
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the interaction mechanism between tiagabine hydrochloride and two serum albumins†
Weiling Zhuoa,
Xilin Pengb and
Xiang Lin *a
*a
aSchool of Chemical Engineering, Sichuan University, Chengdu 610065, People's Republic of China. E-mail: linxiang@scu.edu.cn; Fax: +86 028 85990908; Tel: +86 02885990908
bSinopharm Chuankang Pharmaceutical Co., Ltd., Chengdu 611731, People's Republic of China
First published on 11th July 2018
Abstract
Tiagabine hydrochloride (TGB) is a widely used anticonvulsive drug for the treatment of epilepsy. To better understand the interactions of TGB with plasma proteins, human serum albumin (HSA) and bovine serum albumin (BSA) were selected as model proteins. TGB slightly increased thermal stability of the proteins as confirmed by VP-capillary differential scanning calorimetric (DSC) measurements. Isothermal titration calorimeter (ITC) results showed that TGB could be combined with HSA and BSA moderately, which was also corroborated by fluorescence analysis. Besides, the thermodynamic parameters (ΔH > 0, ΔS > 0) indicated that hydrophobic forces played a major role in the formulation of TGB–HSA and TGB–BSA complexes. Moreover, the main binding sites of TGB to HSA and BSA were also examined by classical fluorescent probe (dansylsarcosine and dansylamide) experiments, showing that TGB and dansylsarcosine competitively interacted with HSA and BSA at the same binding sites. Additionally, TGB had no obvious effect on the conformation change of HSA and BSA as indicated by spectroscopic analyses. This study provides useful information about the interaction mechanism of TGB and serum albumins, which could help to better utilize TGB in biomedical field.
1. Introduction
Tiagabine hydrochloride (Fig. 1) is a classical anticonvulsive drug used for treating epileptics in clinics, and is widely used in the United States, Europe and other countries.1,2 It selectively increased the concentration of γ-aminobutyric acid in presynaptic neurons and glial cells for effective therapy. Besides, it has been utilized to treat anxiety disorders, panic disorders, posttraumatic stress disorders, as well as neuropathic pain. But, the U.S. Food and Drug Administration (FDA) found it has potential risk in causing patients to generate suicidal thoughts or behavior.3,4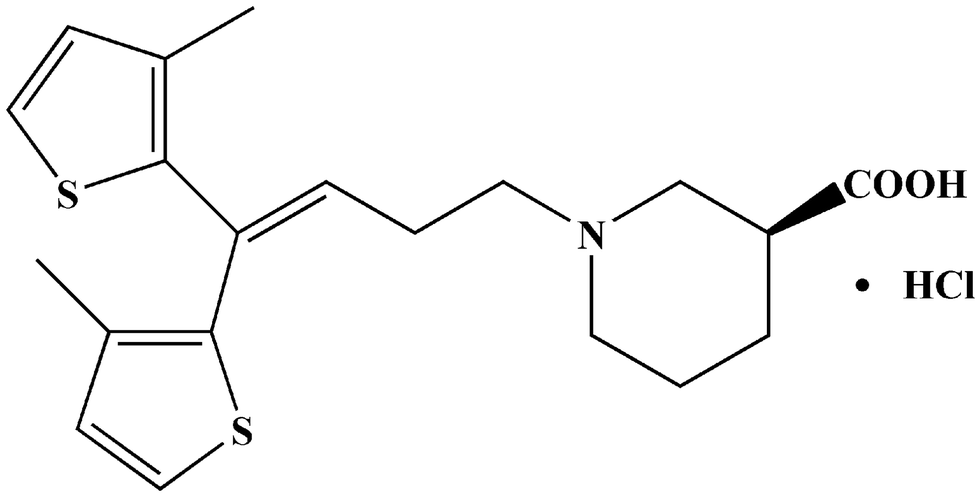 | ||
| Fig. 1 Chemical structure of TGB. | ||
Research shows that tiagabine could be extensively (approximately 96%) bound to plasma proteins. However, there is little information about how tiagabine binds with proteins, for instance, binding mechanism, binding sites, etc.5,6 TGB binding with plasma proteins may affect its absorption, distribution, metabolism, excretion, toxicity, and stability in vivo.7 Serum albumin is the most abundant protein in plasma, and plays an important role in the transportation, deposition, and metabolism of numerous types of exogenous and endogenous compounds.8 This study chose human serum albumin (HSA) and bovine serum albumin (BSA) as model proteins to study their interactions with TGB. The BSA and HSA display approximately 76% sequence homology, and with different numbers and types of amino acids. HSA and BSA contain 585 and 583 amino acid residues, respectively, while HSA has only one tryptophan (Trp-214) residue, and BSA has two tryptophan (Trp-134, 213) residues.9,10
In this study, several advanced analysis methods were performed to investigate TGB–HSA and TGB–BSA interactions. As many physiological functions of proteins depend on their intrinsic structure, a VP-capillary DSC measurement was performed to determine thermal stability of HSA and BSA when they interacted with or without TGB. Later, ITC was used to determine TGB-serum albumin binding ability, and fluorescence spectroscopy was utilized as an alternative method to further analyze their interactions. Site marker competitive experiments were conducted to determine the potential main binding sites of TGB to HSA and BSA. Thereafter, the optimal binding models of TGB–HSA complex and TGB–BSA complex were simulated through molecular docking. Three-dimensional (3D) fluorescence spectroscopy and circular dichroism (CD) spectroscopy were adopted to monitor structural changes of the proteins affected by TGB. This study provides in-depth insights into the mechanism of the TGB–serum albumin binding interaction, which may be helpful for improving applications of TGB in clinical, and standardized screening in pharmaceutical firms and clinical research.11,12
2. Materials and methods
2.1 Chemicals and reagents
HSA (fatty acid-free) was purchased from Sigma-Aldrich Company (St. Louis, USA). BSA (fatty acid-free, nuclease, and protease-free) was obtained from EMD Millipore Company (Billerica, USA). TGB (>98% purity index) was acquired from Tokyo Chemical Industry Company (Tokyo, Japan). Dansylamide and dansylsarcosine were purchased from J&K Scientific Ltd. (Beijing, China). Stock solutions of HSA, BSA, and TGB were prepared in phosphate buffer saline (PBS; 0.1 M, pH 7.40). The concentrations of HSA and BSA in buffer were corrected by the extinction coefficients of 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 M−1 cm−1 and 44720 M−1 cm−1, respectively.13
700 M−1 cm−1 and 44720 M−1 cm−1, respectively.13
2.2 Differential scanning calorimetry measurements
The VP-Capillary DSC measurement was run by means of a MicroCal VP-Capillary DSC (Malvern, Worcestershire, UK). Samples were prepared in PBS (pH 7.4) and concentrations of HSA and BSA were maintained at 40 μM, whereas the molar ratios of TGB to HSA/BSA were 0:1 and 1:1. PBS was also used as a reference buffer. All the solutions were degassed properly under mild vacuum prior to the experiments. Scans were collected from 298 K to 363 K at a scan rate of 200 K h−1. Data were analyzed by using Origin 7.0 software provided by the instrument manufacturer to obtain the temperature at the midpoint of the unfolding transition (Tm).
