
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlteration of vitrified intermediate level nuclear waste in alkaline media: effects of cementitious materials, pH and temperature
Tomo Suzuki-Muresan *a,
A. Abdelouasa,
C. Landesmana,
A. Ait-Chaoua,
Y. El Mendilia,
S. Ribeta,
K. Perrigauda,
D. Shitarab,
C. Martinc and
X. Bourbonc
*a,
A. Abdelouasa,
C. Landesmana,
A. Ait-Chaoua,
Y. El Mendilia,
S. Ribeta,
K. Perrigauda,
D. Shitarab,
C. Martinc and
X. Bourbonc
aSUBATECH (IMT Atlantique, CNRS/IN2P3, Université de Nantes), BP 20722, 44307 Nantes cedex 3, France. E-mail: tomo.suzuki@subatech.in2p3.fr; Fax: +33 2 51 85 84 52; Tel: +33 2 51 85 86 71
bDivision of Energy and Environmental Systems, Graduate School of Engineering, Hokkaido University, Kita 13 Nishi 8, Kita-ku, Sapporo, Hokkaido 060-8628, Japan
cANDRA, 1/7, rue Jean Monnet, Parc de la Croix-Blanche, 92298 Châtenay-Malabry, France
First published on 9th November 2018
Abstract
Alteration experiments involving intermediate level nuclear waste (ILW) glass in contact with hardened cement paste (HCP) were performed to assess its behavior under simulated repository conditions. Batch experiments were conducted at 20 °C and 50 °C in several artificial cement pore water (ACW) samples (pH from 10 to 13), in the presence of HCP (CEM-I, CEM-V and low pH), with a ratio of glass surface to volume of solution of 8000 m−1 and a ratio of mass of HCP to volume of solution of 10 g L−1. Glass alteration rates increase up to ∼4 × 10−2 g m−2 d−1 with pH in contact with HCP, notably with CEM-I. This value decreases by 2 orders of magnitude in low pH cement solution and also for residual alteration rates. The effect of calcium on glass alteration was observed, mainly in Ca(OH)2 saturated solution, with an incubation effect on the release of Si in solution. Experimental data were successfully modeled with the PhreeqC geochemical code. Glass and HCP samples were characterized via SEM/EDX and micro-Raman studies. This work showed that vitrified glass exhibits good performance in terms of low alteration rates (∼10−4 g m−2 d−1), the absence of secondary phases, and the formation of a gel layer at the surface, when in contact with low pH conditions (in the presence or absence of low pH HCP).
1. Introduction
The nuclear industry, like any industry, generates a variety of wastes, which, according to their radioactivity levels, are categorized, oriented to suitable disposal solutions and treated according to specific procedures. In general, radioactive wastes are packaged in solid forms to reduce and optimize significantly their volume, to ensure the best properties of confinement and manipulation, and to meet the requirements of safety assessments.1 Therefore, vitrification is one of the processes largely proven and notably effective in conditioning high level waste (HLW) for several decades.2 Highly exothermic HLW glass is characterized by the presence of residues from the reprocessing of spent nuclear fuel, mainly fission products and minor actinides. In France, the alteration of HLW glass and the understanding of its kinetic and long-term behavior have extensively been studied using simulated inactive SON68 glass and R7T7 doped glass under acidic to alkaline pH conditions (aqueous media), and from 20 °C to 100 °C (aqueous media and relative humidity controlled atmospheres), under repository conditions.3–8 The glass surface evolution has extensively been studied in aqueous media, for example the mechanism of gel layer formation, in relation to the formation of secondary mineral phases and their stability. The experimental data (dissolution kinetics, alteration products) has been successfully modeled using geochemical codes.9–14 A general multi-step mechanism for aqueous glass alteration has been proposed and validated for a large number of glass types: first hydration, then the interdiffusion of protons and alkali and alkali earth ions, and finally the hydrolysis/recondensation of the glass network. A porous-hydrated-gel layer is therefore formed at the surface leading to a drop in alteration rates (residual alteration rate: ∼10−4 g m−2 d−1; 90 °C, deionized water) and acting as a diffusion barrier.3 Secondary alumino-silicate crystalline phases, such as phyllosilicates, precipitate at the glass surface.13–17 However, recent studies by Geisler et al.18 and Hellmann et al.19 have provided another interpretation of glass dissolution. Indeed, it was shown that under some experimental conditions, the gel layer is formed by a dissolution/precipitation mechanism, without evidence at micrometer/nanometer space resolution of interdiffusion-controlled ion-exchange during the glass/gel reaction.The knowledge and understanding acquired regarding HLW glass and the technology developed for making the glass are intended to be transposed to intermediate level waste (ILW). During the decontamination of process equipment following the decommissioning and dismantling of the UP2-400 plant at the reprocessing plant at La Hague in France, liquid wastes will be generated for example:1,20–24 washing reagents (NaOH and HNO3); oxides in rinsing effluents, including transition and platinoid elements (Fe, Cr, Mn, Tc, Ru, Rh and Pd); rare earth and actinide elements (Ce, Gd, U, Pu, Np, Cm and Am); main short life βγ-emitters (137Cs, 137mBa, 90Sr, 90Y, 241Pu, 60Co); main long life βγ-emitters (63Ni); and other elements (Mo, P, S, Ba). Some rinsing effluent wastes from the UP2-800 and UP3 plants of La Hague will also be incorporated. These effluents will be calcined, vitrified by mixing with a glass frit and heated by direct induction in a cold crucible.1,21 The specific requirement of this glass is the incorporation of a higher amount of sodium coming from the waste to be vitrified, which is a limiting factor for the chemical composition and formulation of the glass.1,24 The glass will be poured into a stainless steel container of the same dimension as that used for HLW.23,24 The estimated total activity in the glass will represent less than 1% of the total volume activity compared to HLW and therefore this glass will be classified as ILW glass.22,23,25
