
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceP-Stereodefined phosphorothioate analogs of glycol nucleic acids—synthesis and structural properties†‡
Agnieszka Tomaszewska-Antczak *,
Katarzyna Jastrzębska,
Anna Maciaszek,
Barbara Mikołajczyk and
Piotr Guga
*,
Katarzyna Jastrzębska,
Anna Maciaszek,
Barbara Mikołajczyk and
Piotr Guga
Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Department of Bioorganic Chemistry, Sienkiewicza 112, 90-363 Łódź, Poland. E-mail: atom@cbmm.lodz.pl
First published on 11th July 2018
Abstract
Enantiomerically pure, protected acyclic nucleosides of the GNA type (glycol nucleic acids) (GN′), obtained from (R)-(+)- and (S)-(−)-glycidols and the four canonical DNA nucleobases (Ade, Cyt, Gua and Thy), were transformed into 3′-O-DMT-protected 2-thio-4,4-pentamethylene-1,3,2-oxathiaphospholane derivatives (OTP-GN′) containing a second stereogenic center at the phosphorus atom. These monomers were chromatographically separated into P-diastereoisomers, which were further used for the synthesis of P-stereodefined “dinucleoside” phosphorothioates GNPST (GN = GA, GC, GG, GT). The absolute configuration at the phosphorus atom for all eight GNPST was established enzymatically and verified chemically, and correlated with chromatographic mobility of the OTP-GN′ monomers on silica gel. The GNPS units (derived from (R)-(+)-glycidol) were introduced into self-complementary PS-(DNA/GNA) octamers of preselected, uniform absolute configuration at P-atoms. Thermal dissociation experiments showed that the thermodynamic stability of the duplexes depends on the stereochemistry of the phosphorus centers and relative arrangement of the GN units in the oligonucleotide strands. These results correlate with the changes of conformation assessed from circular dichroism spectra.
Introduction
Nucleic acids play a fundamental role in life. These polyanionic polymers, as well as numerous synthetic analogs, are also used in biochemistry, molecular biology, and other fields.1 There is also growing interest in their application in nanotechnology.2,3 Certain applications require nucleic acids with specific modifications, e.g., at the nucleobases, sugars (ribose or deoxyribose), or inter-nucleotide phosphate linkages.4–6 In phosphorothioate analogs of DNA (PS-DNA) one of the non-bridging phosphate oxygen atoms is replaced with a sulfur atom (Fig. 1A).7–9 PS-DNA oligomers are remarkably resistant towards nucleolytic enzymes. Although phosphorothioate and phosphate diesters are formally isoelectronic, in PS-DNA there is a greater density of negative charge on the sulfur atom.10 A single O → S replacement creates a new stereogenic center, thus a PS-DNA decamer synthesized using a non-stereocontrolled method consists of 29 = 512 isomers (usually of comparable abundance), which may have considerably different properties.§ Over several years P-stereodefined oligonucleotides (up to 15–18 nt in length) of PS-DNA11 and PS-LNA12 series, were obtained using an oxathiaphospholane method.13 In several instances these oligomers showed P-stereodependent biological properties.14–16 | ||
| Fig. 1 Structures of (A) P-stereodefined PS-DNA, (B) C-stereodefined GNA, and (C) P,C-stereodefined PS-(DNA/GNA) oligomers. | ||
Recent advances in nucleic acids chemistry enable synthesis of analogs of DNA or and RNA with improved chemical and biological stability. These polymers (often denoted XNA) usually carry standard nucleobases but have profoundly altered sugar-phosphate backbones.17 One of the most fundamental changes is the replacement of the cyclic ribose (deoxyribose) moiety with an acyclic structure. Because many of those analogs are poor substrates of nucleases, they are explored in modulation of the properties of nucleic acid and in synthetic biology.4,5 The inherent flexibility of acyclic “nucleosides”, combined with specific “contact points” offered by a given scaffold, make them suitable for building of molecular devices.18 Glycol nucleic acid (GNAs; Fig. 1B) attracted our attention because of interesting physico-chemical properties, which make them potentially suitable in biotechnology or nanotechnology.19
The glycol “Gnucleoside” units (GN) can exist in two enantiomeric forms, i.e. (RC)-GN and (SC)-GN (1a and 1b, respectively, B = Ade, Cyt, Gua, Thy).

The GNA oligomers hybridize according to the Watson–Crick's scheme and form highly stable duplexes with a complementary GNA strand.20 Hybridization with natural nucleic acids is only observed for (SC)-GNAs (and not (RC)-GNAs) with complementary ssRNA templates whereas duplexes with ssDNA are much less stable. The reasons of this difference were thoroughly discussed based on X-ray analysis results.21
Because interesting P-stereodependent effects were observed in PS-DNA (e.g., different thermodynamic stability of antiparallel duplexes (PS-DNA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DNA)22 or different propensity for the B → Z conversion23), as well as in conformationally more restricted “locked” PS-LNA,12 we have turned our efforts toward synthesis of P-stereodefined phosphorothioate analogs of conformationally flexible, glycol-based GNA (PS-GNA). Taking into account poor hybridization properties of GNA and the fact that the phosphorothioate modification usually decreases thermal stability of the modified duplexes¶ we assumed that PS-GNA oligomers would not be able to act as antisense or antigene probes specifically interacting with mRNA or genomic DNA strands. Nonetheless, certain acyclic, thermally destabilizing modifications (including GNA) enhance RNAi-mediated gene silencing, because the Ago2 protein favors loading of the 5′-end of the strand from the less stable end of an siRNA duplex.21,24,25 Thus, one cannot reject PS-GNA from future application in the RNAi methodology.
:DNA)22 or different propensity for the B → Z conversion23), as well as in conformationally more restricted “locked” PS-LNA,12 we have turned our efforts toward synthesis of P-stereodefined phosphorothioate analogs of conformationally flexible, glycol-based GNA (PS-GNA). Taking into account poor hybridization properties of GNA and the fact that the phosphorothioate modification usually decreases thermal stability of the modified duplexes¶ we assumed that PS-GNA oligomers would not be able to act as antisense or antigene probes specifically interacting with mRNA or genomic DNA strands. Nonetheless, certain acyclic, thermally destabilizing modifications (including GNA) enhance RNAi-mediated gene silencing, because the Ago2 protein favors loading of the 5′-end of the strand from the less stable end of an siRNA duplex.21,24,25 Thus, one cannot reject PS-GNA from future application in the RNAi methodology.
Theoretical studies on the influence of the phosphorothioate modification on the electronic properties of model single (ss) and double-stranded (ds) dinucleotides revealed that the electron migration process through an ss-PS-DNA molecule may be slowed down or, in extreme cases, quenched.26 Also, interesting differences in the electron transfer potentials of P-stereodefined chimeric DNA decamers d(CGPSGCCGCCGA), as well as their duplexes formed with complementary strands d(TCGGCGPSGCCG), were found by cyclic voltammetry assay.27 The cited above electronic properties of PS-DNA depend on hybridization status and/or stereochemistry of the phosphorothioate linkages.
In this work, we wanted to answer a question whether the stereochemistry of the phosphorus atoms would affect the hybridization and/or structural properties of PS-GNA oligomers. Anticipated differences in hybridization properties between RP- and SP-PS-(DNA/GNA) oligomers and possible P-stereodependent electronic properties may be advantageous for the applications of P-stereodefined PS-GNA units in nanotechnology. Toward this goal, we present synthesis and separation of P-diastereomers of 2-thio-4,4-pentamethylene-1,3,2-oxathiaphospholane derivatives of the 3′-O-DMT protected glycidol-derived “Gnucleosides” (OTP-GN′), and enzymatic and chemical determination of the absolute configuration of the P-atoms in the assembled GN′PST “dinucleotides”. Because of unexpected destructive side-reactions encountered during oligonucleotide assembly, we synthesized chimeric selfcomplementary PS-(DNA/GNA) oligomers (Fig. 1C), which were subjected to melting experiments and circular dichroism analysis.
