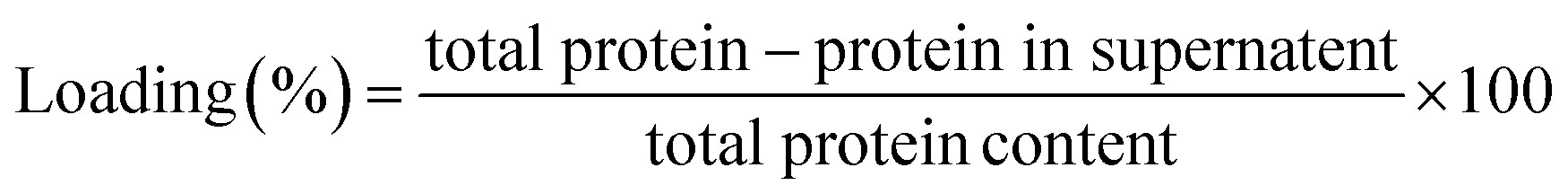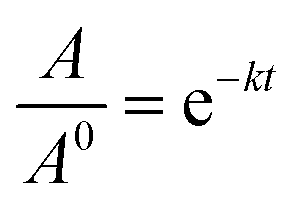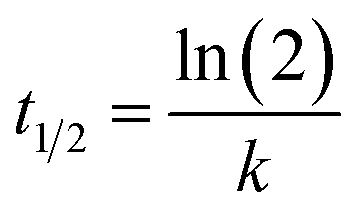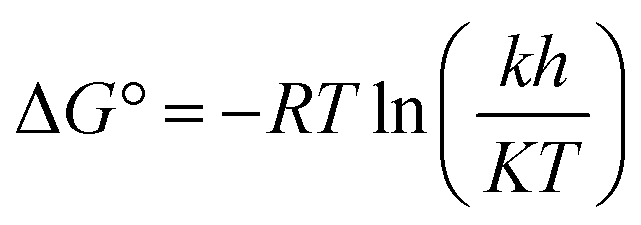DOI:
10.1039/C8RA05463K
(Paper)
RSC Adv., 2018,
8, 27763-27774
Facile fabrication of a recyclable nanobiocatalyst: immobilization of Burkholderia cepacia lipase on carbon nanofibers for the kinetic resolution of a racemic atenolol intermediate†
Received
26th June 2018
, Accepted 25th July 2018
First published on 3rd August 2018
Abstract
Immobilization of surfactant treated Burkholderia cepacia lipase on the surface of carbon nanofibers was performed via two different methods: adsorption and covalent attachment. Simple adsorption of lipase on carbon nanofibers turned out to be a poor strategy, exhibiting an immobilization efficiency of 36%, while covalent coupling using 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide (EDC)/N-hydroxysuccinimide (NHS) showed better immobilization efficiency (56%). The nanobioconjugate fabricated using the latter method showed an eleven-fold increase in enzyme activity towards the hydrolysis of p-nitrophenyl palmitate and enhanced dispersion in organic solvents. At 80 °C, the half-life of lipase in the nanobioconjugate was almost 20 fold higher than that of free lipase, demonstrating its thermal stability. The as-prepared nanobioconjugate was reused for nine consecutive reaction cycles achieving 100% yield in the hydrolysis of p-nitrophenol palmitate but losing almost 50% of the initial activity after seven operational cycles. Finally, this heterogeneous nanobioconjugate was more active and enantioselective [C = 47.8, eep = 97.0 and E = 194] than free lipase [C = 35.4, eep = 97.1 and E = 88] towards the kinetic resolution of a racemic intermediate of atenolol yielding the S enantiomer, which signifies its importance as a nanobiocatalyst.
Introduction
Biocatalysts are emerging as viable alternatives to chemical catalysts offering various advantages over them like high selectivity, specificity, cost effectiveness, eco-friendliness etc.1,2 Although enzymes are noted as ideal catalysts for industrial applications, their use is limited because of poor operational stability and reusability and inefficient separation. Enzyme immobilization has primarily been capitalized to improve the reusability of the enzymes, and if properly performed, may greatly enhance enzyme stability, activity, resistance to inhibitors or chemicals etc.3–7 Different types of support materials having unique characteristics have been fabricated to alleviate the existing limitations in this technology.8,9 In this expansion, nanostructured materials offer advantages over conventional support materials. In several examples, high surface area of the nanostructures favors higher enzyme loading and lower mass transfer resistance yielding higher enzyme activity and stability.10–13 In this category, carbon based nanomaterials attract growing interest for development as heterogeneous catalyst mainly due to (1) high thermal and mechanical stability, (2) high surface area, (3) facile recovery, (4) easy surface modification.14–16 High superficial density of groups on the nanomaterial surface provides multiple points for enzymes attachment leading to an efficient immobilization process.17–20 Many porous materials such as silica, activated carbon etc. have been extensively used for immobilization of enzymes as they provide cavities adequate for this purpose.21–23 Thus immobilization of enzymes on such support materials can modify their catalytic properties.4 However, the morphology, distribution pattern of the pores and chemical groups present on the surface of these support materials also affect the enzyme activity.24,25 In such situation carbon nanofibers present as the most suitable choice. They are cylindrical fibres made up of a series of graphene layers stacked neatly into different shapes. They are a few micrometres in length with a diameter of 80–200 nm and have high mechanical strength.26 The physical properties like size and ordering of the graphite layers can be easily controlled and modified as needed.27 Well defined structural arrangements provide an optimal geometry to the nanomaterial, making it ideal for high enzyme loading. Compared to the alternative carbon based nanomaterials like carbon nanotubes, the open structure of carbon nanofibers are highly accessible to even large enzyme molecules for immobilization.20 High thermal and mechanical stability of carbon nanofibers is another feature that can be emphatically advantageous for their industrial application. Several studies on the analogous carbon based materials like diamond, glassy carbon etc. have revealed extraordinarily high stability upon surface functionalization. Likewise carbon nanofibers could also be subjected to functionalization to improve their thermal stability and thus harnessing their potential for industrial purposes.28,29 The entire surface area of the carbon nanofibers can be activated yielding stable active sites for enzyme immobilization.
The principle of bioimprinting methodology is to “freeze” the enzyme in a presumably active form, generated by binding with the imprint molecules (such as surfactants, natural substrates, substrate analogs etc.) followed by “washing out” with an anhydrous solvent. After this treatment the imprinted conformation of enzyme is expected to be ‘preserved or trapped’. Among the several industrially useful lipases, Burkholderia cepacia lipase (BCL) is a very useful enzyme. It catalyzes a broad range of different reactions in water as well as non-polar solvents under mild conditions.30 Like other lipases Burkholderia cepacia lipase (BCL) also consists of a mobile element at the surface, a lid, which covers the active site.31 The lid opens at a hydrophobic interface, making active site accessible to substrates and enhancing the activity of lipase. Thus, interfacial activation of lipase by addition of imprint molecules has been widely explored and designated as interfacial activation based molecular (bio)-imprinting (IAMI) by Navorra and Braco.32 Bioimprinting strategy has been integrated with immobilization techniques33,34 to improve lipase performance in cross-linked enzyme aggregates (CLEAs),35 cross-linked protein-coated microcrystals (CLPCMCs),36 sol–gel immobilization with methyltrimethoxysilane (MTMS) and tetramethoxysilane (TMOS),37 macroporous resin NKA,38 diatomaceous earth and CM-Sephadex39 etc. in organic media. In addition to this, lipases in solution form tend to aggregate.
Atenolol is a well-known β-adrenergic receptor antagonist (β-blocker) drug used for the management and treatment of acute myocardial infarction, angina pectoris, hypertension, cardiac arrthymia, tachycardia and the symptoms of alcohol withdrawal.40,41 It has been well established that the β-blocker activity of atenolol resides in the (S)-enantiomer which is devoid of the side effects produced by the racemic form of this drug.42 One of the most feasible approaches for synthesis of enantiopure atenolol is kinetic resolution of the drug or its intermediates. This can be achieved either through enantioselective esterification or hydrolysis of the racemate using immobilized enzymes in organic media43,44 or ionic liquid.45 BCL is an industrially useful enzyme that catalyzes a broad range of kinetic resolution reactions in water as well as non-polar solvents under mild conditions.46–48
Using this perspective, the present study is aimed to implement the imprinting strategy on Burkholderia cepacia lipase in combination with immobilization on carbon nanofiber to improve its performance as a nanobiocatalyst. To the best of our knowledge there is no report on the immobilization of surfactant treated Burkholderia cepacia lipase (BCL) on carbon nanofibers. In context of the advantages that carbon nanofibers could offer, objectives for the fabrication of the nanobiocatalyst were: (i) selection of a suitable method for the immobilization of surfactant treated lipase on carbon nanofiber (CNF). (ii) Optimization of the immobilization parameters. (iii) Determination of thermostability, storage stability and reusability of the nanobioconjugate. (iv) Application of the nanobioconjugate in the kinetic resolution of racemic atenolol intermediate.