Similar to vitrified HLW, ILW glass is planned to be placed in a deep geological formation. However, the ILW glass is expected to be conditioned in a cementitious over-pack imposing high alkaline conditions during the water resaturation period. An evaluation of glass alteration under repository conditions has already been performed in contact with deionized water in unsaturated conditions, but alteration studies in cementitious media are scarce.26–28 Countries such as Belgium, the United Kingdom and France are currently involved in such studies.29–31 In Belgium, an engineering barrier system concept is designed to place vitrified HLW in a super-container constituted of a carbon steel over-pack surrounded by a Portland cement concrete buffer, with the overall set-up enveloped by an outer stainless-steel liner. The key point is to induce the corrosion of the over-pack in a highly alkaline environment and to form corrosion products, which will act as a sink for the retention of radionuclides.29,32 In the United Kingdom, alteration studies on ILW glass compositions have been performed in saturated Ca(OH)2 solutions simulating a calcium-rich cement-based environment.30 The results showed that calcium may contribute to the formation of a protective gel layer. In France, similar results were obtained by Mercado-Depierre et al. who identified different mechanisms depending on parameters such as pH, the solid to volume ratio (S/V) and the calcium concentration,28 which controls the calcium complexation at the glass surface, the precipitation of calcium silicate hydrate phases (C–S–H) or calcium incorporation into the altered layer. Chave et al. showed that whether calcium comes from the glass or the solution, it influences the alteration of nuclear glass and is retained within the silica network in the hydrated layer at the glass surface.33 The presence of calcium and the influence of its concentration in solution determine and control the aggregation and gelation processes. On one hand, solutions with high calcium concentrations yield to gels composed of small fractal aggregates and large particles formed just after the addition of calcium. On the other hand, solutions with a low calcium content induce the formation of gels mainly composed of large fractal aggregates coexisting with small primary silicate species. The consequence is an enhancement of the passivating properties of the surface layer. Conversely, according to Jollivet et al.34 and Stockmann et al.,35 calcium has no significant influence on the dissolution rates of nuclear glass. Calcium precipitates with carbonate to form a calcite phase independently of the glass surface. The lack of a structural match between glass and calcium carbonate favors CaCO3 nucleation and growth in the form of discrete crystals.34
This paper aims to provide new contributions to the understanding of ILW glass alteration in contact with hardened cement paste (HCP) under simulated disposal conditions. Ordinary Portland Cement (OPC) CEM-I and CEM-V36 examples have been used and are compared to a ternary formulated low hydration heat/low-pH HCP. This last cementitious material is formulated with a blend of hydraulic and pozzolanic materials (blast furnace slag, fly ash, and silica fume mixed with CEM I), leading to hydrated cement without portlandite (due to the pozzolanic reaction) and C–S–H with a low Ca/Si (C/S) ratio incorporating small amounts of Al.36,37 The hydration of this latter cementitious material generates a low pH pore water solution (pH ≤ 11) compared to those involving OPC (13.2–13.5 (ref. 37–41)). This raises the question of their potential application relating to the disposal of ILW glass. The process of pore water chemistry evolution shows that pore water of pH ≤ 11 can be achieved via strong reduction under alkali concentrations, and the absence of portlandite when hydration is completed. Under these conditions, the pH is controlled by the dissolution of C–S–H phases: the lower the C/S ratio, the lower the pH.
Studies of the effects of low-pH cements on clay have been performed by Dauzères et al., comparing the physico-chemical behavior of a low-pH material with CEM-I cement paste.42 Under aqueous alteration, low-pH HCP was strongly degraded, which results in coarser porosity, whereas the thickness degradation of CEM-I HCP was limited by the precipitation of a magnesium-calcite-silicate crust over the surface, which reduces the exchange of soluble species. Ettringite precipitation occurred, resulting from the dissolution of portlandite in CEM-I HCP, while it dissolved initially in low-pH cements. These authors and Jenni et al.43 indicated that mainly leaching and carbonation occurred for low-pH HCP in contact with a clayey environment. Leaching leads to the partial or total decalcification of C–S–H to amorphous silica, resulting in a decrease in strength and the formation of macroporosity.42,43 Carbonation is linked to the precipitation of calcite and thus the decalcification of C–S–H. Dauzères et al. investigated magnesium perturbation on low-pH cements placed in a clayey environment.44 Magnesium perturbation was observed in any kind of environment containing at least 3 × 10−3 mol L−1 of magnesium in the pore solution, and produced a mineralogical phase identified as magnesium–silicate–hydrate (M–S–H).
There is no literature about the effects of low-pH cement on glass alteration, while the effects of OPC have been presented by many authors. For example, Ferrand et al. conducted glass leach tests at 30 °C in suspensions of OPC.45 The authors showed that the cement appeared to trigger glass dissolution via the consumption of glass matrix components, leading to fast glass dissolution at a constant rate with the formation of a porous gel layer on the glass. The two main reactions are: (1) the reaction of Si released by glass with portlandite, forming C–S–H; and (2) the reaction of Al released by glass with C–S–H, forming C–A–S–H phases. The key mechanism driving the long-term glass dissolution is the formation of secondary phases. After the consumption of portlandite, the glass alteration rate is expected to decrease. Therefore, the present research aims to provide new findings about the effects of low-pH cement on glass dissolution.
2. Experimental setup and methodology
2.1. Alteration experiments on glass and hardened cement pastes
2.1.1.1. Glass. French nuclear glass simulating intermediate level waste (ILW glass) was synthesized by CEA in the form of a rectangular block, whose composition is given in Table 1. ILW glass is an inactive surrogate glass, constituted of network formers (Si, B, and Al), network modifiers (Na, Ca, and Li) and intermediate metals (Zr, Al, and Zn). The particular feature of this type of glass is its Si, Al and Na oxide enrichment, and the depletion of oxides simulating fission products (TeO2, Cs2O, MnO2, La2O3) and minor actinide oxides (Nd2O3, Ce2O3).
| Oxide | ILW glass27 weight% |
|---|---|
| SiO2 | 50.33 |
| B2O3 | 14.44 |
| Na2O | 12.58 |
| Al2O3 | 8.7 |
| CaO | 3.1 |
| Li2O | 2.17 |
| Fe2O3 | 2.84 |
| NiO | 0.33 |
| Cr2O3 | 0.07 |
| SO3 | 0.19 |
| P2O5 | 0.42 |
| ZnO | |
| ZrO2 | 1.99 |
| MoO3 | 0.69 |
| MnO2 | 0.19 |
| CoO | 0.27 |
| BaO | 0.36 |
| La2O3 | |
| Ce2O3 | |
| Nd2O3 | |
| RuO2 | 0.12 |
| Oxides (fission products + actinides) | 0.47 |
| Other | 0.74 |
| Total | 100 |
Two types of glass were used: glass powder and glass monolith plates. Glass monoliths were sent to the PRIMEVERRE Company to be prepared as a powder with a controlled grain size (between 50 and 100 μm). Finally, the glass powder was cleaned in ethanol via ultrasound and dried at 90 °C. The specific surface area was measured by BET methods using Kr-gas (688 cm2 g−1). The powder was used in batch alteration experiments.