Results
Preparation of DMT-protected glycidol “Gnucleosides” (DMT-GN′)
The 3′-O-DMT-protected glycidol “Gnucleosides” (DMT-GN′, 4a–c, Scheme 1) were obtained by a literature method.28 In brief, the commercially available enantiomerically pure (R)-(+)- and (S)-(−)-glycidols 2 were protected with DMT-Cl to yield almost quantitatively the corresponding dimethoxytritylated glycidols 3. The regioselective and stereospecific epoxide ring opening reaction with a nucleobase (thymine, adenine or N-benzoylcytosine) in the presence of NaH afforded DMT-GN′s 4a (B = Thy), 4b* (B = Ade) or 4c (B = CytBz).28 DMT-GA (4b*) was next protected at the exocyclic amino group with benzoyl chloride to yield DMT-GABz (4b). The guanine derivative 4d was obtained via a different method (Scheme 2), since the literature protocol28 didn't work in our hands. In this case we decided to work only with (R)-(+)-glycidol because the related (SC)-GNA has more useful properties than (RC)-GNA.20 The epoxide 2 was opened with 2-amino-6-chloropurine to give compound 6, which was further treated with sodium thiosulfate. The resultant 6-thioguanosine derivative 7 was oxidized to GG (8) using hydrogen peroxide.29 The amino group in 8 was acylated with iso-butyryl chloride to yield diol 9, which was further protected with DMT-Cl to furnish DMT-GGiBu (4d). | ||
| Scheme 1 Synthesis of DMT-GN′ (4a–c) and their conversion into the oxathiaphospholane monomers 5a–d. In the inset, the numbers in parentheses indicate the priority of substituents (in terms of the Cahn–Ingold–Prelog's rules) around the stereogenic centers. The synthesis of DMT-GGiBu (4d) is shown in Scheme 2. | ||
 | ||
| Scheme 2 Synthesis of DMT-GGiBu (4d). | ||
Starting from (R)-(+)- and (S)-(−)-glycidol (2), the corresponding DMT-GN′s (4) of SC and RC absolute configuration, respectively, were obtained (see inset in Scheme 1; note that the methylene group in the oxirane ring (marked as CH2) has priority over the hydroxymethyl group, as shown by the numbers in parentheses).
Preparation of P-diastereomerically pure oxathiaphospholane derivatives of glycol “Gnucleosides” (OTP-GN′)
The enantiomerically pure “Gnucleoside” substrates (DMT-GN′, 4a–d) were phosphitylated with 2-chloro-4,4-pentamethylene-1,3,2-oxathiaphospholane (prepared according to ref. 11) in the presence of DIPEA and elemental sulfur (see Scheme 1). After purification on a silica gel column the derivatives 5a–d (OTP-GN′) were obtained in good yield (78%, 74%, 71%, 79%, respectively) and characterized by 31P NMR, 1H NMR, 13C NMR (for representative spectra see Fig. 1S–3S, ESI‡) and HRMS (for the spectra of 5a–d see Fig. 4S, ESI‡).A molecule of OTP-GN′ (5) has two stereogenic centers (at the carbon atom C-2′ and the phosphorus atom, both marked with asterisks in Scheme 1), therefore, each of 5a–d may exist in four diastereomeric forms. If the absolute configuration at the carbon center is preselected by the use of a particular enantiomer of glycidol, the number of possible forms is reduced to two P-diastereomers. For each compound of 5a–d we were able to separate these P-diastereomers by semi-preparative HPLC on a silica gel column. MALDI-TOF MS spectra for fast- and slow-eluting 5a are shown in Fig. 5S (ESI),‡ and further characterization of fast and slow-eluting isomers is given in Table 1S (ESI).‡
Attempts to prepare crystals of OTP-GN′ for X-ray analysis
Our efforts to obtain crystals of 5a–d for X-ray analysis were unsuccessful. Following the previous studies on crystallization of the oxathiaphospholane derivatives in DNA11 and LNA12 series, we tried to remove the DMT group in 5a–d to obtain 3′-OH OTP-GN′ derivatives 5*a–d (shown in square brackets in Scheme 1) using earlier successful p-toluenesulfonic acid monohydrate, yet with no success. Another protic acid (dichloroacetic acid), Lewis' acids (ZnCl2, SnCl4), tetra-n-butylammonium peroxydisulfate30 and NaHSO4/SiO2 (ref. 31) did not work either. In each case we observed the formation of a very polar compound, and in the case of 5a that polar compound was isolated and identified by 31P NMR and MS (Fig. 6S, ESI‡) as the corresponding Gthymidine cyclic 2′,3′-O,O-phosphorothioate (cGTMPS) bearing a 5-membered ring (Scheme 3, B = Thy). The reasons of this unexpected elimination are unclear. It should be noted that the conversion of the detritylated OTP derivative of dCBz into the corresponding cyclic 3′,5′-O,O-phosphorothioate, which has an entropically favored 6-membered ring, occurred only after DBU (1,4-diazabicyclo[5.4.0]undec-7-ene, an activator of high basicity) was added.32 The possible mechanism of forming this five membered by-product is shown in Scheme 3. | ||
| Scheme 3 A possible mechanism of spontaneous oxathiaphospholane ring-opening/cyclization during detritylation of OTP-GT (5a). | ||
Assignment of the absolute configuration of the P-atom in “dinucleotides” GNPST obtained using OTP-GN′
 | ||
| Scheme 4 Synthesis of “dinucleotides” GNPST (11a–d) using OTP-GN′ (5a–d) and products of their hydrolysis (if successful) with svPDE. | ||
The experiments with svPDE gave more useful results. The HPLC profiles recorded after hydrolysis (Fig. 9S and 10S, ESI‡) showed that compounds 11a–d obtained from slow-eluting (RC)-OTP-GN′ (synthesized from S-(−)-glycidol) or fast-eluting (SC)-OTP-GN′ (obtained from R-(+)-glycidol) were hydrolyzed to yield GN (1, Scheme 4) and thymidine 5′-O-phosphorothiate (TMPS), identified by means of RP HPLC (Fig. 11S, ESI‡). Thus the svPDE-digestable diastereomers were tentatively assigned an RP configuration. Those obtained from fast-eluting (Rc)-OTP-GN′ or slow-eluting (SC)-OTP-GN′ remained intact after 24 h incubation, so were assigned an SP configuration.
Chemical verification of the enzymatic assignment
Because phosphorothioates 11 are much different from svPDE natural substrates, one cannot exclude the possibility that these compounds do not conform the general observation of known stereoselectivity of the enzyme towards PS-DNA. To strengthen our stereochemical assignment, a chemical verification was carried out, based on the “inverted” synthesis of GNPST (11*a–d; the synthesis of 11*b is shown in Scheme 5). We used fast- and slow-eluting P-diastereoisomers of the 5′-O-oxathiaphospholane derivative of 3′-O-camphanoylated thymidine (5′-OTP-TCamph, 12), which are known to be the precursors of (SP)-NPST and (RP)-NPST, respectively.35 In the presented example fast-12 was reacted with (SC + RC)-DMT-GABz (4b, prepared from racemic glycidol) to yield a pair of protected DMT-GABzPSTCamph (13b). By a routine two-step deprotection both isomers of 13b were converted into a pair of SCSP- and RCSP-11*b, but this time the absolute configuration at P-atoms (i.e. SP) was known.|| The second pair of 11*b consisting of SCRP- and RCRP-isomers was obtained using slow-eluting 12. The RP and SP pairs of 11*b were mixed at ca. 1:2 molar ratio and we found RP-HPLC conditions allowing for baseline separation of all four components (Fig. 2, an inset in the panel A).
 | ||
| Scheme 5 The “inverted” synthesis of GAPST (11*b) using DMT-GABz and 5′-OTP-TCamph (12). | ||
 | ||
| Fig. 2 HPLC profiles for the mixture of standards 11*b (an inset in panel A) and co-injections of 11*b and GAPST (11b) obtained from fast- and slow-eluting (RC)-OTP-GABz 5b (panels A and B, respectively). HPLC conditions: a Kinetex column, 250 × 4.6 mm; flow rate 1 mL min−1, buffer A: 0.1 M TEAB, buffer B: 40% CH3CN in 0.1 TEAB, gradient: 0–38% of buffer B over 20 min. | ||
In next HPLC analyses this mixture of standards was spiked with a given 11b and it was found that 11b derived from fast-RC-5b (Fig. 2, panel A) and slow-RC-5b (panel B) (both obtained from S-(−)-glycidol) were precursors of the internucleotide phosphorothioate linkages of SP and RP configuration, respectively.
The analogous analysis indicated (data not shown) that the fast- and slow-eluting SC-5b (obtained from R-(+)-glycidol) were precursors of the internucleotide phosphorothioate linkages of RP and SP configuration, respectively. In conclusion, the experiment shown in Scheme 5, as well as those carried out with 4a, c, d (data not shown) proved that the absolute configurations at the phosphorus atom in 11a–d assigned using svPDE were correct.