Results and discussion
Effect of surfactants on the precipitation of Burkholderia cepacia lipase
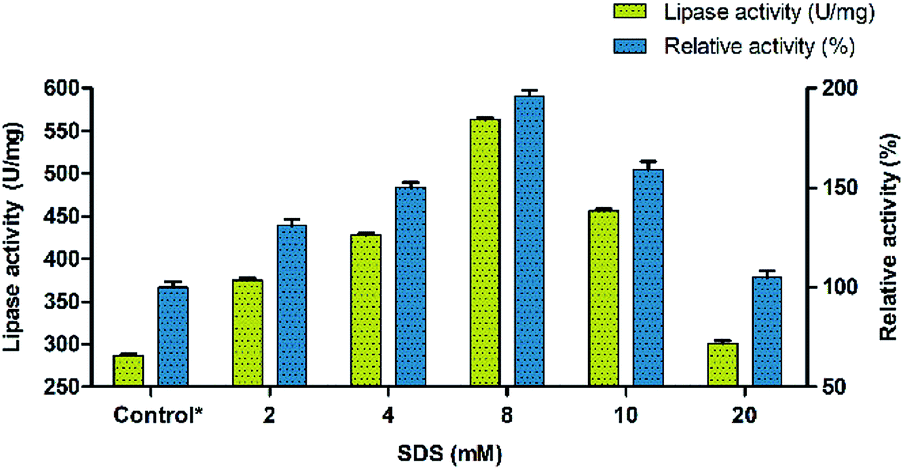
The conformational changes induced by the surfactants in BCL involve mostly the displacement by 13 Å of a so-called lid encompassing 40 amino acid residues and composed of two helical elements (α5- and α9-helices) separated by a short loop.49 Thus, the effect of surfactants was investigated for evaluating their ability to precipitate Burkholderia cepacia lipase. Eight different surfactants with different properties (non-ionic, cationic, anionic and zwitterionic) were used for modulating the lipase activity. Sodium dodecyl sulphate (SDS) was found to have maximum effect on the enhancement of lipase activity followed by PEG 6000. Other surfactants had moderate effect on activity with Triton X-100 showing the least influence. The results indicated that SDS enhanced the activity of BCL towards p-nitrophenyl palmitate (p-NPP) at premicellar concentrations and exhibited the highest activity at around the critical micelle concentration (cmc). During the interfacial activation using surfactants, the catalytic activity of lipase takes a quantum leap at the cmc.50 However, above the cmc, the activity declined but was still significantly higher than in buffer alone indicating that this aggressive detergent did not denature the lipase. This contrasts with the effect of SDS on conventional proteins which unfold above the cmc.51,52 Ionic surfactants such as SDS at low concentrations can form complexes with lipase in solution. In these complexes, the conformational stability of the protein may be altered and the hydrophobicity of the protein surface may also be changed.53,54 The surfactant was used in the range of 2 mM to 20 mM and its effect on lipase activity of BCL was monitored. Thus, the precipitation of BCL with 8 mM of the anionic surfactant (SDS) showed an almost two fold increase in the lipase activity (Fig. 1).
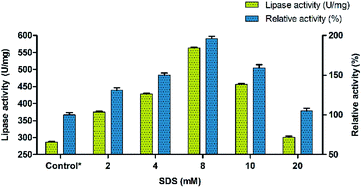 |
| | Fig. 1 Precipitation of lipase (BCL) using different concentrations of SDS. | |
Immobilization of lipase on carbon nanofiber via adsorption
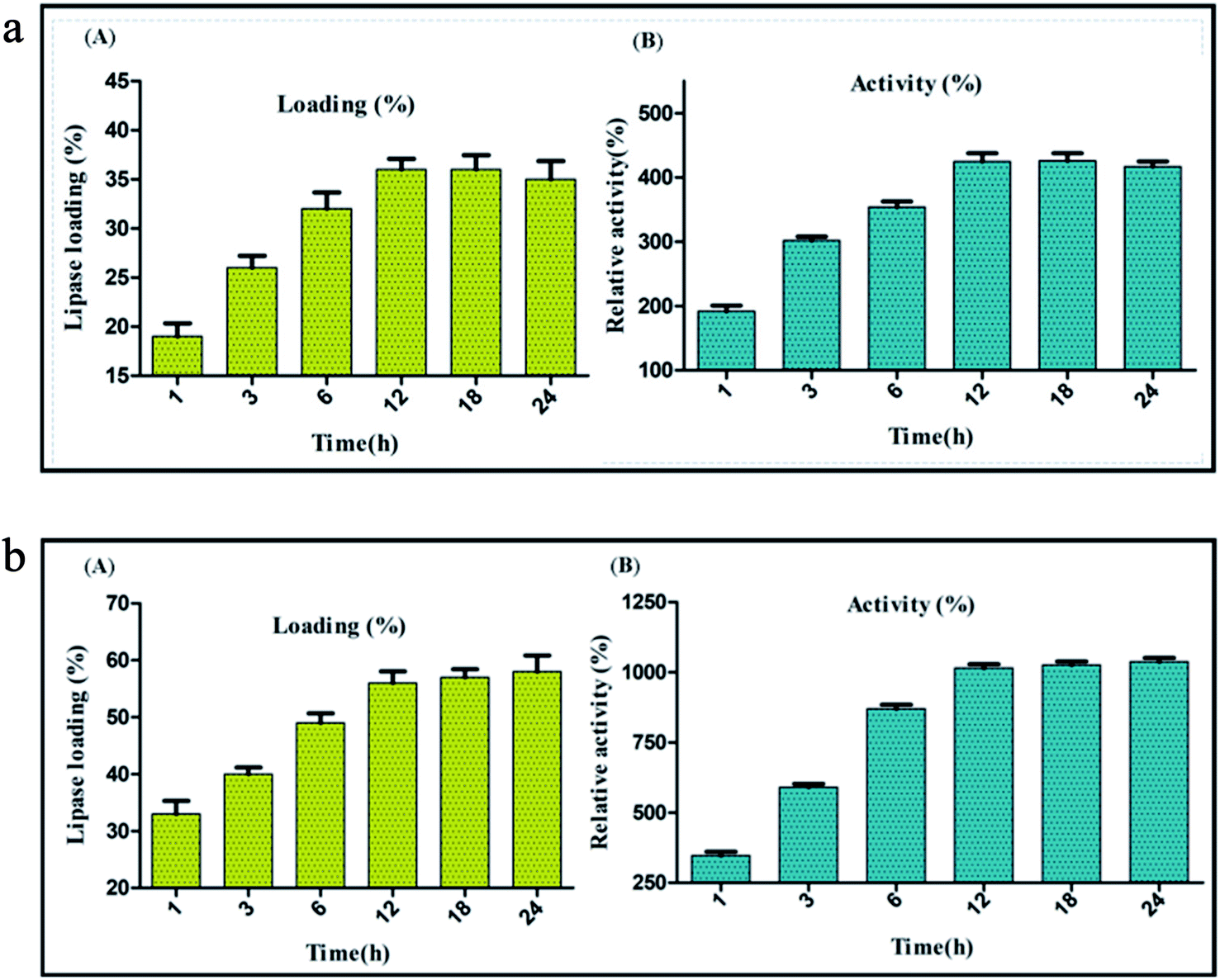
The diameter of graphitized carbon nanofiber (CNF) is generally higher than the carbon nanotubes and lies in the range of 80–200 nm. CNF are very high quality nanofibers; synthesized by heat treatment of the primary nanofibers to a very high temperature (>3000 °C).55 The anaerobic high temperature treatment of the nanofibers induces the engraving of a “reverse saw-toothed” like morphology on the superficial layers of CNF. This yields an ordered assembly of the graphene layers with minimum spacing between them and leads to the generation of a large number of active sites on the surface of CNF.26,56 Thus, the very high surface area etched mainly in the form of active sites on the nanofibers provides a solid ground for immobilization of enzymes through adsorption. These advanced surface characteristics of CNF play an important role in the physical binding of lipase (BCL) for immobilization via adsorption. The evaluation of immobilization efficiency through adsorption method was done by comparing lipase activity and percentage of lipase loading on CNF at different time intervals between 1 to 24 h. Lipase activity of 1219 U mg−1 was observed with lipase loading 36% after 12 h of immobilization. Beyond this time period lipase loading became almost constant and a slight decrease in the lipase activity was recorded (Fig. 2a).
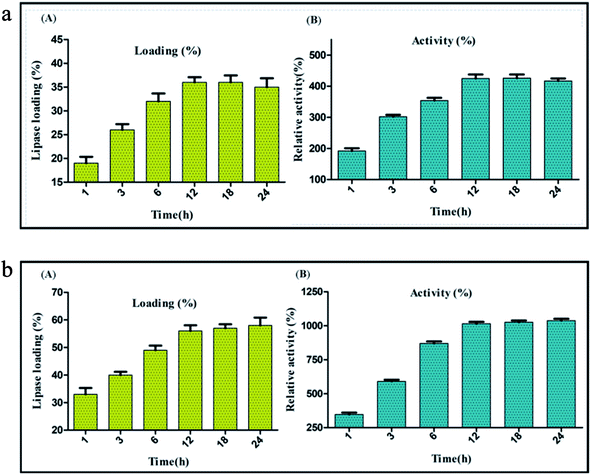 |
| | Fig. 2 Evaluation of (A) lipase loading (%) and (B) relative activity (%) of BCL after immobilization on CNF by adsorption. Evaluation of (A) lipase loading (%) and (B) relative activity (%) of BCL after immobilization on CNF via covalent attachment. | |
Immobilization of lipase on carbon nanofiber via covalent attachment
The carbon based nanomaterials are intrinsically stable. However, several past studies have shown that surface functionalization imparts a substantially high stability and temperature endurance to the carbon nanomaterials28,29 Based on these reports, functionalization of carbon nanofiber via oxidation with a strong acid mixture H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 (3:1 v/v) was performed which led to the addition of free COOH groups and also introduced some structural changes on the surface of the nanofibers. The structural changes were observed mainly due to the twin effect of oxidative treatment which induced either a uniform or a localized etching on the surface of the nanofibers. While the former led to a reduction in nanofiber diameter, the latter reduced the particle length. This reduction in the size of the nanofiber particles promoted their easy dispersion. It also facilitated the incorporation of the –COOH groups to the poorly accessible edges of the nanofibers, thereby increasing the overall density of functionalization fostering the dispersibility of nanofibers.57,58 Also the surface functionalization by acid treatment enhances the dispersibility of nanofibers in aqueous solution by virtue of the increased number of carboxylate anion formation through deprotonation of the newly introduced carboxyl groups in water based buffers. The generation of carboxylate functionality enhances the hydrophilic properties of the CNF adding to their easy dispersion in water.59 Acid treatment was followed by the addition of NHS and EDC, carbodiimide coupling agents that would aid the successful hybridization of lipase on the nanofibers through covalent bond formation. Covalent immobilization was validated by treating immobilized preparation with anionic surfactant, retention of enzymatic activity after treatment confirms covalent immobilization. The immobilization efficiency through covalent attachment method was evaluated by comparing lipase activity and percentage lipase loading on CNF at different time intervals between 1 to 24 h. Optimum lipase activity of 2913 U mg−1 was observed with lipase 56% loading after 12 h of immobilization. At 12 h of covalent immobilization on activated carbon nanofibers, a substantial increase of 1015% in the activity was observed. Beyond this time lipase loading became almost constant and a nominal increase in lipase activity was recorded. Thus, it was concluded that within 12 h of immobilization, optimum lipase loading might occur on the surface of activated-functionalized CNFs and once this level had reached further immobilization of lipase did not take place (Fig. 2b). This trend also indicated that the increase in lipase loading augments the increase in lipase activity. Also, hydrophobic surface of carbon nanofibers act as a catalyst for interfacial activation of lipase resulting into drastic increase in activity after immobilization.21,28,29 A comparative analysis showed the precedence of covalent immobilization over adsorption in improving the lipase activity and enzyme loading. Thus, further experiments were carved to optimize other process variables impacting the immobilization through covalent attachment.
