, G. Szabó, Z. Kónya and Á. Kukovecz, J. Mol. Struct., 2013, 1044, 87–93 CrossRef.
- V. P. Singh, S. B. Rai, H. Mishra and C. Rath, Dalton Trans., 2014, 43, 5309–5316 RSC.
- D. Errandonea, R. S. Kumar, J. Ruiz-Fuertes, A. Segura and E. Haussühl, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 1–6 Search PubMed.
- P. Escribano, M. Marchal, M. Luisa Sanjuán, P. Alonso-Gutiérrez, B. Julián and E. Cordoncillo, J. Solid State Chem., 2005, 178, 1978–1987 CrossRef.
- M. Avdeev, S. Yakovlev, A. a. Yaremchenko and V. V. Kharton, J. Solid State Chem., 2007, 180, 3535–3544 CrossRef.
- C. M. B. Henderson and D. Taylor, Mineral. Mag., 1982, 45, 111–127 CrossRef.
- Q. Wu, Z. Liu and H. Jiao, Phys. B, 2009, 404, 2499–2502 CrossRef.
- A. Douy and M. Capron, J. Am. Ceram. Soc., 2002, 40, 3036–3040 Search PubMed.
- V. P. Singh, S. B. Rai, H. Mishra and C. Rath, Dalton Trans., 2014, 43, 5309–5316 RSC.
- R. E. Rojas-Hernandez, F. Rubio-Marcos, R. H. Gonçalves, M. Á. Rodriguez, E. Véron, M. Allix, C. Bessada and J. F. Fernandez, Inorg. Chem., 2015, 54, 9896–9907 CrossRef PubMed.
- N. Thompson, An Approach to the Synthesis of Strontium Aluminate Based Nanophosphors, School of Applied Science RMIT University, 2012 Search PubMed.
- A. Nag and T. R. N. Kutty, J. Alloys Compd., 2003, 354, 221–231 CrossRef.
- E. Cordoncillo, B. Julian-Lopez, M. Martínez, M. L. Sanjuán and P. Escribano, J. Alloys Compd., 2009, 484, 693–697 CrossRef.
- E. Shafia, M. Bodaghi and M. Tahriri, Curr. Appl. Phys., 2010, 10, 596–600 CrossRef.
- D. Jia, B. Wu and J. Zhu, Chin. Phys., 2000, 9(1), 69–72 CrossRef.
- M. Capron, F. Fayon, D. Massiot and A. Douy, Chem. Mater., 2003, 15, 575–579 CrossRef.
- S. ichi Yanagiya, N. Van Nong, J. Xu and N. Pryds, Materials, 2010, 3, 318–328 CrossRef.
- M. Capron, Synthèse et Caractérisation D’aluminates et D’aluminosilicates de La Température Ambiante Jusqu’au Liquide À Haute Température, PhD thesis, 2001.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 1272–1276 CrossRef.
- R. E. Rojas-Hernandez, F. Rubio-Marcos, M. Á. Rodriguez and F. Fernandez, Renewable Sustainable Energy Rev., 2017, 1–12 Search PubMed.
- F. Clabau, X. Rocquefelte, S. Jobic, P. Deniard, M. Whangbo, A. Garcia and J. Rouxel, Chem. Mater., 2005, 17, 3904–3912 CrossRef.
- R. E. Rojas-Hernandez, F. Rubio-Marcos, A. Serrano, A. Del and J. F. Fernandez, Sci. Rep., 2017, 7, 1–9 CrossRef PubMed.
- R. E. Rojas-Hernandez, F. Rubio-Marcos, E. Enríquez, M. A. De La Rubia and J. F. Fernandez, RSC Adv., 2015, 5, 42559–42567 RSC.
- G. Le Caër and R. A. Brand, J. Phys.: Condens. Matter, 1998, 10, 10715–10774 CrossRef.
- R. M. Ravenelle, J. R. Copeland, W.-G. Kim, J. C. Crittenden and C. Sievers, ACS Catal., 2011, 1, 552–561 CrossRef.
- J. Z. Hu, S. Xu, J. H. Kwak, M. Y. Hu, C. Wan, Z. Zhao, J. Szanyi, X. Bao, X. Han, Y. Wang and C. H. F. Peden, J. Catal., 2016, 336, 85–93 CrossRef.
- L. Kovarik, M. Bowden, A. Genc, J. Szanyi, C. H. F. Peden and J. H. Kwak, J. Phys. Chem. C, 2014, 118, 18051–18058 CrossRef.
- L. Kovarik, M. Bowden, D. Shi, N. M. Washton, A. Andersen, J. Z. Hu, J. Lee, J. Szanyi, J.-H. Kwak and C. H. F. Peden, Chem. Mater., 2015, 27, 7042–7049 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef PubMed.
- J.-P. Amoureux, C. Fernandez and S. Steuernagel, J. Magn. Reson., Ser. A, 1996, 123, 116–118 CrossRef PubMed.
- D. J. States, R. A. Haberkorn and D. J. Ruben, J. Magn. Reson., 1982, 48, 286–292 Search PubMed.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05601c |
| This journal is © The Royal Society of Chemistry 2018 |