Attempts at solid phase synthesis of P-stereodefined PS-GNA oligomers
The diastereomerically pure OTP-GN′ monomers 5a–d were tested in a manually performed, solid-phase synthesis of PS-GNA oligonucleotides. To prevent the DBU-promoted cleavage of the standard lcaa linker, the first nucleoside was attached to the solid support via a DBU-resistant sarcosine linker.36 Even with double coupling used at each condensation step (earlier effectively applied in synthesis of PS-LNA12), only ca. 75% repetitive yield was achieved (based on a DMT cation assay), so synthesis of octamers and decamers would provide only ca. 13% and 7%, respectively, of the theoretical amounts (0.757 = 0.133, 0.759 = 0.07), whereas the amount of actually isolated products would be further reduced at least by half.37 This is unacceptably low efficiency, compared to 92–94% repetitive yield noted for OTP monomers of DNA11 and LNA12 series. To acquire an insight into a possible mechanism contributing to the problem, a PS-GNA oligonucleotide (GUPS)11dA has been synthesized using a modified28 phosphoramidite/sulfurization methodology and the sarcosine linker. A 2-cyanoethyl-N,N-diisopropyl-phosphoramidite derivative of DMT-GU was used (prepared according to ref. 28) and >99.2% repetitive yield was noted (based on a DMT cation assay, Fig. 12S, ESI‡). After the synthesis was complete, the oligomer (still being attached to the support) was treated with piperidine/acetonitrile solution (1 M) to remove the 2-cyanoethyl protecting groups, and the resultant phosphorothioate diester compound was subjected to a DBU/acetonitrile solution (1.5 M) for 1 h. The remaining material was cleaved from the support and isolated by RP-HPLC (a broad peak eluting at 15.65 min, Fig. 13S, ESI‡). A MALDI-TOF MS analysis showed three ladders of ions corresponding to the molecular ion (m/z 3154), the depurinated (due to the acidic matrix) derivative (m/z 3020), and the depurinated/hydroxylated oligomer (m/z 3037), all consecutively truncated by up to 5 residues, each of 264 amu (Fig. 14S, ESI‡). The relative intensities of the consecutive bands related to the most intense band at m/z 3037 are 1:0.69:0.30:0.13:0.09:0.06, and the other two sets basically follow this pattern. Thus one can assume that the cleavage took place at the end of the consecutively truncated oligomer and not randomly inside the chain. The 264 Da residue seems to be Guridine cyclic 2′,3′-O,O-phosphorothioate (cGUMPS, Scheme 3, B = Ura), which is an analog of cGTMPS, although this time it was formed upon intramolecular attack of the sequentially released terminal hydroxyl groups at the phosphorus atom of the nearest internucleotide linkage, with the concomitant release of next primary hydroxyl group, so the cleavage process could go further. Undoubtedly, to avoid that “walking” truncation some mechanistic studies and optimization of the synthetic protocol are necessary.
Solid phase synthesis of P-stereodefined chimeric PS-(DNA/GNA) and PS-DNA oligomers
Because of the unsatisfactory coupling efficiency we decided to synthesize “chimeric” PS-(DNA/GNA) oligonucleotide octamers containing 3 or 4 GN units, using appropriate OTP-GN′ and OTP-N′ monomers.** The syntheses were performed manually at a 1 μmol scale. Selfcomplementary oligomers of a general sequence PS-d(ATGCGCAT) were synthesized in two DNA/GNA variants (each chimeric oligomer in two P-stereoregular forms, Table 1), bearing either purine GNs (PS-GATGGCGGCGAT; RP, 14; SP, 15) or pyrimidine GNs (PS-AGTGGCGGCAT; RP, 16; SP, 17).†† Only SC-OTP-GN′ were used in the syntheses. Decay of the trityl cation absorption after consecutive coupling steps was measured photometrically (Fig. 15S, ESI‡). After each synthesis was complete, the DMT tagged oligomer was cleaved from the support and the N-protecting groups were removed using routine treatment with conc. NH4OH. The oligonucleotides were isolated by RP-HPLC, then detritylated with 50% acetic acid, purified by RP-HPLC (Fig. 16S, ESI‡), and finally characterized by MALDI-TOF MS (Fig. 17S, ESI‡). The syntheses furnished ca. 3 OD260 of each oligomer.| Duplex | P-Chirality | Tma (°C) | ΔA260 (%) | Θ254b [mdeg] |
|---|---|---|---|---|
| a Temperature gradients of 1°C min−1 for annealing and 0.5°C min−1 for melting were applied.b CD spectra were recorded after 15 minutes incubation at 15 °C. | ||||
| 14 5′-(GA TGG CGG CGA T)-3′ | RP | 24 | 2.4 | −1.6 |
| 14 3′-(T GA C GG C GG T GA)-5′ | ||||
| 16 5′-(A GT G GC G GC A T)-3′ | RP | 27 | 5.9 | −2.9 |
| 16 3′-(T A GC G GC G GT A)-5′ | ||||
| 14 5′-(GA T GG C GG C GA T)-3′ | RP | <12 | 2.0 | −1.1 |
| 16 3′-(T A GC G GC G GT A)-5′ | ||||
| 15 5′-(GA TGG CGG CGA T)-3′ | SP | 22 | 7.9 | −2.4 |
| 15 3′-(T GA C GG C GG T GA)-5′ | ||||
| 17 5′-(A GT G GC G GC A T)-3′ | SP | 33 | >7 | −3.7 |
| 17 3′-(T A GC G GC G GT A)-5′ | ||||
| 15 5′-(GA T GG C GG C GA T)-3′ | SP | 20 | 2.0 | −0.8 |
| 17 3′-(T A GC G GC G GT A)-5′ | ||||
| 18 5′-d(ATGCGCAT)-3′ | RP | 29 | 7.6 | −3.1 |
| 18 3′-d(TACGCGTA)-5′ | ||||
| 19 5′-d(ATGCGCAT)-3′ | SP | 25 | 10.7 | −1.7 |
| 19 3′-d(TACGCGTA)-5′ | ||||
The reference oligomers (RP)-PS-5′-d(ATGCGCAT)-3′ (18) and (SP)-PS-5′-d(ATGCGCAT)-3′ (19) were obtained in an analogous way (a single coupling was used).
Thermal stability and circular dichroism analysis of complexes formed by P-stereodefined chimeric PS-(DNA/GNA) octamers 14–17
The P-stereoregular, selfcomplementary chimeric PS-(DNA/GNA) oligomers 14–17, where the GN units in the related duplexes occupy “alternate” positions (see the duplex structures in Table 1), and the reference PS-DNA oligomers 18 and 19 were dissolved in pH 7.2 buffer containing 10 mM Tris–HCl, 100 mM NaCl, and 10 mM MgCl2. The annealing was done from 40 °C to 5 °C with a temperature gradient of 1°C min−1. Then the melting profiles were recorded over a 5 → 40 °C range (0.5°C min−1) and melting temperatures (Tm) were calculated using the first order derivative method. For the reference compounds 18 and 19 we found Tm = 29 °C and 25 °C (Table 1), hyperchromicity of 7.6% and 10.7% (Fig. 18S, ESI‡) and Θ254 = −3.1 and −1.7 mdeg (Fig. 3), respectively. | ||
| Fig. 3 CD spectra for selfcomplementary oligomers 15–19 (dissolved in pH 7.2 buffer containing 10 mM Tris–HCl, 100 mM NaCl, and 10 mM MgCl2) recorded after 15 minutes incubation at 15 °C. | ||
The melting curves for homoduplexes formed by 15–17, showed hyperchromicity (in a range 5.9–7.9%) and cooperativity similar to 18 and 19 (Fig. 18S, ESI‡). However, for 14 only 2.4% hyperchromicity was noted and the cooperativity was very poor (Fig. 19S, ESI‡). This dichotomy was also observed in the corresponding CD spectra, which were recorded after 15 minutes incubation of the samples at 15 °C. The spectra for homoduplexes 15 and 16 were similar to those for 18 and 19, as they have strong negative bands around 254 nm and weak negative signals around 280 nm (Fig. 3). In contrast, for compound 17 a positive band around 294 nm (Θ294 = 0.52 mdeg) had a wide shoulder around 280 nm. Interestingly, the CD spectrum for RP-14 (on contrary to that recorded for the SP-15 counterpart) has a weak signal at 254 nm and a positive signal around 280 nm, so one can assume that the conformation got substantially changed compared to RP-18. Furthermore, the Tm value for SP-17 was by 6 °C higher than for RP-16 and by 8 °C higher than for the reference SP-19. Also, ellipticity Θ254 = −3.7 mdeg found for SP-17 was the highest among all the oligomers studied, including the reference homoduplexes formed by PS-DNA oligomers 18 and 19. These observations indicate that the stereochemistry of the phosphorothioate linkages is important for the mode of hybridization of the oligomers and the resultant conformation of the duplexes.
For the heteroduplexes 14/16 and 15/17, where the GN units occupy the “face-to-face” positions (see Table 1), rather unexpected effects were noted. In both cases very low hyperchromicities (2%) and low Tm values were observed. For 14/16 the Tm value was only roughly assessed at <12 °C, and Tm = 20 °C was noted for 15/17. For those systems one might expect the presence of certain amounts of the duplexes formed by 16 (Tm = 24 °C) or 17 (Tm = 33 °C), with the latter duplex expected to be quite abundant. However, the melting curves did not show the relevant inflection points, as both profiles reached a plateau at much lower temperatures (Fig. 19S,‡ the melting profile for 16 is given as a reference). Also the corresponding CD spectra (Fig. 4) indicated a transition of both heteroduplexes to the conformation already observed for 14, with the absence of bands characteristic for homoduplexes formed by 16 and 17. At that moment we cannot offer any possible reasons for that behavior.
