
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing biomedically relevant cell and lectin type-dependent structure–activity profiles for glycoclusters by using tissue sections as an assay platform†
Herbert Kaltnera,
Joachim C. Manninga,
Gabriel García Caballeroa,
Claudia Di Salvob,
Adele Gabbab,
Laura L. Romero-Hernándezb,
Clemens Knospe c,
Dan Wud,
Harrison C. Dalyd,
Donal F. O'Shead,
Hans-Joachim Gabiusa and
Paul V. Murphy*b
c,
Dan Wud,
Harrison C. Dalyd,
Donal F. O'Shead,
Hans-Joachim Gabiusa and
Paul V. Murphy*b
aInstitute of Physiological Chemistry, Faculty of Veterinary Medicine, Ludwig-Maximilians-University Munich, Veterinärstr. 13, 80539 Munich, Germany
bSchool of Chemistry, National University of Ireland Galway, University Road, Galway, Ireland. E-mail: paul.v.murphy@nuigalway.ie
cInstitute of Anatomy, Histology and Embryology, Faculty of Veterinary Medicine, Ludwig-Maximilians-University Munich, Veterinärstr. 13, 80539 Munich, Germany
dDepartment of Chemistry, Royal College of Surgeons of Ireland (RCSI), 123 St. Stephen's Green, Dublin 2, Ireland
First published on 14th August 2018
Abstract
The increasing realization of the involvement of lectin-glycan recognition in (patho)physiological processes inspires envisioning therapeutic intervention by high-avidity/specificity blocking reagents. Synthetic glycoclusters are proving to have potential for becoming such inhibitors but the commonly used assays have their drawbacks to predict in vivo efficacy. They do not represent the natural complexity of (i) cell types and (ii) spatial and structural complexity of glycoconjugate representation. Moreover, testing lectins in mixtures, as present in situ, remains a major challenge, giving direction to this work. Using a toolbox with four lectins and six bi- to tetravalent glycoclusters bearing the cognate sugar in a model study, we here document the efficient and versatile application of tissue sections (from murine jejunum as the model) as a platform for routine and systematic glycocluster testing without commonly encountered limitations. The nature of glycocluster structure, especially core and valency, and of protein features, i.e. architecture, fine-specificity and valency, are shown to have an influence, as cell types can differ in response profiles. Proceeding from light microscopy to monitoring by fluorescence microscopy enables grading of glycocluster activity on individual lectins tested in mixtures. This work provides a robust tool for testing glycoclusters prior to considering in vivo experiments.
Introduction
The realization of the unsurpassed capacity of glycans to store biological information in a minimum of space enables elucidating the chain of molecular events, from oligosaccharide synthesis and establishing the spatial aspects of their presentation on glycoconjugates and membranes to actual cellular effects, to become a staple of current research.1 Shaping a paradigm for this area, these efforts disclosed that reading of sugar-encoded messages by tissue lectins is an efficient means to facilitate molecular bridging. When occurring between cells and also between cells and the extracellular matrix (in trans), it can lead to adhesion.2–5 Cross-linking of constituents within a membrane (in cis) will form so-called glycoconjugate-lectin lattices that can trigger ensuing signalling.6–10 In each case, the intimate interplay of structural and topological parameters on both sides minimises stabilisation of random contacts, thus underlying the functional pairing of a lectin with not just any glycoconjugate, but with its matching counterreceptor(s). Hereby, glycan-encoded messages are read and then translated into particular post-binding responses. This theme of mutually specific protein(lectin)–glycan recognition appears to have a broad physiological significance.11–14Naturally, these emerging insights give strong incentive to search for compounds that will block this type of interaction if clinically unfavourable, and mimetics of polyvalent glycans termed glycoclusters have already been found to be particularly active.15–18 Besides their design and synthesis, a central challenge to master toward a prospect of biomedical application is the experimental set-up to measure inhibitory capacity. Optimally, it should come as closely as possible to the in vivo situation (in animals and man) with its diversity on the levels of glycans presented by glycoconjugates19–22 and of cell types, at the same time allowing for wide-scale testing in a robust manner with a clear read-out. The experimental approach to meet this challenge presented herein is a logical extension of classical lectin histochemistry.
Given their binding to certain glycans, labelled lectins from plants have acquired popularity as reliable and sensitive tools to map the distribution of their ligands in sections of any type of tissue.23–28 What commonly is a routine specificity control, i.e. the inhibition of lectin binding to cells in a tissue section by a sugar (mono- to oligosaccharide),24,27,29 has the attractive potential to be adapted to an assay for glycocluster efficacy, fulfilling the requirements for physiological relevance listed above. By assembling a toolbox of (i) seven mono- to tetravalent N-acetyl-D-galactosamine (GalNAc)-presenting compounds and (ii) four lectins that share nominal specificity to a monosaccharide (GalNAc), among them a human protein that binds to GalNAc presented by mucins (for an overview on mucins, please see30), and then working with these two sets to systematically monitor staining and intensity profiles in sections of a model tissue, i.e. murine jejunum, by light microscopy, we provide the proof-of-principle case study to document this assay's applicability. In fact, it opens the route to activity grading for any compound and any lectin on standard tissue specimens, offering the possibility for having natural binding partners act on both sides. Considering the occurrence of endogenous lectins in mixtures in vivo, a highly relevant situation for glycocluster testing, we also succeeded in adapting the assay to determine the individual behaviour of lectins when tested in combination. Toward this end fluorescence microscopy at different wavelengths was performed.
Results and discussion
The toolbox: 1. glycoclusters and control compounds
The panel of glycocluster-based inhibitors comprises six bi- to tetravalent compounds (1–6), all presenting GalNAc with α-anomeric linkages as thioglycosides (Fig. 1). The parameters of scaffold structure and valency are independently varied within this group. Compounds 3 and 5 differ in valency but share scaffold structure; the three pairs of compounds, i.e. 1 and 6, 2 and 3 as well as 4 and 5, share valency but differ in scaffold structure. The group of test compounds is completed by adding an the α-GalNAc derivative 7 and two control compounds (8, 9) with the same bulky cores of compound 5 and 6, but presenting a non-cognate sugar, GlcNAc.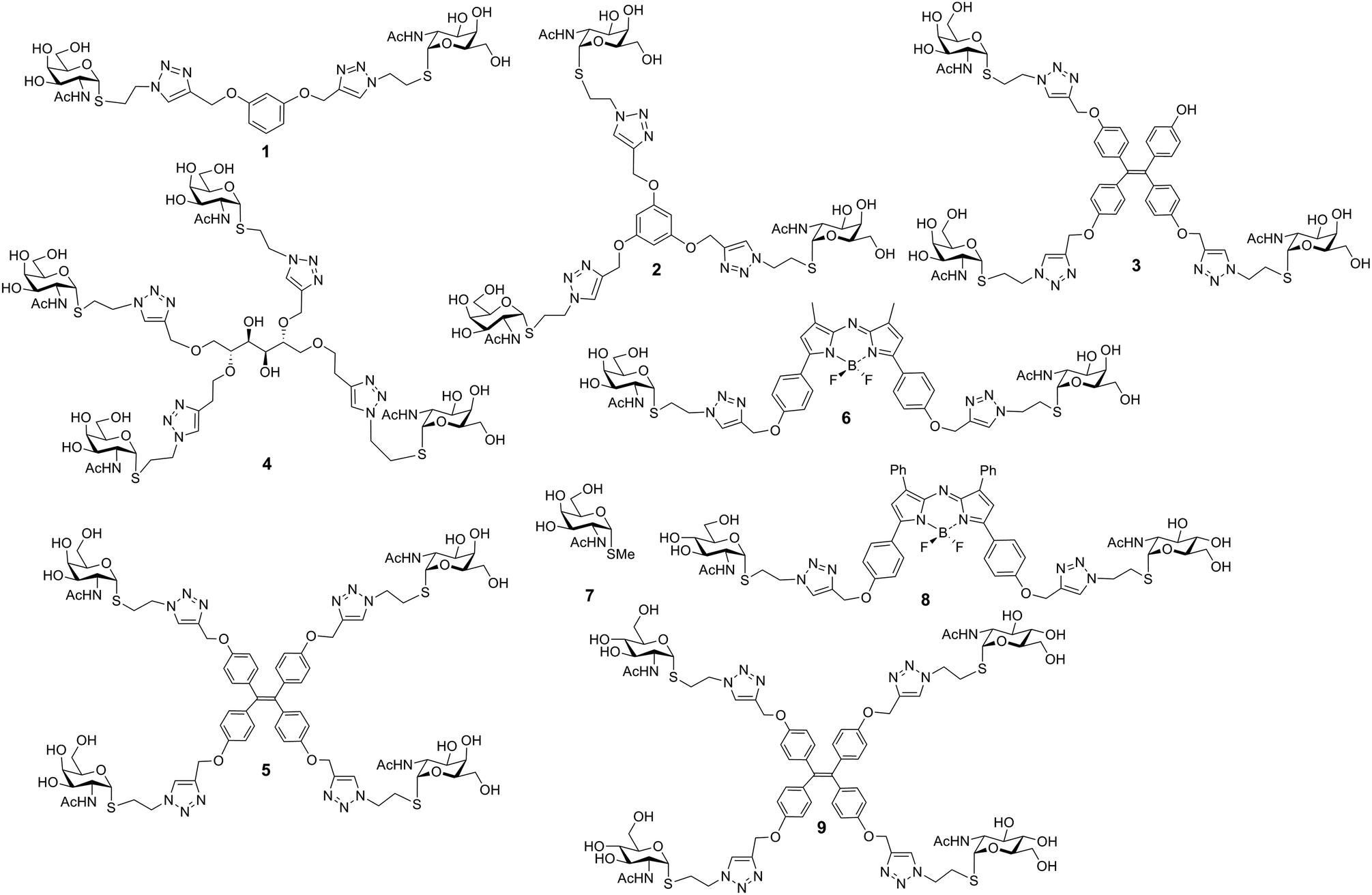 | ||
| Fig. 1 Structures of glycoclusters 1–6 (with GalNAc as headgroup) and 8, 9 (with GlcNAc) as well as of the monovalent α-anomeric thioglycoside derivative 7. | ||
The synthesis of glycoclusters 1, 2 and 5 has been described previously.31 The route to obtaining trivalent compound 3 with its tetraphenylethylene core is shown in Scheme 1. It commenced from the tetraphenylethylene derivative 10.31 Monoacetylation using acetic anhydride in limiting quantity (0.7 equiv.) followed by propargylation of the remaining phenols gave 11. Next, the copper-catalysed azide–alkyne cycloaddition (CuAAC) between the known azide 12 was performed, followed by deacetylation of the product to give 3, in which three of four positions are occupied by a sugar headgroup (in comparison to tetravalent 5).
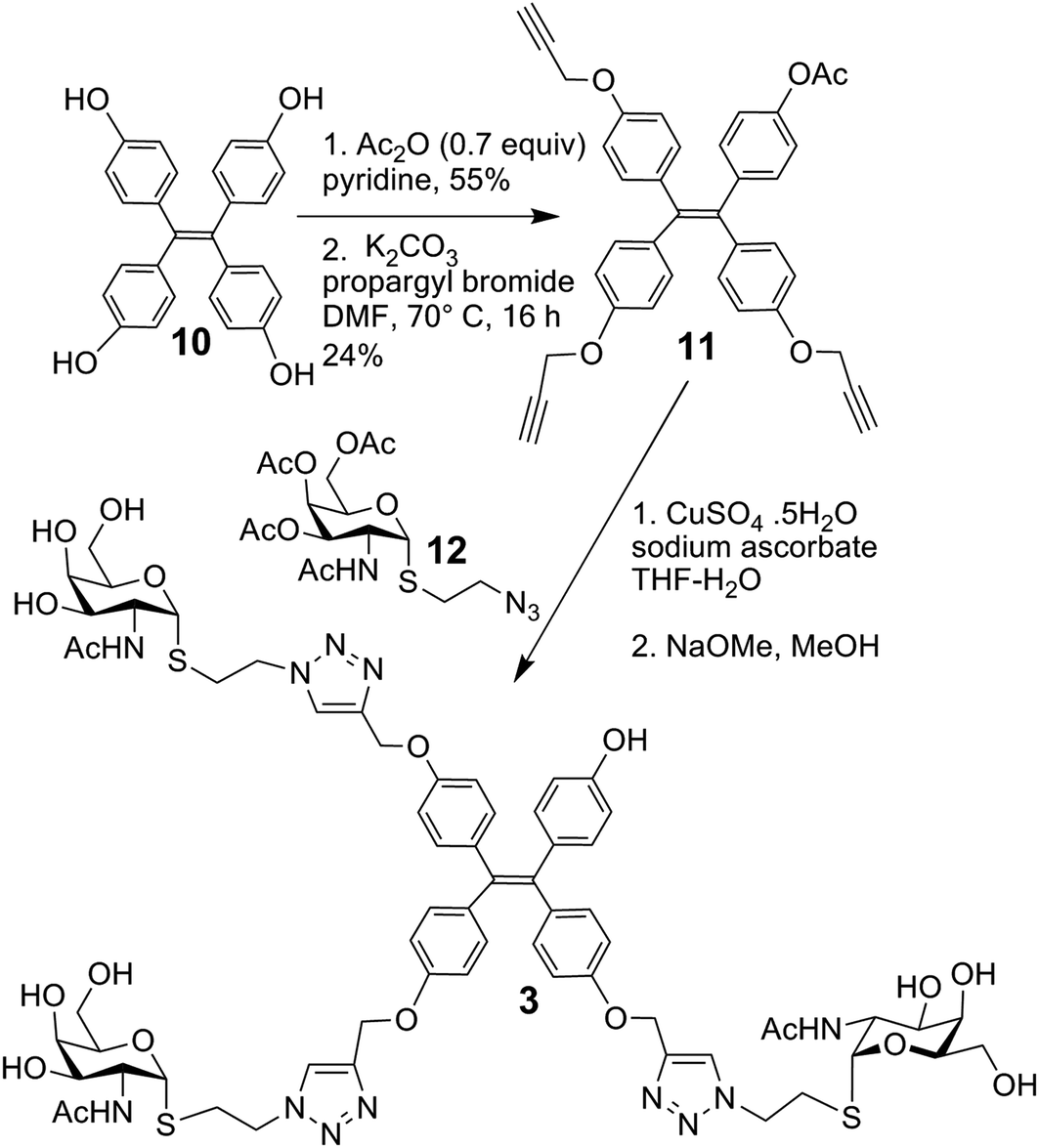 | ||
| Scheme 1 Synthesis of 3. | ||
The synthesis of compound 4, using conjugation sites on D-mannitol to implement tetravalency, started with 13 and is summarised in Scheme 2.32 Tetrapropargylation was followed by removal of the acetonide group, resulting in 14. Next, CuAAC with 12 and subsequent deacetylation generated 4.
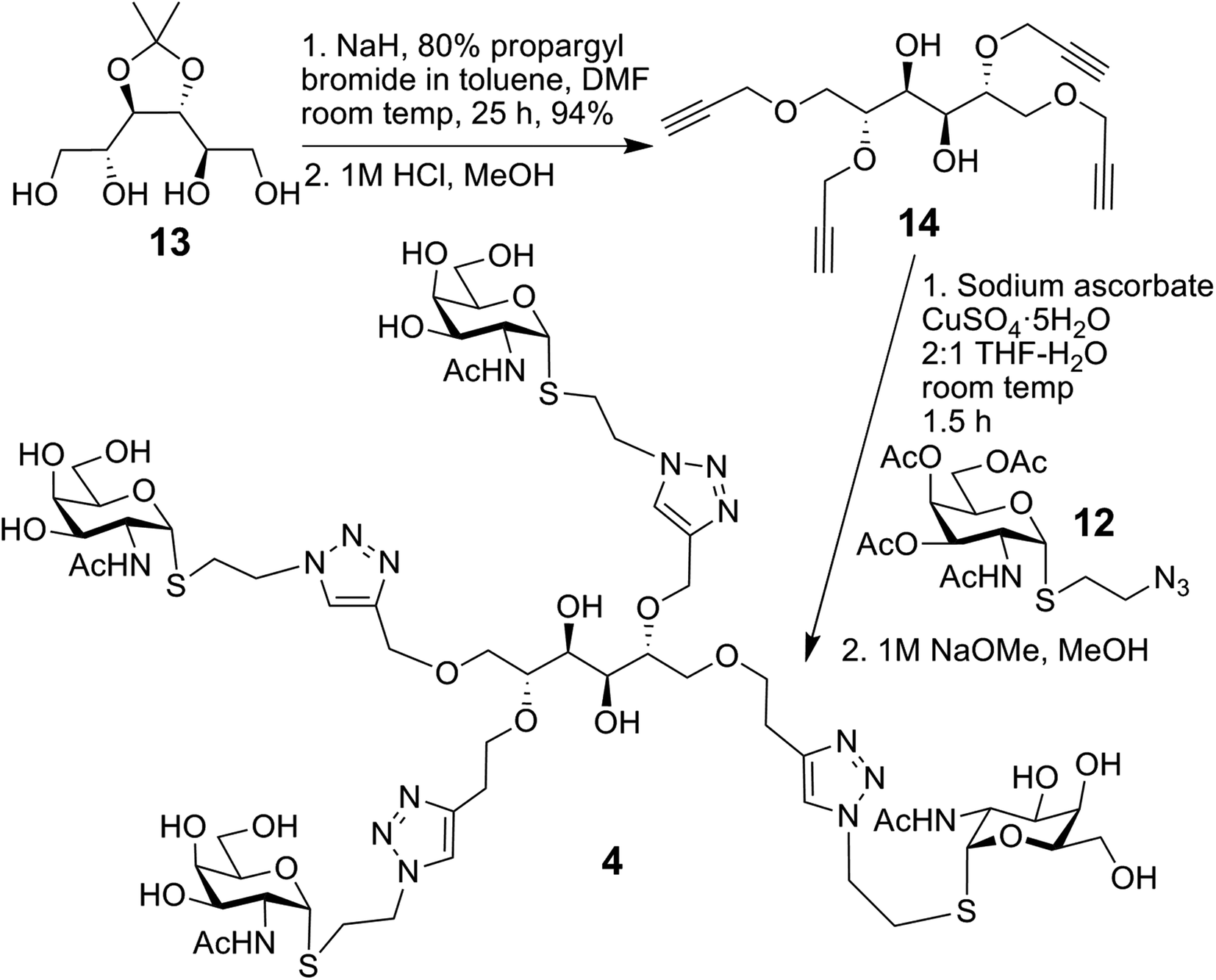 | ||
| Scheme 2 Synthesis of 4. | ||
The bivalent compound 6 was prepared as summarised in Scheme 3. Firstly, 1231 was deacetylated, and then, with heating, the CuAAC reaction using dialkyne 1633 produced 6. The di-alkyne 16 was prepared in three steps from known 15.34 Following the preparation of the GalNAc-presenting glycoclusters 1–6, a monovalent α-anomeric GalNAc derivative was prepared as a probe to measure anomeric preference of lectin binding, when set in relation to results with free GalNAc as standard. In detail, methyl α-thioglycoside 7 was obtained in two steps from glycosyl thiol 1735 (Scheme 4). In addition to this panel of compounds with cognate sugar, an N-acetyl-D-glucosamine (GlcNAc)-bearing bivalent analogue of compound 6 was synthesized. Its availability allows to perform controls that exclude interaction of glycoclusters with bulky core with lectins by carbohydrate-independent molecular stickiness. This bivalent GlcNAc derivative 833 was prepared via 18–20,33 as summarised in Scheme 5.