2.3 Isothermal titration calorimetry measurements
ITC measurements were carried out on a Microcal ITC200 (Malvern, Worcestershire, UK) with an active cell volume of 185 μL. All titrations were performed in PBS at a stirring rate of 750 rpm and using a 50 μL titrating syringe at 298 K. An automated sequence of 20 injections was used, with each of 2 μL ligand (TGB) titration into the sample cell containing serum albumin. The intervals between each injection were spaced at 1.5 min for HSA samples and at 2 min for BSA samples to allow complete equilibration, which was performed with the equivalence point at the area midpoint of the titration. Titrations of TGB into buffer were performed as shown in Fig. S1.† The heat released or absorbed upon each injection was measured, and the data were plotted as integrated quantities. The resulting data were fitted to a one set of sites model and analyzed using MicroCal Origin 7.0 software, which was supplied by the instrument manufacturer.2.4 Fluorescence spectroscopy analyses
Fluorescence analyses were conducted in a 1 cm quartz cell-using a Cary Eclipse fluorescence spectrophotometer (Varian, California, USA) at 298 K. In a TGB–HSA system, the concentration of HSA was maintained at 2.4 μM, whereas the concentrations of TGB varied from 0 μM to 28.8 μM with a 4.8 μM gradient growth based on preliminary experiments. In the TGB–BSA system, the concentration of BSA was 0.7 μM, and the concentration of TGB changed from 0 μM to 16.8 μM with a 2.4 μM gradient growth. Fluorescence spectra were obtained using 5/10 nm (excitation/emission) slit widths. The excitation wavelength was set at 280 nm, and the emission range was from 300 to 450 nm unless specified otherwise. All emission intensities were corrected for inner filter effect, according to the following equation:14
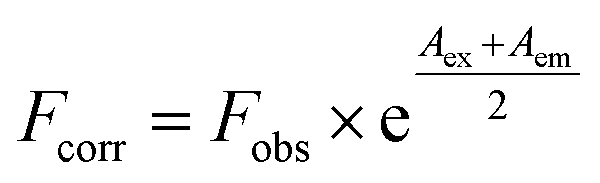 | (1) |
3D fluorescence spectroscopy was performed at an excitation wavelength range from 200 to 400 nm in increments of 5 nm. Emission spectra were monitored between 200 and 450 nm. Concentrations of HSA and BSA were maintained at 2.4 μM and 0.7 μM, respectively. The molar ratios of TGB to HSA/BSA were 0:1 and 6:1, respectively.
Site marker competitive experiments were conducted using dansylsarcosine and dansylamide as probes at 298 K. All solutions were investigated at an exitation wavelength of 350 nm. The emission spectra were recorded in the range of 380–670 nm.
Fluorescence lifetime was measured on a Jobin Yvon Fluorolog-3 spectrofluorometer (Horiba, LesUlis, FRA). Time-resolved HSA/BSA fluorescence quenching by TGB was recorded by maintaining the excitation wavelength and emission wavelength at 280 and 345 nm, respectively. The concentration of HSA was fixed at 2.4 μM, and the concentration of BSA was 0.7 μM. The molar ratios of TGB to HSA/BSA were 0:1, and 12:1, respectively. Data were analyzed through the tail fitting method. The fit qualities were assessed based on χ2 values and residuals. The amplitude-weighted lifetime (〈τ〉) for biexponential iterative fitting was calculated based on decay times and pre-exponential factors (α) in accordance with the following equation:15
| 〈τ〉 = τ1α1 + τ2α2 + τ3α3 | (2) |
2.5 Molecular docking analysis
The molecular docking calculations were adopted with an AutoDock 4.2.5.1 program between ligand (TGB) and receptor (HSA/BSA). The 3D structure of TGB was acquired with Chemidraw 11.0 and optimized by using the Discover Minimization tool of Materials Studio 6.0 software (Accelrys, California, USA). Crystal structures of HSA and BSA were obtained from the Protein Data Bank (PDB), selecting the PDB IDs 2BXG for HSA and 4OR0 for BSA for docking simulation.16,17 Both crystal structures were optimized by adding polar hydrogen atoms, removing water molecules and ions, and adding and assigning Kollman united-atom partial charges. Docking was conducted by setting the grid box size at 126 Å × 126 Å × 126 Å along with the x, y, and z axes and with 0.619 Å grid spacing. The center of the grid was set to 24.165, 8.326, and 17.384. MGLTools 1.5.6 AutoDockTools with AutoGrid 4 and AutoDock 4 were performed to set up and conduct docking calculations of the ligand and receptor. Docking was performed using the Lamarckian genetic algorithm in AutoDock. Global optimization started with 200 runs in 2500000 energy evaluations, with a maximum of 27000 generations. Binding models with the lowest binding free energy and most cluster members were used for further analyses.
2.6 Circular dichroism spectroscopy
CD spectra were recorded on a Chirascan-plus circular dichroism spectrometer (Applied Photophysics, Surrey, UK) at 298 K. Concentrations of HSA and BSA were 2 μM, and that of TGB was 24 μM. Spectra were obtained in a 1 mm quartz cell in a range of 195 to 250 nm with a 1 nm step size, 1 nm band width, and 0.5 s point−1 scanning speed and each spectrum was the average of three scans.3. Results and discussion
3.1 Evaluation of the thermal stability changes of HSA and BSA
VP-DSC is a highly sensitive and high-throughput technique for characterizing thermal stability of proteins and ligand–proteins.18 For an equilibrium in the unfolding process of a protein, changes in its midpoint of the unfolding transition (Tm), as a result of ligand binding, can be used to estimate shifts in thermal stability of the protein.19 Fig. 2 depicts the thermograms of TGB–HSA and TGB–BSA at molar ratios of 0:1 and 1:1. In Fig. 2a, the HSA (40 μM) is unfolded and gives a single endothermic peak with a Tm of 337.1 K. After addition of TGB (40 μM), the Tm increases to 337.5 K. In Fig. 2b, the BSA (40 μM) solution containing TGB (40 μM) exhibits a single endothermic peak with a Tm of 335.8 K, compared with 335.2 K in TGB-free solution. Thus, the Tm increment of HSA and BSA after TGB addition indicated that the thermal stability of proteins slightly increased after binding with TGB.