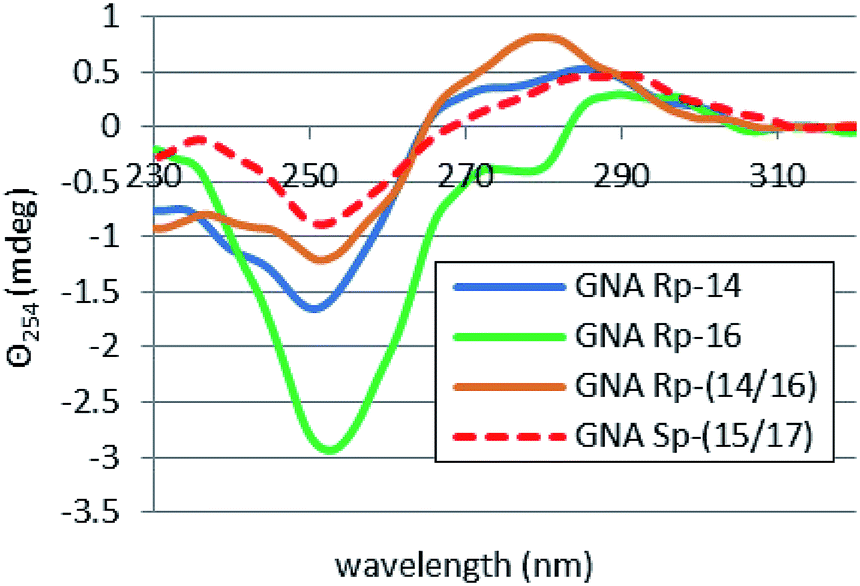 | ||
| Fig. 4 CD spectra for selfcomplementary oligomers 14, 16, 14/16 and 15/17 (dissolved in pH 7.2 buffer containing 10 mM Tris–HCl, 100 mM NaCl, and 10 mM MgCl2) recorded after 15 minutes incubation at 15 °C. | ||
Thermal stability of complexes formed by PS-(DNA/GNA) octamers 14–17 with DNA and RNA templates
The unexpected hybridizing properties of chimeric PS-(GNA/DNA) oligomers prompted us (despite of the most often observed destabilizing effect of the phosphorothioate modification) to investigate the interactions of 14–17 with natural DNA (DNA, (d(ATGCGCAT), isosequential to 18) and (2′-OMe)-RNA ((m)RNA, (2′-OMe)-AUGCGCAU)) templates. However, the results of melting experiments (Table 2S‡) indicate that the Tm value recorded for the reference homoduplex formed by DNA (43 °C) has been only very little reduced (<2 °C) by the presence of equimolar amount of any of the 14–17 oligomer. In the case of (m)RNA (Tm = 62 °C) the reduction was only slightly bigger (up to 4 °C for SP-17). Because the melting curves also showed very good cooperativity (Fig. 20S, ESI‡), one can conclude that the oligomers 14–17 only weakly interact with DNA or (m)RNA, which preferentially form thermodynamically much more stable homoduplexes. This was confirmed by CD spectra (recorded at room temperature, Fig. 21S, ESI‡) which showed that the bands characteristic for the (m)RNA/(m)RNA homoduplex remained virtually unchanged, except for their lowered intensity resulting from the reduced by half concentration of (m)RNA.Conclusions
P-Diastereomers of OTP-GN′s (obtained from enantiomerically pure (R)-(+)- and (S)-(−)-glycidols 2) were chromatographically separated and used in synthesis of “dinucleotide” phosphorothioates GNPST. The absolute configuration of the P-atom in GNPST was established by enzymatic and chemical methods. Then, four self-complementary P-stereodefined chimeric oligomers PS-(DNA/GNA) were synthesized, although even double coupling at the condensation steps utilizing OTP-GN′ led to a significant drop in reaction efficiency. Thermal dissociation experiments showed that the thermodynamic stability of the duplexes depends on the stereochemistry at phosphorus and relative arrangement of the GN units in the oligonucleotide strands. Most interestingly, a duplex formed by 17 (SP)-PS-(AGTGGCGGCAT) was thermally more stable than that formed by the corresponding PS-DNA congener 19 (ΔTm = 8 °C). The results on thermal stability correlate with the changes of overall conformation assessed from circular dichroism spectra.Further studies on the adaptation of the oxathiaphospholane approach to synthesis of P-stereodefined phosphorothioate glycol nucleic acid are underway.
Experimental section
1H NMR and 31P NMR spectra were recorded using a Bruker instrument (AV-200, 200 MHz for 1H). Chemical shift values (δ) are given in ppm, relative to internal tetramethylsilane (TMS) or residual solvent protons for 1H NMR, and external 85% H3PO4 for 31P NMR.Mass spectra (FAB-MS) were recorded on a Finnigan MAT 95 spectrometer, in the positive and negative ion modes. MALDI-TOF MS analyses of oligonucleotides were performed using a Voyager-Elite instrument (PerSeptive Biosystems Inc., Framingham, MA) operating in the reflector mode with the detection of negative ions, unless otherwise stated. HRMS measurements were performed on a qTOF Waters ESI SYNAPT G2-Si high definition mass spectrometer fitted with an atmospheric pressure ionization electrospray source (Waters Corporation, Milford, MA). The instrument was operated in negative-ion mode, with capillary voltage 2.5 kV, cone 40.0 V, and source offset 50 V. Desolvation temperature 150 °C was applied.
Routine UV spectra were recorded on a CINTRA 10e spectrophotometer (GBC, Dandenong, Australia), using a quartz cuvette of 1 cm path length. UV monitored melting experiments were carried out in 1 cm path length cells, using a spectrophotometer CINTRA 4040 (GBC), equipped with a Peltier thermocell.
CD measurements were done on a Jasco J-815 dichrograph using a cuvette with a 5 mm path-length, thermostated (with a Peltier effect accessory) at 15 °C. The spectra were recorded over a 200–400 nm range with a 1.00 nm bandwidth, at a scanning speed 100 nm min−1, and a data pitch of 0.2 nm. After 5 spectra were accumulated, the baseline was subtracted and the resultant spectrum was smoothed with a means-movement algorithm using a 5–25 point filter.
Preparative HPLC separation of the oxathiaphospholane monomers was performed using a Shimadzu Prominence HPLC system consisting of an LC-20AP preparative pump (a 2 mL sample loop in the Rheodyne valve was used) and an SPD-M20A UV detector (set at 260 nm) equipped with a 0.2 mm path length preparative cell. A silica gel column Pursuit XRs (10 μm, 250 × 21.2 mm) was eluted at a flow rate of 25 mL min−1.
In search for the condition suitable for HPLC separation of the P-diastereomers of the oxathiaphospholane monomers a Phenomenex Luna 5u Silica column (100 Å; 250 × 10 mm; flow rate 5 mL min−1) and a binary Varian HPLC system (two PrepStar 210 pumps, 25 mL pump heads, a ProStar 320 UV/VIS detector set at 275 nm) were used.
HPLC solvents and reagents of gradient grade (from Sigma-Aldrich, Baker or ChemPur) were used.
All moisture-sensitive operations were carried out under anhydrous argon.
Note: compounds 3, 4a, 4b*, 4c and 4b were obtained according to ref. 28.
Unmodified DNA d(ATGCGCAT, DNA), and (2′-OMe)-RNA ((2′-OMe)-AUGCGCAU, (m)RNA) oligonucleotides were synthesized at a 0.2 μmol scale, using commercially available LCA-CPG supports (Biosearch Technologies, Inc., Petaluma, CA) and standard DNA and 2′-OMe-RNA phosphoramidite monomers (Glen Research, Sterling, VA).
Dimethoxytritylation of glycidol – synthesis of enantiomeric compounds 3
To a solution of (R)-(+)- or (S)-(−)-glycidol 2 (1.0 mL, 15.1 mmol) and Et3N (5.4 mL, 40.7 mmol) in CH2Cl2 (50 mL) solid DMT-Cl (6.45 g, 19 mmol) was added. After 12 h, the reaction mixture was treated with saturated aqueous NaHCO3 (50 mL). The organic layer was separated, dried with MgSO4 and concentrated. The residue was dissolved in EtOAc and the solution was washed with saturated aq. NaHCO3. The solvent was evaporated and the product was isolated chromatographically on a silica gel column eluted with hexane–EtOAc–Et3N (from 95:5:0.1 to 85:15:0.1, v/v/v) to give compounds 3 (typically 5.4 g, 95% yield).