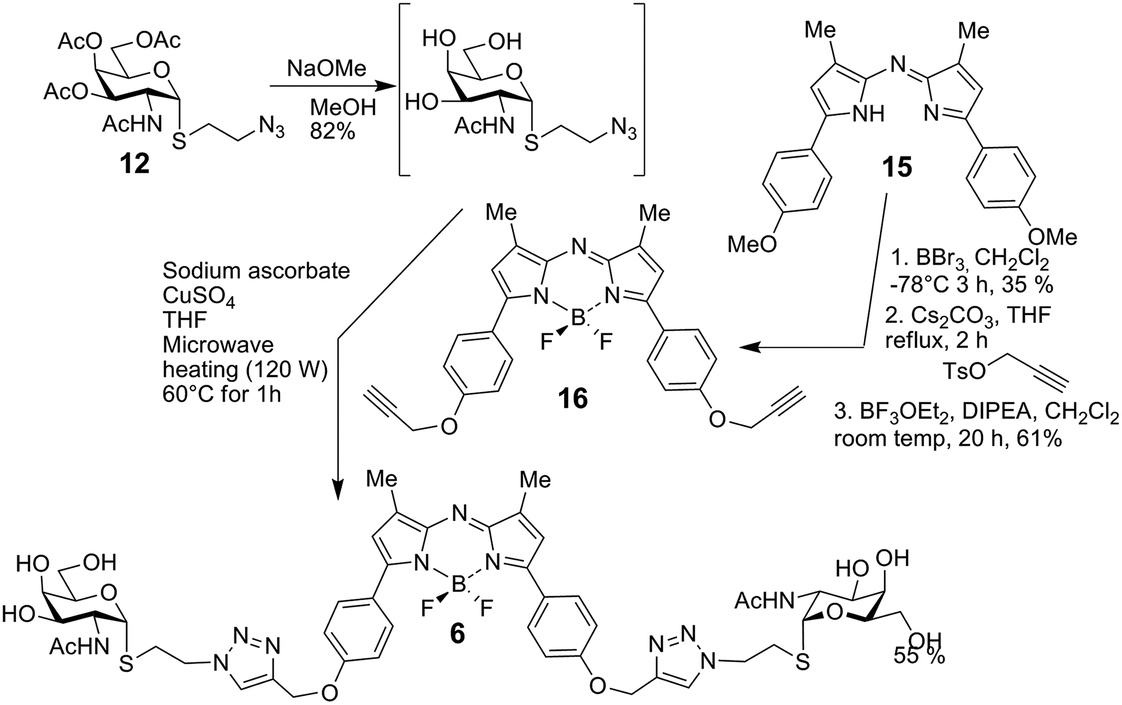 | ||
| Scheme 3 Synthesis of 6. | ||
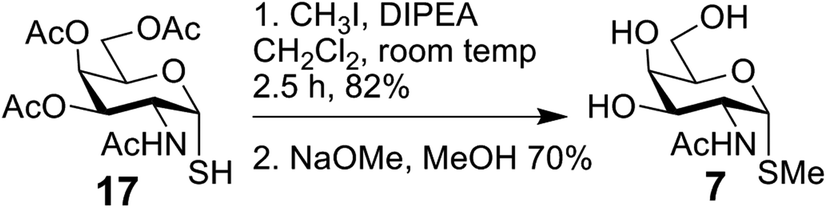 | ||
| Scheme 4 Synthesis of 7. | ||
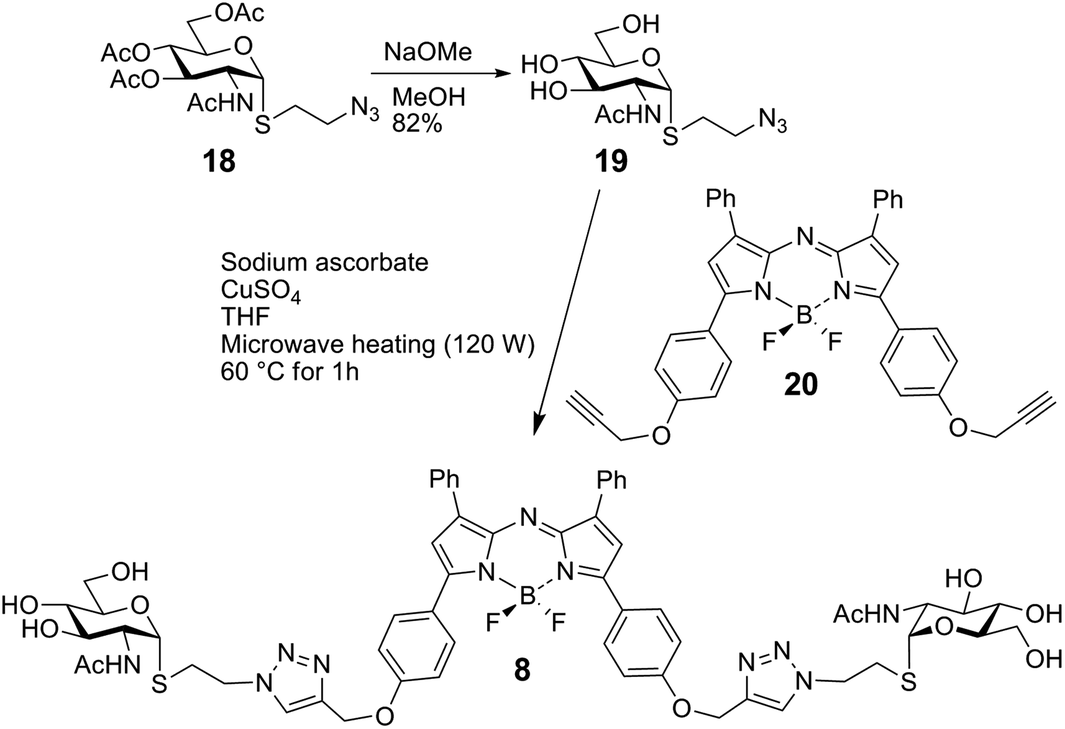 | ||
| Scheme 5 Synthesis of 8. | ||
As further control compound for specificity testing, we included compound 9, a GlcNAc-presenting analogue of compound 5. It had proven to be bioactive as potent inhibitor of two plant lectins with this monosaccharide specificity, i.e. WGA and GSA-II,36 here served as additional control to trace carbohydrate-independent signal generation.
In order to characterize spatial aspects of valency on the tetraphenylethylene, D-mannitol and BF2-azadipyrromethene cores, distances between the hydroxyl groups were calculated by molecular modelling based on crystal structures obtained from the Cambridge Crystallographic Database in three cases34,37 (Fig. 2). In fact, the nature of the core will constrain the maximum spacing possible between GalNAc residues in the most extended conformations. As shown in Fig. 3, these distances in the tetraphenylethylene derivative 3 can likely be larger than for the D-mannitol-based 4 (29 Å vs. 20 Å in Fig. 3). The BF2-azadipyrromethene scaffold facilitates a spacing that could extend to ∼28 Å between its two GalNAc residues, again longer than for the D-mannitol derivative 4 (Fig. 3) and also longer than for 1, the latter estimated at ∼22 Å in our earlier work.31 The D-mannitol- and tetraphenylethylene-based glycoclusters can thus be considered as cornerstones for the spectrum of degree of compactness and of distance between sugar headgroups.
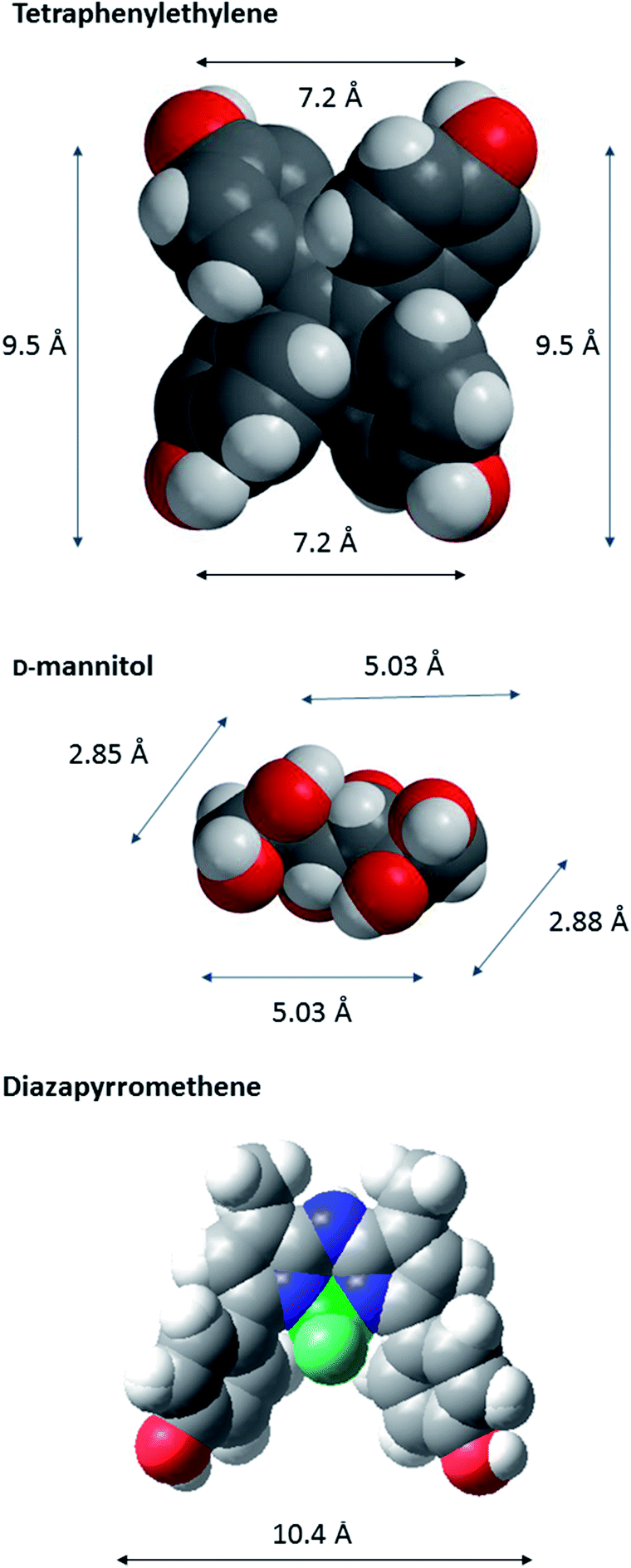 | ||
| Fig. 2 CPK models of the core scaffolds tetraphenylethylene (a), D-mannitol (b), and BF2-azadipyrromethene (c), on which GalNAc-containing glycoclusters studied herein were built. The distances between O-l atoms are shown; those in D-mannitol are between O atoms at C-1, -2, -5 and -6. The models are based on data derived for crystal structures obtained from the Cambridge Crystallographic Database.37 The CPK models were generated in Macromodel (http://www.schrodinger.com). | ||
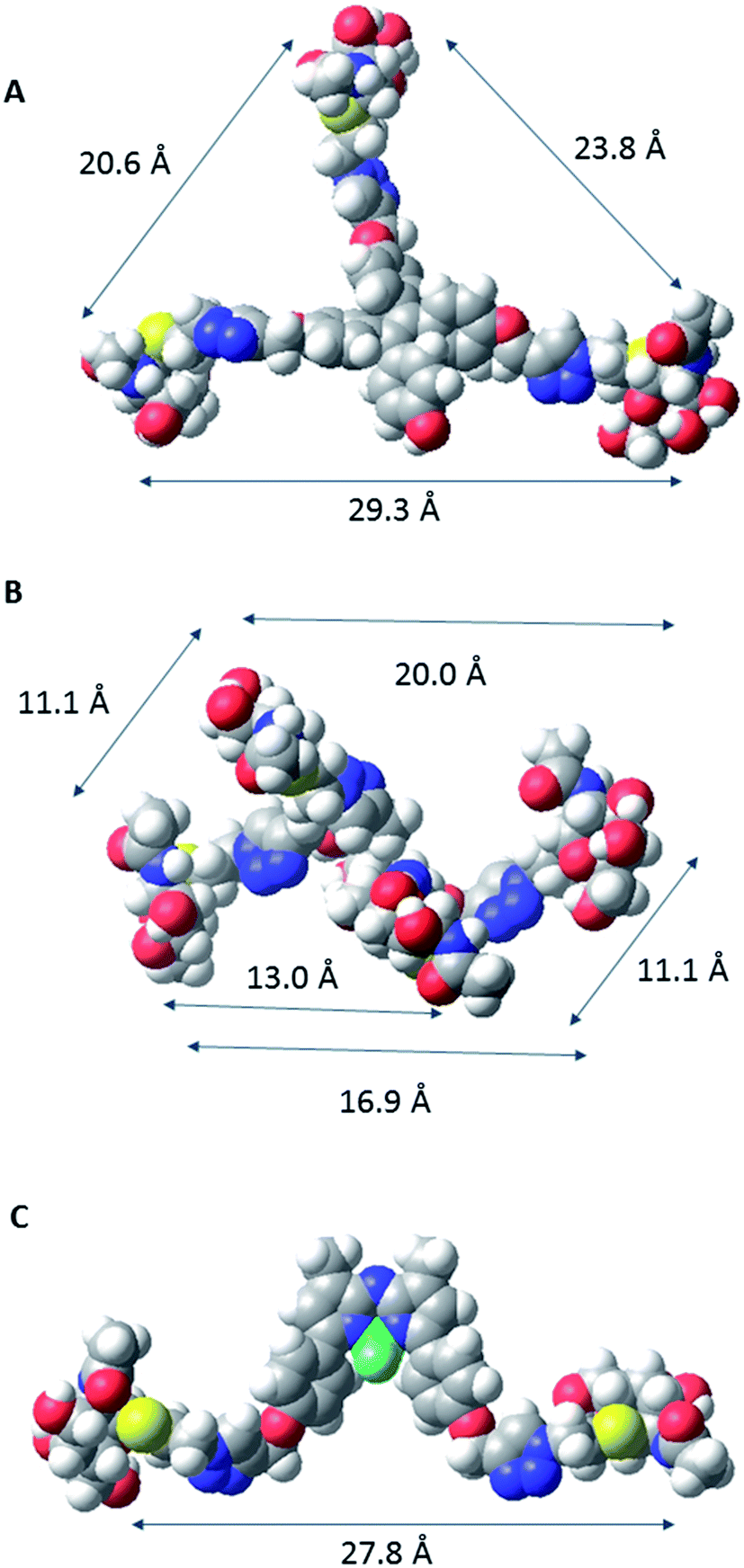 | ||
| Fig. 3 CPK models of selected conformers of compounds 3 (a), 4 (b) and 6 (c). Distances between anomeric carbons of the GalNAc residues presented by each core in these conformers are shown. The CPK models were built in Macromodel. | ||
In summary, the panel of glycoclusters covers different aspects of valency and inter-headgroup spacing for the same sugar used as lectin ligand. The effect of a glycocluster on this activity can likely vary between lectins. As consequence, it will best be measured, if GalNAc-specific receptors with different characteristics in terms of spatial presentation of contact sites for the sugar are selected. Of note, spatial valency on both sides of the recognition process can cooperate in situ so that its complementarity can result in a Lego-like association with beneficial effects on affinity by lowering off-rates38,39 and the inhibitory potency.40,41 Besides spatial aspects, the fine-specificities of lectins may well differ. Since more than a dozen folds with diverse molecular architecture and quaternary structure have developed the capacity to act as lectins, “lectins considered ‘identical’ in terms of monosaccharide specificity possess the ability to recognize fine differences in more complex structures”42 and protein engineering facilitates custom-made tailoring, this is possible: the group of selected lectins is presented in the next section.