:HNO3 (3:1 v/v) was performed which led to the addition of free COOH groups and also introduced some structural changes on the surface of the nanofibers. The structural changes were observed mainly due to the twin effect of oxidative treatment which induced either a uniform or a localized etching on the surface of the nanofibers. While the former led to a reduction in nanofiber diameter, the latter reduced the particle length. This reduction in the size of the nanofiber particles promoted their easy dispersion. It also facilitated the incorporation of the –COOH groups to the poorly accessible edges of the nanofibers, thereby increasing the overall density of functionalization fostering the dispersibility of nanofibers.57,58 Also the surface functionalization by acid treatment enhances the dispersibility of nanofibers in aqueous solution by virtue of the increased number of carboxylate anion formation through deprotonation of the newly introduced carboxyl groups in water based buffers. The generation of carboxylate functionality enhances the hydrophilic properties of the CNF adding to their easy dispersion in water.59 Acid treatment was followed by the addition of NHS and EDC, carbodiimide coupling agents that would aid the successful hybridization of lipase on the nanofibers through covalent bond formation. Covalent immobilization was validated by treating immobilized preparation with anionic surfactant, retention of enzymatic activity after treatment confirms covalent immobilization. The immobilization efficiency through covalent attachment method was evaluated by comparing lipase activity and percentage lipase loading on CNF at different time intervals between 1 to 24 h. Optimum lipase activity of 2913 U mg−1 was observed with lipase 56% loading after 12 h of immobilization. At 12 h of covalent immobilization on activated carbon nanofibers, a substantial increase of 1015% in the activity was observed. Beyond this time lipase loading became almost constant and a nominal increase in lipase activity was recorded. Thus, it was concluded that within 12 h of immobilization, optimum lipase loading might occur on the surface of activated-functionalized CNFs and once this level had reached further immobilization of lipase did not take place (Fig. 2b). This trend also indicated that the increase in lipase loading augments the increase in lipase activity. Also, hydrophobic surface of carbon nanofibers act as a catalyst for interfacial activation of lipase resulting into drastic increase in activity after immobilization.21,28,29 A comparative analysis showed the precedence of covalent immobilization over adsorption in improving the lipase activity and enzyme loading. Thus, further experiments were carved to optimize other process variables impacting the immobilization through covalent attachment.
Optimization of pH for immobilization of lipase on CNF via covalent attachment
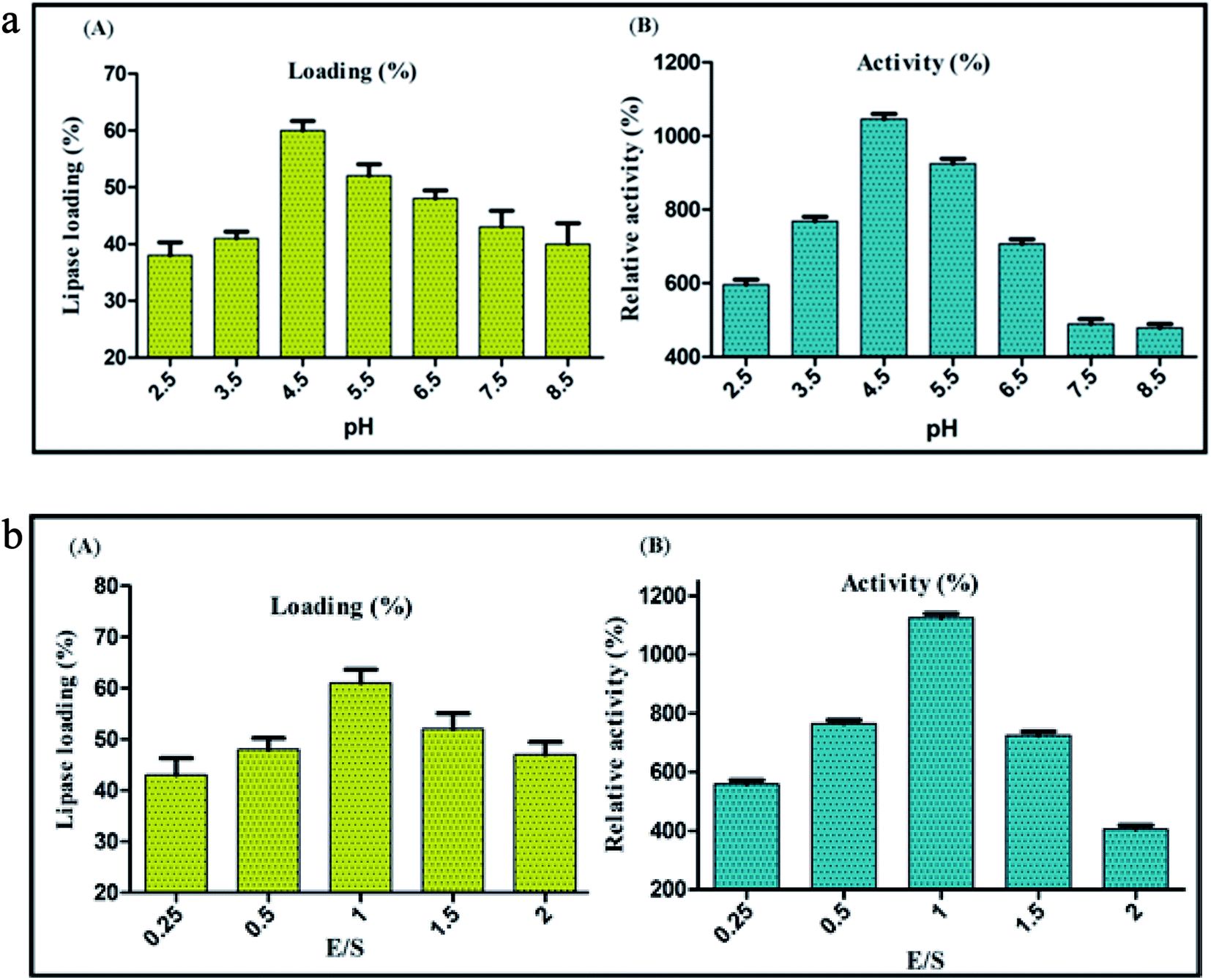
Highly stable nature of CNFs projects it as a promising support material for enzyme immobilization. Since the coupling efficiency of covalent immobilization is susceptible to change in pH,59 immobilization of BCL on carbon nanofibers by covalent attachment was performed at different pH values ranging from 2.5 to 8.5, to decipher the optimum pH of the reaction medium. Maximum lipase activity was detected when covalent conjugation was performed at pH 4.5, with a loading percentage of 60. Under these conditions of immobilization the enzyme activity was 1046% of the activity of soluble lipase (taken as 100%) (Fig. 3a). It is pertinent to mention here that the enzyme activity was estimated under standard protocol. At very low pH value (1 to 3), CNF had a poor dispersibility in the buffer (MES buffer) because of the low extent of ionization of acid groups present on the surface, culminating to the aggregation of nanofibers due to hydrogen bonding between adjacent –COOH groups.60 Shifting of buffer pH to higher range (4–6), yielded higher ionization of –COOH groups generating more carboxylate anions (–COO−). Repulsion between the anions led to the loosening of the graphitized layers in the nanofibers. This broadened the scope of interaction between the water molecules and an individual nanofiber particle, allowing the solvation and ease of dispersion. Towards the upper end of the pH values tested, the lipase loading and activity both started to decline, which could be because of a higher ionic strength of the solution leading to aggregation by ionic bonding.61 These aggregations reduced the overall free surface area available for attachment, barring the nanofibers to reach their maximal loading efficiency. This was clearly evident in the decreased percent loading of lipase on CNF from 60% at pH 4.5 to 40% at pH 8.5. Still higher pH (>9) would interfere further with the process of immobilization as the Na+ (originating from NaOH, added to increase the pH) would create a highly ionic environment thereby aggregating the nanofibers and hindering the immobilization.