Thin glass monoliths, with dimensions of 25 mm × 25 mm × 1 mm, were cut from the block glass, polished to 3 μm and cleaned with ethanol. These samples were specifically prepared for solid characterization after long-term alteration.
2.1.1.2. Hardened cement pastes. Three hardened cement pastes (HCPs) were provided by LMDC (Laboratoire Matériaux et Durabilité des Constructions, INSA-Toulouse, France): (1) ordinary Portland Cement (CEM I, Val d'Azergue, Lafarge, France) cured over 34 months with a water to cement ratio (w/c) of 0.43; (2) blast furnace slag/fly ash cement (CEM V, Airvault, Calcia, France) cured over 35 months (w/c = 0.43); and (3) low-pH HCP, a ternary mixture of OPC (37.5% w/w), silica fume (32.5% w/w) and fly ash (30% w/w), cured over 33 months (w/c = 0.50). CEM-I and CEM-V are two HCPs considered by the French radioactive waste management agency (Andra) in their long-term behavior surveys. They are used in this work as reference materials to compare their performances to that of low-pH HCP. The HCPs were crushed manually in an agate mortar and sieved between 100 and 160 μm. All solid preparations were performed in a glove box under inert gas (argon) to avoid any contamination by atmospheric CO2(g) gas, which is deleterious for HCP (the precipitation of calcite).
| mol L−1 | S1b | S2b | S5 | |||
|---|---|---|---|---|---|---|
| 20 °C | 50 °C | 20 °C | 50 °C | 20 °C | 50 °C | |
| Na+ | 6.55 × 10−2 | 6.55 × 10−2 | — | — | 4.7 × 10−3 | 4.7 × 10−3 |
| K+ | 0.1611 | 0.161 | — | — | 1.5 × 10−3 | 1.5 × 10−3 |
| Mg2+ | — | — | — | — | 2 × 10−6 | 1.95 × 10−6 |
| Ca2+ | 2.2 × 10−3 | 1.2 × 10−3 | (20 ± 1) × 10−3 | (15 ± 1) × 10−3 | 4.7 × 10−3 | 4.87 × 10−3 |
| Si | 3 × 10−6 | 2 × 10−6 | — | — | 6 × 10−4 | 7.8 × 10−4 |
| SO42− | — | — | — | — | 6.7 × 10−3 | 6.7 × 10−3 |
| Al3+ | — | — | — | — | 4 × 10−6 | 3.89 × 10−6 |
| OH− | 0.231 | 0.229 | — | — | 1 × 10−3 | 1 × 10−3 |
| pH | 13.2 ± 0.1 | 12.6 | 12.4 ± 0.1 | 11.6 | 10.8 ± 0.2 | 10.3 |
|
||||
|---|---|---|---|---|
| Binary system | Materials | Solution | ||
| S1b | S2b | S5 | ||
| Experiment 1 | Glass powder (20 °C, 50 °C) | ✗ | ✗ | ✗ |
| Experiment 2 | CEM-I, CEM-V powder (20 °C, 50 °C) | ✗ | ✗ | |
| Experiment 3 | Low-pH powder (20 °C, 50 °C) | ✗ | ||
| Experiment 4 | Glass monolith (50 °C) | ✗ | ✗ | ✗ |
| Experiment 5 | Glass powder + glass monolith (50 °C) | ✗ | ✗ | ✗ |
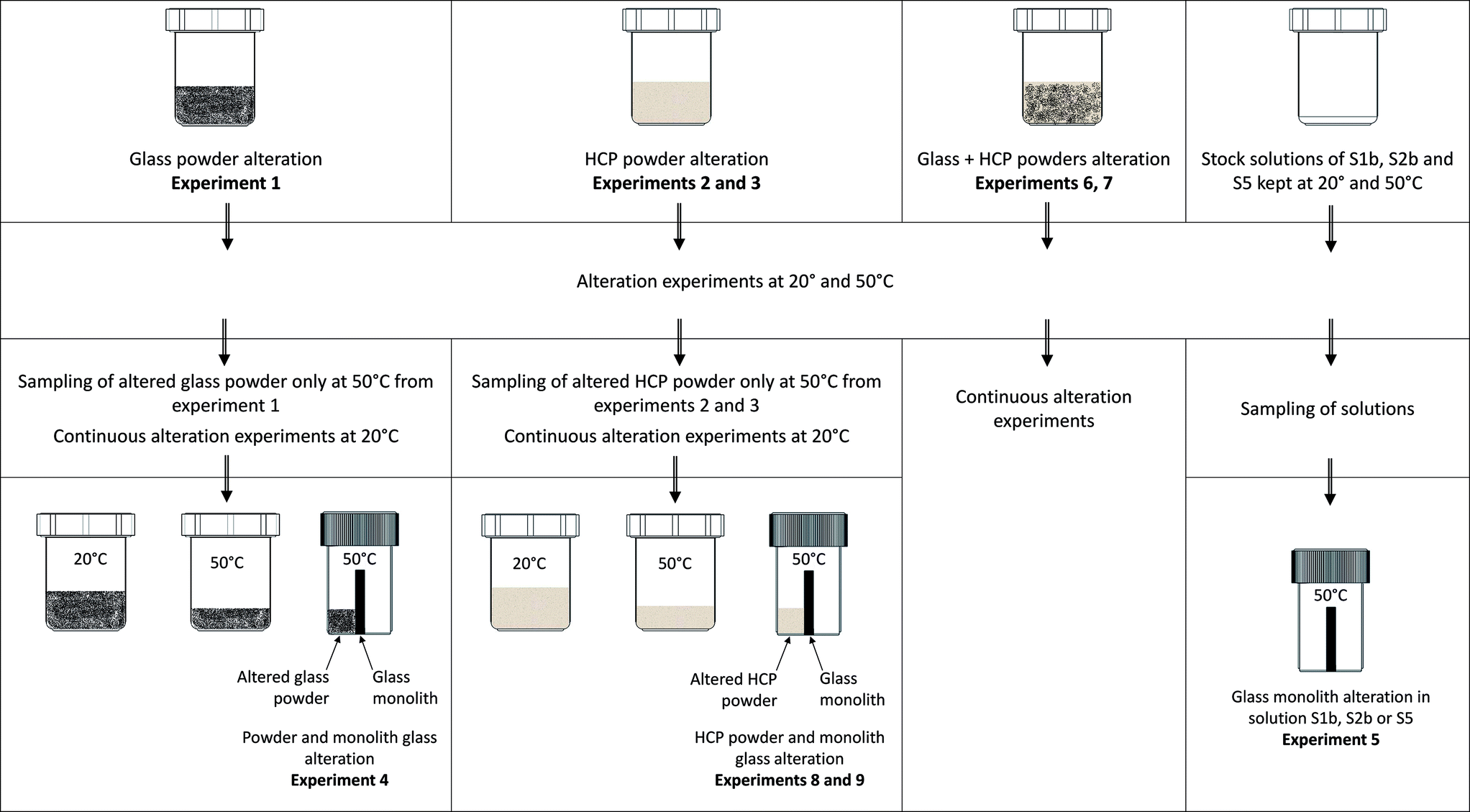
| Ternary system | Material | Solution/HCP | ||||
|---|---|---|---|---|---|---|
| S1b | S2b | S5 | ||||
| CEM-I | CEM-V | CEM-I | CEM-V | Low-pH | ||
| Experiment 6 | Glass powder (20 °C, 50 °C) | ✗ | ✗ | ✗ | ✗ | |
| Experiment 7 | Glass powder (20 °C, 50 °C) | ✗ | ||||
| Experiment 8 | Glass monolith (only at 50 °C) | ✗ | ✗ | ✗ | ✗ | |
| Experiment 9 | Glass monolith (only at 50 °C) | ✗ | ||||
• Experiment (1): the alteration of glass powder in S1b, S2b or S5 solution (116 g L−1);
• Experiment (2): the alteration of CEM-I and CEM-V HCP in S1b or S2b solution (10 g L−1);
• Experiment (3): the alteration of low-pH HCP in solution S5 (10 g L−1);
• Experiment (4): the alteration of glass powder (90 g L−1) + a glass monolith (65 g L−1) in S1b, S2b or S5 solution;