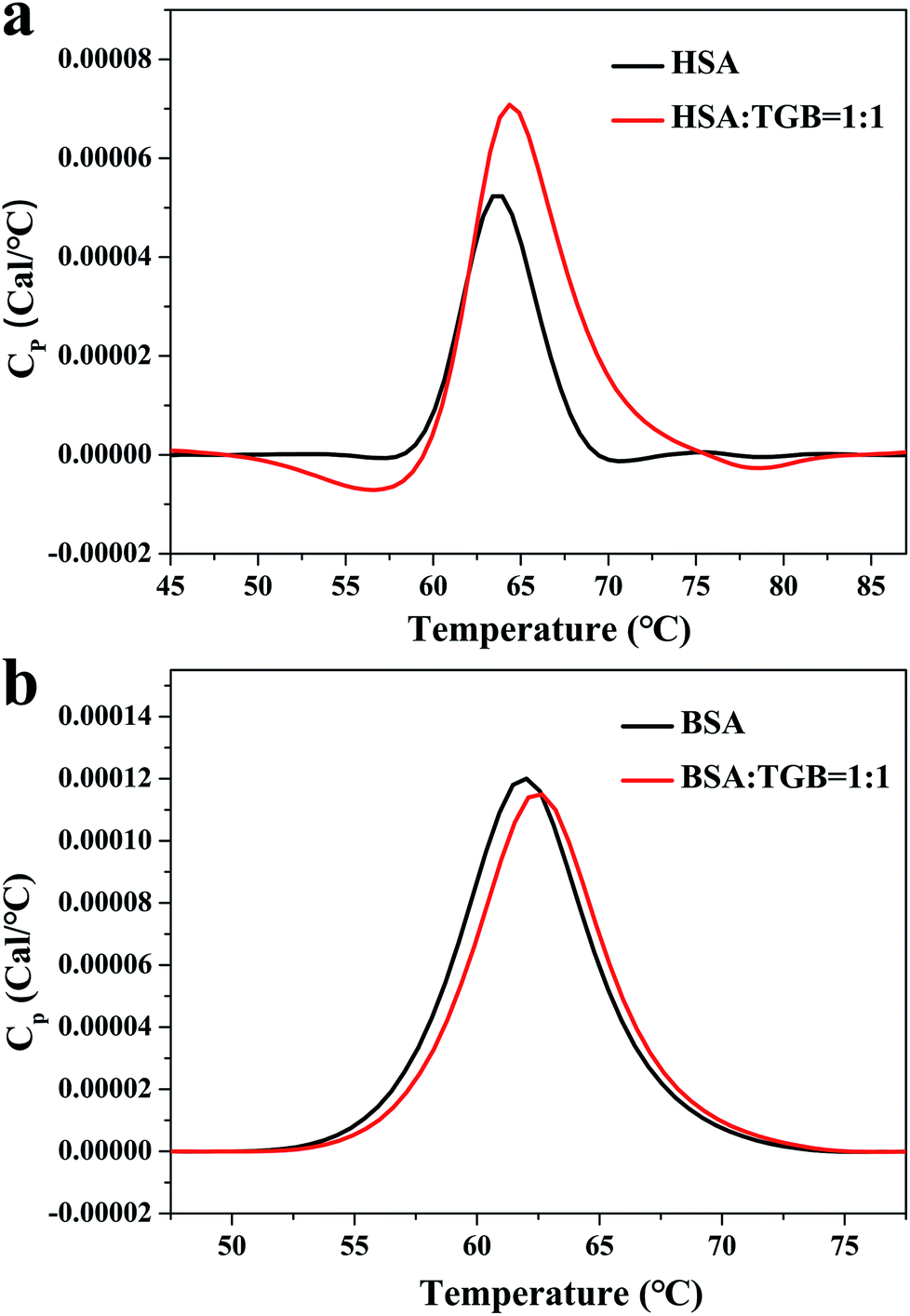 | ||
| Fig. 2 DSC thermograms of TGB–HSA (a) and TGB–BSA (b) systems at pH 7.40 and 298 K, respectively. The concentrations of HSA and BSA are both 40 μM, and the molar ratios of TGB to HSA/BSA are 0:1 and 1:1. | ||
3.2 Characterization of the TGB–HSA and TGB–BSA binding
ITC is a direct and common technique for investigating biomolecular interactions, evaluating thermodynamics of ligand–protein interactions and studying the binding constant.20,21 Here, the ITC has been used to study the combination of TGB–HSA and TGB–BSA, respectively. The calorimetric titration data at pH 7.4 and 298 K are shown in Fig. 3. In Fig. 3, the upper panel shows the raw ITC results, where each of the heat burst peaks corresponded to a single injection of TGB into a HSA/BSA solution in the calorimeter cell. The lower panel shows an integrated heat profile that has been appropriately corrected by subtracting the corresponding dilution heats.22,23 In the lower panel, heat data are plotted against the corresponding molar ratio and fitted to a one set of sites model using MicroCal Origin 7.0 software, which was supplied by the instrument manufacturer.24 These data can be utilized to calculate the binding constant (K), and the thermodynamics of binding, i.e., enthalpy changes (ΔH) and entropy changes (ΔS). The fitting curve is shown as a red curve in the lower panel of Fig. 3, whose amplitude is proportional to the change of enthalpy (ΔH), and where the slope of inflection point yields the binding constant (K) in case of a sigmoidal titration curve.25 Obtained from ITC analyses, it revealed that the values of K were 6.35 ± 1.4 × 104 M−1 for the TGB–HSA system and 3.67 ± 2.89 × 104 M−1 for TGB–BSA system, implying that binding abilities of the two systems were both moderate compared to that of other strong ligand–protein interactions with binding constants larger than 106 M−1.26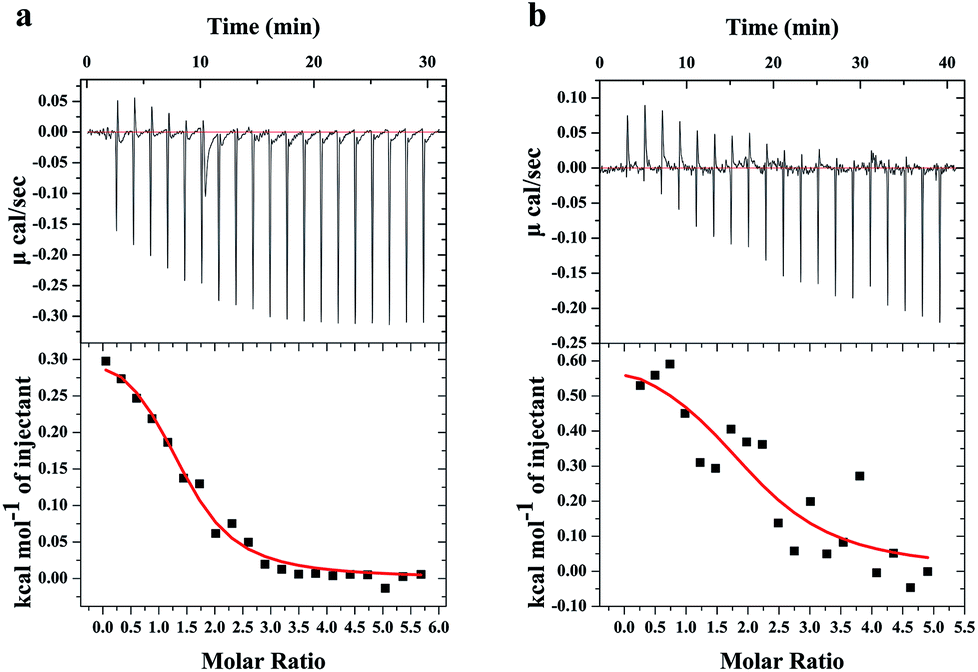 | ||
| Fig. 3 ITC profiles for the titration of TGB with HSA (a) and BSA (b) at pH 7.40 and 298 K, respectively. The upper panel represents raw signals for the calorimetric titration of TGB with HSA/BSA, with a concentration of 2 mM for TGB, 73.1 μM for has, and 80.0 μM for BSA. The lower panel shows integrated heat data after correcting for heat of TGB dilution. The solid line represents the best nonlinear least-squares fit to a one set of sites model. | ||
The fluorescence spectroscopy method that is usually used for studying ligand–protein interactions,27–29 has also been applied to study binding ability between TGB and HSA/BSA. TGB can quench the intrinsic fluorescence of HSA and BSA to varying degrees, respectively (Fig. 4). This fluorescence quenching could reflect any process that decreases the fluorescence intensity of a fluorophore. Molecular interactions, including collisional quenching, ground-state complex formation, excited-state reactions, molecular rearrangements, and energy transfer, can induce fluorescence quenching.30 In this study, the fluorescence quenching mechanism of TGB with HSA and BSA were both static quenching, as the fluorescence lifetimes 〈τ〉 of HSA and BSA were nearly unchanged in the presence of TGB (Fig. 4, Table 1).