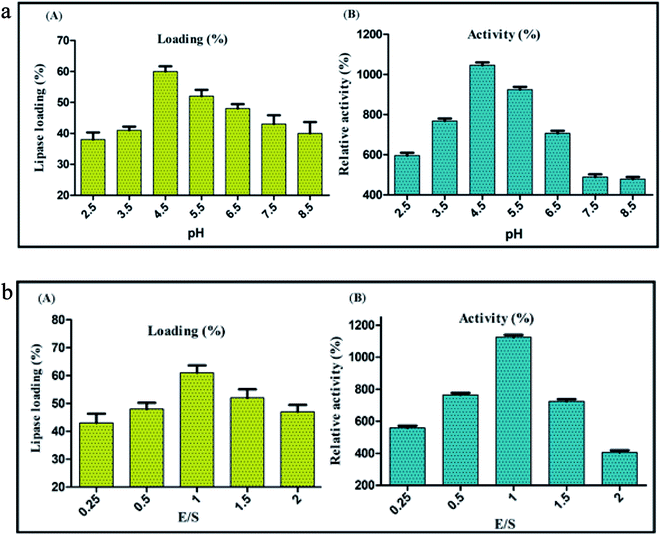 |
| | Fig. 3 Evaluation of (A) lipase loading (%) and (B) relative activity (%) of BCL after immobilization on CNF via covalent attachment at different pH values. Evaluation of (A) lipase loading (%) and (B) relative activity (%) of BCL after immobilization on CNF via covalent attachment at different enzyme support ratio (E/S). | |
Optimization of enzyme-nanosupport ratio for immobilization of lipase on CNF via covalent attachment
The fine control of enzyme-nanosupport ratio is a crucial factor accountable to the success of immobilization. An inaccurate enzyme-nanosupport ratio could result in a dramatic loss of enzyme activity over time. In this context, immobilization via covalent attachment was performed at different enzyme-nanosupport ratio to decipher its optimum value. The maximum lipase activity was detected at enzyme support ratio, E/S = 1, with a lipase loading of 60% and relative activity of 1125% (Fig. 3b). It has been reported that an optimum congruence between the enzyme and nanosupport is necessary for an intense enzyme-nanosupport binding.62 At first glance it appears appropriate to use a higher concentration of the nanosupport with the conception that the entire enzyme might get immobilized at once. However, a large number of nanosupport particles in the same volume may result in unwanted interactions among themselves, decreasing their dispersibility.14 Conversely, a higher concentration of enzyme might result in protein–protein interactions caused by the lack of availability of sufficient immobilization sites. Thus, an optimum proportion of both of the components should be chosen that would ensure a subtle distance between two adjacent immobilized entities. This would prevent any unwanted interactions between the neighbouring nanobioconjugates.63,64
CD-spectroscopy of free lipase, carbon nanofiber and the nanobioconjugate
CD spectroscopy was used to investigate the secondary structure of free enzyme and the nanobioconjugate (SDS-BCL@CNF). The immobilization support material has no contribution to the CD spectrum, which was clearly indicated by the straight line in the spectrum. CD data of SDS-BCL@CNF revealed a decrease in the α-helix content of BCL from 27.6% in native form to 20.5% after immobilization on activated CNF. The decrease in the α-helix, β-sheet, random coil and increase in the β-turn content after immobilization on activated CNF indicated a change in the secondary structure. The “lid” in lipases is mainly composed by the α-helix. The hydrophobic nature of CNF might provide synergistic effect favouring the “lid opening” which is a necessary step for interaction of the catalytic centre of enzyme and substrate.15 The decrease in the α-helix content is an indicative of this effect. A considerable shift in the CD spectrum of SDS-BCL@CNF compared to free BCL depicted conformational changes after the covalent attachment on nanosupports and explained enhancement in lipase activity.
FTIR spectroscopy of free lipase, functionalized carbon nanofiber and the nanobioconjugate
FTIR spectral analysis verified the chemical oxidation of carbon nanofiber (CNF) and formation of an amide bond between BCL and carboxyl group of functionalized carbon nanofiber (CNF-COOH). O–H stretching vibration of the carboxylic acid group appears as a broad band in the range of 3000 to 2500 cm−1. In the FTIR spectrum of functionalized carbon nanofiber, the characteristic band of O–H starching vibrations was observed between 2648 and 2324 cm−1. Also functionalized CNF preparation showed a typical carboxylic acid C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration band at 1777 cm−1 which indicated the incorporation of COOH groups in the carbon nanofiber. The spectrum of conjugated system (SDS-BCL@CNF) also showed the O–H stretching vibration band of carboxylic acid at 2916 cm−1 and 2648 cm−1. SDS-BCL@CNF also showed amide I band at approximately 1628 cm−1 due to CO stretching vibrations and amide II band at 1550 cm−1 indicating a conformational change of secondary structure after covalent immobilization. The N–H stretching vibrations of amide functional groups which occur around 3200–3400 cm−1 were visible at 3181 cm−1 predicting the formation of an amide bond owing to the covalent coupling of BCL with activated functionalized carbon nanofiber.
O stretching vibration band at 1777 cm−1 which indicated the incorporation of COOH groups in the carbon nanofiber. The spectrum of conjugated system (SDS-BCL@CNF) also showed the O–H stretching vibration band of carboxylic acid at 2916 cm−1 and 2648 cm−1. SDS-BCL@CNF also showed amide I band at approximately 1628 cm−1 due to CO stretching vibrations and amide II band at 1550 cm−1 indicating a conformational change of secondary structure after covalent immobilization. The N–H stretching vibrations of amide functional groups which occur around 3200–3400 cm−1 were visible at 3181 cm−1 predicting the formation of an amide bond owing to the covalent coupling of BCL with activated functionalized carbon nanofiber.
Transmission electron microscopy and EDX analysis of the nanobioconjugate
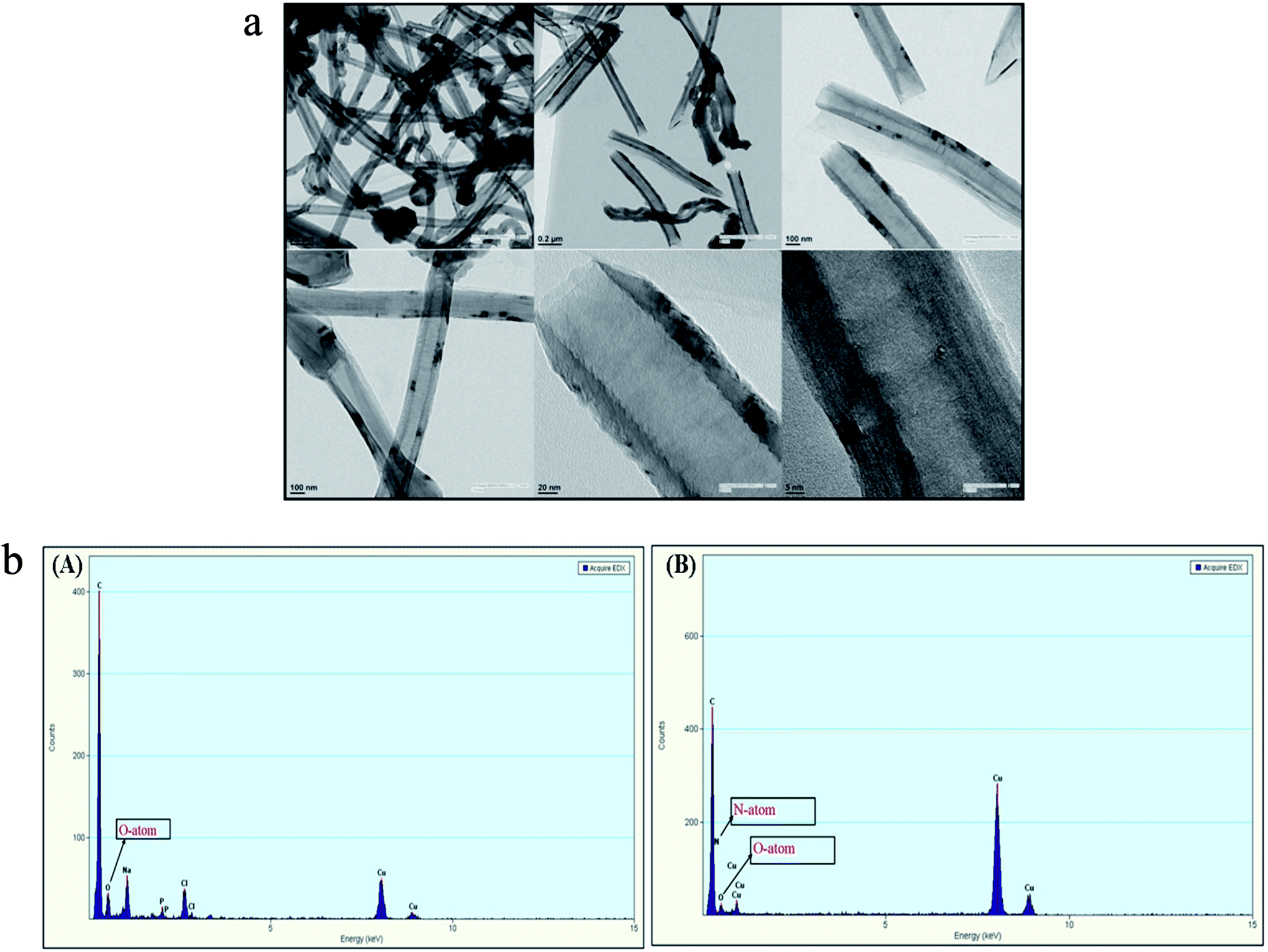
SDS-BCL@CNF nanobioconjugate was further characterized by TEM and EDX analysis. TEM image of functionalized carbon nanofiber (CNF-COOH) depicted fibrous structure of the nanofiber. In TEM image of SDS-BCL@CNF, surface texture of carbon nanofiber did not show any significant structural changes upon immobilization of SDS-BCL on carbon nanofibers (Fig. 4a). The functionalization and immobilization of enzyme on CNFs was also confirmed by an EDX diagram. The appearance of a peak of O atom validated the introduction of –COOH groups on the surface of the carbon nanofibers (CNF-COOH) [Fig. 4b(A)]. Similarly, appearance of N atom in EDX of immobilized enzyme preparation confirmed immobilization of enzyme on CNFs [Fig. 4b(B)].
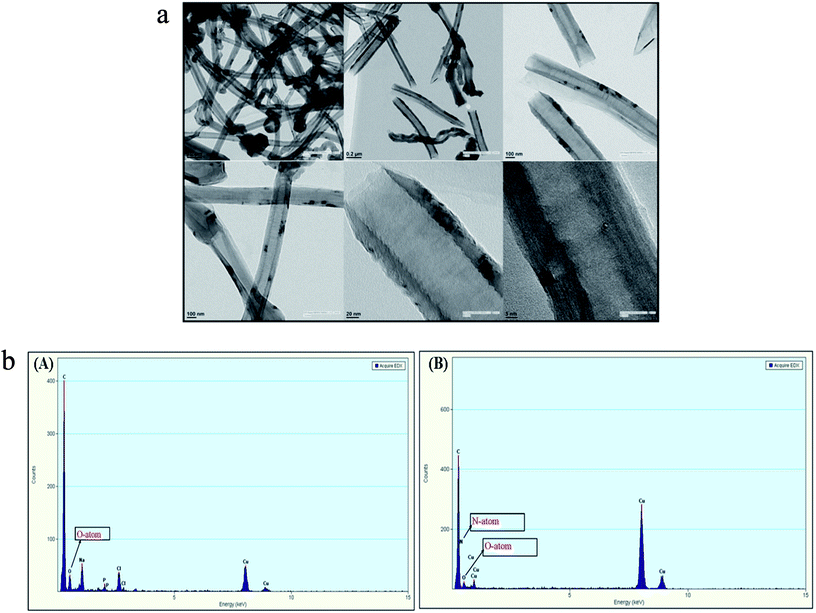 |
| | Fig. 4 Transmission electron microscopy (TEM) images of SDS-BCL@CNF nanobioconjugate. Energy dispersive X-ray diagram (EDX) of: (A) CNF-COOH and (B) SDS-BCL@CNF nanobioconjugate. | |
Thermodynamic study for free lipase and the nanobioconjugate (SDS-BCL@CNF)
Thermostability study was carried out for free and immobilized lipase by incubation at different temperatures ranging from 40 to 100 °C. The results showed that high temperatures led to almost total inactivation of free lipase due to disruption of the non-covalent bonds and hydrophobic interactions causing denaturation of the enzyme.65 In the range of 40–100 °C, the deactivation constant (k) for free lipase (BCL) was higher than lipase immobilized on carbon nanofibers (SDS-BCL@CNF), indicating that immobilization increased the thermal stability of lipase. The half-life of both free and immobilized lipase decreased with the increasing temperature, which showed that enzyme activity decreases at higher temperature. At 80 °C, the half-life (t1/2) of free lipase decreased substantially from 1386 min to 2 min and at 40 °C from 2310 min to 40 min for the immobilized lipase.