• Experiment (5): the alteration of a glass monolith in contact with S1b, S2b or S5 solution (65 g L−1);
• Experiment (6): the alteration of glass powder (116 g L−1) in contact with CEM-I and CEM-V HCP (10 g L−1) in S1b or S2b solution;
• Experiment (7): the alteration of glass powder (116 g L−1) in contact with low-pH HCP (10 g L−1) in S5 solution;
• Experiment (8): the alteration of a glass monolith (65 g L−1) in contact with CEM-I and CEM-V HCP (80 g L−1) in S1b or S2b solution;
• Experiment (9): the alteration of a glass monolith (65 g L−1) in contact with low-pH HCP (80 g L−1) in S5 solution.
Experiments 4, 8 and 9 were conducted in two steps: (1) the first step consisted of the equilibration of glass powder or HCP with the solution (for example: CEM-I HCP in solution S1b, low-pH HCP in solution S5) at 50 °C; and (2) the equilibrated solution was thereafter put in contact with the glass monolith and kept at 50 °C. The monolith separates the vial into two compartments, in which one side (the front side) is in contact with glass powder (experiment 4) or HCP (experiments 8 and 9) and the other side (the rear side) is free of solids. For experiment 5, both sides of the monolith are in contact with solution only. These specific experiments (Exp. 4, 5, 8 and 9) were dedicated to studying the effects of solids in contact with the glass monolith and to allow surface characterization via EDX-SEM and micro-Raman studies. Table 3 presents the general experimental setup. Aliquots were sampled and filtered at 0.45 μm in a glove box for ICP-MS and ion chromatography analyses and pH measurements. The pH electrode was calibrated at room temperature using buffer solutions between pH 7 and 12.45 (Radiometer pH buffer). For all experiments conducted at 50 °C, the solutions were cooled down at room temperature before measurements.
2.2. Solution analysis and solid characterization
All solutions were stored in a refrigerator at 4 °C until analyses were carried out. Li, B, Mo, Si, and Al concentrations were analyzed via inductively coupled plasma mass spectrometry (Quadrupole ICP-MS Xseries 2, THERMOELECTRON). The limits of quantification (LQs) are 0.3 μg L−1 for Li, 1.3 μg L−1 for B, 7.5 μg L−1 for Si, and 0.2 μg L−1 for Al, with an analytical uncertainty of 5–10%. Prior to analysis, samples were filtered (Minisart 0.45 μm cellulose filters) and diluted in 2% vol. high purity HNO3 solution.Ca, K, Na, and SO4 concentrations were analyzed via ion chromatography (DIONEX ICS 2500). The LQs are 0.1 mg L−1, with an error of 5%. Prior to analysis, samples were filtered (Millex PTFE 0.45 μm filters). The samples are diluted in ultra-pure water or in eluent for anion or cation analysis, respectively.
Crystallographic structures were recorded via X-ray diffractometry (BRUKER-AXS D5000, Bragg–Brentano geometry, copper anticathode, Cu, at λ = 1.5406 Å, room temperature) for pristine and reacted glass powder, as well as for HCPs. Data were collected in the 2θ range of 5° to 90° with a step size of 0.02° and a scan-speed of 10 s per step. Phases were identified using the DIFFRACPlus EVA software package (BRUKER).