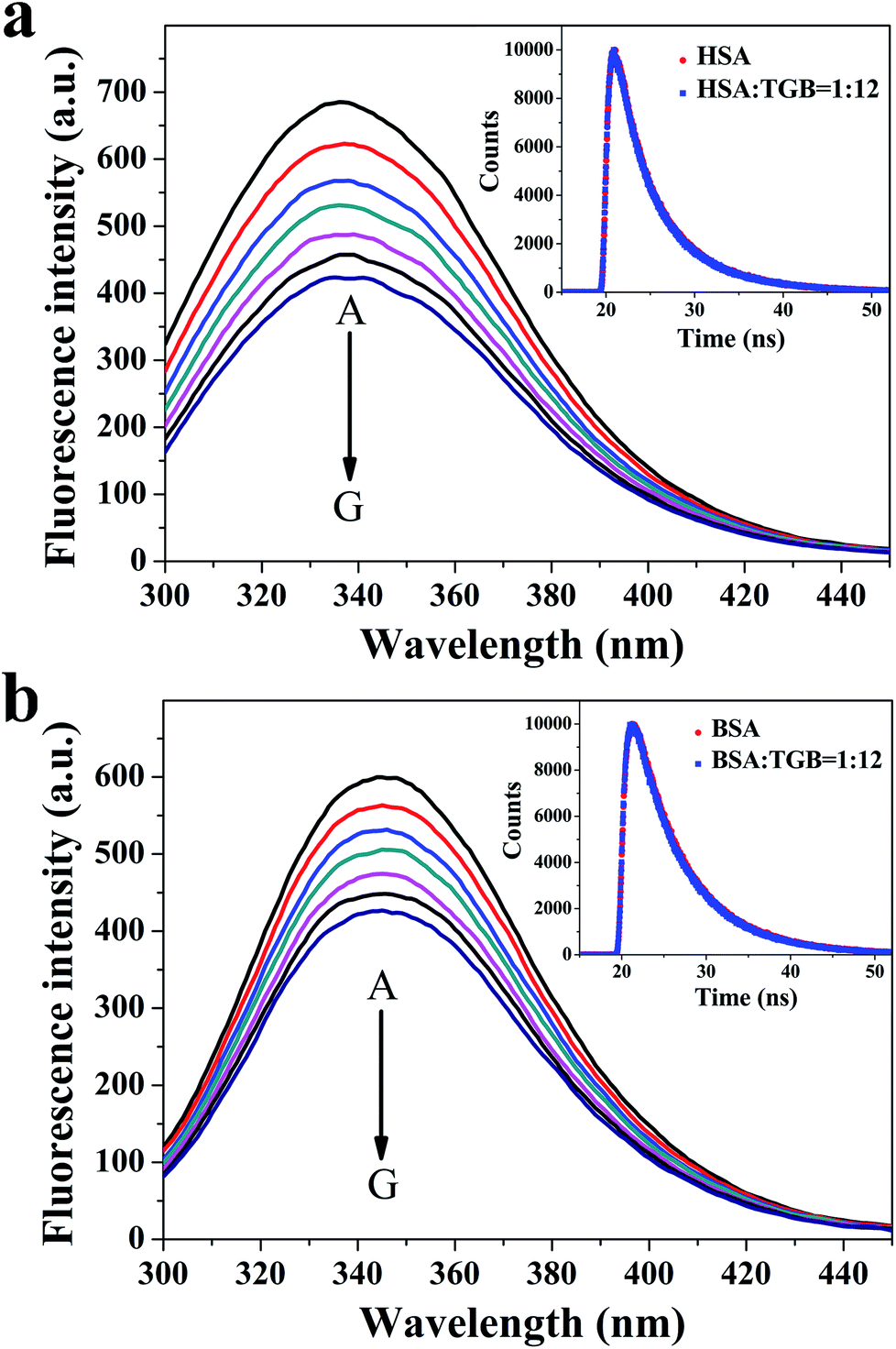 | ||
| Fig. 4 Fluorescence spectra of HSA (a) and BSA (b) in the absence and presence of TGB at pH 7.40 and 298 K, respectively. The concentration of HSA is 2.4 μM (A), and the concentrations of TGB are 4.8 (B), 9.6 (C), 14.4 (D), 19.2 (E), 24.0 (F), and 28.8 μM (G). The concentration of BSA is 0.7 μM (A), and the concentrations of TGB are 2.8 (B), 5.6 (C), 8.4 (D), 11.2 (E), 14.0 (F), and 16.8 μM (G). The insets are the time-resolved fluorescence decay of HSA (a) and BSA (b) in the absence and presence of TGB. The concentrations of HSA and BSA are 2.4 μM and 0.7 μM, respectively, and the molar ratios of TGB to HSA/BSA are 0:1 and 12:1. | ||
| System | Protein:drug |
τ1 | τ2 | τ3 | α1 | α2 | α3 | 〈τ〉 | χ2 |
|---|---|---|---|---|---|---|---|---|---|
| TGB–HSA | 0:1 |
3.030 | 0.491 | 6.853 | 0.328 | 0.046 | 0.626 | 5.305 | 1.055 |
| 12:1 |
2.994 | 0.440 | 6.906 | 0.338 | 0.045 | 0.617 | 5.292 | 1.083 | |
| TGB–BSA | 0:1 |
0.208 | 3.871 | 6.994 | 0.005 | 0.209 | 0.786 | 6.309 | 1.040 |
| 12:1 |
0.701 | 3.970 | 6.980 | 0.011 | 0.224 | 0.765 | 6.237 | 0.993 |
Then, fluorescence quenching was analyzed according to the well-known Stern–Volmer equation:
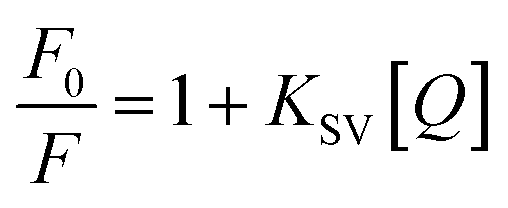 | (3) |
In addition to the binding constant, thermodynamic parameters can also be obtained from ITC analysis.33 The positive values of ΔH (331.0 ± 17.9 cal mol−1) and ΔS (23.1 ± 0.4 cal mol−1 K−1) indicated that hydrophobic interactions played major roles in the formation of the TGB–HSA complex.34 Moreover, the Gibbs free energy change (ΔG) can be calculated by the van't Hoff equation (ΔG = ΔH − TΔS). The negative ΔG (−6.6 ± 0.1 kcal mol−1) implied that the binding process of TGB with HSA was spontaneous. In the TGB–BSA system, ΔH and ΔS were 647.0 ± 139.7 cal mol−1 and 23.1 ± 1.6 cal mol−1 K−1, respectively and the ΔG was calculated to be −6.2 ± 0.5 kcal mol−1. Thus, the binding force between TGB and HSA/BSA was dominant with a hydrophobic interaction, and the binding process was spontaneous.