However, the half-life of the lipase in nanobioconjugate, at 80 °C was almost 20 folds higher than in the free form. Carbon nanofibers are robust nanomaterial and covalent attachment of the lipase on the carbon nanofibers decreases the vibration of the atoms caused by higher temperature, thereby preventing changes in the three dimensional structure of the lipase and maintaining the enzyme in active form.66 The activation energy of BCL in free and immobilized form was evaluated using the Arrhenius plot. The activation energy decreased from 150.8 kJ mol−1 in free lipase to 134.2 kJ mol−1 in SDS-BCL@CNF. Decrease in the activation energy for the nanopreparation is indicative of higher thermostability. For free lipase the decrease in the ΔG° value at higher temperature showed progressive destabilization of the enzyme molecules with temperature. For immobilized preparation, a gradual increase in the ΔG° value was observed upto 70 °C indicating better stabilization of the lipase in SDS-BCL@CNF. Moreover, high values (100.0 kJ mol−1) for free energy of thermal denaturation (ΔG°) at 70 °C indicated that the immobilized preparation was stable enough to prevent the thermal unfolding at higher temperatures. The enthalpy (ΔH) of deactivation did not show considerable variation within the range of temperature studied each, for free and immobilized lipase, respectively. The thermal denaturation of enzymes is accompanied with a concomitant increase in the enthalpy of activation (ΔH).67 Lowering of ΔH value from 148.2 kJ mol−1 in free lipase to 131.6 kJ mol−1 in lipase immobilized on carbon relates increase in the thermostability upon immobilization. The high value of entropy of activation for free enzyme revealed the opening up of enzyme structure and an increased disorder as compared to the nanobioconjugate, which showed comparatively lower values of entropy.68 Thermostability of enzymes in their native form can be increased by preventing the breakage of noncovalent bonds including hydrogen bonds, salt bridges, hydrophobic interactions or by decreasing the entropy of unfolding.67,69 Thus we observed that the nanobioconjugate was more thermostable than free lipase.
Reusability of the nanobioconjugate (SDS-BCL@CNF)
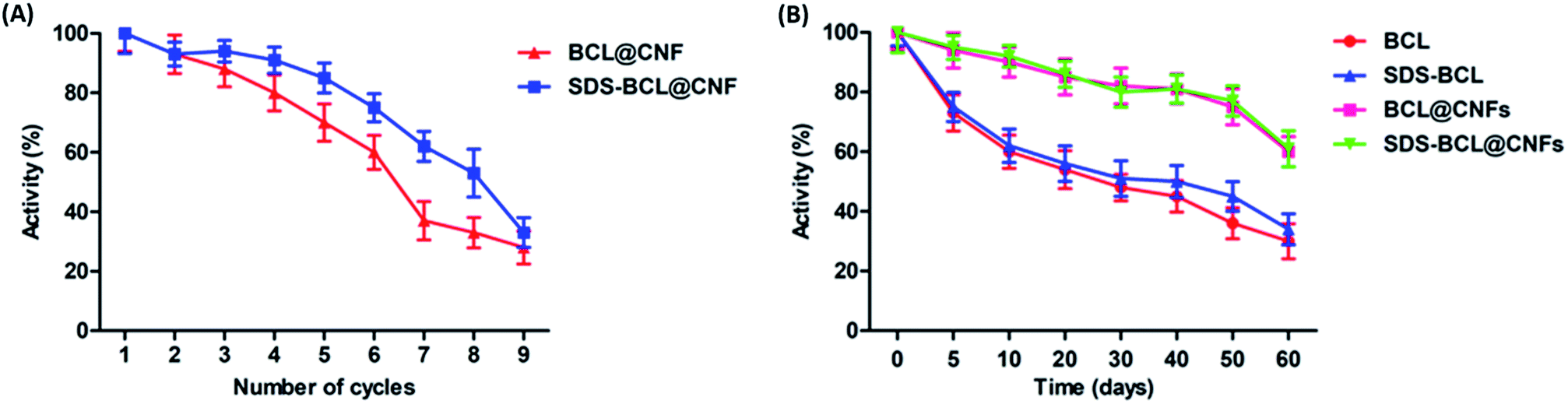
For any industrial application, the reusability of immobilized enzymes is a key factor for its cost-effective use. Thus we demonstrated the reusability of the SDS-BCL@CNF based on the hydrolysis of the p-NPP as a substrate. As shown in Fig. 5A, SDS-BCL@CNF retained 30% of the initial activity after nine cycles of use. After each cycle, activity in supernatant was checked where no enzymatic activity was observed. This study proves that loss in activity is because of other factors not due to desorption. The possible explanation could be unfavourable morphological changes and reaction environment lead to gradual decrease in activity. Apart from this fact, reuse of immobilized preparation upto 9 cycles with considerable activity is more economical rather than using free enzyme for single use.
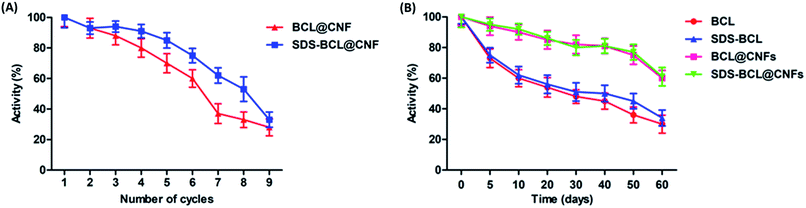 |
| | Fig. 5 (A) Reusability of the nanobioconjugates (BCL@CNF, SDS-BCL@CNF); (B) storage stability of free lipase and the nanobioconjugates (BCL@CNF, SDS-BCL@CNF). | |
Storage stability of free lipase and the nanobioconjugate
The storage stability of the free and immobilized lipase was investigated and the results are presented in Fig. 5B. The nanobioconjugate was stored at 4 °C for 60 days and samples withdrawn at regular intervals showed that the nanobioconjugate retained almost 60% of the initial activity. The free lipase showed a fast decrease in activity with time, on the other hand, decrease in activity of the nanobioconjugate was gradual and slower than the free lipase. Certainly the nanobioconjugates (BCL@CNF and SDS-BCL@CNF) were superior to the free lipase in the process of long storage. Covalent immobilization of the lipase on activated carbon nanofibers imparted robustness which appeared in the storage stability.
Kinetic properties of free lipase and lipase immobilized on activated carbon nanofibers (SDS-BCL@CNF)
The kinetic study of free and immobilized lipase was based on the evaluation of the kinetic parameters, KM and Vmax obtained from the initial reaction rates of p-nitrophenyl palmitate hydrolysis at different substrate concentrations, using free and immobilized lipase. The plot of p-nitrophenyl palmitate hydrolysis versus substrate concentration was hyperbolic, following the classical Michaelis–Menten model. This point was confirmed by the linearity of the Lineweaver–Burk plot. The kinetic parameters of the free and immobilized lipase are given in Table 1. The KM and Vmax values were calculated as 2.07 mM and 1.73 mmol min−1 g−1 for free enzyme and 0.15 mM and 1.77 mmol min−1 g−1 for immobilized lipase, respectively. These results depicted distinct behaviour of free and immobilized lipase. A 14-fold decrease in KM was obtained for immobilized lipase, which showed the increased affinity of the enzyme for the substrate and hence the increased enzyme activity upon immobilization. The catalytic constant (kcat) was estimated from the Vmax values using a molecular mass of 33 kDa. The specificity constant, kcat/KM increased about five times. The possible explanations for the decrease of KM could be because of favourable conformational change in the lipase upon immobilization on carbon nanofibers resulting in an increased substrate concentration in the microenvironment.
Table 1 Kinetic parameters for BCL and SDS-BCL@CNF nanoconjugate for the hydrolysis of p-nitrophenyl palmitate (p-NPP)
| S. No. |
Kinetic parameter |
Free lipase |
Immobilized lipase |
| 1 |
KM (mM) |
2.07 |
0.15 |
| 2 |
Vmax (mmol min−1 g−1) |
1.73 |
1.77 |
| 3 |
kcat (s−1) |
0.95 |
0.97 |
| 4 |
kcat/KM (s−1 mM−1) |
0.45 |
6.47 |
Application of the nanobioconjugate in kinetic resolution of (RS)-2-(4-(3-chloro-2-hydroxypropoxy) phenyl) acetamide: racemic atenolol intermediate
The SDS-BCL@CNF nanobioconjugate was successfully applied in the kinetic resolution of an important racemic secondary alcohol which is the intermediates of atenolol, a well-known β-blocker drug. SDS-BCL@CNF nanobioconjugate mediated kinetic resolution led to the formation of the (S)-form of the intermediate alcohol, while the (R)-form was acylated (Scheme 1). The same reaction was performed using BCL and surfactant treated BCL (SDS-BCL). BCL was slower than SDS-BCL@CNF and the reaction occurred with 35.4% conversion. The surfactant treated BCL (SDS-BCL) was also slower than SDS-BCL@CNF and the reaction occurred with 40.7% conversion, in contrast to the highest conversion rate of 47.8% obtained with the nanobioconjugate (SDS-BCL@CNF). Nevertheless, the ee value was comparable for all three lipase preparations remaining at 97%. The value of enantioselectivity of SDS-BCL@CNF mediated reaction increased about two times than free enzyme which captivated the usefulness of the immobilized preparation (Table 2). The selectivity of the preparations towards racemic alcohol was similar, where all of them favoured conversion of the R isomer to its corresponding ester. The result demonstrated that BCL immobilized on activated carbon nanofibers via covalent attachment was more active than soluble enzyme.