Opening of the oxirane ring in 3 with nucleobases – synthesis of compounds 4a, 4b*, and 4c
A mixture of nucleobase (1 mmol, 126 mg of thymine, 135 mg of adenine, or 215 mg of N-benzoylcytosine) and NaH (60% suspension in mineral oil, ∼10 mg, 0.2 mmol) in DMF (2 mL) was stirred at room temperature for 2 h. A solution of dimethoxytritylated glycidol 3 (340 mg, 0.9 mmol) in DMF (2–3 mL) was added and the resulting mixture was heated at 110 °C for 20 h. The solvent was evaporated under reduced pressure (an oil pump), and the residue was dissolved in EtOAc and loaded on a silica gel column, initially eluted with hexane–EtOAc–Et3N (1:2:0.01, v/v/v), then with EtOAc–Et3N (100:1, v/v).
:1, v/v); FAB MS calc. for C29H30N2O6: 502.57, found: 502.4 (+VE), 501.3 (−VE).:1, v/v); FAB MS calc. for C29H29N5O4: 511.59, found: 512.3 (+VE), 510.3 (−VE).:1, v/v); FAB MS calc. for C35H33N3O6: 591.67, found: 592.1 (+VE), 590.3 (−VE).N-Benzoylation of DMT-GA – synthesis of compound 4b
To a solution of 4b* (511 mg, 1 mmol) in anhydrous pyridine (10 mL) trimethylsilyl chloride (507 μL, 4 mmol) was added. After 2 h stirring at room temperature the mixture was cooled to 0 °C and benzoyl chloride (174 μL, 1.5 mmol) was added dropwise. The mixture was allowed to warm up slowly and was then stirred for 2 h at room temperature. The mixture was cooled to 0 °C and the reaction was quenched with H2O (5 mL, at 0 °C with stirring), and after 15 min conc. aq. NH4OH (10 mL) was added. After 40 min stirring, the mixture was extracted with CH2Cl2 (2 × 50 mL). The organic layer was collected, the solvent was evaporated and the residue was purified chromatographically on a silica gel column eluted with a CHCl3–iPrOH mixture (gradient from 100:0 to 95:5, v/v) containing 0.1% of pyridine. Appropriate fraction (vide infra) was collected and the solvent was evaporated under reduced pressure to yield DMT-GABz: 387 mg (63%, colorless foam); Rf = 0.47 (CHCl3–MeOH 9:1, v/v); FAB MS calc. for C36H33N5O5: 615.7, found: 616.3 (+VE), 614.2 (−VE).
General procedure for synthesis of compounds 5a–d
To a magnetically stirred solution of 4 (1 mmol; 502 mg for 4a, 615 mg for 4b, 591 mg for 4c or 597 mg for 4d) dried overnight under high vacuum and anhydrous N,N-diisopropylethylamine (20% molar excess, 208 μL, 1.2 mmol) in anhydrous methylene chloride (20 mL), 2-chloro-4,4-pentamethylene-1,3,2-oxathiaphospholane (20% molar excess, 178 μL, 1.2 mmol) was added dropwise (under dry argon) at room temperature using a gas-tight Hamilton syringe. The reaction was complete in 5 h and elemental sulfur (64 mg, 2-fold molar excess) was added. Stirring was continued for 24 h and excess sulfur was filtered off. After evaporation of the solvent, the residue was dissolved in chloroform (3 mL) and loaded on a silica gel column. The column was eluted with a mixture of CHCl3 and iPrOH (a gradient from 100:0 to 95:5, v/v) containing 0.1% of pyridine. Appropriate fractions (vide infra) were collected and the solvent was evaporated under reduced pressure.
:1, v/v); HRMS for C36H40N2O7PS2: calc. 707.2015, found 707.2015 (Fig. 4SA, ESI‡); 31P NMR: δ 105.8 ppm, δ 105.5 ppm (no deuterated solvent).:1, v/v); HRMS for C43H43N5O6PS2: calc. 820.2392, found 820.2400 (Fig. 4SB, ESI‡); 31P NMR: δ 105.9 ppm, δ 105.2 ppm (no deuterated solvent).:1, v/v); HRMS for C42H44N3O7PS2: calc. 796.2280, found 796.2287 (Fig. 4SC, ESI‡); 31P NMR: δ 105.6 ppm, δ 105.1 ppm (no deuterated solvent).:1, v/v); HRMS for C40H45N5O7PS2: calc. 802.2498, found 802.2514 Fig. 4SD, ESI;‡ 31P NMR: δ 106.1 ppm, δ 106.5 ppm (no deuterated solvent).Synthesis of (6-chloropurine) Gnucleoside (6)
A mixture of 2-amino-6-chloropurine (169 mg, 1 mmol), glycidol (73 μL, 1.1 mmol) and K2CO3 (23.5 mg, 0.17 mmol) in DMF (5 mL) was stirred at 100 °C for 18 h. The reaction mixture was concentrated in vacuo, the residue was dissolved (not easily) in pyridine and loaded on a silica gel column. Initially the column was eluted with CHCl3 and later with CHCl3–MeOH (1:1, v/v). Fractions containing (6-chloropurine) Gnucleoside (6, Rf = 0.41 CHCl3–MeOH 9:1, v/v) were collected and evaporated to yield 90 mg (37%). FAB MS (CI): calc. for C8H10ClN5O2 243.6, found: 244,1 (+VE).
Conversion of (6-chloropurine) Gnucleoside into the 6-thio derivative 7
To a solution of (6-chloropurine) Gnucleoside 6 (243 mg, 1 mmol) in water (25 mL) Na2S2O3 × 5H2O (1.24 g, 5 mmol) was added and the mixture was refluxed with stirring (ca. 16 h) until no starting Gnucleoside was detected by TLC. The mixture was cooled down to room temperature, and extracted with ethyl acetate (2 × 50 mL). The aqueous phase was concentrated under reduced pressure and the residue was chromatographed on a reverse phase silica gel column using water as eluent to yield 180 mg of 7 (75%); Rf = 0.55 (i-PrOH/conc. NH4OH/H2O 7:1:1, v/v/v); FAB MS (CI): calc. for C8H11N5O2S 241.3 found: 242.0 (+VE).
Conversion of (6-thiopurine) Gnucleoside into compound 8
A solution of 7 (241 mg, 1 mmol) in NH4OH – 30% H2O2 (5:1, v/v, 8 mL) was allowed to stand at room temperature for 5 h. The solvents were removed under reduced pressure. The obtained crude residue was purified by crystallization from H2O to give the corresponding Gnucleoside 8 as white solid material (155 mg, 69%). Rf = 0.42 (CHCl3–MeOH 9:1, v/v); FAB MS: calc. for C8H11N5O3 225.2, found: 226.2 (+VE), 224.1 (−VE).
Iso-butyrylation of GG – synthesis of compound 9
To a solution of 8 (225 mg, 1 mmol) in anhydrous pyridine (30 mL) trimethylsilyl chloride (1.01 mL, 8 mmol) was added at 0 °C. The reaction mixture was stirred at 0 °C for 15 min, followed by 40 min at room temperature. The mixture was cooled to 0 °C and iso-butyryl anhydride (415 μL, 2.5 mmol) was added dropwise. The mixture was allowed to warm slowly to room temperature and was stirred for 2 h. The reaction was stopped by addition of H2O (2 mL) at 0 °C. After 10 min, conc. NH4OH (2 mL) was added and the mixture was left for 15 min. The volatile components were removed in vacuo. The residue was dissolved in MeOH, loaded onto a silica gel column and the column was eluted with EtOAc, EtOAc–MeOH (9:1, v/v), and finally with EtOAc–MeOH (9:2, v/v) to yield 239 mg (81%) of GGiBu. Rf = 0.10 (CHCl3–MeOH 9:1, v/v); FAB MS: calc. for C12H17N5O4 295.3, found: 296.1 (+VE), 294.2 (−VE).
Dimethoxytritylation of 9 – synthesis of DMT-GG (compound 4d)
To a solution of 9 (295 mg, 1 mmol) in anhydrous pyridine (4 mL) solid DMT-Cl (406 mg, 1.2 mmol) was added at room temperature. After 2 h, the reaction mixture was concentrated in vacuo. The residue was purified by chromatography on a silica gel column, first eluted with hexane–EtOAc–Et3N (1:2:0.01, v/v/v), then with EtOAc–Et3N (100:1, v/v), and finally with EtOAc–MeOH–Et3N (25:1:0.01, v/v/v). The fraction containing DMT-GGiBu (Rf = 0.46 (CHCl3–MeOH 9:1, v/v)) was concentrated in vacuo to yield 430 mg (colorless foam, 72%); FAB MS: calc. for C33H35N5O6 597.7 found: 296,1 ([M-DMT]+; +VE).