Crystallographic reports revealed that serum albumin, a heart-shaped and highly helical structure protein, possesses multiple binding sites. Both HSA and BSA consist of a single polypeptide chain in three structurally homologous α-helical domains (I–III), and each domain is divided into two subdomains (A and B). Two main binding sites named Sudlow site I and II, in subdomains IIA and IIIA, respectively, are highly adaptable for binding many widely accepted drugs.35 To investigate whether TGB could bind to HSA/BSA at the two primary binding sites or not, site marker competitive experiments were performed.
3.3 Identification of the main binding sites of TGB to HSA and BSA
As early as the 1970s, Sudlow et al. used fluorescent probes to identify the above two high-affinity binding sites, confirming that dansylsarcosine and ibuprofen were shown to bind specifically to Sudlow site II, while dansylamide, warfarin, and phenylbutazone were bound specifically to Sudlow site I.36,37 At the excitation wavelength of 350 nm, the HSA, BSA, or TGB cannot emit fluorescence, while dansylsarcosine or dansylamide only exclusively exhibit weak fluorescence, but the fluorescent intensity becomes stronger in the presence of HSA/BSA (Fig. 5). Here, dansylsarcosine and dansylamide were selected as specific fluorescent probes for the two distinct binding sites of TGB to HSA and BSA. Any combination of the single, binary, and ternary systems of the probe (dansylsarcosine/dansylamide), the protein (HSA/BSA), and the drug (TGB) were performed using a fluorescence spectrometry method (λex = 350 nm). In the present study, the molar ratios of protein:probe:TGB were 1:4:24.
 | ||
| Fig. 5 At the excitation wavelength of 350 nm, pH 7.40, and 298 K, the fluorescence emission spectra for (a) dansylsarcosine-HSA–TGB, (b) dansylamide-HSA–TGB, (c) dansylsarcosine-BSA–TGB, and (d) dansylamide-BSA–TGB. C(HSA) = C(BSA) = 2.4 μM, C(Dansylsarcosine) = C(Dansylamide) = 9.6 μM, C(TGB) = 57.6 μM. | ||
In Fig. 5a, the fluorescence intensity of dansylsarcosine-HSA–TGB is weaker than that of dansylsarcosine-HSA. We hypothesized that the original balance of the binary complex was interrupted by TGB, and a competitive binding with HSA might occur between dansylsarcosine and TGB, which causes liberation of some dansylsarcosine molecules from the dansylsarcosine-HSA complex in the presence of TGB, while TGB and HSA formed a ground-state complexation without fluorescence. Thus, the fluorescence intensity of dansylsarcosine-HSA was reduced after addition of TGB. These results indicated that TGB and dansylsarcosine could bind to the same binding site (Sudlow site II) in HSA, while the addition of TGB could partially replace dansylsarcosine that is bound to HSA. By contrast, as shown in Fig. 5b, the emission intensity of dansylamide-HSA–TGB is nearly the same as that of dansylamide-HSA. This indicated that TGB, which exerted limited influence on the dansylamide-HSA complex, could not compete with dansylamide. Given the above, the TGB could bind to Sudlow site II (subdomain IIIA) of HSA.
Similarly, in the site maker competitive experiment of dansylsarcosine/dansylamide-BSA–TGB (Fig. 5c and d), the addition of TGB can reduce the fluorescence intensity of dansylsarcosine-BSA but it has no obvious influence on the emission intensity of dansylamide-BSA, indicating that TGB could compete with dansylsarcosine in BSA. However, there is no obvious competition between TGB and dansylamide. Therefore, TGB and dansylsarcosine could bind to the same site (Sudlow site II) of BSA, and addition of TGB could partially replace the dansylsarcosine that bound to BSA.
Subsequently, AutoDock software was used to present the optimal binding model for further binding-orientation analyses of the TGB–HSA complex and TGB–BSA complex (Fig. 6).