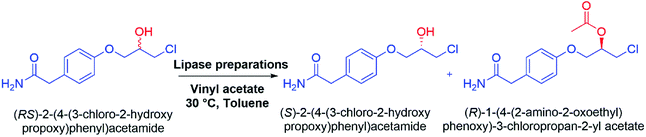 |
| | Scheme 1 Kinetic resolution of (RS)-2-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide: racemic atenolol intermediate using lipase preparations. | |
Table 2 Kinetic resolution of (RS)-2-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide: racemic atenolol intermediate using lipase preparations
| S. No. |
Lipase preparation |
Ca (%) |
eepb |
E-valuec |
| % Conversion were calculated from the enantiomeric excess (ee) of (S)-4 and (R)-5 as follows: conversion (C) = ees/(ees + eep). Enantiomeric excess of product was determined by HPLC analysis (Daicel Chiralcel OD-H column) 90:10; hexane:IPA, flow rate of 1.0 mL min−1, detected at 254 nm. E value were calculated using the formula: E = ln[(eep (1 − ees)/(eep + ees)]/ln[(eep (1 + ees)/(eep + ees)]. |
| 1 |
BCL |
35.4 |
97.1 |
88 |
| 2 |
SDS-BCL |
40.1 |
96.8 |
95 |
| 3 |
SDS-BCL@CNFs |
47.8 |
97.0 |
194 |
Reusability of the nanobioconjugate in the kinetic resolution of (RS)-2-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide: racemic atenolol intermediate
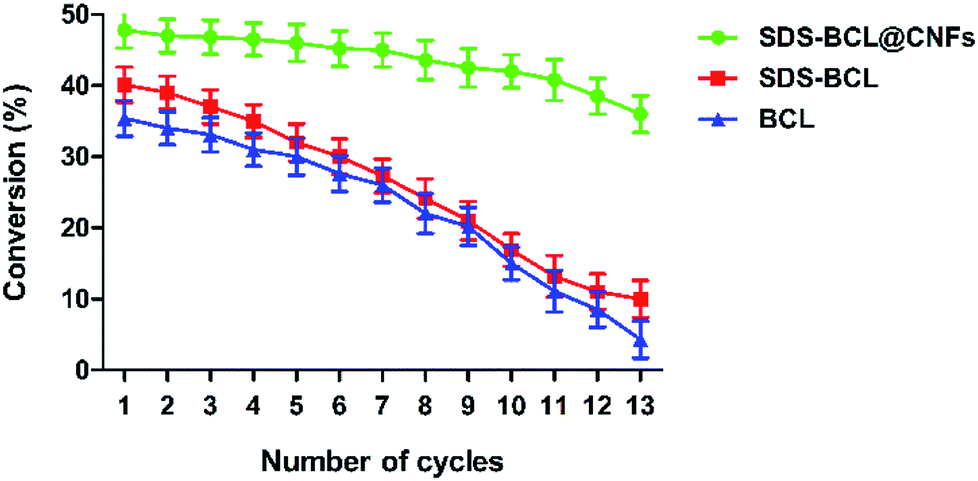
The reusability of the SDS-BCL@CNF nanobioconjugate was demonstrated in the kinetic resolution of (RS)-2-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide, which is a racemic inter-mediate of atenolol. As shown in Fig. 6, the reusability of SDS-BCL@CNF was demonstrated for thirteen cycles showing a good rate of conversion compared to the surfactant treated lipase and untreated lipase which depicted a gradual loss in the conversion rate. After reusing for thirteen cycles the nanobioconjugate showed a conversion rate of 40% which is an indicator of the increased robustness and operational stability that the immobilized preparation, thereby proving its utility as a nanobiocatalyst.
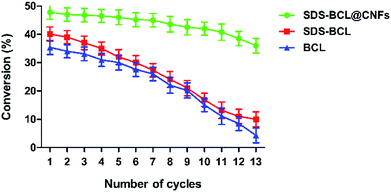 |
| | Fig. 6 Reusability of the lipase preparations (SDS-BCL@CNFs, SDS-BCL and BCL) in the kinetic resolution of (RS)-2-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide. | |
Comparative analysis of lipase activity in different preparations
A comparative study of the lipase activity presented by BCL in different forms revealed that under optimal conditions lipase immobilization through covalent attachment exhibited higher enzymatic activity. The method of adsorption also increased the enzymatic activity, but it was far less than the covalent attachment method (Table 3).
Table 3 Comparative analysis of lipase activity in different preparations
| S. No. |
Enzyme preparation |
Relative activity (%) |
Fold increase |
| 1 |
Untreated BCL |
100 |
1.0 |
| 2 |
SDS-BCL |
123 |
1.2 |
| 3 |
BCL@CNF (adsorption) |
425 |
4.3 |
| 4 |
SDS-BCL@CNF (covalent attachment) |
1125 |
11.3 |
Experimental
Materials
Carbon nanofiber (CNF), Burkholderia cepacia lipase (BCL), nitric acid, sulphuric acid, vinyl acetate, toluene, surfactants viz. [4-(1,1,3,3-tetramethylbutyl)phenyl-polyethylene glycol (Triton X-100), polyoxyethylene (20) sorbitan monooleate, n-octyl-β-D-glucoside (OG), polyethylene glycol 6000 (PEG6000), sodium dodecyl sulphate (SDS), dioctyl sulfosuccinate (DOSS), cetyl trimethyl ammonium bromide (CTAB), merpol], p-nitro-phenyl palmitate (p-NPP), 2-ethanesulfonic acid (MES), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS) were obtained from Sigma-Aldrich Corporation (St. Louis, MO, USA). HPLC grade solvents were obtained from Merck Ltd.
Surfactant treatment of Burkholderia cepacia lipase
Burkholderia cepacia lipase (10 mg) was dissolved in 1 mL Tris HCl (100 mM, pH 7.4) containing different surfactants (1% w/v). They were precipitated in 5 mL chilled ethanol for 30 minutes at 4 °C (200 rpm). The precipitate obtained was centrifuged at 12000 × g for 5 minutes. The supernatant was discarded and precipitate was then washed with chilled propanol to remove the surfactant.
Functionalization of carbon nanofiber
The pristine CNF were functionalized using the reported method with few modifications.14,70,71 In brief, a strong oxidizing agent H2SO4:HNO3 (3:1 v/v) was used for the generation of carboxylic acid groups on the surface of carbon nanofiber. Carbon nanofiber (50 mg) were dissolved in 100 mL acidic solution [H2SO4:HNO3 (75:25)] and sonicated for 4 h in a bath sonicator (40 kHz). The suspension was then diluted with distilled water, filtered through a polycarbonate membrane (0.45 μm) and washed with distilled water for several cycles until the complete removal of acids. Finally the samples were dried under vacuum at 50 °C and characterized through FTIR spectroscopy (Perkin-Elmer Spectrum GX IR spectrometer) to confirm surface functionalization.
Activation of functionalized carbon nanofiber (CNF-COOH)
For effective immobilization, a stable amide bond linkage is required between the enzyme and support material. EDC (a carbodiimide coupling agent) and NHS are extensively used for this purpose as coupling agents. In a typical process of activation, carboxylic acid group containing CNF (CNF-COOH, 10 mg) were dissolved in MES buffer (5 mL, pH 5.5) and sonicated with NHS (30 mg mL−1) for 1 h at 25 °C. It was followed by the addition of EDC (5 mg mL−1) and incubation for 1 h at 25 °C (200 rpm). Finally the suspension was washed with distilled water, filtered through a polycarbonate membrane (0.45 μm) and vacuum dried at 50 °C.14,70,71
Immobilization of lipase on carbon nanofibers for preparation of the nanobioconjugate
Two separate methods were employed for the immobilization of lipase on CNF. First, a simple adsorption was done in which pristine CNF (non-functionalized/non-activated) were dispersed in MES buffer (pH 5.5, 5 mL) and sonicated (40 kHz) for 1 h at 25 °C. Lipase solution (5 mL) was added after sonication and the suspension was incubated under stirring at 25 °C (150 rpm). Thereafter, the suspension was centrifuged at 10000 × g for 10 min, supernatant was separated and the nanobioconjugate was washed thrice with the same buffer to remove the loosely bound or unbound lipase. The nanobioconjugate was stored at 4 °C.
For the second method of covalent immobilization, freshly prepared activated-functionalized carbon nanofibers were dispersed in MES buffer (pH 5.5, 5 mL). Lipase solution (5 mL) was added right after activation and the suspension was incubated under stirring for given period of time at 25 °C (150 rpm). Thereafter, the suspension was centrifuged at 10000 × g for 10 min, supernatant was separated and the nanobioconjugate was washed thrice with the same buffer to remove the loosely bound or unbound lipase. The nanobioconjugate was stored at 4 °C.14,70,71 Both immobilization methods were carried out at different time period (1 h, 3 h, 6 h, 12 h, 18 h and 24 h) and enzyme support ratio (E/S = 0.25, 0.5, 0.75 and 1.5) to get optimized condition.
Determination of lipase loading on the nanobioconjugate
The amount of lipase bound on the surface of pristine CNF (through adsorption) and functionalized CNF (through EDC/NHS mediated covalent coupling) was determined. In a typical experiment the immobilized preparations were centrifuged and the supernatant was separated. After three subsequent washings, the supernatants were subjected to Bradford assay.72 All the experiments were carried out in triplicate.