The toolbox: 2. lectins
The panel of lectins comprises plant, invertebrate and human proteins that differ in fine-specificity and valency (Table 1). In detail, two tetravalent leguminous lectins (Dolichos biflorus agglutinin (DBA) and soybean agglutinin (SBA)), a hexameric lectin from the albumen gland of the edible (or vineyard) snail (Helix pomatia (HPA)) and the human macrophage galactose(-binding C)-type lectin (MGL) in two engineered forms, i.e. as free monomeric carbohydrate recognition domain (CRD) and as CRD extended by the α-helical coiled-coil stalk of the transmembrane receptor that confers its trimerisation. Table 1 presents schemes of the quaternary structures and listings of specificity profiles (for details on fine-specificity, please see;43–54 for details on structural aspects, please see48,50,55–60). As documented in this table, the relative degrees of affinity to natural GalNAc-containing glycans are not identical. As a consequence, extents of cell binding are likely to differ for these lectins, and this has for example been indeed revealed by array analysis in the cases of Chinese hamster ovary (CHO) cells and three of its glycosylation mutants (Lec1, Lec2, Lec8) for DBA, SBA and HPA.61,62| Acronym | Quaternary structure (number of binding sites)a | Glycoligand | |
|---|---|---|---|
| a Estimated/measured distances between contact sites for the ligand: DBA, SBA about 55 Å or 68 Å; HPA, hexamer arranged as two flat surfaces about 100 Å apart, each formed by a trimer with inter- contact site distance of 25 Å; MGL trimer about 50 Å. | |||
| DBA | Tetramer (4) |  |
α-GalNAc, GalNAcα3GalNAcα3Galβ4Galβ4Glc >A-tetrasaccharide; GalNAcα4Gal/GalNAcβ3Gal (P-like)/GalNacβ4Gal (asialo CAD)/T(F) disaccharide only weakly active |
| SBA | Tetramer (4) |  |
α/β-GalNAc, GalNAcα3Gal(β6Glc); GalNAcα4Gal, GalNAcβ3/4Gal more active than Tn antigen, α2fucosylation of A-disaccharide reduces activity, T(F) disaccharide only weakly active |
| HPA | Hexamer (6) |  |
α-GalNAc > β-GalNAc/α-GlcNAc, GalNAcα3GalNAc/GalNAcα3/4Gal/GalNAcβ4Gal as active as Tn (GalNAcβ3 only weakly active), (s/su)T(F) antigens active, GlcNAcα3/4-terminated LacNAc; highly potent precipitinogen/sensor for low-density presentation of Tn antigen |
| MGL | CRD monomer (1) |  |
Tn and s/suTn antigens (Tn presentation by Tyr active), core 5/6 mucin-type O-glycans, β4-linked GalNAc in LacdiNAc or in chains of GD2/GM2 (not GD3/GM3) gangliosides |
| (CRD + stalk) trimer (3) |  |
||
In summary, the panel of lectins covers five proteins with well-characterised biochemical characteristics (Table 1). A human lectin is deliberately included to document feasibility to work with endogenous (tissue) lectins. The effect of glycocluster presence on ligand binding in a lectin histochemical model study will best be measured in sections of a tissue, to which all tested proteins can bind, and this then yielding strong signals, most preferably for more than a single cell type. Known binding properties of gastrointestinal tissue to this group of proteins when used as histochemical probes63–67 guided us to select murine jejunum.
The toolbox: 3. murine jejunum
Main aspects of the structure of jejunal villi and crypts (of Lieberkuehn) are illustrated in Fig. 4, where a schematic drawing is combined with microphotographs of sections at two levels of magnification. Running the lectin histochemical protocol without the incubation step with a labelled sensor for GalNAc-containing glycoconjugates led to no generation of a signal, that is to no change in appearance of the haemalaun-stained tissue (Fig. 5a). If generated, the signals will thus be entirely dependent on the presence of a labelled lectin.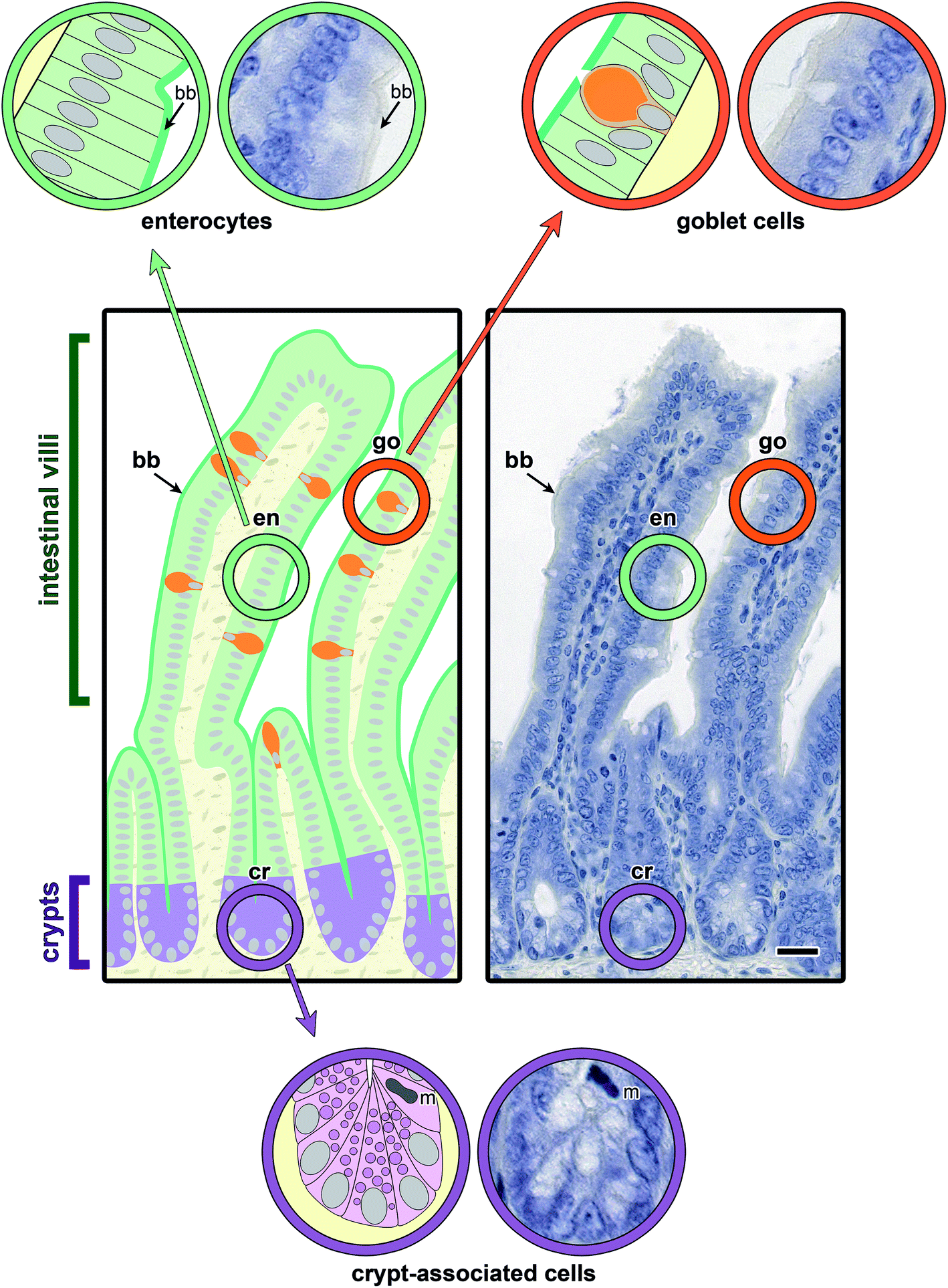 | ||
| Fig. 4 Illustration of the morphological aspects in a longitudinal section of fixed murine jejunum by a combination of schematic drawings with colour coding and microphotographs at two levels of magnification. Representative regions in intestinal villi (en, enterocytes; go, goblet cells; bb, brush border) and also crypt-associated cells (incl. Paneth cells, enteroendocrine cells, precursors of enterocytes and goblet cells and mitotic cells) at the basis of the crypts (cr) were marked by colour-coded circles, these areas shown in enlarged representation. | ||
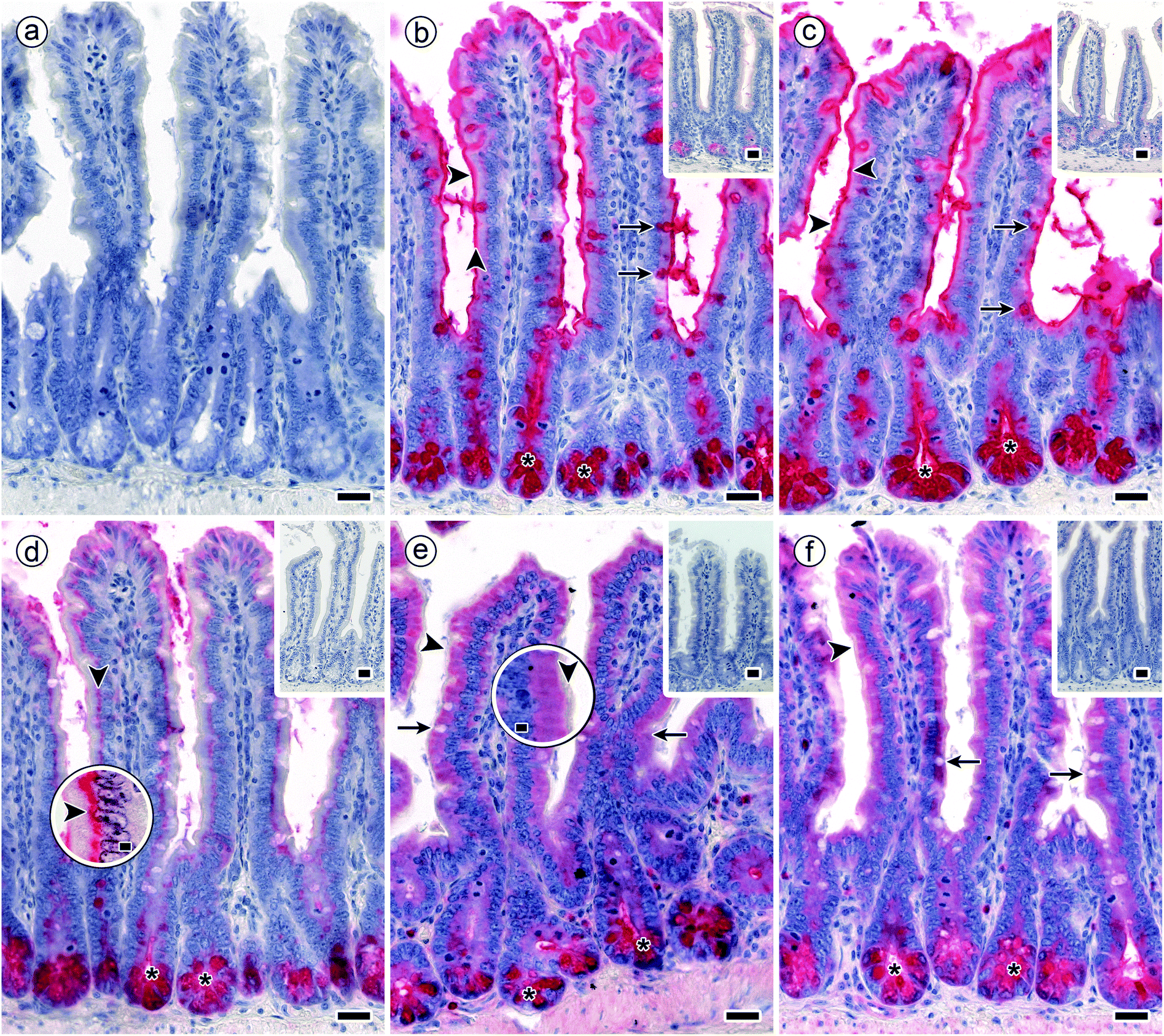 | ||
| Fig. 5 Staining profiles by biotinylated DBA, SBA, HPA and the two forms of MGL in longitudinal sections through fixed murine jejunum. Incubations were done with concentrations of lectins that lead to a strong signal with minimal background (100% level). (a) Negative control by omission of the incubation step with first-step reagent (labelled lectin) with no evidence for lectin-independent signal generation (blank value as shown in Fig. 4) (b and c) strong reactivity for DBA (b) and SBA (c) was found in the brush border of surface enterocytes from intestinal villi (arrowheads), goblet cells (arrows) and in crypt-associated cells, especially in the deep parts of the crypts (asterisks). (d) Binding of biotinylated HPA was detected supranuclearly in surface enterocytes from intestinal villi (arrowheads) (high-level magnification of this region is shown in an inserted circle above the respective area) and in the deep parts of the crypts (asterisks). (e and f) MGL-dependent positivity (similar for the two protein forms) in crypt-associated cells, most prominently in the crypts' deep parts (asterisks) and comparatively weaker in the cytoplasm of surface enterocytes from intestinal villi. No staining was seen in the brush border (arrowheads; higher magnification is shown in an inserted circle above the respective area). Insets in panels b to e show extent of blocking lectin binding by co-incubation of the lectins with free GalNAc at 200 mM. Concentration of biotinylated lectins used were 1 μg mL−1 for DBA, 3 μg mL−1 for SBA, 1.5 μg for HPA and 4 μg mL−1 for both engineered forms of MGL. Scale bars are 20 μm (a–f) and 5 μm (circles in d and e). | ||
In order to record stainings for each protein and to determine optimal conditions for applying glycoclusters as inhibitors for lectin binding systematic titrations of lectin concentrations were performed for each lectin. Staining was routinely assessed as the intensity of the signal and as percentage of positive cells. An example for the correlation between lectin concentration and staining intensity/number of positive cells is shown in ESI Fig. S1† for SBA. For each lectin, signal increases reached plateau values, revealing that binding was saturable (for details on titration ranges, please see Experimental section). These titration series thus led to defining experimental conditions to reach signals of strong intensity as starting point for testing glycoclusters.
The signals were present in different regions of the tissue sections. As summarized in Table 2, surface enterocytes and crypt-associated cells together with goblet cells, here primarily apically targeting the mucus, are the main sites for lectin binding. Binding to intestinal villi exhibited differences, whereas signal intensity of crypt-associated cells was rather uniform (Table 2). Exemplary microphotographs illustrate the staining profile for each type of lectin (Fig. 5b–f). They also document (by insets) reduction of staining by inhibition of lectin binding by the cognate monosaccharide used at high concentration (Fig. 5b–f). In sum, staining was dependent on the lectin, saturable, inhibitable by cognate sugar and different with respect to cell and lectin types.
| DBA | SBA | HPA | MGL | ||
|---|---|---|---|---|---|
| CRD | CRD + stalk | ||||
| a Capillaries of the lamina propria.b Incl. Paneth cells, enteroendocrine cells, precursor of enterocytes and goblet cells, and mitotic cells.c For details on categories of semiquantitative assessments in terms of intensity of lectin-dependent signal (− to ++++)/number of positive cells (0–100%), please see section on lectin histochemistry in Experimental. | |||||
| Intestinal villi | |||||
| Surface enterocytes | |||||
| Cytoplasm | +(3)c | +(3) | − | +++(8) | +++(8) |
| Subapical cytoplasm | +(3) | +(3) | − | +++(8) | +++(8) |
| Supranuclear cytoplasm | − | ++(4) | ++++(8) | +++(8) | +++(8) |
| Brush (striated) border | ++++(8) | ++++(8) | +/−(2) | − | − |
| Goblet cells | ++++(8) | ++++(8) | − | − | − |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Lamina propria | |||||
| Immune cells | − | − | − | − | − |
| Fibroblasts | − | − | − | − | − |
| Smooth muscle cells | − | − | − | − | − |
| Endothelial cellsa | − | − | − | − | − |
|
|||||
| Crypts of Lieberkuehn | |||||
| Crypt-associated cellsb | |||||
| Cytoplasm | ++++(8) | ++++(8) | ++++(8) | ++++(8) | ++++(8) |
| Subapical cytoplasm | ++++(8) | ++++(8) | ++++(8) | ++++(8) | ++++(8) |
| Supranuclear cytoplasm | ++++(8) | ++++(8) | ++++(8) | ++++(8) | ++++(8) |
| Goblet cells | ++++(8) | ++++(8) | − | − | − |
Having equipped the toolbox with panels of synthetic glycoclusters and lectins, also selected a tissue for this model study, whose sections will present carbohydrate-inhibitable signals in distinct cell types, and defined parameters of the histochemical protocol, systematic titrations with the panel of glycocompounds shown in Scheme 1 were performed at a certain lectin concentration to determine relative activities in each case for each type of lectin. Specimens of four animals were processed, in each case at least with duplicates obtained as serial sections in parallel and examined independently by two observers.