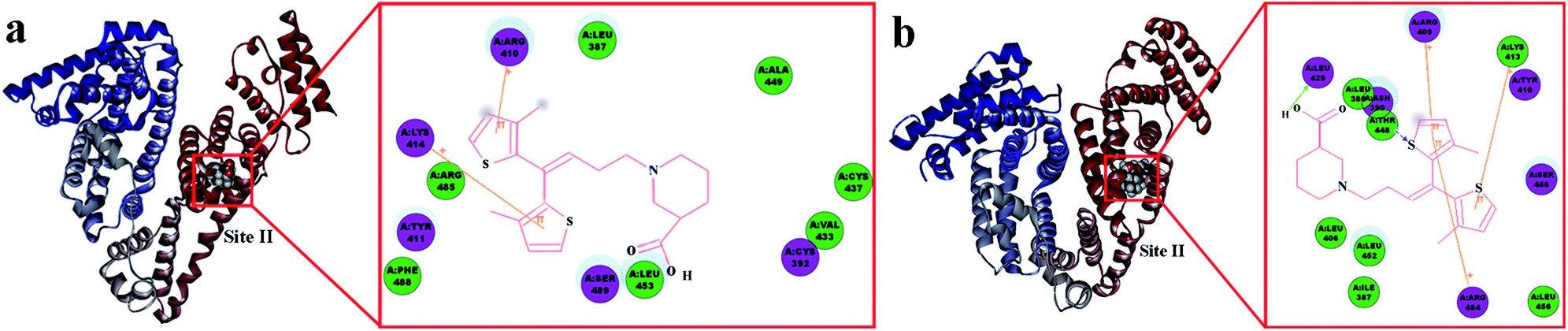 | ||
| Fig. 6 Surface around TGB in Sudlow site II of HSA (a) and BSA (b), respectively. The inset is the 2D docking modes between TGB and HSA or BSA. Only important interacting residues are shown and represented as sticks. Residues involved in hydrogen bonding, charge, and polar interactions are represented by magenta circles. Residues involved in van der Waals interactions are represented by green circles. The solvent-accessible surfaces of atoms are represented by a blue halo around the atom. The diameter of the circle is proportional to the solvent accessible surface. Cation–π interactions are represented by an orange line, whereby connecting positive ions of amino acid residues of HSA or BSA with the thiophene rings of TGB. | ||
In Fig. 6a, the ligand TGB is located inside the protein structure of HSA, particularly, in the hydrophobic cavity in Sudlow site II. It consists of the following amino acids: Lys-414, Arg-485, Tyr-411, Phe-488, Arg-410, Leu-387, Ser-489, Leu-453, Ala-449, Cys-437, Val-433, and Cys-392. TGB and HSA undergo changes with the influence of cation–π interaction, which is a noncovalent molecular interaction between a positively charged molecule and a π system, to minimize the space between them.38 Panel 2D displays the cation–π interactions between HSA and TGB at pH 7.40, as the Lys-414 and Arg-410 took part in forming cation–π binding when the cations on these residues were separately incorporated into the π system of the two thiophene rings of TGB. Fig. 6b shows the lowest energy molecular modelling of TGB docking to site II of BSA. The following amino acids are around the binding site: Leu-429, Leu-386, Asn-390, Thr-448, Leu-406, Leu-452, Ile-387, Arg-484, Leu-456, Arg-409, Lys-413, Tyr-410, and Ser-488. Especially, there are cation–π interactions between the thiophene ring of TGB and the Arg-409, Arg-484, and Lys-413 of BSA, and a hydrogen bonding force also exists.
In combination with site marker competitive experiments and molecular docking simulation results, the TGB could steadily bind to Sudlow site II (subdomain IIIA) of HSA and BSA, and the hydrophobic interactions between the thiophene rings of TGB and the hydrophobic amino acid residues of proteins played a major role in the TGB–protein complexation.
3.4 Evaluation of the conformational changes of HSA and BSA induced by TGB
:1 and 6:1 (drug to protein) are shown in Fig. 7a–d, and their parameters are illustrated in Table 2. Peak 1 (λex = 225 nm, λem = 335 nm for HSA and λex = 225 nm, λem = 342 nm for BSA) mainly exhibits fluorescence characteristics of polypeptide backbone structures. Peak 2 (λex = 280 nm, λem = 338 nm for HSA and λex = 280 nm, λem = 340 nm for BSA) mainly reveals the spectral behavior of tryptophan and tyrosine residues and reflects the polarity of the HSA microenvironment.40,41 By comparing and analyzing the position of the excitation and emission wavelength, and the Stokes shift (Table 2) of TGB–HSA complex in the absence and presence of TGB, it could be seen that both peak 1 and 2 were nearly unchanged, which indicated that the molecular microenvironment around the fluorophore residues of HSA was almost unaffected by TGB. Similarly, the microenvironment of BSA was not obviously perturbed by TGB either, as the Stokes shift of BSA had no significant change.