Determination of lipase activity of the nanobioconjugate
The free lipase activity, as well as lipase immobilized on carbon nanofiber was evaluated using p-nitrophenyl palmitate (p-NPP) as a substrate using the standard assay (Winkler Stackmann method).73 The reaction mixture consisting of 90 μL of p-nitrophenyl palmitate (p-NPP) in 810 μL solution B [solution of 0.11% (w/v) gum arabic and 0.4(v/v) triton X-100], 1.5 mL Tris–HCl buffer (0.05 M, pH 7.0) and 500 μL calcium chloride (75 mM) solution, was incubated at 60 °C for 5 minutes followed by the addition of 0.1 mL lipase preparation. Reaction was incubated for 10 min at 60 °C and stopped by addition of 0.5 N Na2CO3 followed by centrifugation at 10000 × g for 10 min. Absorbance of the supernatant was measured at 410 nm in a UV-spectrophotometer (Hitachi U-3010, Japan). Enzyme blank and substrate blank study was performed along with control to nullify the effects of other factors. Exact concentration of enzyme preparation (free/immobilized) was taken for activity measurement.
Determination of kinetic parameters for free and immobilized preparation
To determine the kinetic parameters of free lipase, enzymatic activity was measured for concentrations of p-nitrophenyl palmitate ranging from 0.2 to 2.0 mM, according to the standard assay method. Similarly, the kinetic parameters for immobilized lipase were determined measuring the enzymatic activity, for the catalysis of p-nitrophenyl palmitate at concentrations ranging from 0.2 to 2.0 mM. Lineweaver–Burk plot was used to determine the kinetic constants, viz. Vmax the maximum rate of the reaction, and KM, the Michaelis–Menten constant.74
Characterization of the nanobioconjugate
Fourier-transform infrared spectroscopy (FTIR) spectra were measured using a Perkin-Elmer Spectrum spectrometer in the range 4000–600 cm−1, in the ATR mode with 10 times accumulation. The UV-CD spectra (190–260 nm) were recorded on a JASCO J-810 CD instrument (JASCO) with a bandwidth of 0.5 nm and a scan speed of 50 nm min−1. All CD measurements were performed at 25 °C. All the spectra were corrected by subtracting a blank spectrum (without lipase). Each scan was repeated three times, and the spectra were then averaged out. Spectra manager software was used to analyze the protein CD spectra for determining the secondary structure fractions. Transmission electron microscopy (TEM, Tecnai 20, USA) was used to observe the changes in the surface texture of CNF before and after functionalization. For this, CNF samples were dispersed in demineralized water and a drop was placed on a copper grid and observed after drying.
Thermodynamic study for free lipase and the nanobioconjugate
The thermostability of lipase in free and immobilized form was investigated by incubating 10 mg of each preparation in Tris buffer (0.05 M, pH 8) at different temperatures ranging from 40 to 100 °C for 6 h. A sample was taken regularly from each reaction and after cooling the enzymatic activity was determined under standard assay conditions. The inactivation rate constant (k) was calculated as a function of temperature in the range 40–100 °C, described as the following equation:
where, where A/A0 is the residual enzyme activity at time t (min), and k (min−1) is the inactivation rate constant at a determined temperature.75
Half-life (t1/2) of inactivation is calculated as:
The Arrhenius plot was utilized to determine the energetic barrier (activation energy, Ea) for the thermal enzyme denaturation process using the following equation:
where,
k0 is the Arrhenius constant,
Ea is the activation energy,
T is the temperature (in kelvin) and
R is the universal gas constant (8.314 J mol
−1 K
−1). The
Ea was estimated from the slope of linear regression analysis of the natural logarithm of rate constant,
k versus the reciprocal of absolute temperature (1/
T).
66 The rate constant of thermal inactivation,
k was also used to determine the free energy of deactivation (Δ
G°) at different temperatures, using the following equation:
where,
R is the universal gas constant (8.314 J mol
−1 K
−1),
T is the temperature (in kelvin),
h is the Planck's constant (6.626176 × 10
−34 J s),
K is the Boltzmann's constant (1.38064852 × 10
−23 J K
−1) and
k (min
−1) the inactivation rate constant of each temperature.
65 The activation enthalpy (Δ
H) at different temperature was calculated from the activation energy using the following equation:
The activation entropy (ΔS) was calculated from the enthalpy and standard free energy as:
Determination of reusability of the nanobioconjugate
The reusability of SDS-BCL@CNF was assessed after the repeated cycles of typical lipase reaction (1 mg mL−1 lipase SDS-BCL@CNF in Tris buffer) and 3 mg mL−1 solution of p-nitrophenyl palmitate in 2-propanol for 10 min at 60 °C). The nanobioconjugate was recovered by centrifugation at 12000 × g for 5 min, and excessive washing with Tris–HCl buffer (0.05 M, pH 8.0). After each cycle, the nanobioconjugate was separated out from the reaction mixture by centrifugation and rinsed with Tris–HCl buffer (0.05 M, pH 8.0). For the next cycle the washed nanobioconjugate was resuspended in fresh buffer and added to fresh solution of p-nitrophenyl palmitate in 2-propanol. Relative activity (%) was calculated as the ratio of residual lipase activity to initial lipase activity.
Storage stability of free lipase and the nanobioconjugate
The storage stability of free lipase (BCL), surfactant treated lipase (SDS-BCL), BCL@CNF and SDS-BCL@CNF was determined by incubating 10 mg of each preparation in Tris-buffer (0.05 M, pH 8.0) and determining the activity for 60 days at regular time intervals.
Kinetic resolution of racemic atenolol intermediate
The racemic alcohol (20 mM, 1 equivalent) was subjected to transesterification with vinyl acetate (acyl donor, 100 μL) in toluene (900 μL) using free lipase (BCL), surfactant treated lipase (SDS-BCL) and the nanobioconjugate (SDS-BCL@CNF). All the lipase preparations were individually added; flasks were tightly capped and placed in shaker at 30 °C (200 rpm). After 24 h, the reaction mixture was extracted with toluene, centrifuged at 10000 × g for 5 min and the supernatant was evaporated under vacuum. Samples were analysed by HPLC (Shimadzu, Japan), enantiomeric excess and enantiomeric ratio was determined. The substrate and product were detected at 254 nm. Enantioselectivity, expressed in terms of E value; substrate enantiomeric excess (ees); rate of conversion (C) were calculated.
(S)-2-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide. 1H NMR (400 MHz; DMSO-d6; Me4Si) δ (ppm) 3.27 (s, 2H, –CH2Cl), 3.68 (m, 2H, –COCH2), 3.95 (d, J = 4.7 Hz, 2H, –OCH2), 4.01 (d, J = 5.1 Hz, 1H, –CHOH), 5.6 (d, J = 5.2 Hz, 1H, =COH), 6.84 (d, J = 8.5 Hz, 2H, ArH), 7.16 (d, J = 8.6 Hz, 2H, ArH), 7.43 (2H, br s, −NH2). Chiral HPLC analysis: OD-H column (0.46 mm dia., 250 mm long, 5 μm, Chiralcel), tS = 40.0 min, hexane:2-propanol (83:17).
(R)-1-(4-(2-amino-2-oxoethyl)phenoxy)-3-chloropropan-2-yl-acetate. 1H NMR (400 MHz; CDCl3; Me4Si) δ (ppm) 2.12 (m, 3H, −COCH3), 3.50 (s, 2H, –CH2Cl), 3.84 (m, 2H, −COCH2), 4.15 (m, 2H,–OCH2), 5.32 (m, 1H, −CHOAc), 5.56 (s, br, 1H, NH), 6.02 (s, br, 1H, NH), 6.91 (m, 2H, ArH), 7.22 (m, 2H, ArH). Chiral HPLC analysis: OD-H column (0.46 mm dia., 250 mm long, 5 μm, Chiralcel). tR = 16.6 min, hexane:2-propanol (83:17).
Conclusions
Immobilization of surfactant treated Burkholderia cepacia lipase on carbon nanofibers was attempted for the first time. A comparative analysis of the results obtained from both the immobilization studies (adsorption and covalent attachment) stated the precedence of covalent attachment over adsorption. The as-prepared nanobioconjugate allowed for both multiple cycles of operation (>nine cycles) and increased thermal stabilization (80 °C) of lipase preparation. The nanobioconjugate showed improved kinetic resolution of racemic intermediates of atenolol with a substantial reusability upto 13 cycles. Carbon nanofibers appear very promising immobilization matrix for the development of series of highly stable and industrially applicable nanobioconjugates using different class of industrially important enzymes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
B. P. D. acknowledges NIPER, Mohali for sponsoring his graduate research fellowship. S. S. acknowledges the Department of Science and Technology (DST, Govt. of India) for providing research fellowship under DST-INSPIRE fellowship.
References
- R. N. Patel, ACS Catal., 2011, 1, 1056–1074 CrossRef
 .
. - B. M. Nestl, B. A. Nebel and B. Hauer, Curr. Opin. Chem. Biol., 2011, 15, 187–193 CrossRef PubMed .
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef .
- R. C. Rodrigues, C. Ortiz, Á. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC .
- S. Cantone, V. Ferrario, L. Corici, C. Ebert, D. Fattor, P. Spizzo and L. Gardossi, Chem. Soc. Rev., 2013, 42, 6262 RSC .
- R. DiCosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437 RSC .
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC .
- A. Liese and L. Hilterhaus, Chem. Soc. Rev., 2013, 42, 6236 RSC .
- R. C. Rodrigues, Á. Berenguer-Murcia and R. Fernandez-Lafuente, Adv. Synth. Catal., 2011, 353, 2216–2238 CrossRef .
- K. Min and Y. J. Yoo, Biotechnol. Bioprocess Eng., 2014, 19, 553–567 CrossRef .
- E. T. Hwang and M. B. Gu, Eng. Life Sci., 2013, 13, 49–61 CrossRef .
- E. P. Cipolatti, A. Valério, R. O. Henriques, D. E. Moritz, J. L. Ninow, D. M. G. Freire, E. A. Manoel, R. Fernandez-Lafuente and D. de Oliveira, RSC Adv., 2016, 6, 104675–104692 RSC .
- H. Vaghari, H. Jafarizadeh-Malmiri, M. Mohammadlou, A. Berenjian, N. Anarjan, N. Jafari and S. Nasiri, Biotechnol. Lett., 2016, 38, 223–233 CrossRef PubMed .