HPLC separation of P-diastereomers of GNA oxathiaphospholane monomers 5a–d
A preparative HPLC silica gel column (Pursuit XRs 10 μm, 250 × 21.2 mm) was equilibrated with a given eluent mixture (see Table 1) at a flow rate 25 mL min−1 for 20 minutes. A solution of 5 (100 mg; 300 mg for 5a) in ethyl acetate (1.5 mL, with 1% TEA) was filtered through a membrane filter (0.2 μm pores, organic solvent resistant) and the filtrate was applied onto the column. The column was eluted at the flow rate of 25 mL min−1 and baseline separation of P-diastereomers was achieved. The collected fractions were concentrated using a rotary evaporator with a “cold finger” condenser filled with isopropyl alcohol/dry ice coolant, with the temperature of a water bath not exceeding 30 °C. Pure P-diastereomers were recovered in ca. 90% and their diastereomeric purity was confirmed by 31P NMR analysis (CDCl3, an estimated limit of detection of a minor component 0.2%). They gave satisfactory 1H NMR, 31P NMR, 13C NMR and MALDI-TOF mass spectra.5a OTP-GT fast: Rf = 0.68 (CHCl3–MeOH 9:1, v/v); Rt = 19.2 min (EtOAc–hexane 50:50, v/v); MALDI TOF MS: calc. for C36H41N2O7PS2 708.8 found: 747.6 (+); 708.8 + K+); 31P NMR δ (CDCl3, ppm) 105.6; 1H NMR δ (CDCl3, ppm) 8.36 (1H, N3–H), 7.43 (1H, C6–H), 7.40–6.79 (13H, DMT), 5.13–5.06 (1H, C2′–H), 4.16–4.15 (2H, C1′–H), 4.08–4.05 (2H, P–O–CH2C–S), 3.75 (6H, 2 × OCH3), 3.33–3.25 (2H, C3′–H), 1.89–1.20 (3H, C5–CH3; 10H, –(CH2)5–“spiro”); 13C NMR δ (CDCl3, ppm) 162.6, 157.3, 143.0, 140.0, 134.2, 128.7, 126.7, 126.6, 125.7, 111.9, 78.0, 76.4, 75.7, 75.1, 62.2, 53.9, 36.0, 35.3, 23.9, 22.7, 22.2, 11.0. 5a OTP-GT slow: Rf = 0.60 (CHCl3–MeOH 9:1, v/v); Rt = 21.5 min (EtOAc–hexane 50:50, v/v); MALDI TOF MS: calc. for C36H41N2O7PS2 708.8 found: 747.6 ((+); 708.8 + K+); 31P NMR δ (CDCl3, ppm) 106.1; 1H NMR δ (CDCl3, ppm) 8.37 (1H, N3–H), 7.40 (1H, C6–H), 7.32–6.79 (13H, DMT), 5.09–5.02 (1H, C2′–H), 4.16–4.11 (2H, C1′–H), 4.08–4.05 (2H, P–O–CH2C–S), 3.77 (6H, 2 × OCH3), 3.38–3.19 (2H, C3′–H), 1.86–1.20 (3H, C5–CH3; 10H, –(CH2)5–“spiro”); 13C NMR δ (CDCl3, ppm) 162.6, 157.3, 149.5, 139.8, 128.7, 126.8, 126.7, 111.9, 76.4, 75.7, 75.1, 53.9, 35.7, 23.9, 22.4, 1.0. 5b OTP-GABz fast: Rf = 0.72 (CHCl3–MeOH 9:1, v/v); Rt = 18 min (EtOAc–hexane 45:55, v/v); MALDI TOF MS calc. 821.9 found 822.6 (+); 31P NMR δ (CDCl3, ppm) 105.8; 1H NMR δ (CDCl3, ppm) 8.95 (1H, NHCO), 8.75 (1H, C8–H), 8.02 (1H, C2–H), 7.54–6.78 (18H, DMT, Bz), 5.22–5.15 (1H, C2′–H), 4.57–4.54 (2H, C1′–H), 4.08–3.98 (2H, P–O–CH2C–S), 3.77 (6H, 2 × OCH3), 3.32–3.30 (2H, C3′–H), 1.61–1.23 (10H, –(CH2)5–“spiro”); 13C NMR δ (CDCl3, ppm) 155.0, 140.5, 128.8, 126.3, 124.2, 113.7, 109.4, 75.8, 51.2, 21.2, 20.0, 19.6. 5b OTP-GABz slow: Rf = 0.58 (CHCl3–MeOH 9:1, v/v); Rt = 23 min (EtOAc–hexane 45:55, v/v); MALDI TOF MS calc. 821.9 found 822.6 (+); 31P NMR δ (CDCl3, ppm) 106.6; 1H NMR δ (CDCl3, ppm) 8.95 (1H, NHCO), 8.75 (1H, C8–H), 8.03 (1H, C2–H), 7.54–6.79 (18H, DMT, Bz), 5.23–5.19 (1H, C2′–H), 4.56–4.55 (2H, C1′–H), 4.09–4.03 (2H, P–O–CH2C–S), 3.77 (6H, 2 × OCH3), 3.31–3.30 (2H, C3′–H), 1.56–1.21 (10H, –(CH2)5–“spiro”); 13C NMR δ (CD3CN, ppm) 157.5, 143.4, 142.9, 134.1, 128.8, 126.7, 116.0, 111.8, 78.1, 62.1, 53.6, 34.7, 23.5, 22.3. 5c OTP-GCBz fast: Rf = 0.70 (CHCl3–MeOH 9:1, v/v); Rt = 27 min (EtOAc–hexane 55:45, v/v); MALDI TOF MS calc. 797.9 found 799.0 (+); 31P NMR δ (CDCl3, ppm) 105.1; 1H NMR δ (CDCl3, ppm) 8.70 (1H, NHCO), 7.88–7.85 (2H, C6–H, C5–H), 7.60–6.79 (18H, DMT, Bz), 5.26–5.22 (1H, C2′–H), 4.20–4.15 (2H, C1′–H), 4.10–4.04 (2H, P–O–CH2C–S), 3.76 (6H, 2 × OCH3), 3.43–3.28 (2H, C3′–H), 1.61–1.20 (10H, –(CH2)5–“spiro”); 13C NMR δ (CDCl3, ppm) 161.0, 157.3, 148.6, 143.0, 134.2, 134.0, 131.9, 128.7, 127.8, 126.8, 126.6, 112.0, 85.1, 78.1, 67.7, 62.2, 59.1, 53.9, 50.9, 36.2, 35.2, 23.9, 22.7, 22.2, 12.9. 5c OTP-GCBz slow: Rf = 0.58 (CHCl3–MeOH 9:1, v/v); Rt = 31.2 min (EtOAc–hexane 55:45, v/v); MALDI TOF MS calc. 797.9 found 799.1 (+); 31P NMR δ (CDCl3, ppm) 105.7; 1H NMR δ (CDCl3, ppm) 8.70 (1H, NHCO), 7.84–7.82 (2H, C6–H, C5–H), 7.60–6.80 (18H, DMT, Bz), 5.30–5.21 (1H, C2′-H), 4.33–4.26 (2H, C1′–H), 4.18–4.08 (2H, P–O–CH2C–S), 3.77 (6H, 2 × OCH3), 3.43–3.21 (2H, C3′–H), 1.61–1.20 (10H, –(CH2)5–“spiro”); 13C NMR δ (CDCl3, ppm) 172.2, 157.3, 148.4, 131.9, 128.8, 127.7, 126.8, 126.6, 126.2, 111.9, 77.9, 76.4, 75.7, 75.1, 53.9, 50.8, 35.5, 23.9, 22.5, 22.4. 5d OTP-GGiBu fast: Rf = 0.65 (CHCl3–MeOH 9:1, v/v); Rt = 13.5 min (EtOAc–hexane 85:15, v/v); MALDI TOF MS calc. 803.9 found 804.6 (+); 31P NMR δ (CDCl3, ppm) 106.1; 1H NMR δ (CDCl3, ppm) 11.89 (1H, NHCO), 8.34 (1H, N1–H), 7.58 (1H, C8–H), 7.24–6.76 (13H, DMT), 5.17–5.10 (1H, C2′–H), 4.41–4.31 (2H, C1′–H), 4.08–3.94 (2H, P–O–CH2C–S), 3.76 (6H, 2 × OCH3), 3.17–3.15 (2H, C3′–H), 2.53–2.50 (1H, COCH), 2.02–1.50 (10H, –(CH2)5–“spiro”), 1.22–1.19 (6H, 2 × CH3); 13C NMR δ (CDCl3, ppm) 177.0, 157.4, 145.8, 143.0, 138.2, 134.1, 128.7, 126.7, 126.6, 125.7, 111.9, 85.3, 76.4, 75.7, 75.1, 67.6, 61.3, 54.0, 35.2, 23.8, 22.4, 17.7. 5d OTP-GGiBu slow: Rf = 0.50 (CHCl3–MeOH 9:1, v/v); Rt = 17 min (EtOAc–hexane 85:15, v/v); MALDI TOF MS calc. 803.9 found 804.6 (+); 31P NMR δ (CDCl3, ppm) 106.5; 1H NMR δ (CDCl3, ppm) 11.83 (1H, NHCO), 7.82 (1H, N1–H), 7.58 (1H, C8–H), 7.30–6.80 (13H, DMT), 4.18–5.16 (1H, C2′–H), 4.41–4.31 (2H, C1′–H), 4.08–3.94 (2H, P–O–CH2C–S), 3.78 (6H, 2 × OCH3), 3.19–3.16 (2H, C3′-H), 2.61–2.48 (1H, COCH), 2.03–1.58 (10H, –(CH2)5–“spiro”), 1.25–1.22 (6H, 2 × CH3); 13C NMR δ (CDCl3, ppm) 177.0, 157.4, 154.2, 147.2, 145.8, 143.0, 138.2, 134.0, 128.7, 126.7, 125.7, 111.9, 85.3, 78.1, 76.3, 75.7, 75.1, 54.0, 35.2, 23.8, 22.3, 17.7.