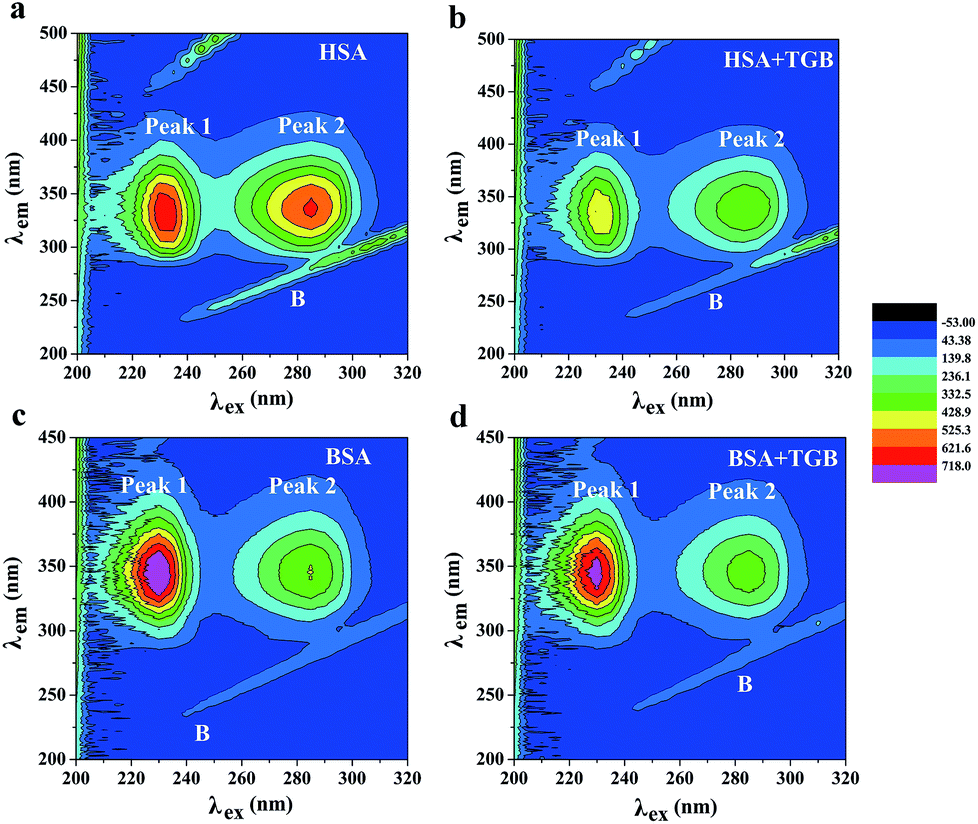 | ||
| Fig. 7 At pH 7.40 and 298 K, the contour spectra of HSA (a and b) and BSA (c and d) in the absence and presence of TGB. The concentrations of HSA and BSA are 2.4 μM and 0.7 μM, respectively, and the molar ratios of TGB to HSA or BSA are 0:1 and 6:1. | ||
| System | Peaks | Peak position [λex/λem (nm nm−1)] | Stokes shift [Δλ (nm)] | Intensity |
|---|---|---|---|---|
| HSA | B | 235/235 → 320/320 | 0 | 71.289 → 364.174 |
| 1 | 225/335 | 110 | 716.917 | |
| 2 | 280/338 | 58 | 649.896 | |
| TGB–HSA | B | 240/240 → 320/320 | 0 | 72.360 → 412.557 |
| 1 | 225/336 | 111 | 526.065 | |
| 2 | 280/339 | 59 | 420.584 | |
| BSA | B | 240/240 → 320/320 | 0 | 56.465 → 124.164 |
| 1 | 225/342 | 117 | 872.205 | |
| 2 | 280/340 | 60 | 434.241 | |
| TGB–BSA | B | 245/245 → −320/320 | 0 | 58.981 → 136.431 |
| 1 | 225/343 | 118 | 790.332 | |
| 2 | 280/341 | 61 | 385.896 |
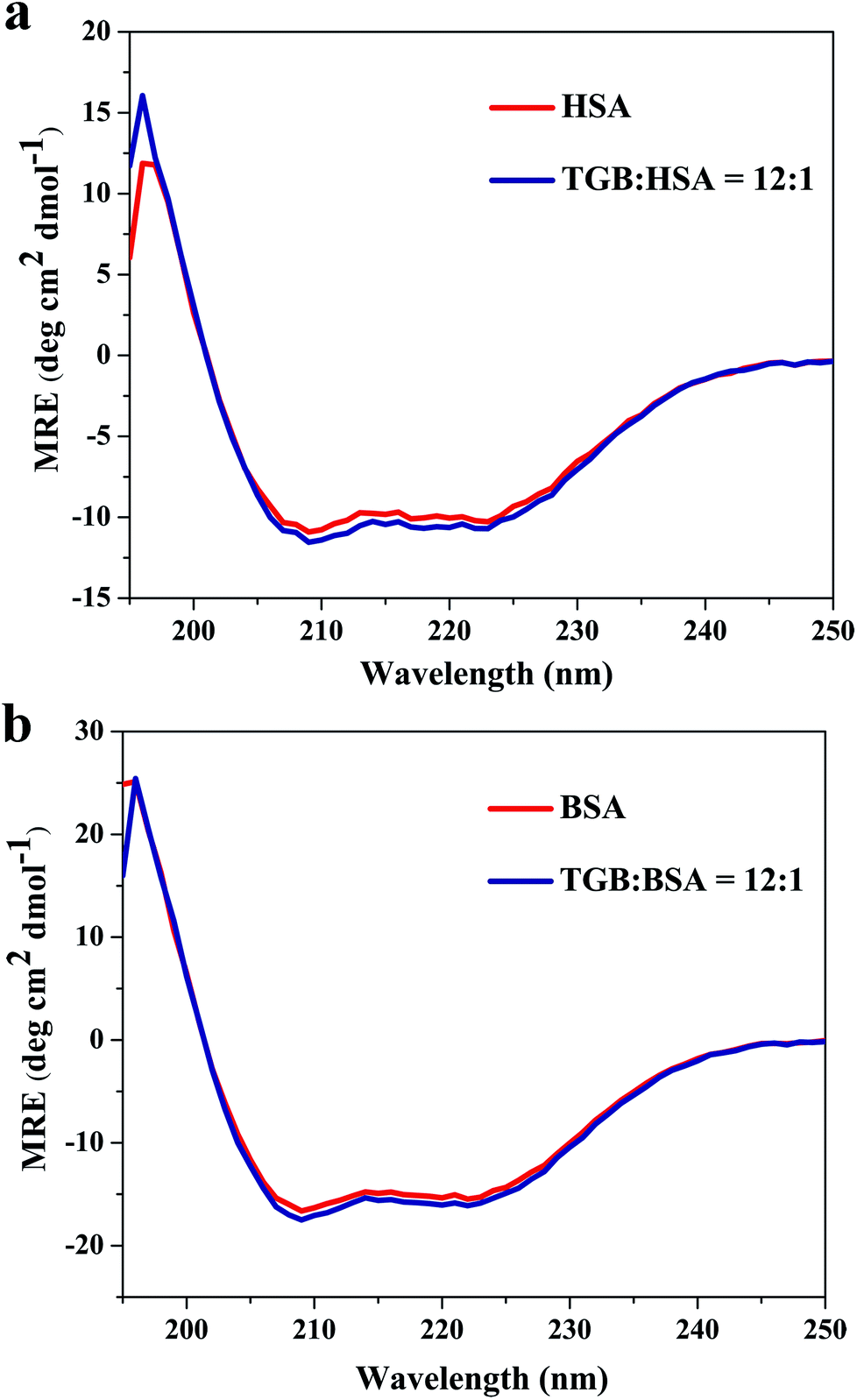 | ||
| Fig. 8 CD spectra of HSA (a) and BSA (b) with or without TGB at pH 7.40 and 298 K, respectively. The concentrations of HSA and BSA are both 2 μM, and the molar ratios of TGB to HSA or BSA are 0:1 and 12:1. | ||
4. Conclusion
In all, this study presented a systematic investigation of the interaction mechanism between TGB and serum albumins, including HSA and BSA, using different calorimetric and spectroscopic techniques. There were no obvious differences between TGB–HSA and TGB–BSA systems. TGB could steadily combine with the proteins and the thermal stabilities of the two serum albumins were slightly enhanced by complexation with TGB. The binding abilities of both the TGB–HSA complex and TGB–BSA complex were moderate. TGB and dansylsarcosine competitively interact with Sudlow site II of HSA and BSA. Hydrophobic interactions were dominant in the formation of TGB–HSA and TGB–BSA complexes. The results of molecular docking intuitively showed that the TGB molecule entered the hydrophobic cavity in Sudlow site II of HSA and BSA and formed specific cation–π bonds with the surrounding amino acids. Additionally, the conformations of HSA and BSA were almost unaffected by TGB. This study has provided insights into the interaction mechanism between TGB and serum albumins, which could provide useful information for better utilization of TGB.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was supported by the Large-scientific Instruments Sharing Platform Ability Construction of Sichuan Province (Grant No. 2016KJTS0037). Thanks to the State Key Laboratory of Biotherapy and the center of Growth, Metabolism and Aging, Sichuan University for the VP-Capillary differential scanning calorimetric (DSC) and isothermal titration calorimeter (ITC) technical assistance. Thanks for the help of Engineering Experimental Teaching Center, School of Chemical Engineering, Sichuan University.Notes and references
- W. Froestl, Future Med. Chem., 2011, 3, 163–175 CrossRef PubMed.
- K. Salat, A. Podkowa, N. Malikowska, F. Kern, J. Pabel, E. Wojcieszak, K. Kulig, K. T. Wanner, B. Strach and E. Wyska, Neuropharmacology, 2017, 113, 331–342 CrossRef PubMed.
- K. Salat, A. Podkowa, P. Kowalczyk, K. Kulig, A. Dziubina, B. Filipek and T. Librowski, Pharmacol. Rep., 2015, 67, 465–472 CrossRef PubMed.