- I. V. Pavlidis, T. Tsoufis, a. Enotiadis, D. Gournis and H. Stamatis, Adv. Eng. Mater., 2010, 12, 179–183 CrossRef .
- P. Serp, M. Corrias and P. Kalck, Appl. Catal., A, 2003, 253, 337–358 CrossRef .
- N. Chaniotakis, V. Vamvakaki, K. Tsagaraki and N. Chaniotakis, Anal. Chem., 2017, 78, 5538–5542 Search PubMed .
- T. Raghavendra, A. Basak, L. M. Manocha, A. R. Shah and D. Madamwar, Bioresour. Technol., 2013, 140, 103–110 CrossRef PubMed .
- S. N. Patel, M. Sharma, K. Lata, U. Singh, V. Kumar, R. S. Sangwan and S. P. Singh, Bioresour. Technol., 2016, 216, 121–127 CrossRef PubMed .
- D. Muñoz Solano, P. Hoyos, M. J. Hernáiz, A. R. Alcántara and J. M. Sánchez-Montero, Bioresour. Technol., 2012, 115, 196–207 CrossRef PubMed .
- C. Lei, Y. Shin, J. Liu and E. J. Ackerman, J. Am. Chem. Soc., 2002, 124, 11242–11243 CrossRef PubMed .
- B. P. Dwivedee, J. Bhaumik, S. K. Rai, J. K. Laha and C. Banerjee, Bioresour. Technol., 2017, 239, 464–471 CrossRef PubMed .
- L. A. DeLouise and B. L. Miller, Anal. Chem., 2005, 76, 6915–6920 CrossRef PubMed .
- J. Wang, Q. Chen, C. L. Renschler and C. White, Anal. Chem., 1994, 66, 1988–1992 CrossRef .
- I. V. Pavlidis, T. Vorhaben, T. Tsoufis, P. Rudolf, U. T. Bornscheuer, D. Gournis and H. Stamatis, Bioresour. Technol., 2012, 115, 164–171 CrossRef PubMed .
- S. Sotiropoulou, V. Ga, V. Vam and N. A. Chaniotakis, Biosens. Bioelectron., 2003, 18, 211–215 CrossRef PubMed .
- M. Endo, Y. A. Kim, T. Hayashi, Y. Fukai, K. Oshida, M. Terrones, T. Yanagisawa, S. Higaki and M. S. Dresselhaus, Appl. Phys. Lett., 2002, 80, 1267–1269 CrossRef .
- V. I. Merkulov, D. K. Hensley, a V. Melechko, M. a Guillorn, D. H. Lowndes and M. L. Simpson, J. Phys. Chem. B, 2002, 106, 10570–10577 CrossRef .
- W. Yang, O. Auciello, J. Butler, W. Cai, J. Carlisle, J. Gerbi, D. Gruen, T. Knickerbocker, T. Lasseter, J. J. Russell, L. Smith and R. Hamers, Nat. Mater., 2002, 1, 253–257 CrossRef PubMed .
- M. Lu, T. Knickerbocker, W. Cai, W. Yang, R. J. Hamers and L. M. Smith, Biopolymers, 2004, 73, 606–613 CrossRef PubMed .
- D. A. Sánchez, G. M. Tonetto and M. L. Ferreira, Biotechnol. Bioeng., 2018, 115, 6–24 CrossRef PubMed .
- P. Trodler, R. D. Schmid and J. Pleiss, BMC Struct. Biol., 2009, 9, 38 CrossRef PubMed .
- H. González-Navarro and L. Braco, J. Mol. Catal. B: Enzym., 1997, 3, 111–119 CrossRef .
- M. G. Pereira, S. Velasco-Lozano, S. Moreno-Perez, A. M. Polizeli, P. R. Heinen, F. D. A. Facchini, A. C. Vici, M. Cereia, B. C. Pessela, G. Fernandez-Lorente and J. M. Guisan, Molecules, 2018, 22, 1448 CrossRef PubMed .
- R. C. Rodrigues, C. Ortiz, Á. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC .
- F. Kartal, Biotechnol. Prog., 2016, 32, 899–904 CrossRef PubMed .
- J. Gao, L. Yin, K. Feng, L. Zhou, L. Ma, Y. He, L. Wang and Y. Jiang, Ind. Eng. Chem. Res., 2016, 55, 11037–11043 CrossRef .
- X. Cao, J. Yang, L. Shu, B. Yu and Y. Yan, Process Biochem., 2009, 44, 177–182 CrossRef .
- T. Liu, Y. Liu, X. Wang, Q. Li, J. Wang and Y. Yan, J. Mol. Catal. B: Enzym., 2011, 71, 45–50 CrossRef .
- E. Yilmaz, World J. Microbiol. Biotechnol., 2002, 18, 621–625 CrossRef .
- B. Anroop, B. Ghosh, V. Parcha, A. Kumar and J. Khanam, J. Drug Delivery Sci. Technol., 2005, 15, 187–190 CrossRef .
- S. R. Mehta, B. M. Bhawal, V. H. Deshpande and M. K. Gurjar, Process for Producing Atenolol of High Optical Purity, US Pat. 6982349B1, 2006 .
- R. A. McCoy, G. D. Clifton, W. A. Clementi, M. D. Smith, T. Q. Garvey, D. P. Wermeling and S. E. Schwartz, J. Clin. Pharmacol., 1994, 34, 816–822 CrossRef PubMed .
- O. Barbosa, C. Ortiz, R. Torres and R. Fernandez-Lafuente, J. Mol. Catal. B: Enzym., 2011, 71, 124–132 CrossRef .
- S. V. Darnle, P. N. Patil and M. M. Salunkhe, Synth. Commun., 1999, 29, 3855–3862 CrossRef .
- B. P. Dwivedee, S. Ghosh, J. Bhaumik, L. Banoth and U. Chand Banerjee, RSC Adv., 2015, 5, 15850–15860 RSC .
- R. D. Schmid and R. Verger, Angew. Chem., Int. Ed., 1998, 37, 1608–1633 CrossRef PubMed .
- R. J. Kazlauskas and H. K. Weber, Curr. Opin. Chem. Biol., 1998, 2, 121–126 CrossRef PubMed .
- K. Jaeger and T. Eggert, Curr. Opin. Biotechnol., 2002, 13, 390–397 CrossRef PubMed .
- S. Barbe, V. Lafaquière, D. Guieysse, P. Monsan, M. Remaud-Siméon and I. André, Proteins: Struct., Funct., Bioinf., 2009, 77, 509–523 CrossRef PubMed .
- I. Mingarro, C. Abad and L. Braco, Proc. Natl. Acad. Sci. U.S.A, 1995, 92, 3308–3312 CrossRef .
- M. Martinelle, M. Holmquist and K. Hult, Biochim. Biophys. Acta, 1995, 1258, 272–276 CrossRef .
- D. E. Otzen, Biophys. J., 2002, 83, 2219–2230 CrossRef PubMed .
- A. S. Antipova, M. G. Semenova, L. E. Belyakova and M. M. Il’in, Colloids Surf., B, 2001, 21, 217–230 CrossRef PubMed .
- F. Secundo, Chem. Soc. Rev., 2013, 42, 6250 RSC .
- L. Ci, Y. Li, B. Wei, J. Liang, C. Xu and D. Wu, Mater. Lett., 2000, 43, 291–294 CrossRef .
- T. Beechem and K. Lafdi, Carbon, 2006, 44, 1548–1559 CrossRef .
- I. D. Rosca, F. Watari, M. Uo and T. Akasaka, Carbon, 2005, 43, 3124–3131 CrossRef .
- G.-W. Lee, J. Kim, J. Yoon, J.-S. Bae, B. C. Shin, I. S. Kim, W. Oh and M. Ree, Thin Solid Films, 2008, 516, 5781–5784 CrossRef .
- Y. T. Shieh, G. L. Liu, H. H. Wu and C. C. Lee, Carbon, 2007, 45, 1880–1890 CrossRef .
- Y.-T. Shieh, J.-Y. Chen, Y.-K. Twu and W.-J. Chen, Polym. Int., 2012, 61, 554–559 CrossRef .
- W. Zhao, C. Song and P. E. Pehrsson, J. Am. Chem. Soc., 2002, 124, 12418–12419 CrossRef PubMed .
- C. Z. Dinu, G. Zhu, S. S. Bale, G. Anand, P. J. Reeder, K. Sanford, G. Whited, R. S. Kane and J. S. Dordick, Adv. Funct. Mater., 2010, 20, 392–398 CrossRef .
- P. Asuri, S. S. Karajanagi, H. Yang, T. J. Yim, R. S. Kane and J. S. Dordick, Langmuir, 2006, 22, 5833–5836 CrossRef PubMed .
- A. S. Campbell, C. Dong, J. S. Dordick and C. Z. Dinu, Process Biochem., 2013, 48, 1355–1360 CrossRef .
- M. H. Rashid and K. S. Siddiqui, Process Biochem., 1998, 33, 109–115 CrossRef .
- A. P. M. Tavares, C. G. Silva, G. Drazic, A. M. T. Silva, J. M. Loureiro and J. L. Faria, J. Colloid Interface Sci., 2015, 454, 52–60 CrossRef PubMed .
- R. M. Daniel, Enzyme Microb. Technol., 1996, 19, 74–79 CrossRef .
- C. Vieille and J. Zeikus, Trends Biotechnol., 1996, 14, 183–190 CrossRef .
- M. Saleem, M. H. Rashid, A. Jabbar, R. Perveen, A. M. Khalid and M. I. Rajoka, Process Biochem., 2005, 40, 849–855 CrossRef .
- M. Holzinger and A. Hirsch, J. Am. Chem. Soc., 2002, 124, 760–761 CrossRef .
- S. Li, Y. Qin, J. Shi, Z. Guo and Y. Li, Chem. Mater., 2005, 17, 130–135 CrossRef .
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef PubMed .
- N. Gupta, P. Rathi and R. Gupta, Anal. Biochem., 2002, 311, 98–99 CrossRef PubMed .
- G. Pencreach, M. Leullier and J. C. Baratti, Biotechnol. Bioeng., 1997, 56, 181–189 CrossRef .
- V. Sant'Anna, F. Cladera-Olivera and A. Brandelli, Food Chem., 2012, 130, 84–89 CrossRef .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05463k |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b