Morphologies and elemental chemical compositions were determined using electron dispersion spectroscopy (EDS) coupled with scanning electron microscopy (SEM, JEOL 5800 LV, 15 kV). Samples were coated by a thin layer of carbon, allowing for the detection of elements (Z > 11). Flat glass surfaces and polished cross sections were prepared in epoxy-resin (Epofix, Struers) for analysis.
Secondary mineral phases were characterized with a confocal micro-Raman spectrometer (T64000 Jobin-Yvon/Horiba, room temperature). Raman spectra were recorded under a microscope (Olympus Bx41) in backscattering geometry with a 100× objective focusing the 647.1 nm and 514 nm beam lines from an argon–krypton ion laser (Spectrum Coherent, 2.5 W). The Raman experiments were performed using a spectrometer equipped with a 600 lines per mm diffraction grating and a nitrogen cooled CCD detector. The spectral resolution is 2 cm−1. The acquisition and basic treatment of spectra were performed with LabSpec V5.25 (Jobin Yvon-Horiba) and Origin 8.6 software. Raman measurements were carried out at a very low laser power to minimize possible sample deterioration or phase transitions during the operating time.
2.3. Data processing
The glass elemental normalized mass loss (NLi, g m−2) is calculated using the equation:
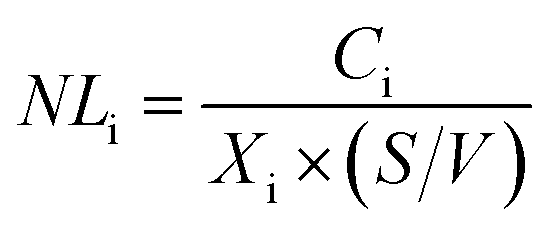 | (1) |
The glass alteration rate (NLRi, g m−2 d−1) is then calculated via the following equation:
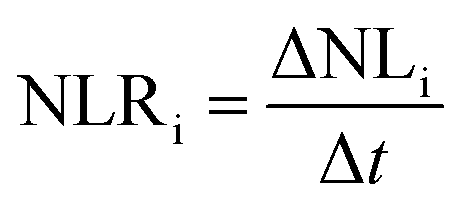 | (2) |
From the normalized mass loss values, the equivalent altered thickness (EEeq, μm) was determined as follows:
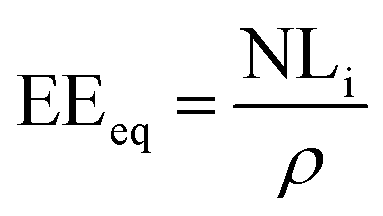 | (3) |
2.4. Modelling
The experimental data were modelled with the geochemical code PhreeqC (version 2.17)47 and the THERMOCHIMIE (version 9 (2014))48 database. Modelling calculations were conducted for binary and ternary systems, such as glass powder alone (Exp. 1), glass powder + CEM-I/CEM-V HCP (Exp. 6), and glass powder + low-pH HCP (Exp. 7) at 20 °C and 50 °C.The glass composition is defined according to the following molar proportions: SiO2 = 0.551; Na2O = 0.1335; B2O3 = 0.136; Al2O3 = 0.056; CaO = 0.0364; Li2O = 0.0478; Fe2O3 = 0.0117; MoO3 = 0.00315; and SO3 = 0.0011. For the cement, the following main phases: portlandite; C–S–H; ettringite; and monosulfoaluminate, were considered to dissolve or precipitate instantaneously to reach thermodynamic equilibrium in solution.42 The compositions and phase distributions of CEM-I and CEM-V HCP are recalculated from Trotignon et al.49 For experiments in contact with HCP, thermodynamic equilibria with portlandite, ettringite, monosulfoaluminate, and different C–S–H values (Ca/Si ratios equal to 0.8, 1.2, and 1.6) were considered.
3. Results and discussion
3.1. Solid characterization
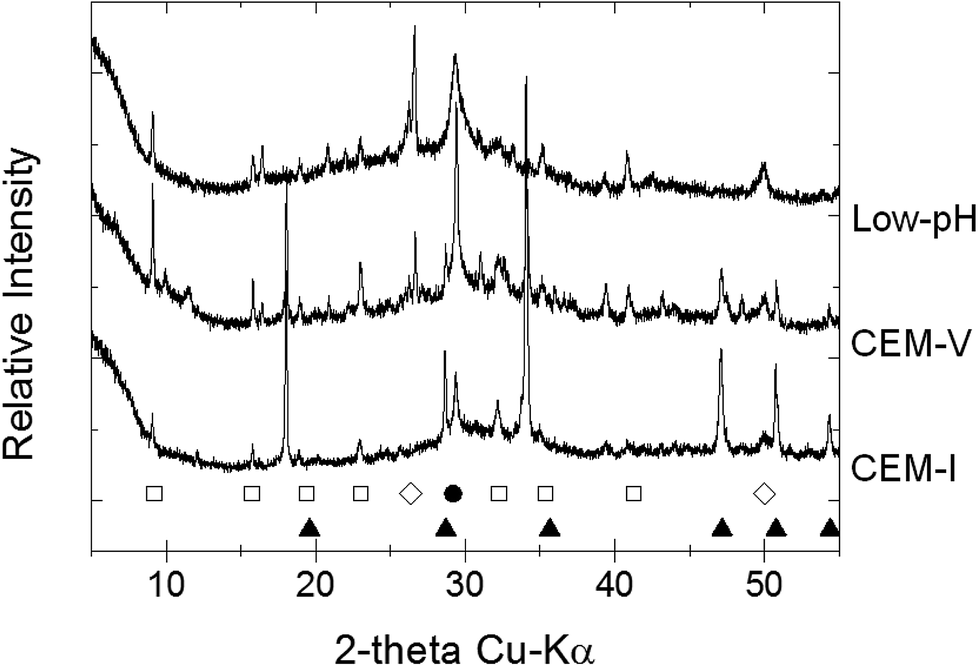 | ||
| Fig. 1 X-ray diffraction patterns of initial CEM-I, CEM-V and low-pH HCP powder (Exp. 2 and 3): (□) ettringite; (●) calcite; (◊) quartz; (●) portlandite. | ||
| Phase | CEM-I | CEM-V | Low-pH | |||
|---|---|---|---|---|---|---|
| Pristine | Altered | Pristine | Altered | Pristine | Altered | |
| Portlandite | ✓ | ✓ | ||||
| Ettringite | ✓ | ✓ | ✓ | |||
| Calcite | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Quartz | ✓ | ✓ | ✓ | ✓ | ||
| C–S–H | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
After the alteration stage at 50 °C, water molecules may be released from the structure of ettringite, reducing its crystallinity or converting it into an amorphous structure.50–52 Moreover, ettringite becomes more soluble with an increase in pH, notably in solution S1b for CEM-I and CEM-V HCP.53 Therefore, the pH effect is coupled with the temperature effect. Conversely, the dissolution of ettringite in low-pH HCP may not be attributable to pH change, but to temperature. The destabilization of ettringite can be explained by the fact that the stability range of ettringite diminishes with temperature in favor of the formation of calcium monosulfo-aluminate hydrate. This result is supported in the literature in an example from Damidot et al., who mentioned the range of ettringite stability in terms of pH-range:54 10.43 < pH < 12.52 (25 °C); 10.52 < pH < 12.41 (50 °C); and 10.87 < pH < 12.25 (85 °C).