Glycoclusters as inhibitors of tissue staining by lectins: 1. light microscopy
The systematic titrations of glycocompound concentration at the constant concentration of a lectin revealed bioactivity of GalNAc, as free monosaccharide or as part of glycoclusters. In other words, lectin binding to GalNAc-containing glycans of cellular glycoconjugates was impaired by the presence of GalNAc in the solution, as free monosaccharide or derivative thereof (compound 7) or as part of a glycocluster (compounds 1–6), in a concentration-dependent manner, as seen in the insets of Fig. 5b–f. The extent of distinctly non-uniform responses depended on the nature of the test compound and the lectin. Exemplary illustrations of how staining profiles are affected by presence of increasing concentrations of the inhibitor are presented for the pairs 3/DBA and 2/SBA (Fig. 6). As shown in this figure, staining (in terms of signal intensity and percentage of positive cells) was reduced by the presence of a GalNAc-presenting compounds. Conjugation of the cognate sugar did obviously not impair its lectin-binding activity. Expressed in terms of the concentration that caused 50% inhibition (IC50) of signal intensity and cell positivity, respective values for inhibitory activity are compiled for each compound and tissue constituent in Table 3.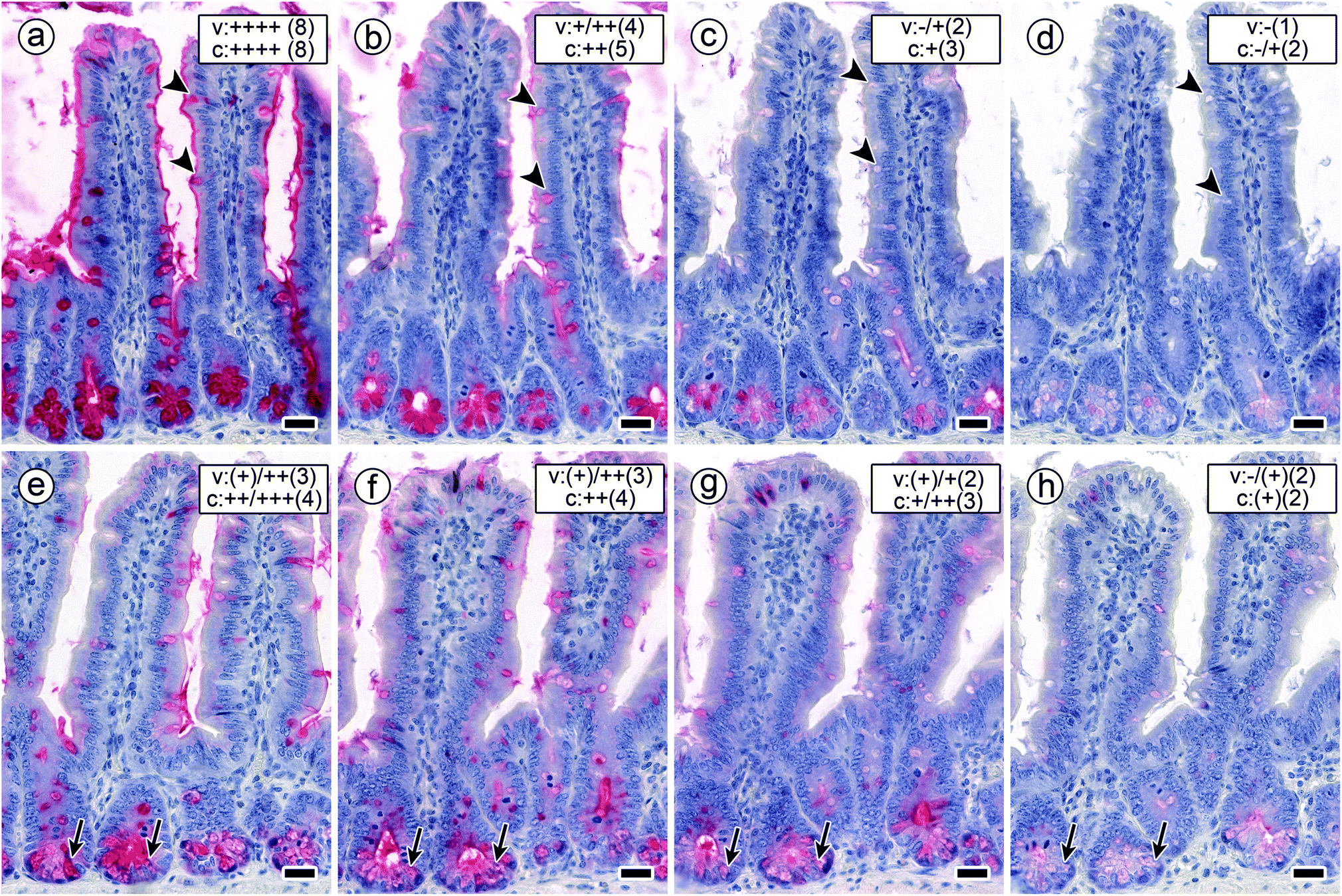 | ||
| Fig. 6 Staining profiles by biotinylated DBA (a–d, arrowheads mark staining of goblet cells in villi) and SBA (e–h, arrows mark staining of crypt-associated cells) in sections of fixed murine jejunum in the presence of increasing concentrations of cognate sugar (GalNAc) assessed in terms of staining intensity (percentage of positive cells). The signal for DBA binding remained at the 100% level in the presence of 1 mM GalNAc presented by compound 3 (a). When raising the sugar concentration stepwisely to 2 mM (b), 5 mM (c) and 10 mM (d), respectively, scaffold-presented GalNAc (by compound 3) led to a notable degree of inhibition. SBA-dependent staining intensity and percentage of positive cells were seen to be reduced more strongly at comparatively low concentrations of 0.001 mM (e), 0.05 mM (f), 0.1 mM (g) and 0.5 mM (h) of GalNAc-presenting compound 2. Semiquantitative grading of staining intensity/percentage of positive cells is given in the rectangular box in the top-right area of each microphotograph (v, surface enterocytes of intestinal villi; c, crypt-associated cells). Intensity of staining in sections is grouped into the following categories: −, no staining; (+), weak but significant staining; ++ medium staining; +++, strong staining; ++++, very strong staining. Percentage of positive cells is expressed in the following eight categories: (1) 0% (no positive cells), (2) < 5% (few positive cells), (3) 5–20%, (4) 21–40%, (5) 41–60%, (6) 61–80%, (7) 81–95% (few negative cells), (8) up to 100% (no negative cells). Concentration of biotinylated DBA/SBA: 1 μg mL−1/3 μg mL−1. Scale bars are 20 μm. | ||
| Compound/cell type | DBA | SBA | HPA | MGL | |
|---|---|---|---|---|---|
| CRD | CRD + stalk | ||||
| a n.i.: not inhibitory; tested up to 20 mM.b Numbers in parentheses denote relative inhibitory potency using activitiy of free GalNAc as standard set to 1. | |||||
| 1 | |||||
| Surface enterocytes | n.i.a | 10.0 (0.05)b | >10.0 (<0.5) | >20.0 (<1.25) | >20.0 (<1.25) |
| Crypt-associated cells | n.i.a | 20.0 (0.1) | >20.0 (<0.25) | >20.0 (<2.5) | >20.0 (<2.5) |
|
|||||
| 2 | |||||
| Surface enterocytes | 1.0 (25) | 0.02/0.01 (50/100) | 5.0 (1.0) | 1.0 (25) | 1.0 (25) |
| Crypt-associated cells | 2.0 (25) | 0.01 (200) | 5.0 (1.0) | 1.0 (50) | 2.0 (25) |
|
|||||
| 3 | |||||
| Surface enterocytes | 2.0 (12.5) | 0.005/0.002 (200/500) | 0.1/0.05 (50/100) | 0.05 (500) | 0.1 (250) |
| Crypt-associated cells | 5.0 (10) | 0.005/0.002 (200/500) | 0.5/0.2 (10/25) | 0.05 (1000) | 0.1 (500) |
|
|||||
| 4 | |||||
| Surface enterocytes | 1.0 (25) | 0.005 (200) | 1.0 (5.0) | 0.05/0.02 (500/1250) | 0.1/0.05 (250/500) |
| Crypt-associated cells | 5.0 (5) | 0.01 (200) | 5.0 (1.0) | 0.05/0.02 (1000/2500) | 0.1/0.05 (500/1000) |
|
|||||
| 5 | |||||
| Surface enterocytes | 2.0 (12.5) | 0.001/0.0005 (1000/2000) | 0.05/0.02 (100/250) | 0.01/0.005 (2500/5000) | 0.005 (5000) |
| Crypt-associated cells | 5.0 (10) | 0.001 (2000) | 0.02 (250) | 0.01/0.005 (5000/10000) |
0.005/0.002 (10000/25000) |
|
|||||
| 6 | |||||
| Surface enterocytes | 20 (1.4) | 0.1 (10) | 0.1 (50) | 0.2 (125) | 0.1 (250) |
| Crypt-associated cells | >20 (<2.5) | 0.2 (10) | 0.2 (25) | 0.2 (250) | 0.2/0.1 (250/500) |
|
|||||
| 7 | |||||
| Surface enterocytes | 20 (1.25) | 0.02 (50) | 0.5 (10.0) | 5.0 (5) | 10.0 (2.5) |
| Crypt-associated cells | >20 (<2.5) | 0.05 (40) | 2.0 (2.5) | 1.0 (50) | 2.0 (25) |
|
|||||
| D-GalNAc | |||||
| Surface enterocytes | 25.0 | 1.0 | 5.0 | 25.0 | 25.0 |
| Crypt-associated cells | 50.0 | 2.0 | 5.0 | 50.0 | 50.0 |
This summary teaches a series of lessons, starting with the broad range of concentrations of sugar needed to diminish the signal within this lectin panel. Proceeding from the arrest induced by the α-S-glycoside 7, the chemical design of bi- to tetravalent glycoclusters with α-GalNAc as headgroup is revealed to be capable to lead to up to marked increases (up to 10000 fold for MGL) of inhibitory capacity of GalNAc (Table 3). Graphically, this point is underscored by comparatively documenting staining profiles by the five labelled proteins in the presence of a constant concentration of tetravalent compound 5 and of bivalent compound 6 (Fig. 7). Since these two types of core are especially bulky and prone to engage in hydrophobic interactions, we tested β-GlcNAc-presenting homologues 8 and 9 to exclude the concern of carbohydrate-independent (non-specific) binding. By substituting cognate GalNAc by its non-cognate epimer GlcNAc, the resulting glycoclusters should lose their capacity to inhibit binding of the lectins. As shown in ESI Fig. S2,† the two compounds with this non-cognate sugar have no inhibitory capacity.
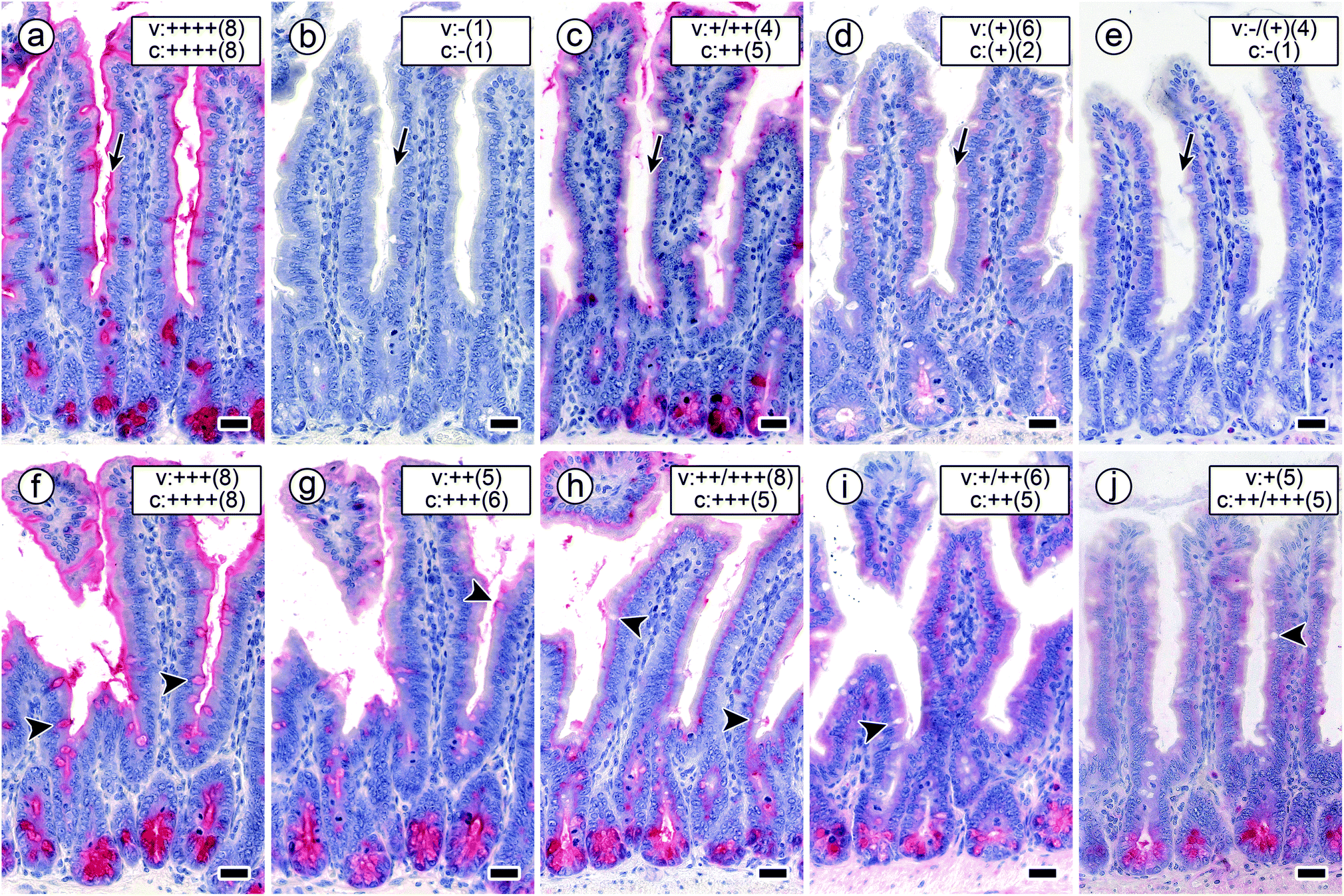 | ||
| Fig. 7 Staining profiles by the four labelled lectins in longitudinal sections of fixed mouse jejunum in the presence of a constant concentration of cognate sugar (0.02 mM in compound 5; arrows mark staining of the brush border). a–e Signal intensity and percentage of positive cells remained at the 100% level after applying biotinylated DBA (a), whereas both parameters dropped to zero levels when using biotinylated SBA (b). This set-up reduced HPA-dependent binding to nearly 50% level (c). Binding of both forms of MGL was strongly inhibited (d and e). The concentration of 0.2 mM GalNAc presented by the bivalent compound 6 (f–j; arrowheads mark staining of goblet cells) had a slight effect on DBA staining in surface enterocytes (f). This concentration and this type of GalNAc presentation significantly reduced the intensity of the staining patterns of the other lectins applied (for details, please see Table 3). Binding of SBA (g) and HPA (h) was subject to a reduction by about 50% in surface enterocytes of intestinal villi (SBA: brush border, HPA: supranuclearly) and by a less diminished extent of staining intensity in crypt-associated cells. (i and j) The degree of inhibition was strongest for both forms of MGL so that the remaining staining intensity in surface enterocytes of intestinal villi and crypt-associated cells was categorized as weak to medium (for details on categories, please see legend to Fig. 6). Concentration of biotinylated lectins used were 1 μg mL−1 for DBA, 3 μg mL−1 for SBA, 1.5 μg for HPA and 4 μg mL−1 for the two engineered forms of MGL. Scale bars are 20 μm. | ||
Considering inhibitory activity, compound core structure (for 1 vs. 6, 2 vs. 3 and 4 vs. 5), valency of the core and the protein, also – but less so – valency of the protein in the two different forms of MGL appear to matter. Already bivalency brings about marked activity increases related to free GalNAc, as for example the case of HPA blocking by compound 6 (but not 1) attests. Overall, structural design of each type of glycocluster accounts for differences in inhibitory potency on this hexavalent lectin (Table 3), indicative of the assumed mutual complementarity. The mechanism(s) underlying the inhibition of lectin binding will involve intra- and/or intermolecular bridging, and, “depending on the size and geometry of proteins and ligands, several binding modes may be possible and effective in parallel and in varying proportions.”68
The comparison of the data sets for the two leguminous lectins discloses large disparities in affinity already at the monosaccharide level, fully in line with published data.43,45 These results emphasise that simple extrapolations between related proteins are not possible, a strong caveat when considering activity on members of families of tissue lectins such as C-type lectins or galectins. Since they are found to be co-expressed in tissues by immunohistochemical monitoring with non-cross-reactive antibodies, as for example accomplished for galectins,69–71 the individual testing of each protein will in the first step be mandatory, as documented herein, then taking the methodological step to testing lectins in mixtures.
In addition to glycocluster and lectin parameters, the assay also provides information on cell types. As shown in Fig. 7 (listed in more detail in Table 3), the reactivity profiles of two cell populations, i.e. surface enterocytes and crypt-associated cells, exhibit disparities. In common binding assays using a certain glycoprotein as ligand, occurrence of such varying response profiles would remain undetectable, because cell type-dependent glycome differences are not established. Having glycome complexity present in this assay is thus certainly an advantage, and the detection of cell type-dependent disparities underscores the merit of this system. What is more, tissue sections as test platform also offer a means to address the mentioned issue that tissue lectins can be co-expressed in situ.
Of note, these lectins can exhibit overlapping carbohydrate specificities, as known from several lectin families.72,73 When co-expressed, they can obviously cooperate with additive or antagonistic impact on the functional outcome, as (first) case studies on galectins are revealing.74–77 Thus, being able to determine binding profiles of individual lectins and effect of a glycocluster on them at the same time in mixtures of tissue lectins would be ideal, and there is a histochemical way to accomplish this. Different modes of protein labelling facilitate to perform these measurements. In detail, different fluorescent dyes can be used for protein labelling (SBA, MGL) or a two-step procedure with a fluorescent probe to detect biotinylated proteins (DBA, HPA) in the tissue is performed.
Glycoclusters as inhibitors of tissue staining by lectins: 2. fluorescence microscopy
Both procedures led to the typical staining profiles of the tested lectins, as shown for each lectin in ESI Fig. S3† together with the essential specificity control, here presence of compound 5. The comparison of the staining distribution in the absence (panels a–d) and presence of compound 5 (panels e–h) unveils a rather similar signal pattern, as obtained by processing with light microscopical assessment. This comparative analysis built the basis to proceed to perform double and triple staining.When testing mixtures, competition between lectins for binding sites can ensue, as is the case (patho)physiologically. The resulting staining profiles then inform about the extent of this competition. An example of staining after testing a binary mixture is presented in ESI Fig. S4† in the case of the two leguminous lectins. Presentation of the microphotographs recorded in each channel and of the (photo)merged files documents the merits of this procedure, in the absence of inhibitor (Fig. S4a–c†) and in the presence of compound 5 (Fig. S4d–f†). When adding fluorescein isothiocyanate (FITC)-labelled MGL to this binary mixture, a solution containing three lectins is established. Binding of each of these lectins to GalNAc-containing glycoconjugates can now be monitored independently at three wavelengths, and the effect of presence of an inhibitor on each lectin in the mixture is readily accessible.