Attempt at detritylation of compound 5a
To a solution of 5a (100 mg) in CH3CN (10 mL) a catalytic amount of NaHSO4/SiO2 (prepared according to ref. 38) was added. The suspension was stirred for 30 minutes and TLC analysis showed no substrate remaining. The product did not migrate on a TLC plate when a mixture CHCl3:MeOH (9:1) was applied as an eluent. It was isolated using a CHCl3:MeOH gradient 6:4→3:7 (v/v). MS analysis revealed the presence of a band at m/z 277. (Fig. 6S, ESI‡).
General procedure for synthesis of compound 11
A given P-diastereoisomer (fast or slow eluting) of OTP-GN′ 5 (0.04 mmol; 28 mg for 5a, 33 mg for 5b, 32 mg for 5c, or 32 mg for 5d) and 3′-O-acetyl-thymidine (2-fold molar excess; 23 mg, 0.08 mmol), were dried over P2O5 in a vacuum desiccator for 24 h, and dissolved in anhydrous acetonitrile (∼1 mL) under dry argon. To the solution 1,8-diazabicyclo-[5.4.0]undec-7-ene (DBU, 1.1 molar equivalent) was added and after 2 h the reaction was complete to give GN′PSTAc 10. The formation of 10 was confirmed by 31P NMR (in CH3CN, no deuterated solvent – the recorded chemical shifts are not precisely reproducible); SC-5 substrates: DMT-GTPSTAc, δ 54.6 ppm (from fast 5a), δ 54.9 ppm (from slow); DMT-GABzPSTAc, δ 55.6 ppm (from fast 5b), δ 55.9 ppm (from slow); DMT-GCBzPSTAc, δ 56.2 ppm (from fast 5c), δ 56.8 ppm (from slow); DMT-GGPSTAc, δ 55.6 ppm (from fast 5d), δ 56.7 ppm (from slow); RC-5 substrates: DMT-GTPSTAc, δ 56.5 ppm (from fast 5a), δ 56.3 ppm (from slow); DMT-GABzPSTAc δ 55.8 ppm (from fast 5b), δ 56.0 ppm (from slow); DMT-GCBzPSTAc δ 56.7 ppm (from fast 5c), δ 56.3 ppm (from slow). The mixture was concentrated under reduced pressure and treated (in a tightly closed vessel) with concentrated ammonia solution (2 mL; 10a for 2 h at room temperature, 10b, 10c and 10d for 15 h at 55 °C). After evaporation, the DMT moiety was removed with 50% aqueous acetic acid for 1.5 h at room temperature, followed by evaporation under reduced pressure. The resultant GNPST 11a–d were isolated using RP-HPLC, a gradient of 0.1 M TEAB, pH 7.3 to 38% CH3CN in 0.1 M TEAB over 20 minutes, elution at 1.0 mL min−1: 11a, b, d: an Alltima C18 column, 250 × 4.6 mm, 5 μm; 11c: a Clarity C18 column, 250 × 4.6 mm, 5 μm. Isolated 11 were characterized by MALDI TOF MS and Rt.From SC-5a–d substrates: 11a GTPST: Rt1 = 18.5 min, Rt2 = 19.5 min; MALDI TOF MS: calc. for C18H24N4O10PS 519.45, found 519.0 and 519.0. 11b GAPST: Rt1 = 16.5 min, Rt2 = 17.0 min; MALDI TOF MS: calc. for C18H25N7O8PS 530.48, found 528.0 and 528.0. 11c GCPST: Rt1 = 15.5 min, Rt2 = 19.0 min; MALDI TOF MS: calc. for C17H23N5O9PS 504.44, found 504.2 and 504.1. 11d GGPST: Rt1 = 18.7 min, Rt2 = 18.5 min; MALDI TOF MS: calc. for C18H23N7O9PS 544.46, found 544.2 and 544.1. From RC-5a–c substrates: 11a GTPST: Rt1 = 19.9 min, Rt2 = 21.0 min; MALDI TOF MS: calc. for C18H24N4O10PS 519.45, found 519.3 and 519.0. 11b GAPST: Rt1 = 17.9 min, Rt2 = 17.6 min; MALDI TOF MS: calc. for C18H25N7O8PS 530.48, found 528.0 and 528.0. 11c GCPST: Rt1 = 15.6 min, Rt2 = 18.2 min; MALDI TOF MS: calc. for C17H23N5O9PS 504.44, found 504.1 and 506.2 (in a positive ions mode).
Hydrolysis of 11 with svPDE
The reaction mixture (20 μL) containing 25 mM Tris–Cl (pH 8.5), 5 mM MgCl2 buffer, 10 μL of the svPDE suspension, and GNPST (11, 0.2 OD260) was incubated for 24 h at 37 °C. Then, the sample was heat-denatured (for 1 min at 95 °C) and analyzed by HPLC, using the conditions specified above.Hydrolysis of 11 with nuclease P1
The reaction mixture (20 μL) containing 100 mM Tris–Cl (pH 7.2), 1 mM ZnCl2, 10 μL of nuclease P1 suspension, and GNPST (11, 0.2 OD) was incubated for 24 h at 37 °C. Then, the sample was heat-denatured (for 1 min at 95 °C) and analyzed by HPLC, using the conditions specified above.Chimeric oligonucleotides 14, 15, 16 and 17 – synthesis and purification
The chimeric oligonucleotides 14–17 were synthesized manually at a 1 μmol scale. A published protocol for synthesis utilizing OTP monomers was used,37 except that the coupling of the OTP-GN′ monomers was extended to 20 min. The first nucleoside unit was anchored to the support by a sarcosinyl linker.36 In the cycles where OTP-GN′ monomers 5 were delivered, two consecutive coupling steps were executed (20 mg + 20 mg), separated by washing with acetonitrile and drying with a stream of argon. The coupling efficiency was controlled by measurement of the DMT cation absorption decay at 504 nm. All oligomers were purified by reverse phase HPLC (conditions as above) and analyzed by MALDI-TOF MS. 14: (RP)-PS-GATGGCGGCGAT; MALDI TOF MS calc. 2353 found 2352.8. 15: (SP)-PS-GATGGCGGCGAT; calc. 2353 found 2352.9. 16: (RP)-PS-AGTGGCGGCAT; calc. 2395 found 2394.5. 17: (SP)-PS- AGTGGCGGCAT; calc. 2395 found 2394.1.Sample preparation and melting UV profile recording
The concentration of oligomers was determined by UV absorbance at their λmax in water, using the extinction coefficients calculated by the standard method.39 The samples were then lyophilized and re-dissolved in pH 7.2 buffer containing 10 mM Tris–HCl, 100 mM NaCl, and 10 mM MgCl2. For melting profiles, the oligonucleotides were mixed at 2.0 μM concentration each. (This concentration is also suitable for CD measurements). First, an annealing step was performed from 40 °C to 5 °C with a temperature gradient of 1°C min−1, followed by a melting step, carried out to 40 °C (or to 85 °C for DNA, (m)RNA and for mixtures DNA/(m)RNA, 14/(m)RNA and 16/(m)RNA) with a gradient of 0.5°C min−1. The measurements were done at 260 nm, using a 60 s cycle time and a 2 s integration time. The melting temperatures (see Tables 1 and 2S‡) were calculated using the first order derivative method.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Centre of Science, Poland, grant UMO-2011/03/B/ST5/02670 (to A. T.-A.) and by statutory funds of CMMS PAS, Łódź, Poland. The authors are grateful to Dr Ewelina Wielgus of CMMS PAS for the recording of mass spectra.Notes and references
- O. Khakshoor and E. T. Kool, Chem. Commun., 2011, 47, 7018 RSC.
- N. Michelotti, A. Johnson-Buck, A. Manzo and N. Walter, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 139 Search PubMed.
- N. C. Seeman, Nature, 2003, 421, 427 CrossRef PubMed.
- D. H. Appella, Curr. Opin. Chem. Biol., 2009, 13, 687 CrossRef PubMed.
- V. B. Pinheiro and P. Holliger, Curr. Opin. Chem. Biol., 2012, 16, 245 CrossRef PubMed.