 | ||
| Fig. 2 SEM pictures of glass monoliths in the absence/presence of glass powder (GP) in S1b, S2b and S5 solution at 50 °C (Exp. 4 and 5). | ||
| Solution S1b | Solution S2b | Solution S5 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| % CaO | % SiO2 | Ca/Si | % CaO | % SiO2 | Ca/Si | % CaO | % SiO2 | Ca/Si | ||
| Experiment 4 | Monolith | 12.8 ± 0.6 | 17.8 ± 0.1 | 0.72 ± 0.04 | 17.5 ± 0.8 | 15.2 ± 0.2 | 1.15 ± 0.05 | Not analyzed | ||
| Experiment 5 | Monolith + powder front side | 36.3 ± 5.9 | 10.6 ± 5.9 | 0.3 ± 0.2 | Quantification not available | Quantification not available | ||||
| Monolith + powder rear side | 26.9 ± 3.3 | 30.5 ± 1.1 | 1.1 ± 0.1 | |||||||
| Experiment 8 | Monolith + CEM-I front side | 26.2 ± 0.5 | 30.6 ± 0.2 | 1.17 ± 0.02 | 21.5 ± 0.4 | 15.5 ± 0.2 | 1.39 ± 0.03 | |||
| Monolith + CEM-I rear side | 19.9 ± 0.7 | 15.3 ± 0.3 | 1.30 ± 0.06 | 22.6 ± 0.2 | 15.3 ± 0.1 | 1.48 ± 0.02 | ||||
| Monolith + CEM-V front side | Quantification not available | 19.9 ± 1.8 | 17.6 ± 1.5 | 0.9 ± 0.1 | ||||||
| Monolith + CEM-V rear side | 21.4 ± 0.4 | 18.5 ± 0.2 | 0.87 ± 0.02 | |||||||
| Experiment 9 | Monolith + low-pH front side | Quantification not available | ||||||||
| Monolith + low-pH rear side | 25.1 ± 1.8 | 39.1 ± 6.8 | 0.6 ± 0.1 | |||||||
In S1b solution, the surface presents a desquamated form, probably due to the combined effects of temperature (50 °C) and high pH (13.2). Magnification of the surface reveals the presence of a gel that is, from EDX analyses, enriched in calcium relative to the initial glass composition, which could be attributed to Ca diffusion through the gel.
In S2b solution, the surface is characterized by the presence of a gel and precipitates. EDX analysis shows that the gel is composed of silicon and calcium with a Ca/Si ratio of 1.1; however, micro-Raman analysis indicates a lower ratio, corresponding to a phase close to a tobermorite-like phase. Analyses shows that the precipitates, clustered together, are enriched in calcium and were identified as calcite via micro-Raman analysis.
In S5 solution, the glass has a homogeneous surface with the presence of an alteration gel. Analysis of this surface via micro-Raman studies indicates the presence of Si, Al, Ca and Na. A phase search seems to indicate the presence of a gyrolite-like phase with a low Ca/Si ratio.
Glass powder in contact with a glass monolith (Exp. 4, Tables 3 and 6). Altered glass powder was put into contact with a fresh glass monolith. The binary glass powder/glass monolith system was thereafter altered in S1b, S2b and S5 solution at 50 °C. After 380 days of alteration, the systems were dismantled and the front (presence of glass powder) and rear (absence of glass powder) sides of the glass monolith were analyzed via SEM/EDX and micro-Raman analysis (Fig. 2).