The obtained results of this test case with the three lectins using three-colour staining the tetravalent compound 5 as inhibitor are presented in Fig. 8. Its panels show measurements without/with inhibitor at each wavelength (Fig. 8a–c and d–f) and the staining profile after (photo)merging the three individual files (Fig. 8g and h). Should it be the aim to determine individual staining profiles separately, in this setting precluding competition among lectins for binding sites, then processing of serial sections is advised. Here, the inherent slight changes of spatial presentation of cellular constituents, as given in Fig. S5,† needs to be taken into account.
 | ||
| Fig. 8 Three-colour fluorescence staining profiles by DBA, SBA and MGL (CRD + stalk) in longitudinal sections of fixed murine jejunum in the absence or presence of cognate sugar (GalNAc) presented by compound 5 and their (photo)merging. Accessible binding sites for the three lectins were visualised by applying a mixture of biotinylated DBA, Alexa Fluor®-555-labelled SBA (colour assignment to blue) and FITC-labelled MGL (CRD + stalk) (green). Localization of biotinylated DBA became detectable by the second-step reagent Alexa Fluor®-647-conjugated streptavidin (5 μg mL−1; colour assignment to red). DAPI was used for staining of nuclei (colour assignment to white). (a–c) Binding of DBA (a) and SBA (b), respectively, was seen in the brush border (arrows) of intestinal enterocytes, in goblet cells (arrowheads) and crypt-associated cells (asterisks). MGL binding was mainly confined to the cytoplasm of surface enterocytes in intestinal villi (c). (d–f) No inhibition of DBA-dependent staining (d) was seen with scaffold (compound 5)-presented GalNAc (0.1 mM), whereas binding of labelled SBA (e) and of MGL (f) was completely inhibited. The three staining patterns were (photo)merged so that binding sites for each lectin in the same section were visualised in the absence (g) and in the presence of cognate sugar (h). The regionally similar DBA- and SBA-dependent signal presence led to overlap (colour change to magenta) in the brush border of intestinal villi, goblet cells and crypt-associated cells (g). No significant overlap, in contrast, was seen with labelled MGL (g). In the presence of compound 5 applied at 0.1 mM GalNAc, DBA-dependent staining was expectably rather insensitive to inhibitor presence, when compared to SBA and MGL (h). Concentration of biotinylated DBA was 1 μg mL−1, that of Alexa Fluor®-555-labelled SBA was 2 μg mL−1 and FITC-labelled MGL was 16 μg mL−1. DAPI was used at 0.5 μg mL−1. Scale bars are 20 μm. | ||
Using the test platform of the tissue sections with their glycome complexity, any combination of lectins hereby becomes amenable to assaying, that is proteins of the same family and from different families and especially tissue lectins such as C-type lectins, galectins or siglecs. Since the distributions of binding sites can well overlap when comparing this property of activity between lectins, this assay is a means to assess the extent of overlap, in itself biomedically relevant information. The described fluorescence microscopical monitoring goes beyond measuring inhibition of lectin binding to a glycoprotein. It facilitates detection of cell type-dependent differences, thus extends in the tissue context what array analysis with printed vesicles or cultured cells can offer.
Conclusions
In conclusion, the emerging broad significance of protein(lectin)–glycan recognition is fueling the interest in developing glycoclusters as inhibitors of lectin binding. Since spatial and structural parameters are being disclosed to matter for achieving this type of functional pairing, the target of a therapeutic glycocluster, assaying of these synthetic products should cover these factors of cellular glycomes to the highest possible extent. Taking steps toward this aim is thus timely.We here introduce a versatile platform for routine and rigorously controlled application, i.e. tissue sections, and illustrate the influence of parameters on the side of glycoclusters (especially core structure and valency) and lectins, especially architecture, fine-specificity and valency, on the experimental read-out, and this on two cell populations. The presented data strongly argue against the possibility for simple extrapolations. The pertinent question on glycocluster activity on lectins in mixtures, a common situation for tissue lectins, is answered by fluorescence microscopical monitoring using differently labelled proteins.
Whereas chemical engineering of nanoparticle surfaces and of cells programs tools for measuring inhibition of their bridging by lectins,78–80 our assay examines all aspects of lectin binding. It takes advantage of having cell and matrix glycomes available in tissue sections, and their processing can for example ensure to maintain glycolipid presence.81,82 In comparison to cultured cells, the tissue ensures analysis of cells within their microenvironment. This favourable aspect is also a part of the in vivo studies with labelled neoglycoproteins presenting natural and synthetic glycans up to custom-made microheterogeneity used for targeting and imaging.83–89 On the side of the proteins, mimicking the natural occurrence of lectins in mixtures, in terms of constituents and relative concentrations, will enable to trace currently not well-defined consequences of competition for binding sites and cross-reactivity of glycoclusters among lectins. Given the assay's reliability and sensitivity as well as its use with any type of tissue, its application is advised, prior to considering testing glycoclusters in vivo, for example to minimise such overlaps in target binding by iterative structural refinements including headgroup chemistry.
Experimental
General methods for synthesis
1H-NMR spectra were recorded (25 °C) at 400 MHz or 500 MHz; 13C-NMR spectra were recorded at 100 MHz or 125 MHz. Chemical shift (δ) data in ppm are report with multiplicities indicated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet); coupling constants (J) given in hertz (Hz). Chemical shifts (δ) are reported relative to internal standard Me4Si in CDCl3 (δ 0.0) for 1H and CDCl3 (δ 77.0) for 13C. 1H NMR spectral signals were assigned with the aid of COSY, 13C NMR spectral signals using DEPT, gHSQCAD and/or gHMBCAD. NMR spectroscopic data for known compounds was in good agreement with those reported in the literature, as referenced. High resolution ESI mass spectra were measured in positive and/or negative mode using a Waters LCT Mass Spectrometer or were acquired using a microTOF-Q spectrometer interfaced to a Dionex UltiMate 3000 LC in positive and negative modes as required. MicroTof control 3.2 and HyStar 3.2 software were used to carry out the analysis for data obtained with the microTOF-Q. TLC was performed on aluminium sheets precoated with silica gel and spots visualized by UV and/or charring with H2SO4–EtOH (1:20) or cerium molybdate, unless otherwise stated. Chromatography was carried out with silica gel 60 (0.040–0.630 mm) and using a stepwise solvent polarity gradient correlated with TLC mobility, unless otherwise stated. CH2Cl2, MeOH, toluene and THF reaction solvents were used as obtained from a Pure Solv™ Solvent Purification System. Unless it is specified, all reagents were used as received without further purification. All reactions involving air-sensitive reagents were performed under nitrogen in oven-dried glassware using syringe-septum cap technique. Optical rotations were determined at the sodium D line at 20 °C using a Schmidt and Haensch UniPol L1000. The IR spectra were recorded using thin film with a PerkinElmer Spectrum 100 FT-IR Spectrometer with an ATR attachment.
4-(1,2,2-Tris(4-(prop-2-yn-1-yloxy)phenyl)vinyl)phenyl acetate 11
To a solution of 10 (1.37 g, 3.45 mmol) at 0 °C in pyridine (27.5 mL), Ac2O (228 μL, 2.41 mmol) was added over 2 h. The reaction mixture was allowed to attain room temp and then stirred for a further 3 h. The solvent was removed under diminished pressure and chromatography (CH2Cl2–MeOH, 96.6:3.4) gave the intermediate monoacetate (827 mg, 55%); 1H NMR (500 MHz, methanol-d4) δ 6.99 (d, J = 8.8 Hz, 2H, o-Ar), 6.85–6.76 (m, 8H), 6.55–6.47 (m, 6H), 2.22 (s, 3H, OAc), 13C NMR (126 MHz, methanol-d4) δ = 169.8(C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 155.6(C), 155.5 (C), 155.4 (C), 148.8 (C), 142.5 (C), 139.9 (C), 137.3 (C), 135.5 (C), 135.3 (C), 132.3 (CH), 132.2 (CH, 2×), 132.0 (CH), 120.3 (CH), 114.1 (CH), 110.0 (C), 19.5(CH3, OAc); ESI-HRMS calcd for C28H21O5 437.1389, found m/z 437.1391 [M − H]−. To this intermediate (1.7 g, 3.9 mmol) in dry DMF (78 mL) was added anhydrous potassium carbonate (6.42 g, 46.4 mmol); propargyl bromide (80% in toluene, 2.72 mL, 31.0 mmol) was then added to the mixture and it was stirred for 14 h at 70 °C under an inert atmosphere. The reaction was then cooled to room temp and diluted with CH2Cl2 (300 mL) and was then washed with satd NH4Cl. The aqueous layer was again washed with further portions of CH2Cl2 (3 × 150 mL). The combined organic layers were washed with water and dried over Na2SO4. The solvent was removed under diminished pressure and column chromatography (cyclohexane-EtOAc, 85:15 to 91:9) gave the title compound (520 mg, 24%); 1H NMR (500 MHz, chloroform-d) δ 7.00 (d, J = 8.7 Hz, 2H, m-Ar), 6.96–6.91 (m, 6H, Ar), 6.83 (d, J = 8.7 Hz, 2H, o-Ar), 6.74–6.68 (m, 6H, Ar), 4.61 (t, J = 2.6 Hz, 6H, CH2), 2.50 (t, J = 2.5 Hz, 3H, propargylic), 2.25 (s, 3H, OAc),13C NMR (126 MHz, chloroform-d) δ = 169.2(C, CO), 156.1 (C), 156.1 (C), 156.0 (C), 148.9 (C), 141.6 (C), 139.5 (C), 138.2 (C), 137.1 (C), 137.0 (C), 137.0 (C), 132.5 (CH), 132.5 (CH), 132.3 (CH), 120.7 (CH), 114.1 (CH), 114.1(CH), 114.0(CH), 75.5 (CH), 75.4 (CH), 75.4 (CH), 55.8 (CH2, 2×), 55.8 (CH2), 21.2 (CH3, OAc); ESI-HRMS calcd for C37H28O5Na 575.1834, found m/z 575.1832 [M + H]+. Note: the tetrapropargylated compound was the main by-product as a result of partial deacetylation during the propargylation reaction.
O), 155.6(C), 155.5 (C), 155.4 (C), 148.8 (C), 142.5 (C), 139.9 (C), 137.3 (C), 135.5 (C), 135.3 (C), 132.3 (CH), 132.2 (CH, 2×), 132.0 (CH), 120.3 (CH), 114.1 (CH), 110.0 (C), 19.5(CH3, OAc); ESI-HRMS calcd for C28H21O5 437.1389, found m/z 437.1391 [M − H]−. To this intermediate (1.7 g, 3.9 mmol) in dry DMF (78 mL) was added anhydrous potassium carbonate (6.42 g, 46.4 mmol); propargyl bromide (80% in toluene, 2.72 mL, 31.0 mmol) was then added to the mixture and it was stirred for 14 h at 70 °C under an inert atmosphere. The reaction was then cooled to room temp and diluted with CH2Cl2 (300 mL) and was then washed with satd NH4Cl. The aqueous layer was again washed with further portions of CH2Cl2 (3 × 150 mL). The combined organic layers were washed with water and dried over Na2SO4. The solvent was removed under diminished pressure and column chromatography (cyclohexane-EtOAc, 85:15 to 91:9) gave the title compound (520 mg, 24%); 1H NMR (500 MHz, chloroform-d) δ 7.00 (d, J = 8.7 Hz, 2H, m-Ar), 6.96–6.91 (m, 6H, Ar), 6.83 (d, J = 8.7 Hz, 2H, o-Ar), 6.74–6.68 (m, 6H, Ar), 4.61 (t, J = 2.6 Hz, 6H, CH2), 2.50 (t, J = 2.5 Hz, 3H, propargylic), 2.25 (s, 3H, OAc),13C NMR (126 MHz, chloroform-d) δ = 169.2(C, CO), 156.1 (C), 156.1 (C), 156.0 (C), 148.9 (C), 141.6 (C), 139.5 (C), 138.2 (C), 137.1 (C), 137.0 (C), 137.0 (C), 132.5 (CH), 132.5 (CH), 132.3 (CH), 120.7 (CH), 114.1 (CH), 114.1(CH), 114.0(CH), 75.5 (CH), 75.4 (CH), 75.4 (CH), 55.8 (CH2, 2×), 55.8 (CH2), 21.2 (CH3, OAc); ESI-HRMS calcd for C37H28O5Na 575.1834, found m/z 575.1832 [M + H]+. Note: the tetrapropargylated compound was the main by-product as a result of partial deacetylation during the propargylation reaction.
4-(1,2,2-Tris(4-((1-(2-(2-deoxy-2-acetamido-D-galactopyranosylthio)ethyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)vinyl)phenol 3
To 11 (124 mg, 0.22 mmol) in degassed THF-H2O (1:1, 17.2 mL) were added azide 12 (300 mg, 0.69 mmol), sodium ascorbate (39 mg, 0.198 mmol) and Cu2SO4·5H2O (4942 mg, 0.198 mmol). The reaction mixture was stirred under an inert atmosphere in a microwave reactor (120 W) at 50 °C for 30 min. Tetrahydrofuran was then removed under diminished pressure followed by the dilution with CH2Cl2. This mixture was then washed with water and the aqueous layer was re-extracted with a further portion of CH2Cl2. The combined organic layers were dried over Na2SO4 and the solvent was removed under diminished pressure. Chromatography of the residue (CH2Cl2–MeOH, 96.5:3.5 to 95.5:4.5) gave the fully acetylated intermediate (411 mg, 70%) as a colourless solid; 1H NMR (500 MHz, chloroform-d) δ 7.67 (s, 1H, triaz.), 7.67 (s, 1H, triaz.), 7.66 (s, 1H, triaz.), 7.00 (d, J = 8.7 Hz, 2H, m-Ar), 6.93–6.91 (m, 6H, Ar), 6.83 (d, J = 8.7 Hz, 2H, o-Ar), 6.73–6.68 (m, 6H, Ar), 5.97 (d, J = 7.8 Hz, 1H, NH), 5.92 (d, J = 8.1 Hz, 1H, NH), 5.71 (d, J = 8.3 Hz, 1H, NH), 5.68–5.63 (m, 3H, H-1), 5.39 (dd, J = 3.2, 1.2 Hz, 3H, H-4), 5.14 (s, 2H, CH2), 5.14 (s, 2H, CH2), 5.13 (s, 2H, CH2), 5.01 (dt, J = 11.8, 3.3 Hz, 3H, H-3), 4.78–4.69 (m, 3H, H-2), 4.65–4.55 (m, 6H, CH2), 4.52 (t, J = 6.8 Hz, 3H, H-5), 4.20–4.06 (m, 6H, CH2), 3.25–3.12 (m, 3H, CH2), 3.12–3.00 (m, 3H, CH2), 2.25 (s, 3H, ArOAc), 2.16 (s, 3H, NHCH3), (s, 6H NHCH3), 2.01 (s, 3H, OAc), 2.00 (s, 6H, OAc), 1.99 (s, 6H, OAc), 1.99 (s, 3H, OAc), 1.97 (s, 3H, OAc), 1.97 (s, 6H, OAc). To a suspension of this intermediate (145 mg, 0.078 mmol), which was precooled to 0 °C, in dry methanol (7 mL), was added freshly prepared NaOMe solution in MeOH until the solution pH was 9. The resulting mixture was stirred for 2 h at room temp and then Amberlite IR-120 H+, which had been washed with methanol, was added and the mixture stirred gently for 10 min until the solution was neutralized. The solution containing the product was separated from the solid materials by decanting and the resin was washed with a warm mixture of water-acetonitrile which was added to the methanol solution. The volatile solvents were removed under diminished pressure until a minimal amount of water remained. Reverse phase column chromatography gave compound 3 (84 mg, 75%) as a colourless solid. 1H NMR (500 MHz, DMSO-d6) δ 9.33 (s, 1H, ArOH) 8.19 (s, 3H, triazo.), 7.80 (d, J = 6.9 Hz, 3H, NH), 6.85 (m, 6H, Ar), 6.79 (m, 6H, Ar), 6.73 (d, J = 8.4 Hz, 2H, m-Ar), 6.51 (d, J = 8.4 Hz, 2H, o-Ar), 5.51 (d, J1-2 = 5.2 Hz, 3H, H-1), 5.02 (s, 6H, CH2), 4.80–4.65 (m, 12H,OH), 4.55 (ddt, J = 27.1, 13.9, 6.8 Hz, 6H,CH2), 4.16 (ddd, J2-3,2-NH,2-1 = 11.8, 6.9,5.2 Hz, 3H, H-2), 3.91 (t, J5-6,5-6′ = 6.0 Hz, 3H,H-5), 3.72 (brs, 3H, H-4), 3.53 (t, J6-5,6′-6 = 5.9 Hz, 6H, H-6,H-6′), 3.49 (dd, J3-2,3-4 = 11.8, 2.9 Hz, 3H, H-3), 3.02 (dt, J = 13.8, 6.9 Hz, 3H, CH2), 2.92 (dt, J = 13.7, 6.8 Hz, 3H), 1.79 (s, 9H, NHAc).13C NMR (126 MHz, dmso) δ = 170.3(C, NHAc), 156.8 (C, Ar), 156.1 (C,m-Ar), 142.9 (C, triaz.), 142.8 (C), 139.0 (C), 137.1 (C), 132.5 (CH, m-Ar), 132.5 (CH,Ar), 125.2 (CH,triaz.), 114.4(CH, o-Ar), 114.3(CH, Ar), 84.8 (CH, H-1), 72.7(CH, H-5), 68.5(CH, H-4), 67.9(CH, H-3), 61.3(CH2), 61.2 (CH2, C-6), 50.6(CH, C-2), 49.8 (CH2), 30.3(CH2), 23.1(CH3, NHAc); ESI-HRMS calcd for C65H79 N12O19S3 1427.4747, found m/z 1427.4752 [M − H]−. Note: for the reverse phase chromatography, a minimum volume of the aqueous residue was loaded onto the column and elution involved passing three column volumes of 1:4 CH3CN–H2O followed by 1:1 CH3CN–H2O.