- A. J. A. Cobb, Org. Biomol. Chem., 2007, 5, 3260 RSC.
- F. Eckstein, Tetrahedron Lett., 1967, 1157 CrossRef.
- J. Micklefield, Curr. Med. Chem., 2001, 8, 1157 CrossRef PubMed.
- R. Corradini, S. Sforza, T. Tedeschi and R. Marchelli, Chirality, 2007, 19, 269 CrossRef PubMed.
- P. A. Frey and R. D. Sammons, Science, 1985, 228, 541 CrossRef PubMed.
- W. J. Stec, B. Karwowski, M. Boczkowska, P. Guga, M. Koziołkiewicz, M. Sochacki, M. W. Wieczorek and J. Błaszczyk, J. Am. Chem. Soc., 1998, 120, 7156 CrossRef.
- K. Jastrzębska, A. Maciaszek, R. Dolot, G. Bujacz and P. Guga, Org. Biomol. Chem., 2015, 13, 10032 RSC.
- W. J. Stec, A. Grajkowski, M. Koziołkiewicz and B. Uznański, Nucleic Acids Res., 1991, 19, 5883 CrossRef PubMed.
- L. Benimetskaya, J. L. Tonkinson, M. Koziołkiewicz, B. Karwowski, P. Guga, R. Zelser, W. J. Stec and C. A. Stein, Nucleic Acids Res., 1995, 23, 4239 CrossRef PubMed.
- A. Krieg, P. Guga and W. J. Stec, Oligonucleotides, 2003, 13, 491 CrossRef PubMed.
- T. Inagawa, H. Nakashima, B. Karwowski, P. Guga, W. J. Stec, H. Takeuchi and H. Takaku, FEBS Lett., 2002, 528, 48 CrossRef PubMed.
- V. B. Pinheiro and P. Holliger, Trends Biotechnol., 2014, 32, 321 CrossRef PubMed.
- V. Kumar and V. Kesavan, RSC Adv., 2013, 3, 19330 RSC.
- I. Anosova, E. A. Kowal, M. R. Dunn, J. C. Chaput, W. D. Van Horn and M. Egli, Nucleic Acids Res., 2016, 44, 1007 CrossRef PubMed.
- E. Meggers and L. Zhang, Acc. Chem. Res., 2010, 43, 1092 CrossRef PubMed.
- M. K. Schlegel, D. J. Foster, A. V. Kel'in, I. Zlatev, A. Bisbe, M. Jayaraman, J. G. Lackey, K. G. Rajeev, K. Charissé, J. Harp, P. S. Pallan, M. A. Maier, M. Egli and M. Manoharan, J. Am. Chem. Soc., 2017, 139, 8537 CrossRef PubMed.
- M. Boczkowska, P. Guga and W. J. Stec, Biochemistry, 2002, 41, 12483 CrossRef PubMed.
- M. Boczkowska, P. Guga, B. Karwowski and A. Maciaszek, Biochemistry, 2000, 39, 11057 CrossRef PubMed.
- M. B. Laursen, M. M. Pakula, S. Gao, K. Fluiter, O. R. Mook, F. Baas, N. Langklær, S. L. Wengel, J. Wengel, J. Kjems and J. B. Bramsen, Mol. BioSyst., 2010, 6, 862 RSC.
- A. Alagia, M. Terrazas and R. Eritja, Molecules, 2015, 20, 7602 CrossRef PubMed.
- B. T. Karwowski, Phys. Chem. Chem. Phys., 2015, 17, 21507 RSC.
- W. Lan, Z. Hu, J. Shen, C. Wang, F. Jiang, H. Liu, D. Long, M. Liu and C. Cao, Sci. Rep., 2016, 6, 25737 CrossRef PubMed.
- L. Zhang, A. Peritz, P. J. Carroll and E. Meggers, Synthesis, 2006, 4, 645 Search PubMed.
- Houben-Weyl Methods of Organic Chemistry, ed. E. Schaumann, 4th edn, Supplement, 1997, vol. E 9b/2, p. 485 Search PubMed.
- S. G. Yang, D. H. Lee and Y. H. Kim, Heteroat. Chem., 1997, 8, 435 CrossRef.
- B. Das, G. Mahender, V. S. Kumar and N. Chowdhury, Tetrahedron Lett., 2004, 45, 6709 CrossRef; V. Janardhana Rao, K. Mukkanti, R. Shankar, N. A. Vekariya and A. Islam, Rasayan J. Chem., 2013, 6, 15 Search PubMed.
- P. Guga, B. Karwowski, D. Błaziak, M. Janicka, A. Okruszek, B. Rębowska and W. J. Stec, Tetrahedron, 2006, 62, 2698 CrossRef.
- F. Eckstein, Annu. Rev. Biochem., 1958, 54, 367 CrossRef PubMed.
- A. D. Griffiths, B. V. L. Potter and I. C. Eperon, Nucleic Acids Res., 1987, 15, 4145 CrossRef PubMed.
- A. Tomaszewska, P. Guga and W. J. Stec, Chirality, 2011, 23, 237 CrossRef PubMed.
- T. Brown, C. E. Pritchard, G. Turner and S. A. Salisbury, J. Chem. Soc., Chem. Commun., 1989, 891 RSC.
- P. Guga and W. J. Stec, in Current Protocols in Nucleic Acid Chemistry, ed. S. L. Beaucage, D. E. Bergstrom, G. D. Glick and R. A. Jones, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2003, p. 4.17.1 Search PubMed.
- R. K. Kannasani, V. V. Satyanarayana Peruri and S. R. Battula, Chem. Cent. J., 2012, 6, 136 Search PubMed.
- T. Brown and D. J. S. Brown, in Oligonucleotides and Analogues: A Practical Approach, ed. F. Eckstein, IRL Press, Oxford, 1991, p. 20 Search PubMed.
- Y.-C. Zhang, J. Liang, P. Lian, Y. Han, Y. Chen, L. Bai, Z. Wang, J. Liang, Z. Deng and Y.-L. Zhao, J. Phys. Chem. B, 2012, 116, 10639 CrossRef PubMed.
- P. Guga, M. Boczkowska, M. Janicka, A. Maciaszek, B. Nawrot, S. Antoszczyk and W. J. Stec, Pure Appl. Chem., 2006, 78, 993 Search PubMed.
- P. Guga, M. Janicka, A. Maciaszek, B. Rebowska and G. Nowak, Biophys. J., 2007, 93, 3567 CrossRef PubMed.
- P. Guga, M. Boczkowska, M. Janicka, A. Maciaszek, S. Kuberski and W. J. Stec, Biophys. J., 2007, 92, 2507 CrossRef PubMed.
- A. P. Guzaev and M. Manoharan, J. Org. Chem., 2001, 66, 1798 CrossRef PubMed.
Footnotes |
| † Dedicated to the memory of our great friend, Dr Andrzej Okruszek. |
| ‡ Electronic supplementary information (ESI) available: Analytical data for 5a–d, MS data for cGTMPS, preparation of 11c and RP-HPLC profiles recorded after its hydrolysis, RP HPLC profiles and MALDI-TOF MS data for (GUPS)11dA treated with DBU and for 17. See DOI: 10.1039/c8ra05568h |
| § Theoretical studies on the PS-DNA energy alteration suggest that RP-phosphorothioate units destabilize the B-helical structure by 2.7 ± 3.4 kcal mol−1, while SP-counterparts stabilize it by −1.4 ± 2.4 kcal mol−1, compared to the reference B-DNA helix.40 |
| ¶ There is only one known exception, where homopurine (RP-PS)-DNA oligomers form thermally highly stable parallel triplexes (RNA:PS-DNA:RNA)41 and parallel duplexes (PS-DNA:RNA)42. They are supposed to be stabilized not only by the Hoogsteen interactions but also by water bridges, which span the O-2 atom in pyrimidines and the sulfur atom of the phosphorothioate moiety.43 |
| || Note, that priority of substituents around the stereogenic phosphorus atom in GAPST and APST (generally in GNPSN and NPSN) is the same. |
| ** Because the coupling step performed with an oxathiaphospholane monomer leads to the phosphodiester product (see Scheme 4), this methodology cannot be combined with the most frequently used phosphoramidite method of synthesis of DNA, because the mandatory oxidation step (typically with I2/H2O/pyridine) would destroy the phosphorothioate linkages present in a growing oligomer. Also the phosphoramidite based elongation of oligomer with the intended safe use of sulfurizing reagents (to convert the phosphite intermediate into the phosphorothioate triester) is difficult because the already assembled anionic phosphorothioate backbone interferes with the very sensitive acid/base equilibrium operating during the 1H-tetrazole activation step.44 |
| †† For simplicity, in the sequences of chimeric DNA/GNA oligonucleotides 14–17 the letters A, C and G are used instead of dA, dC and dG, respectively. |
| This journal is © The Royal Society of Chemistry 2018 |