| T °C | Glass alone (Exp. 1) | Glass + HCP (Exp. 6 and 7) | ||||||
|---|---|---|---|---|---|---|---|---|
| Type of solution | pH | NLRinit in g m−2 d−1 (duration) | NLRlt in g m−2 d−1 (duration) | Type of HCP | pH | NLRinit in g m−2 d−1 short-term alteration (duration) | NLRlt in g m−2 d−1 long-term alteration (duration) | |
| (*) Value calculated before the resumption of glass alteration; (**) value calculated after the resumption of glass alteration. | ||||||||
| 20 °C | S1b | 13.0 ± 0.3 | 5.5 × 10−3 (0–50 days) | 6.7 × 10−5 (231–854 days) | CEM-I | 13.0 ± 0.3 | 6.4 × 10−3 (0–100 days) | 5.8 × 10−4 (576–859 days) |
| CEM-V | 13.1 ± 0.1 | 5.7 × 10−3 (0–21 days) | 2.8 × 10−4 (388–730 days) | |||||
| S2b | 12.1 ± 0.4 | 2.9 × 10−3 (0–41 days) | 5.7 × 10−5 (236–722 days) | CEM-I | 12.3 ± 0.3 | 5.2 × 10−3 (0–81) | 5.1 × 10−4 (520–859 days) | |
| CEM-V | 11.9 ± 0.1 | 3.2 × 10−3 (0–81 days) | 2.0 × 10−4 (388–833 days) | |||||
| S5 | 10.2 ± 0.5 | 9.8 × 10−5 (0–204 days) | 2.8 × 10−5 (478–817 days) | Low-pH | 10.3 ± 0.2 | 1.0 × 10−4 (0–478 days) | 2.3 × 10−5 (478–817 days) | |
| 50 °C | S1b | 13.0 ± 0.4 | 3.5 × 10−2 (0–7 days) | 1.3 × 10−4 (*) (381–583 days) | CEM-I | 12.6 ± 0.5 | 3.1 × 10−2 (0–21 days) | 1.5 × 10−2 (387–783 days) |
| 4.0 × 10−2 (**) (700–833 days) | CEM-V | 12.7 ± 0.4 | 2.2 × 10−2 (0–14 days) | 2.1 × 10−2 (545–776 days) | ||||
| S2b | 10.8 ± 0.4 | 1.1 × 10−2 (0–50 days) | 9.6 × 10−5 (381–713 days) | CEM-I | 11.8 ± 0.1 | 1.1 × 10−2 (0–50 days) | 1.2 × 10−3 (387–652 days) | |
| CEM-V | 11.7 ± 0.1 | 9.3 × 10−3 (0–21 days) | 4.1 × 10−4 (422–663 days) | |||||
| S5 | 10 ± 1 | 4.5 × 10−4 (0–50 days) | 7.4 × 10−5 (472–826 days) | Low-pH | 9.8 ± 0.6 | 6.9 × 10−4 (0–15 days) | 2.8 × 10−5 (359–638 days) | |
In S1b solution, the area of the monolith surface in contact with glass powder shows the formation of phyllosilicate-like phases, which are commonly formed during borosilicate glass alteration.12,55 EDX analysis of the globular precipitates indicates the presence of Si, Al and Ca. For a post-mortem characterization of the glass monolith via SEM, the glass powder was separated from the surface of the monolith, showing bright network-shaped lines. These lines are related to an electrically charged zone, typically observed with this technique. Within this network, the formation of a honeycomb-shaped gel is observed. Micro-Raman analyses of both sides of the monolith surface indicate the presence of a phase with a low Ca/Si ratio close to a chabazite-like phase, Ca2Al4Si8O24·13H2O. The low Ca/Si ratio of 0.25 was confirmed via EDX analysis. In the absence of glass powder, EDX analysis reveals a composition similar to pristine glass, suggesting a very small thickness of glass alteration.
In solution S2b, SEM analysis on both sides of the monolith shows again the formation of a phyllosilicate-like alteration phase, but the thinness of the alteration layer precluded any obtaining of good EDX micro-analysis. Similar observations were found by Utton et al. on an ILW glass surface altered in same solution but at higher temperature (70–90 °C).31 Micro-Raman analysis of the front side shows the formation of a tobermorite-like phase. In the absence of glass powder, calcite was identified via micro-Raman spectroscopy (an artefact due to probable contamination with atmospheric CO2 during analysis).
In S5 solution, an alteration layer consisting of phyllosilicates has been also observed via SEM on both sides. Micro-Raman analyses on both sides indicate a very low Ca/Si ratio, with a structure close to a gyrolite-like phase. These results are similar to those observed in the absence of glass powder.
Glass powder in contact with HCP (Exp. 6 and 7, Tables 3 and 5). Glass powder before and after alteration, in the presence and absence of HCP was characterized via X-ray diffraction after experiments at 50 °C (data not shown). In the absence of HCP, the glass powder altered in the three solutions S1b, S2b and S5 was characterized via XRD at the end of the experiments under the same conditions as those for HCP alone. The diffraction data show a typical glass signature with a wide peak between 2θ values of 13° and 38°. Crystalline phases were not detected. In contact with the HCPs, only a calcite phase was identified in all X-ray diffraction patterns, probably due to sample preparation and potential contamination from atmospheric CO2 during analysis. The glass powder and HCP were probably not dried enough prior to analysis, promoting the formation of calcite during storage before analysis. Portlandite, ettringite and analcime phases have not been detected, but their existence at trace levels cannot be excluded, as suggested by PhreeqC modelling. Finally, it can be noted that in S2b solution, ettringite disappears in the presence of glass powder, whereas this phase is present in CEM I and CEM V pastes only at 50 °C.
Glass monoliths in contact with HCP (Exp. 8 and 9, Fig. 3, Tables 3 and 5). Fig. 3 shows the characteristics of the front and rear sides of a monolith surface in contact with HCP.