1,2,5,6-Tetra-O-(propargyl)-D-mannitol 14
Sodium hydride (1.0 g, 26.3 mmol, 60% in mineral oil) was added to a cooled solution of 1332 (0.49 g, 2.2 mmol) in dry DMF (30 mL). The suspension was stirred for 15 min and then 80% propargyl bromide in toluene (2.9 mL, 26.3 mmol) was added. The resulting mixture was stirred at room temp for 25 h, and after this time MeOH was added at 0 °C. The mixture was diluted with EtOAc, then washed with brine (3 × 20 mL), dried over Na2SO4, filtered and the solvent was removed. Column chromatography (9:1 cyclohexane–EtOAc) gave the propargylated intermediate as a yellow oil (0.77 g, 94%); [α]20D = +6 (c 0.8, CHCl3); 1H NMR (500 MHz, CDCl3) δ 4.38 (dd, 2H, Jgem = 16.0 Hz, JH,H = 2.4 Hz, CH2aC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH-2), 4.35 (dd, 2H, Jgem = 16.0 Hz, JH,H = 2.4 Hz, CH2bCCH-2), 4.20 (dd, 2H, Jgem = 15.9 Hz, JH,H = 2.4 Hz, CH2aCCH-1), 4.15 (dd, 2H, Jgem = 15.9 Hz, JH,H = 2.4 Hz, CH2bCCH-1), 4.14 (dd, 2H, J3,2 = 3.6 Hz, J3,1 = 1.5 Hz, H-3), 3.88 (dddd, 2H, J2,3 = 3.6 Hz, J2,1b = 6.3 Hz, J2,1a = 3.2 Hz, JH,H = 1.5 Hz, H-2), 3.84 (dd, 2H, J1a,1b = 10.6 Hz, J1a,2 = 3.2 Hz, H-1a), 3.64 (dd, 2H, J1b,1a = 10.6 Hz, J1b,2 = 6.3 Hz, H-1b), 2.44 (t, 2H, JH,H = 2.4 Hz, CH2CCH-2), 2.42 (t, 2H, JH,H = 2.4 Hz, CH2CCH-1), 1.39 (s, 6H, CH3); 13C NMR (126 MHz, CDCl3) δ 110.1 (C(CH3)2), 80.1 (CH2CCH-2), 79.7 (CH2CCH-1), 78.1 (C-2), 78.0 (C-3), 74.8 (CH2CCH-1), 74.7 (CH2CCH-2), 70.0 (C-1), 58.7 (CH2CCH-1), 58.0 (CH2CCH-2), 27.2 (CH3); ESI-HRMS calcd for C21H26NaO6 397.1627, found: 397.1663 [M + Na]+: hydrochloric acid (0.3 mL of 1.0 M) was added to this intermediate (0.12 g, 0.32 mmol) in MeOH (2.7 mL). The resulting mixture was heated at reflux for 2.5 h and then allowed to attain room temperature. Excess solid NaHCO3 was added and the mixture stirred for a further 15 min. The solid was filtered off and the solvent was evaporated and column chromatography (cyclohexane–EtOAc, 7:3) gave 14 as a pale yellow oil (0.1 g, 94%); 1H NMR (500 MHz, CDCl3) δ 4.37 (dd, 2H, Jgem = 16.0 Hz, JH,H = 2.4 Hz, CH2aCCH-2), 4.32 (dd, 2H, Jgem = 16.0 Hz, JH,H = 2.4 Hz, CH2bCCH-2), 4.20 (d, 4H, JH,H = 2.4 Hz, CH2CCH-1), 3.91 (m, 2H, H-3), 3.88 (m, 2H, H-1a), 3.85 (m, 2H, H-2), 3.75 (dd, J1b,1a = 10.1 Hz, J1b,2 = 4.8 Hz, 2H, H-1b), 2.84 (d, 2H, JOH,3 = 6.2 Hz, OH), 2.46 (t, 2H, JH,H = 2.4 Hz, CH2CCH-2), 2.45 (t, 2H, JH,H = 2.4 Hz, CH2CCH-1); 13C NMR (126 MHz, CDCl3) δ 80.0 (CH2CCH-2), 79.5 (CH2CCH-1), 78.2 (C-2), 75.0 (CH2CCH-1), 74.8 (CH2CCH-2), 69.8 (C-1), 69.4 (C-3), 58.8 (CH2CCH-1), 58.1 (CH2CCH-2). ES-HRMS calcd for C18H22NaO6: 357.1314, found: 357.1295 [M + Na]+.
CH-2), 4.35 (dd, 2H, Jgem = 16.0 Hz, JH,H = 2.4 Hz, CH2bCCH-2), 4.20 (dd, 2H, Jgem = 15.9 Hz, JH,H = 2.4 Hz, CH2aCCH-1), 4.15 (dd, 2H, Jgem = 15.9 Hz, JH,H = 2.4 Hz, CH2bCCH-1), 4.14 (dd, 2H, J3,2 = 3.6 Hz, J3,1 = 1.5 Hz, H-3), 3.88 (dddd, 2H, J2,3 = 3.6 Hz, J2,1b = 6.3 Hz, J2,1a = 3.2 Hz, JH,H = 1.5 Hz, H-2), 3.84 (dd, 2H, J1a,1b = 10.6 Hz, J1a,2 = 3.2 Hz, H-1a), 3.64 (dd, 2H, J1b,1a = 10.6 Hz, J1b,2 = 6.3 Hz, H-1b), 2.44 (t, 2H, JH,H = 2.4 Hz, CH2CCH-2), 2.42 (t, 2H, JH,H = 2.4 Hz, CH2CCH-1), 1.39 (s, 6H, CH3); 13C NMR (126 MHz, CDCl3) δ 110.1 (C(CH3)2), 80.1 (CH2CCH-2), 79.7 (CH2CCH-1), 78.1 (C-2), 78.0 (C-3), 74.8 (CH2CCH-1), 74.7 (CH2CCH-2), 70.0 (C-1), 58.7 (CH2CCH-1), 58.0 (CH2CCH-2), 27.2 (CH3); ESI-HRMS calcd for C21H26NaO6 397.1627, found: 397.1663 [M + Na]+: hydrochloric acid (0.3 mL of 1.0 M) was added to this intermediate (0.12 g, 0.32 mmol) in MeOH (2.7 mL). The resulting mixture was heated at reflux for 2.5 h and then allowed to attain room temperature. Excess solid NaHCO3 was added and the mixture stirred for a further 15 min. The solid was filtered off and the solvent was evaporated and column chromatography (cyclohexane–EtOAc, 7:3) gave 14 as a pale yellow oil (0.1 g, 94%); 1H NMR (500 MHz, CDCl3) δ 4.37 (dd, 2H, Jgem = 16.0 Hz, JH,H = 2.4 Hz, CH2aCCH-2), 4.32 (dd, 2H, Jgem = 16.0 Hz, JH,H = 2.4 Hz, CH2bCCH-2), 4.20 (d, 4H, JH,H = 2.4 Hz, CH2CCH-1), 3.91 (m, 2H, H-3), 3.88 (m, 2H, H-1a), 3.85 (m, 2H, H-2), 3.75 (dd, J1b,1a = 10.1 Hz, J1b,2 = 4.8 Hz, 2H, H-1b), 2.84 (d, 2H, JOH,3 = 6.2 Hz, OH), 2.46 (t, 2H, JH,H = 2.4 Hz, CH2CCH-2), 2.45 (t, 2H, JH,H = 2.4 Hz, CH2CCH-1); 13C NMR (126 MHz, CDCl3) δ 80.0 (CH2CCH-2), 79.5 (CH2CCH-1), 78.2 (C-2), 75.0 (CH2CCH-1), 74.8 (CH2CCH-2), 69.8 (C-1), 69.4 (C-3), 58.8 (CH2CCH-1), 58.1 (CH2CCH-2). ES-HRMS calcd for C18H22NaO6: 357.1314, found: 357.1295 [M + Na]+.
(2R,3S,4S,5R)-1,2,5,6-Tetrakis((1-(2-(2-deoxy-2-acetamido-α-D-galactopyranosylthio)ethyl)-1H-1,2,3-triazol-4-yl)methoxy)-hexane-3,4-diol 4 to 14
(0.16 g, 0.37 mmol) and 12 (43 mg, 0.13 mmol) in THF–H2O (10 mL, 2:1), sodium ascorbate (26 mg, 0.13 mmol) and CuSO4·5H2O (33 mg, 0.13 mmol) were added. The resulting mixture was stirred for 1.5 h at room temp. After this time, the solvent was removed and the residue dissolved in CH2Cl2, then washed with brine (2 × 10 mL), dried over Na2SO4, filtered and the solvent was removed. Column chromatography of the residue (CH2Cl2–MeOH, 15:1) gave the protected intermediate as a white solid (0.11 g, 57%); [α]20D: +81 (c 1.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.79 (s, 2H, H-triazole 1), 7.78 (s, 2H, H-triazole 2), 6.82 (d, 2H, JNH,2 Gal = 7.6 Hz, NH), 6.76 (d, 2H, JNH,2 Gal = 7.6 Hz, NH), 5.75 (d, 2H, J1,2 = 4.6 Hz, H-1 Gal), 5.74 (d, 2H, J1,2 = 4.6 Hz, H-1 Gal), 5.41 (s, 4H, H-4 Gal), 5.0 (dd, 2H, J3,2 = 11.8 Hz, J3,4 = 3.2 Hz, H-3 Gal), 4.99 (dd, 2H, J3,2 = 11.8 Hz, J3,4 = 3.2 Hz, H-3 Gal), 4.80 (d, 2H, Jgem = 12.8 Hz, CH2-triazole 2), 4.72 (m, 4H, H-2 Gal), 4.71 (d, 2H, Jgem = 12.8 Hz, CH2-triazole 2), 4.71 (d, 2H, Jgem = 12.8 Hz, CH2-triazole 1), 4.62 (d, 2H, Jgem = 12.8 Hz, CH2-triazole 1), 4.61 (m, 8H, SCH2CH2), 4.55 (dd, 4H, J5,6b = 7.0 Hz, J5,6a = 5.4 Hz, H-5 Gal), 4.15 (dd, 4H, J6a,6b = 11.3 Hz, J6a,5 = 5.4 Hz, H-6a Gal), 4.09 (dd, 4H, J6b,6a = 11.3 Hz, J6b,5 = 7.0 Hz, H-6b Gal), 3.87 (dd, 2H, J3,2 = J3,OH = 6.4 Hz, H-3), 3.80 (m, 2H, H-1a), 3.71 (m, 2H, H-2), 3.69 (m, 2H, H-1b), 3.65 (d, 2H, JOH,3 = 6.4 Hz, OH), 3.18 (m, 4H, SCH2aCH2), 3.08 (m, 4H, SCH2bCH2), 2.16 (s, 12H), 2.0 (s, 6H), 1.99 (s, 6H), 1.99 (s, 12H), 1.98 (s, 6H) (each OCOCH3), 1.98 (s, 6H, NHCOCH3); 13C NMR (126 MHz, CDCl3) δ 171.1, 171.1 (each NHCOCH3), 170.9, 170.9, 170.6, 170.6, 170.4, 170.4 (each OCOCH3), 145.3 (C-4 triazole-2), 145.0 (C-4 triazole-1), 123.7 (×2) (C-5 triazole-1, C-5 triazole-2), 85.2, 84.9 (each C-1 Gal), 78.6 (C-2), 70.0 (C-1), 68.9 (C-3), 68.3 (×2) (C-3 Gal), 67.5 (×2) (C-5 Gal), 67.2 (×2) (C-4 Gal), 64.7 (CH2-triazole 1), 63.7 (CH2-triazole 2), 62.2, 62.2 (each C-6 Gal), 49.8, 49.6 (each SCH2CH2), 48.4 (×2) (C-2 Gal), 31.3, 31.2 (each SCH2CH2), 23.2 (×2) (NHCOCH3), 20.9, 20.9, 20.8 (×2), 20.8 (×2) (each OCOCH3); ESI-HRMS calcd for C82H118N16NaO38S4: 2085.6574, found: 2085.6567 [M + Na]+. To this protected intermediate (42 mg, 0.02 mmol) in dry MeOH (2 mL), freshly prepared NaOMe solution in MeOH (0.35 mL of 1 M) was added and the mixture was stirred at room temp for 4 h. Then Amberlite IR-120 H+ was added and stirred gently until the pH was 7. The resin was filtered off and the solvent was removed. Reverse phase column chromatography (MeCN–H2O, 0:1 to 1:1) gave the title compound 4 as a white solid (24 mg, 76%); [α]20D: +106 (c 0.8, DMSO); 1H NMR (500 MHz, DMSO-d6) δ 8.10 (d, 2H, H-triazole 1), 8.0 (d, 2H, H-triazole 2), 7.77 (d, 4H, JNH,2 Gal = 6.5 Hz, NH), 5.51 (d, 4H, J1,2 = 5.1 Hz, H-1 Gal), 4.70 (m, 6H, OH, CH2-triazole 2), 4.68 (m, 4H, OH), 4.61 (m, 2H, OH), 4.58 (m, 6H, CH2-triazole 2, OH), 4.56 (m, 4H, CH2-triazole 1), 4.53 (m, 8H, (SCH2CH2), 4.19 (ddd, 4H, J2,3 = 11.8 Hz, J2,NH = 6.5 Hz, J2,1 = 5.1 Hz, H-2 Gal), 3.92 (m, 4H, H-5 Gal), 3.85 (m, 2H, H-1a), 3.73 (m, 4H, H-4 Gal), 3.60 (m, 4H, H-2, H-3), 3.58 (m, 2H, H-1b), 3.55 (m, 8H, H-6 Gal), 3.50 (m, 4H, H-3 Gal), 3.02 (m, 4H, SCH2aCH2), 2.93 (m, 4H, SCH2bCH2), 1.81 (s, 6H), 1.80 (s, 6H) (each NHCOCH3). 13C NMR (126 MHz, DMSO-d6) δ 169.9, 169.9 (NHCOCH3), 144.4 (C-4 triazole-2), 144.0 (C-4 triazole-1), 124.0 (C-5 triazole-1), 123.8 (C-5 triazole-2), 84.4 (×2) (C-1 Gal), 78.1 (C-2), 72.2, 72.2 (each C-5 Gal), 71.1 (C-1), 68.1 (×3) (C-4 Gal, C-3), 67.5 (×2) (C-3 Gal), 64.0 (CH2-triazole 1), 63.4 (CH2-triazole 2), 60.9, 60.9 (each C-6 Gal), 50.0 (×2) (C-2 Gal), 49.3 (×2) (SCH2CH2), 29.9, 29.8 (each SCH2CH2), 22.7 (×2) (NHCOCH3). ESI-HRMS calcd for C58H94N16NaO26S4: 1581.5306, found: 1581.5365 [M + Na]+.
BF2 chelated 3-methyl-N-(3-methyl-5-(4-(prop-2-yn-1-yloxy)phenyl)-1H-pyrrol-2-yl)-5-(4-(prop-2-yn-1-yloxy)phenyl)-2H-pyrrol-2-imine 16
To 1534 (500 mg, 1.35 mmol) in dry CH2Cl2 (20 mL) at −78 °C, was added dropwise BBr3 (1 M in CH2Cl2, 5.4 mmol) and the mixture stirred at −78 °C for 3 h. The reaction mixture was quenched by the slow addition of saturated sodium bicarbonate (30 mL), the organic layer was then separated and the aqueous layer was extracted with CH2Cl2 (2 × 40 mL). The combined organic layers were washed with saturated sodium bicarbonate, deionized H2O, brine, dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. Silica gel chromatography (cyclohexane–EtOAc, 3:1) gave the demethylated intermediate as a dark blue solid (169 mg, 35%, mp 121–122 °C); 1H NMR (400 MHz, CD3OD-d4): δ 7.72 (dd, J = 8.8, 1.9 Hz, 4H), 6.89 (dd, J = 8.8, 1.9 Hz, 4H), 6.71 (s, 2H), 4.57 (br, 2H), 2.26 (s, 6H) ppm; 13C NMR (100 MHz, CD3OD-d4): δ 159.4, 154.4, 150.6, 140.3, 127.7, 123.8, 115.7, 115.7, 9.9 ppm; ESI-HRMS [M + H]+: 358.1541, C22H20N3O2 requires 358.1556. To this intermediate (160 mg, 0.45 mmol) in dry THF (25 mL) cesium carbonate (439 mg, 1.35 mmol) was added, and the mixture was stirred at 0 °C under a nitrogen atmosphere for 10 min. Propargyl p-toluenesulfonate (473 mg, 2.25 mmol) was then added via syringe and the solution was heated at reflux, whilst stirring, for 2 h. The reaction mixture was cooled to room temp and the mixture was partitioned between CH2Cl2 (100 mL) and PBS buffer (100 mL, pH 7). The organic phase was washed with water (3 × 100 mL), brine (50 mL), dried over Na2SO4, filtered and evaporated to dryness. The crude product was purified by silica gel chromatography, eluting with CH2Cl2 to yield the propargylated intermediate compound (107 mg, 55%, mp 133–135 °C) as a dark green solid; 1H NMR (400 MHz, CDCl3): δ 7.80 (d, J = 7.5 Hz, 1H), 7.08 (d, J = 7.4 Hz, 1H), 6.66 (s, 1H), 4.78 (s, 1H), 2.57 (s, 1H), 2.34 (s, 2H) ppm. 13C NMR (100 MHz, CDCl3): δ 158.7, 154.0, 150.0, 141.0, 127.8, 126.2, 116.3, 115.4, 78.2, 76.0, 55.9, 11.4 ppm; ESI-HRMS calcd for C28H24N3O2 434.1869, found m/z 434.1869 [M + H]+.To this intermediate (100 mg, 0.23 mmol) in dry CH2Cl2 (20 mL) DIPEA (0.4 mL, 2.3 mmol) was added and BF3·OEt2 (0.4 mL, 3.22 mmol), and the mixture was stirred at room tem under N2 for 20 h. The reaction mixture was washed with water (3 × 50 mL), brine, dried over Na2SO4, filtered and the solvent was removed under reduced pressure. Chromatography (cyclohexane–EtOAc, 4:1) gave the title compound 16 as red metallic solid (67 mg, 61%); 1H NMR (400 MHz, DMSO-d6): δ 8.05 (d, J = 9.0 Hz, 4H), 7.14 (d, J = 9.0 Hz, 4H), 7.01 (s, 2H), 4.93 (d, J = 2.4 Hz, 4H), 3.65 (t, J = 2.4 Hz, 2H), 2.33 (s, 6H) ppm; 13C NMR (100 MHz, DMSO-d6): δ 160.0, 157.8, 146.0, 143.3, 131.8, 124.3, 122.6, 115.6, 79.3, 79.2, 56.2, 11.6 ppm; ESI-HRMS calcd for C28H23BF2N3O2 482.1851, found m/z 482.1849 [M + H]+.