- J. Liu, D. Huang, J. Xu, J. Tong, Z. Wang, L. Huang, Y. Yang, X. Bai, P. Wang, H. Suo, Y. Ma, M. Yu, J. Fei and F. Huang, Sci. Rep., 2015, 5, 15720 CrossRef PubMed.
- P. A. Rutecki and B. E. Gidal, Epilepsy Behav., 2002, 3, 24 CrossRef PubMed.
- H. A. Spiller, D. Wiles, J. L. Russell and M. J. Casavant, Hum. Exp. Toxicol., 2016, 35, 109–113 CrossRef PubMed.
- S. Fatima, P. Sen, P. Sneha and C. G. Priyadoss, Appl. Biochem. Biotechnol., 2017, 182, 82–96 CrossRef PubMed.
- A. Bujacz, J. A. Talaj, K. Zielinski, A. J. Pietrzyk-Brzezinska and P. Neumann, Acta Crystallogr., Sect. D, 2017, 73, 896–909 CrossRef PubMed.
- J. H. Shi, D. Q. Pan, X. X. Wang, T. T. Liu, M. Jiang and Q. Wang, J. Photochem. Photobiol., B, 2016, 162, 14–23 CrossRef PubMed.
- K. Gałęcki, K. Hunter, G. Daňková, E. Rivera, L. W. Tung and K. Mc Sherry, J. Lumin., 2016, 177, 235–241 CrossRef.
- G. A. McMillin and M. D. Krasowski, Therapeutic Drug Monitoring of Newer Antiepileptic Drugs, Clinical Challenges in Therapeutic Drug Monitoring, Elsevier Inc., 2016, pp. 101–134 Search PubMed.
- G. Rabbani, M. H. Baig, E. J. Lee, W. K. Cho, J. Y. Ma and I. Choi, Mol. Pharm., 2017, 14, 1656–1665 CrossRef PubMed.
- E. L. Gelamo, C. H. T. P. Silva, H. Imasato and M. Tabak, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2002, 1594, 84–99 CrossRef.
- K. Mikael, S. Robert, E. Svante and A. Bo, Analyst, 1994, 119, 417–419 RSC.
- K. Ray, R. Badugu and J. R. Lakowicz, RSC Adv., 2015, 5, 54403–54411 RSC.
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353, 38–52 CrossRef PubMed.
- A. Bujacz, K. Zielinski and B. Sekula, Proteins, 2014, 82, 2199–2208 CrossRef PubMed.
- C. J. Layton and H. W. Hellinga, Protein Sci., 2011, 20, 1439–1450 CrossRef PubMed.
- S. Yasmeen and Riyazuddeen, J. Chem. Thermodyn., 2017, 110, 79–86 CrossRef.
- S. A. Kantonen, N. M. Henriksen and M. K. Gilson, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 485–498 CrossRef PubMed.
- M. Braia, D. Loureiro, G. Tubio, M. E. Lienqueo and D. Romanini, Colloids Surf., B, 2017, 155, 507–511 CrossRef PubMed.
- A. Khammari, A. A. Saboury, M. H. Karimi-Jafari, M. Khoobi, A. Ghasemi, S. Yousefinejad and O. K. Abou-Zied, J. Phys. Chem. C, 2017, 19, 10099–10115 Search PubMed.
- G. Rabbani, M. H. Baig, E. J. Lee, W. K. Cho, J. Y. Ma and I. Choi, Mol. Pharm., 2017, 14, 1656–1665 CrossRef PubMed.
- E. Kaspchak, L. I. Mafra and M. R. Mafra, Food Chem., 2018, 252, 1–8 CrossRef PubMed.
- O. Callies and A. Hernandez Daranas, Nat. Prod. Rep., 2016, 33, 881–904 RSC.
- X. Li and S. Wang, New J. Chem., 2015, 39, 386–395 RSC.
- H. Yang, P. Tang, B. Tang, Y. Huang, X. Xiong and H. Li, RSC Adv., 2017, 7, 10242–10251 RSC.
- Y. Chen, Y. Zhou, M. Chen, B. Xie, J. Yang, J. Chen and Z. Sun, Food Chem., 2018, 258, 393–399 CrossRef PubMed.
- J. C. N. Santos, I. M. da Silva, T. C. Braga, A. de Fatima, I. M. Figueiredo and J. C. C. Santos, Int. J. Biol. Macromol., 2018, 113, 1032–1040 CrossRef PubMed.
- J. R. Lakowics, J. Biomed. Opt., 2008, 13, 277–289 Search PubMed.
- M. Chakraborty, S. Paul, I. Mitra, M. Bardhan, M. Bose, A. Saha and T. Ganguly, J. Photochem. Photobiol., B, 2018, 178, 355–366 CrossRef PubMed.
- V. Dahiya, B. Chaubey, A. K. Dhaharwal and S. Pal, Pestic. Biochem. Physiol., 2017, 139, 92–100 CrossRef PubMed.
- E. Morgunova, S. Saller, I. Haase, M. Cushman, A. Bacher, M. Fischer and R. Ladenstein, J. Biol. Chem., 2007, 282, 17231–17241 CrossRef PubMed.
- P. D. Ross and S. Subramanian, Biochemistry, 2002, 20, 3096–3102 CrossRef.
- F. Bou-Abdallah, S. E. Sprague, B. M. Smith and T. R. Giffune, J. Chem. Thermodyn., 2016, 103, 299–309 CrossRef.
- G. Sudlow, D. J. Birkett and D. N. Wane, Mol. Pharmacol., 1975, 11, 824–832 Search PubMed.
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1976, 12, 1052–1061 Search PubMed.
- G. U. Nienhaus, Protein-ligand interactions: methods and applications, Humana Press, 2005, pp. 451–492 Search PubMed.
- Z. Chen, H. Xu, Y. Zhu, J. Liu, K. Wang, P. Wang, S. Shang, X. Yi, Z. Wang, W. Shao and S. Zhang, RSC Adv., 2014, 4, 25410–25419 RSC.
- M. S. Ali, M. Amina, H. A. Al-Lohedan and N. M. Al Musayeib, J. Photochem. Photobiol., B, 2018, 182, 9–17 CrossRef PubMed.
- M. S. Ali and H. A. Al-Lohedan, Colloids Surf., B, 2015, 134, 392–400 CrossRef PubMed.
- S. Ikhlas, A. Usman and M. Ahmad, J. Biomol. Struct. Dyn., 2018, 1–11 CrossRef PubMed.
- J. Kaur, L. Katopo, A. Hung, J. Ashton and S. Kasapis, Food Chem., 2018, 252, 163–170 CrossRef PubMed.
- A. Maji, M. Beg, A. K. Mandal, S. Das, P. K. Jha, A. Kumar, S. Sarwar, M. Hossain and P. Chakrabarti, J. Mol. Struct., 2017, 1141, 584–592 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04153a |
| This journal is © The Royal Society of Chemistry 2018 |