3-Methyl-N-(3-methyl-5-(4-((1-(2-(2-acetamido-2-deoxy-α-D-galactopyranosylthio)ethyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-2H-pyrrol-2-ylidene)-5-(4-((1-(2-(2-acetamido-2-deoxy-α-D-galactopyranosylthio)ethyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-1H-pyrrol-2-amine, BF2 chelate 6
Compound 16 (5.1 mg, 0.0168 mmol) was dissolved in H2O (1 mL) and de-O-acetylated 12 (3.7 mg, 7.7 μmol), prepared as described for 7 (below) from 12, was dissolved in THF (1 mL) and both solutions were added to a microwave vial and to this sodium ascorbate (7.65 mg, 0.0386 mmol) and copper sulphate pentahydrate (4.19 mg, 0.0168 mmol) were added. The mixture was heated in a microwave reactor (120 W) at 60 °C, for 1 h. The THF was then removed under vaccum and the remaining water was diluted with CH3CN and reverse phase chromatography (H2O–CH3CN, 1:1) gave the title compound 6 as a green solid (5.4 mg, 64%); 1H NMR (500 MHz, DMSO-d6) δ 8.21 (s, 2H, triazole CH), 8.03–7.98 (m, 4H, Ar–H), 7.70 (d, J = 7.0 Hz, 2H, NH), 7.18–7.11 (m, 4H), 6.97 (s, 2H, Ar–H), 5.46 (d, J = 5.3 Hz, 2H, H-1), 5.21 (s, 4H, CH2OAr), 4.68–4.61 (m, 4H, OH-4, OH-6), 4.60–4.45 (m, 6H, SCH2CH2, OH-3), 4.13 (dt, J = 11.9, 6.3 Hz, 2H, H-2), 3.87 (t, J = 6.0 Hz, 2H, H-5), 3.68 (t, J = 4.0 Hz, 2H, H-4), 3.50 (t, J = 5.8 Hz, 4H, H-6), 3.44 (ddd, J = 10.8, 6.7, 3.0 Hz, 2H. H-3), 3.00 (dt, J = 14.0, 6.9 Hz, 2H SCH2CH2), 2.90 (dt, J = 13.9, 6.9 Hz, 2H, SCH2CH2), 2.28 (s, 6H, CH3), 1.76 (d, J = 1.4 Hz, 6H, NHAc); 13C NMR (126 MHz, dmso) δ 170.21 (CO), 160.95, 157.70, 155.31, 145.95, 143.04, 142.42 (C), 131.91 (CH), 125.45 (triazole CH), 123.91, 115.48 (each Ar-CH), 84.77 (C-1), 72.67 (C-5), 68.53 (C-4), 67.91 (C-3), 61.85 (CH2OAr), 61.34 (C-6), 50.43 (C-2), 49.79 (SCH2CH2), 30.27 (SCH2CH2), 23.06 (NHAc), 11.49 (CH3); ESI-HRMS: calcd for C48H58BN11O12 F2S2 Na 1116.3667; found 1116.3676 [M + Na]+
Methyl 2-acetamido-2-deoxy-1-thio-α-D-galactopyranoside 7
The thiol 1635 (77 mg, 0.199 mmol) was dissolved in dichloromethane (3 mL) and DIPEA (0.4 mmol, 70 μL) and methyl iodide (0.9 mmol, 56 μL) was added and the mixture was stirred for 2.5 h at room temp. The solvent was removed and flash chromatography (toluene–acetone (6:4)) gave the protected intermediate (62 mg, 82%); 1H NMR (500 MHz, chloroform-d) δ 5.64 (d, J = 8.8 Hz, 1H, NH), 5.44 (d, J = 5.3 Hz, 1H, H-1), 5.38 (dd, J = 3.3, 1.3 Hz, 1H, H-4), 5.09 (dd, J = 11.7, 3.3 Hz, 1H, H-3), 4.80–4.74 (m, 1H, H-2), 4.55–4.50 (m, 1H, H-5), 4.17–4.06 (m, 2H, H-6), 2.16 (s, 3H, OAc), 2.12 (s, 3H, CH3), 2.04 (s, 3H, OAc), 2.00 (s, 3H, OAc), 1.97 (s, 3H, NHAc); 13C NMR (126 MHz, CDCl3) δ 171.06, 170.37, 170.26, 170.15(each CO), 85.53 (C-1), 68.44 (C-3), 67.35 (C-4), 67.11 (C-5), 61.92 (C-6), 48.38 (C-2), 23.30 (NHAc), 20.74, 20.69, 20.68 (each OAc), 13.15 (CH3). To this intermediate (55 mg, 0.14 mmol) dissolved in MeOH (2 mL) was added 1 M NaOMe in MeOH and the mixture was left to stir until the pH was ∼10 and stirring was continued for 20 min. Amberlite H+ resin was then added until the pH was ∼6 and was stirred for another 20 min. The mixture was then filtered and solvent removed under reduced pressure to give the title compound as a white solid (25 mg, 70%); 1H NMR (500 MHz, methanol-d4) δ 5.44 (d, J = 5.4 Hz, 1H, H-1), 4.41 (dd, J = 11.2, 5.4 Hz, 1H, H-2), 4.14 (ddd, J = 6.7, 5.2, 1.3 Hz, 1H, H-5), 3.88 (dd, J = 3.3, 1.4 Hz, 1H, H-4), 3.76–3.70 (m, 3H, H-3, H-6a, H-6b), 2.06 (s, 3H, CH3), 1.96 (s, 3H, NHAc); 13C NMR (126 MHz, methanol-d4) δ 172.51 (CO), 84.82 (C-1), 71.29 (C-5), 68.91 (C-4), 68.08 (C-3), 61.38 (C-6), 50.55 (C-2), 21.14 (NHAc), 11.55 (CH3); ESI-HRMS calcd for C9H17NO5SNa 274.0725, found m/z 274.0729 [M + Na]+.
2-Azidoethyl 2-acetamido-2-deoxy-1-thio-α-D-glucopyranoside 18
Thioglycoside 17 (40 mg, 0.092 mmol) was deacetylated as described in the preparation of 7 to give the title compound (24 mg, 85%); 1H NMR (500 MHz, methanol-d4) δ 5.56 (d, J = 5.3 Hz, 1H, H-1), 4.03 (dd, J = 11.1, 5.3 Hz, 1H, H-2), 3.96 (ddd, J = 10.0, 5.8, 2.4 Hz, 1H, H-5), 3.83 (dd, J = 12.0, 2.3 Hz, 1H, H-6a), 3.71 (dd, J = 12.0, 5.8 Hz, 1H, H-6b), 3.58 (dd, J = 11.1, 8.7 Hz, 1H, H-3), 3.55–3.49 (m, 1H, SCH2CH2N3), 3.45 (dt, J = 12.8, 6.7 Hz, 1H, SCH2CH2N3), 3.37–3.31 (m, 1H, H-4), 2.89–2.81 (m, 1H, SCH2CH2N3), 2.73 (dt, J = 13.7, 6.6 Hz, 1H, SCH2CH2N3), 1.97 (s, 3H, NHAc); 13C NMR (126 MHz, methanol-d4) δ 172.31 (CO), 83.98 (C-1), 73.05 (C-5), 71.19 (C-3), 71.08 (C-4), 61.20 (C-6), 54.41 (C-2), 50.78 (SCH2CH2N3), 29.77 (SCH2CH2N3), 21.12 (NHAc); ESI-HRMS: calcd. For C10H19N4O5S 307.1076; found 307.1070 [M + H]+.
3-Phenyl-N-(3-phenyl-5-(4-((1-(2-(2-acetamido-2-deoxy-α-D-glucoopyranosylthio)ethyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-2H-pyrrol-2-ylidene)-5-(4-((1-(2-(2-acetamido-2-deoxy-α-D-glucopyranosylthio)ethyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-1H-pyrrol-2-amine, BF2 chelate 8
Glucopyranoside 18 (5.56 mg, 0.0181 mmol) in H2O (1 mL) and 1933 (5 mg, 0.082 mmol) in THF (1 mL) were mixed and added to a microwave vial and to this mixture sodium ascorbate (8.2 mg, 0.041 mmol) and copper sulphate pentahydrate (8.2 mg, 0.041 mmol) were added. The reaction was performed in the microwave (120 W) at 60 °C for 1 h. The THF was then removed under vacuum to concentrate the mixture with some water remaining. This was diluted with CH3CN and reverse phase chromatography (H2O–CH3CN, 1:1) gave 8 as a green solid (8.6 mg, 86%); 1H NMR (500 MHz, DMSO-d6) δ 8.29 (s, 2H, triazole CH), 8.19 (dd, J = 11.3, 8.1 Hz, 7H, Ar–H), 7.87 (d, J = 7.0 Hz, 2H, NH), 7.64 (s, 2H, Ar–H), 7.55 (t, J = 7.6 Hz, 4H, Ar–H), 7.49 (t, J = 7.3 Hz, 2H, Ar–H), 7.26 (dd, J = 7.9, 5.5 Hz, 4H, Ar–H), 7.19–7.14 (m, 1H, Ar–H), 5.47 (d, J = 5.3 Hz, 2H, H-1), 5.30 (s, 4H, CH2OAr), 5.15 (d, J = 5.6 Hz, 2H, 4-OH), 4.85 (d, J = 5.7 Hz, 2H, 3-OH), 4.68 (t, J = 5.7 Hz, 2H, 6-OH), 4.58 (ddt, J = 27.7, 13.9, 7.0 Hz, 4H, SCH2CH2 triazole), 3.81 (ddd, J = 11.8, 6.9, 5.3 Hz, 2H, H-2), 3.78–3.68 (m, 4H, H-5, H-6a), 3.51 (dt, J = 11.9, 6.2 Hz, 2H, H-6b), 3.41–3.35 (m, 2H, H-3), 3.18–3.04 (m, 4H, H-4, SCH2CH2), 3.00 (dt, J = 13.7, 6.8 Hz, 2H, SCH2CH2), 1.83 (s, 6H, NHAc); 13C NMR (126 MHz, dmso) δ 170.07 (CO), 170.07 (CO), 161.27, 157.84, 148.14, 145.00, 142.46, 142.60 (each Ar–C), 132.27, 130.05, 129.58, 129.35, 129.17, 128.65 (each Ar–H), 125.45, 123.93, 115.60 (each Ar–H), 84.49 (C-1), 74.21(C-5), 71.42 (C-4), 71.08 (C-3), 61.96 (CH2OAr), 61.34 (C-6), 54.63 (C-2), 49.82 (SCH2CH2), 30.56 (SCH2CH2), 23.05 (NHAc); ESI-HRMS: calcd for C58H62BN11O12F2S2Na 1240.3980; found 1240.3395 [M + Na]+.
Lectins
The two recombinant MGL proteins (CRD and CRD extended by the stalk) were produced, purified and subjected to quality controls as described in detail previously.31 SBA was purchased from Sigma-Aldrich (Munich, Germany). Biotinylated DBA was obtained from Enzo Life Sciences (Lörrach, Germany), biotinylated HPA from Sigma-Aldrich. Biotinylation of the MGL proteins and SBA was performed under activity-preserving conditions with the N-hydroxysuccinimide ester derivative of biotin (Sigma-Aldrich) using the protocol used for human galectin-1.83,90 Fluorescent labeling was performed using the Alexa Fluor® 555 or the Alexa Fluor® 488 Protein Labeling Kits (Molecular Probes, Thermo Fisher Scientific, Darmstadt, Germany) following the recommendations of the supplier. Maintained lectin activity was ascertained by carbohydrate-inhibitable cell binding in cytofluorometric assays using CHO wild-type and Lec8 mutant cells31,84,91 and in the histochemical protocol (please see below).Tissue samples
Specimens of jejunum obtained from four six-week-old C57BL/6 mice were thoroughly washed, then cut into small pieces and fixed in Bouin's solution (71% (v/v) saturated picric acid, 24% (v/v) formaldehyde solution (37% w/v) and 5% (v/v) glacial acetic acid) for 24 h. Dehydration of specimens was carried out in a series of aqueous ethanol solutions of increasing alcohol concentrations (70%, 80% and 99%) and then in isopropanol, which finally was replaced by xylene. Embedding of the pieces of tissue in paraffin wax at 61 °C followed, thereafter each specimen was cut into serial sections of about five μm thickness that were mounted on Superfrost® plus glass slides (Menzel, Braunschweig, Germany). Tissue specimen of murine jejunum were a kind gift of Dr E. Wolf of the Faculty of Veterinary Medicine, Ludwig-Maximilians-University Munich, Germany; all further processing was done by the authors as described.Lectin histochemistry
The protocols for processing tissue sections by lectin histochemistry and monitoring staining profiles by light and fluorescence microscopy had been optimized previously.69,85,92,93 Routinely, systematic titrations with each labelled lectin were carried out to determine the concentrations, which generate strong signals combined with minimal background staining. The tested concentration ranges for light/fluorescence microscopy were 0.25–1.5/0.5–1 μg mL−1 for DBA, 0.5–4/0.25–4 μg mL−1 for SBA, 0.5–2/1–2 μg mL−1 for HPA, 1–8 μg mL−1 for MGL (CRD) and 1–8/8–32 μg mL−1 for MGL (CRD + stalk), respectively. Assessment of inhibitory capacity of glycocompounds was determined after a pre-incubation of the mixture containing a glycocompound and lectin for one hour at room temperature. Aliquots of these mixtures were then applied to deparaffinized tissue sections for an overnight incubation at 4 °C. The processing of sections used for light or fluorescence microscopy differed slightly.When running the protocol for light microscopy, sections had been washed in 10 mM 4-(2-hydroxyethyl)-1-piperazine-ethanesulfonic acid buffer (HEPES, pH 7.5) containing 0.1 mM CaCl2 and then treated with this buffer containing 1% (w/v) carbohydrate-free bovine serum albumin (BSA), for 1 h at room temperature to saturate sites for non-specific binding of proteins. Following washing steps and after the incubation with lectin-containing solution in the absence/presence of glycoinhibitors, visualisation of signals was accomplished by applying commercial Vectastain® ABC kit and Vector Red® reagents (Biozol, Eching, Germany).
The route to obtaining profiles of fluorescent staining started by incubating deparaffinized sections in a humid chamber with a 1% (w/v) solution of BSA in HEPES buffer containing 0.1 mM CaCl2 and 0.1% (w/v) Tween-20 (blocking solution) to saturate non-specific binding of protein. Subsequently, sections were exposed to labelled lectins dissolved in blocking solution, e.g. Alexa Fluor®-555-labelled SBA, biotinylated DBA or HPA, or a ternary mixture containing the fluorescent SBA, biotinylated DBA and FITC-labelled MGL (CRD + stalk), in a humid chamber overnight at 4 °C. After two thorough washing steps (each for 5 min) in HEPES buffer/Tween 20 and a washing step in PBS/Tween-20 (PBS-T) to remove the labelled probes, Alexa Fluor®-647-conjugated streptavidin (Invitrogen, Thermo Fisher Sciences, Darmstadt, Germany) (solutions of 2–10 μg mL−1 were tested, 5 μg mL−1 was found to be optimal) in 1% BSA/PBS-T was applied for 1 h, then 4′,6-diamidino-2-phenylindole (DAPI; 0.5 μg mL−1) in PBS-T was added for 5 min at room temperature in a humid chamber in the dark.
Processed sections for light microscopy were counterstained with Mayer's haemalaun, dehydrated and mounted with Eukitt® (Kindler, Freiburg, Germany). For fluorescence microscopy, a mounting medium containing an anti-fading agent (Dako Fluorescence Mounting Medium; Agilent DAKO, Waldbronn, Germany) was used. Controls to exclude lectin-independent staining were performed by omission of the incubation step with lectin-containing solution. Controls of sugar specificity were carried out by using the cognate sugar in titrations. Lack of inhibition by carbohydrate-independent binding of the bulky scaffolds in bivalent compound 6 and tetravalent compound 5 by comparing the activity of GalNAc-presenting compounds 5 and 6 to interfere with lectin binding in parallel with testing the corresponding GlcNAc-presenting compounds 8 and 9 (0.05–2 mM of sugar) on MGL (CRD + stalk) and SBA.
Microphotographs were recorded using an AxioImager.M1 microscope (Carl Zeiss MicroImaging, Göttingen, Germany) equipped with an AxioCam MRm digital camera and the software AxioVision version 4.9. Semi-quantitative analysis by two independent observers was based on independently examining at least ten high-power fields per section. Signal intensity is expressed in the following grading system: −, no staining; (+), very weak but significant staining; +, weak staining; ++, medium staining; +++, strong staining; ++++, very strong staining. The percentage of positive cells is grouped in the categories: (1) 0% (no positive cells), (2) < 5% (few positive cells), (3) 5–20%, (4) 21–40%, (5) 41–60%, (6) 61–80%, (7) 81–95% (few negative cells), (8) up to 100% (no negative cells).
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Inspiring discussions with Drs B. Friday and A. Leddoz are gratefully acknowledged. Research in this paper was funded in part by Science Foundation Ireland (Grant Number 12/IA/1398) and the Irish Research Council (Grant Number GOIPG/2016/858). L. L. R.-H. thanks CONACYT-México (Grant 290936) for financial support.Notes and references
- H.-J. Gabius and J. Roth, Histochem. Cell Biol., 2017, 147, 111–117 CrossRef PubMed.
- F. L. Harrison and C. J. Chesterton, FEBS Lett., 1980, 122, 157–165 CrossRef PubMed.
- W. M. Gallatin, I. L. Weissman and E. C. Butcher, Nature, 1983, 304, 30–34 CrossRef PubMed.
- D. Vestweber and J. E. Blanks, Physiol. Rev., 1999, 79, 181–213 CrossRef PubMed.
- R. P. McEver, Cardiovasc. Res., 2015, 107, 331–339 CrossRef PubMed.
- G. L. Nicolson, Int. Rev. Cytol., 1974, 39, 89–190 Search PubMed.
- L. Bhattacharyya, M. I. Khan, J. Fant and C. F. Brewer, J. Biol. Chem., 1989, 264, 11543–11545 Search PubMed.
- C. Boscher, J. W. Dennis and I. R. Nabi, Curr. Opin. Cell Biol., 2011, 23, 383–392 CrossRef PubMed.
- H. Kaltner, S. Toegel, G. García Caballero, J. C. Manning, R. W. Ledeen and H.-J. Gabius, Histochem. Cell Biol., 2017, 147, 239–256 CrossRef PubMed.
- K.-i. Kasai, Trends Glycosci. Glycotechnol., 2018, 30, E221–E228 CrossRef.
- A. Varki, J. Clin. Invest., 1997, 99, 158–162 CrossRef PubMed.
- E. P. McGreal, J. L. Miller and S. Gordon, Curr. Opin. Immunol., 2005, 17, 18–24 CrossRef PubMed.
- H.-J. Gabius, J. C. Manning, J. Kopitz, S. André and H. Kaltner, Cell. Mol. Life Sci., 2016, 73, 1989–2016 CrossRef PubMed.
- S. Mayer, M. K. Raulf and B. Lepenies, Histochem. Cell Biol., 2017, 147, 223–237 CrossRef PubMed.
- Y. C. Lee and R. T. Lee, Acc. Chem. Res., 1995, 28, 321–327 CrossRef.
- Y. M. Chabre and R. Roy, in The Sugar Code. Fundamentals of glycosciences, ed. H.-J. Gabius, Wiley-VCH, Weinheim, Germany, 2009, ch. 4, pp. 53–70 Search PubMed.
- G. C. Daskhan, N. Berthet, B. Thomas, M. Fiore and O. Renaudet, Carbohydr. Res., 2015, 405, 13–22 CrossRef PubMed.
- R. Roy, P. V. Murphy and H.-J. Gabius, Molecules, 2016, 21, 629 CrossRef PubMed.
- A. P. Corfield and M. Berry, Trends Biochem. Sci., 2015, 40, 351–359 CrossRef PubMed.
- R. W. Ledeen and G. Wu, Trends Biochem. Sci., 2015, 40, 407–418 CrossRef PubMed.
- A. P. Corfield, Histochem. Cell Biol., 2017, 147, 119–147 CrossRef PubMed.
- J. Kopitz, Histochem. Cell Biol., 2017, 147, 175–198 CrossRef PubMed.
- J. Roth, Exp. Pathol., 1978, 3(suppl 1), 1–186 Search PubMed.
- J. Schrével, D. Gros and M. Monsigny, Prog. Histochem. Cytochem., 1981, 14(2), 1–269 CrossRef.
- J. Roth, Biochim. Biophys. Acta, 1987, 906, 405–436 CrossRef.
- S. S. Spicer and B. A. Schulte, J. Histochem. Cytochem., 1992, 40, 1–38 CrossRef PubMed.
- J. Roth, Histochem. Cell Biol., 2011, 136, 117–130 CrossRef PubMed.
- J. C. Manning, A. Romero, F. A. Habermann, G. García Caballero, H. Kaltner and H.-J. Gabius, Histochem. Cell Biol., 2017, 147, 199–222 CrossRef PubMed.
- R. Roy, Y. Cao, H. Kaltner, N. Kottari, T. C. Shiao, K. Belkhadem, S. André, J. C. Manning, P. V. Murphy and H.-J. Gabius, Histochem. Cell Biol., 2017, 147, 285–301 CrossRef PubMed.
- A. P. Corfield, Biochim. Biophys. Acta, 2015, 1850, 236–252 CrossRef PubMed.
- S. André, S. O'Sullivan, C. Koller, P. V. Murphy and H.-J. Gabius, Org. Biomol. Chem., 2015, 13, 4190–4203 RSC.
- G. J. Miller, K. R. Broberg, C. Rudd, M. R. Helliwell, G. C. Jayson and J. M. Gardiner, Org. Biomol. Chem., 2015, 13, 11208–11219 RSC.
- D. Wu, S. Cheung, R. Daly, H. Burke, E. M. Scanlan and D. F. O'Shea, Eur. J. Org. Chem., 2014, 6841–6845 CrossRef.
- D. Wu and D. F. O'Shea, Org. Lett., 2013, 15, 3392–3395 CrossRef PubMed.
- S. Knapp and D. S. Myers, J. Org. Chem., 2002, 67, 2995–2999 CrossRef PubMed.
- S. André, S. O'Sullivan, H.-J. Gabius and P. V. Murphy, Tetrahedron, 2015, 71, 6867–6880 CrossRef.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., 2016, B72, 171–179 Search PubMed.
- G. R. Gunther, J. L. Wang, I. Yahara, B. A. Cunningham and G. M. Edelman, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 1012–1016 CrossRef.
- D. K. Mandal and C. F. Brewer, Biochemistry, 1993, 32, 5116–5120 CrossRef PubMed.
- D. Schwefel, C. Maierhofer, J. G. Beck, S. Seeberger, K. Diederichs, H. M. Moller, W. Welte and V. Wittmann, J. Am. Chem. Soc., 2010, 132, 8704–8719 CrossRef PubMed.
- T. Matsushita, K. Tsuchibuchi, T. Koyama, K. Hatano and K. Matsuoka, Bioorg. Med. Chem. Lett., 2018, 28, 1704–1707 CrossRef PubMed.
- H. Debray, D. Decout, G. Strecker, G. Spik and J. Montreuil, Eur. J. Biochem., 1981, 117, 41–55 CrossRef PubMed.
- M. E. Pereira, E. A. Kabat and N. Sharon, Carbohydr. Res., 1974, 37, 89–102 CrossRef PubMed.
- S. Hammarström, L. A. Murphy, I. J. Goldstein and M. E. Etzler, Biochemistry, 1977, 16, 2750–2755 CrossRef.
- D. A. Baker, S. Sugii, E. A. Kabat, R. M. Ratcliffe, P. Hermentin and R. U. Lemieux, Biochemistry, 1983, 22, 2741–2750 CrossRef PubMed.
- B. V. Torres, D. K. McCrumb and D. F. Smith, Arch. Biochem. Biophys., 1988, 262, 1–11 CrossRef PubMed.
- V. Piller, F. Piller and J.-P. Cartron, Eur. J. Biochem., 1990, 191, 461–466 CrossRef PubMed.
- S.-i. Iida, K. Yamamoto and T. Irimura, J. Biol. Chem., 1999, 274, 10697–10705 CrossRef PubMed.
- S. J. van Vliet, E. van Liempt, E. Saeland, C. A. Aarnoudse, B. Appelmelk, T. Irimura, T. B. H. Geijtenbeek, O. Blixt, R. Alvarez, I. van Die and Y. van Kooyk, Int. Immunol., 2005, 17, 661–669 CrossRef PubMed.
- J.-F. Sanchez, J. Lescar, V. Chazalet, A. Audfray, J. Gagnon, R. Alvarez, C. Breton, A. Imberty and E. P. Mitchell, J. Biol. Chem., 2006, 281, 20171–20180 CrossRef PubMed.
- O. Oyelaran, Q. Li, D. Farnsworth and J. C. Gildersleeve, J. Proteome Res., 2009, 8, 3529–3538 CrossRef PubMed.
- N. Mortezai, H. N. Behnken, A. K. Kurze, P. Ludewig, F. Buck, B. Meyer and C. Wagener, Glycobiology, 2013, 23, 844–852 CrossRef PubMed.
- G. Artigas, J. T. Monteiro, H. Hinou, S. I. Nishimura, B. Lepenies and F. Garcia-Martin, J. Med. Chem., 2017, 60, 9012–9021 CrossRef PubMed.
- S. Hadjialirezaei, G. Picco, R. Beatson, J. Burchell, B. T. Stokke and M. Sletmoen, PLoS One, 2017, 12, e0175323 CrossRef PubMed.
- A. Imberty, F. Casset, C. V. Gegg, M. E. Etzler and S. Perez, Glycoconjugate J., 1994, 11, 400–413 CrossRef PubMed.
- A. Dessen, D. Gupta, S. Sabesan, C. F. Brewer and J. C. Sacchettini, Biochemistry, 1995, 34, 4933–4942 CrossRef PubMed.
- M. H. Dao-Thi, T. W. Hamelryck, J. Bouckaert, F. Korber, V. Burkow, F. Poortmans, M. Etzler, G. Strecker, L. Wyns and R. Loris, Acta Crystallogr., 1998, D54, 1446–1449 CrossRef.
- T. W. Hamelryck, R. Loris, J. Bouckaert, M. H. Dao-Thi, G. Strecker, A. Imberty, E. Fernandez, L. Wyns and M. E. Etzler, J. Mol. Biol., 1999, 286, 1161–1177 CrossRef PubMed.
- P. J. Coombs, R. Harrison, S. Pemberton, A. Quintero-Martinez, S. Parry, S. M. Haslam, A. Dell, M. E. Taylor and K. Drickamer, J. Mol. Biol., 2010, 396, 685–696 CrossRef PubMed.
- S. A. Jégouzo, A. Quintero-Martínez, X. Ouyang, A. dos Santos, M. E. Taylor and K. Drickamer, Glycobiology, 2013, 23, 853–864 CrossRef PubMed.
- H. Tateno, N. Uchiyama, A. Kuno, A. Togayachi, T. Sato, H. Narimatsu and J. Hirabayashi, Glycobiology, 2007, 17, 1138–1146 CrossRef PubMed.
- K. T. Pilobello, D. E. Slawek and L. K. Mahal, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11534–11539 CrossRef PubMed.
- M. Watanabe, Z. Takeda, Y. Urano and T. Muramatsu, in Teratocarcinoma and Embryonic Cell Interactions, ed. T. Muramatsu, G. Gachelin, A. A. Moscona and Y. Ikawa, Academic Press/Japan Scientific Societies' Press, Tokyo, 1982, pp. 217–228 Search PubMed.
- M. Sato and T. Muramatsu, Differentiation, 1985, 29, 29–38 CrossRef PubMed.
- Y. Kamada, H. Muramatsu, Y. Arita, T. Yamada and T. Muramatsu, J. Biochem., 1991, 109, 178–183 CrossRef PubMed.
- U. Brinck, R. Bosbach, M. Korabiowska, A. Schauer and H.-J. Gabius, Histol. Histopathol., 1995, 10, 61–70 Search PubMed.
- E. Saeland, S. J. van Vliet, M. Bäckström, V. C. M. van den Berg, T. B. H. Geijtenbeek, G. A. Meijer and Y. van Kooyk, Cancer Immunol. Immunother., 2007, 56, 1225–1236 CrossRef PubMed.
- P. Rohse and V. Wittmann, Chem. –Eur. J., 2016, 22, 9724–9733 CrossRef PubMed.
- J. C. Manning, G. García Caballero, C. Knospe, H. Kaltner and H.-J. Gabius, J. Anat., 2017, 231, 23–37 CrossRef PubMed.
- J. C. Manning, G. García Caballero, F. M. Ruiz, A. Romero, H. Kaltner and H.-J. Gabius, Trends Glycosci. Glycotechnol., 2018, 30, SE11–SE20 CrossRef.
- J. Nio-Kobayashi, Trends Glycosci. Glycotechnol., 2018, 30, SE89–SE96 CrossRef.
- T. Angata and E. C. M. Brinkman-van der Linden, Biochim. Biophys. Acta, 2002, 1572, 294–316 CrossRef.
- J. Iwaki and J. Hirabayashi, Trends Glycosci. Glycotechnol., 2018, 30, SE137–SE153 CrossRef.
- J. Kopitz, C. von Reitzenstein, S. André, H. Kaltner, J. Uhl, V. Ehemann, M. Cantz and H.-J. Gabius, J. Biol. Chem., 2001, 276, 35917–35923 CrossRef PubMed.
- H. Sanchez-Ruderisch, C. Fischer, K. M. Detjen, M. Welzel, A. Wimmel, J. C. Manning, S. André and H.-J. Gabius, FEBS J., 2010, 277, 3552–3563 CrossRef PubMed.
- D. Weinmann, K. Schlangen, S. André, S. Schmidt, S. M. Walzer, B. Kubista, R. Windhager, S. Toegel and H.-J. Gabius, Sci. Rep., 2016, 6, 39112 CrossRef PubMed.
- D. Weinmann, M. Kenn, S. Schmidt, K. Schmidt, S. M. Walzer, B. Kubista, R. Windhager, W. Schreiner, S. Toegel and H.-J. Gabius, Cell. Mol. Life Sci. DOI:10.1007/s00018-018-2856-2.
- B. Belardi, G. P. O'Donoghue, A. W. Smith, J. T. Groves and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 9549–9552 CrossRef PubMed.
- H.-J. Gabius, Folia Biol. (Praha), 2017, 63, 121–131 Search PubMed.
- Q. Xiao, A.-K. Ludwig, C. Romano, I. Buzzacchera, S. E. Sherman, M. Vetro, S. Vértesy, H. Kaltner, E. H. Reed, M. Moller, C. J. Wilson, D. A. Hammer, S. Oscarson, M. L. Klein, H.-J. Gabius and V. Percec, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E2509–E2518 CrossRef PubMed.
- A. Schwarz and A. H. Futerman, J. Histochem. Cytochem., 1997, 45, 611–618 CrossRef PubMed.
- J. W. Celie, R. H. J. Beelen and J. van den Born, J. Immunol. Methods, 2005, 298, 155–159 CrossRef PubMed.
- J. C. Rogers and S. Kornfeld, Biochem. Biophys. Res. Commun., 1971, 45, 622–629 CrossRef PubMed.
- S. Kojima, M. Ishido, A. Kubota, T. Kubodera, T. Hellmann, B. Kohnke-Godt, B. Wosgien and H.-J. Gabius, Biol. Chem. Hoppe-Seyler, 1990, 371, 331–338 CrossRef PubMed.
- S. André, C. Unverzagt, S. Kojima, X. Dong, C. Fink, K. Kayser and H.-J. Gabius, Bioconjugate Chem., 1997, 8, 845–855 CrossRef PubMed.
- C. Unverzagt, S. André, J. Seifert, S. Kojima, C. Fink, G. Srikrishna, H. Freeze, K. Kayser and H.-J. Gabius, J. Med. Chem., 2002, 45, 478–491 CrossRef PubMed.
- A. Ogura, T. Tahara, S. Nozaki, K. Morimoto, Y. Kizuka, S. Kitazume, M. Hara, S. Kojima, H. Onoe, A. Kurbangalieva, N. Taniguchi, Y. Watanabe and K. Tanaka, Sci. Rep., 2016, 6, 21797 CrossRef PubMed.
- L. Latypova, R. Sibgatullina, A. Ogura, K. Fujiki, A. Khabibrakhmanova, T. Tahara, S. Nozaki, S. Urano, K. Tsubokura, H. Onoe, Y. Watanabe, A. Kurbangalieva and K. Tanaka, Adv. Sci., 2017, 4, 1600394 CrossRef PubMed.
- A. Ogura, S. Urano, T. Tahara, S. Nozaki, R. Sibgatullina, K. Vong, T. Suzuki, N. Dohmae, A. Kurbangalieva, Y. Watanabe and K. Tanaka, Chem. Commun., 2018, 54, 8693–8696 RSC.
- H.-J. Gabius, B. Wosgien, M. Hendrys and A. Bardosi, Histochemistry, 1991, 95, 269–277 Search PubMed.
- J. Kopitz, Q. Xiao, A.-K. Ludwig, A. Romero, M. Michalak, S. E. Sherman, X. Zhou, C. Dazen, S. Vértesy, H. Kaltner, M. L. Klein, H.-J. Gabius and V. Percec, Angew. Chem., Int. Ed., 2017, 56, 14677–14681 CrossRef PubMed.
- H. Kaltner, G. García Caballero, F. Sinowatz, S. Schmidt, J. C. Manning, S. André and H.-J. Gabius, Biochim. Biophys. Acta, 2016, 1860, 2298–2312 CrossRef PubMed.
- J. C. Manning, G. García Caballero, C. Knospe, H. Kaltner and H.-J. Gabius, Ann. Anat., 2018, 217, 66–81 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05382k |
| This journal is © The Royal Society of Chemistry 2018 |