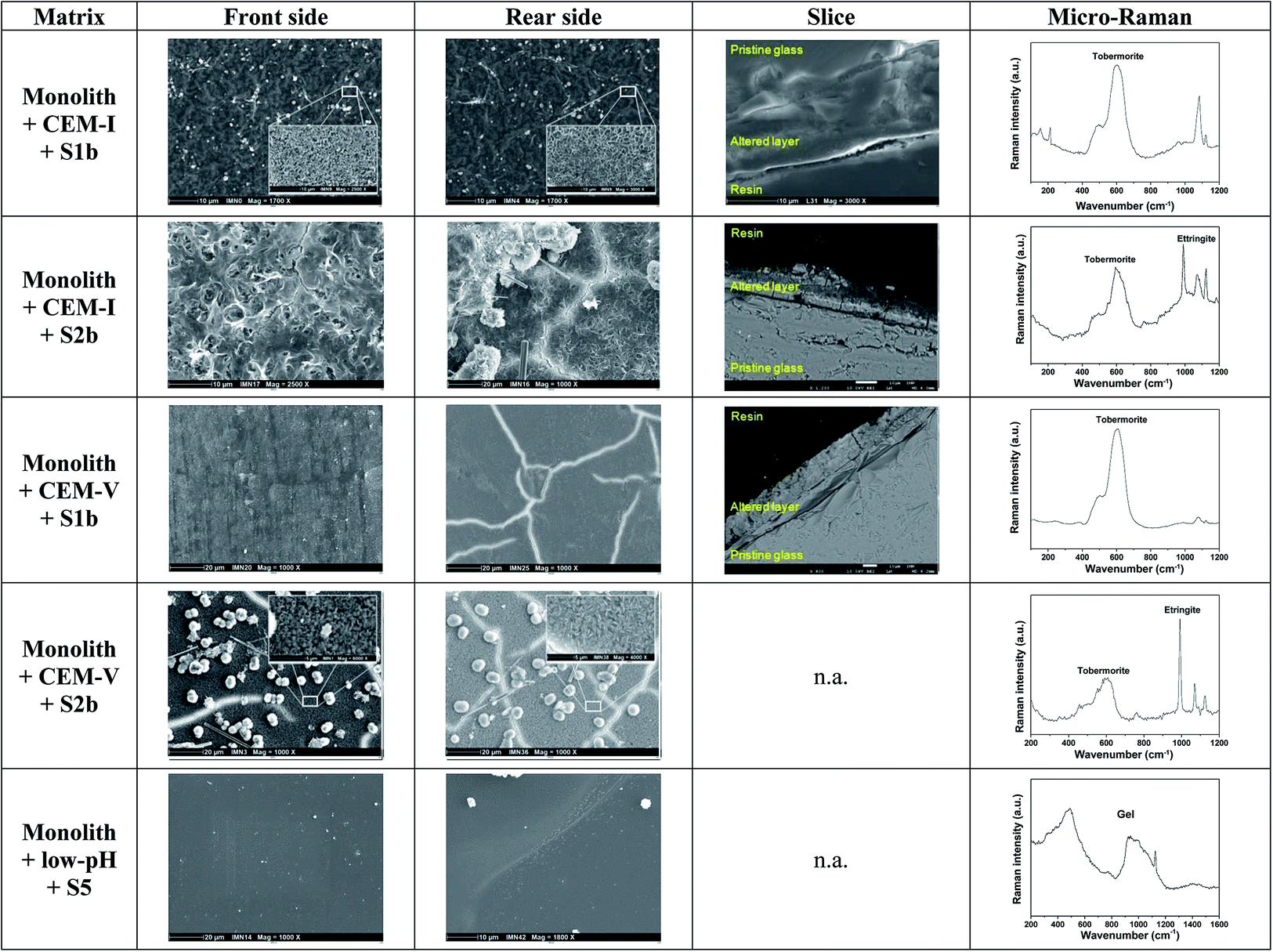 | ||
| Fig. 3 Surface characterization of glass monoliths in contact with CEM-I, CEM-V and low-pH HCP in S1b, S2b and S5 solution at 50 °C. n.a.: not available (Exp. 8 and 9). | ||
When in contact with CEM-I HCP powder in S1b solution, the surface of the monolith presents an altered layer made of phyllosilicate-like phases enriched with Si and Ca. Characterization by micro-Raman indicates the presence of a tobermorite-like phase (Ca/Si: ∼0.83). On the front side, direct contact with HCP powder promotes the formation of a supplementary hastingsite-like phase, NaCa2(Fe42+Fe3+)(Si6Al2)O22(OH)2. At the rear side, a C–S–H phase with a high Ca/Si ratio of ∼1.7 (ref. 31) is characterized via micro-Raman.56 In solution S2b, an altered layer is formed on both sides of the monolith, with a high Ca/Si ratio of 1.4–1.5 determined via EDX. The high concentration of Ca in S2b solution may be responsible for the increase in the Ca/Si ratio. Micro-Raman analysis shows an additional C–S–H phase with a low Ca/Si ratio close to a tobermorite-like phase. On the rear side, the needle-like morphology of ettringite is observed via SEM and is confirmed from EDX and micro-Raman analyses.57
In contact with CEM-V HCP in S1b solution, an altered layer is observed on both sides via SEM with the presence of a tobermorite-like phase. The chemical composition determined via EDX is similar to pristine glass, indicating that the layer formed is very thin to be detected by EDX. In solution S2b, for both sides, the surface exhibits an altered layer with a phyllosilicate-like phase, with spherical precipitates and the needle-like morphology of ettringite. Analysis of the altered layer demonstrates a low Ca/Si ratio tobermorite-like phase, confirmed via EDX and micro-Raman. These morphologies are reminiscent of those found by Utton on glass altered at 70 °C and 90 °C in Ca(OH)2 solution.31 The presence of CEM-V seems to accelerate the process of formation of these phases.
In contact with low-pH HCP in S5 solution, no altered layer is observed via SEM, however analysis via micro-Raman shows the signature of a gel layer. On the rear side, the glass shows precipitates enriched with Si and Ca, with a low Ca/Si ratio, close to a tobermorite-like phase.
Surface characterization of the monoliths via SEM and micro-Raman analysis is in good agreement with the results of PhreeqC modeling. It is of note that the ettringite exists at elevated temperature (50 °C) in a solution enriched with calcium (typically, S2b solution) and in the presence of a tobermorite type phase, showing that in this particular environment ettringite may remain stable even at temperatures higher than 40 °C:52 the formation and stabilization of ettringite seem to be dependent on the presence of tobermorite. The mechanism of tobermorite formation generally occurs during the hydration processes of anhydrous calcium silicate phases such as C3S (3CaO·SiO2) and C2S (2CaO·SiO2),58,59 and is initiated by the presence of a C–S–H gel with a low Ca/Si ratio (<1), sustained by the presence of Si and Al, alkaline media and sulfate compounds.60,61 In addition, the ageing of the C–S–H gel gives rise to a final product, which is tobermorite at a low Ca/Si ratio (<1) or jennite at a high Ca/Si ratio (>1), and intermediate phases, which are afwillite, gyrolite and xonotlite.40,61 In this present study, the formation of tobermorite was observed as the main C–S–H phase formed during alteration due to the chemical conditions of the systems: Si was released by glass and HCP alteration; Ca from solution (S1b, S2b); and Al by CEM-I and CEM-V HCPs and glass dissolution.
3.2. Glass alteration
3.3. Effects of the solution composition of S5 solution versus S1b and S2b solution
The influence of solution composition related to the pH effect on glass powder alteration was assessed using the binary system (glass/solution). The tracers of glass alteration, Li, B and Mo, are monitored in S1b, S2b and S5 solution at 20 °C and 50 °C (Exp. 1). Fig. 4(a)–(c) show the evolution of the normalized mass loss of selected elements in S1b, S2b and S5 solution at 20 °C. Similar results are obtained at 50 °C (not shown here).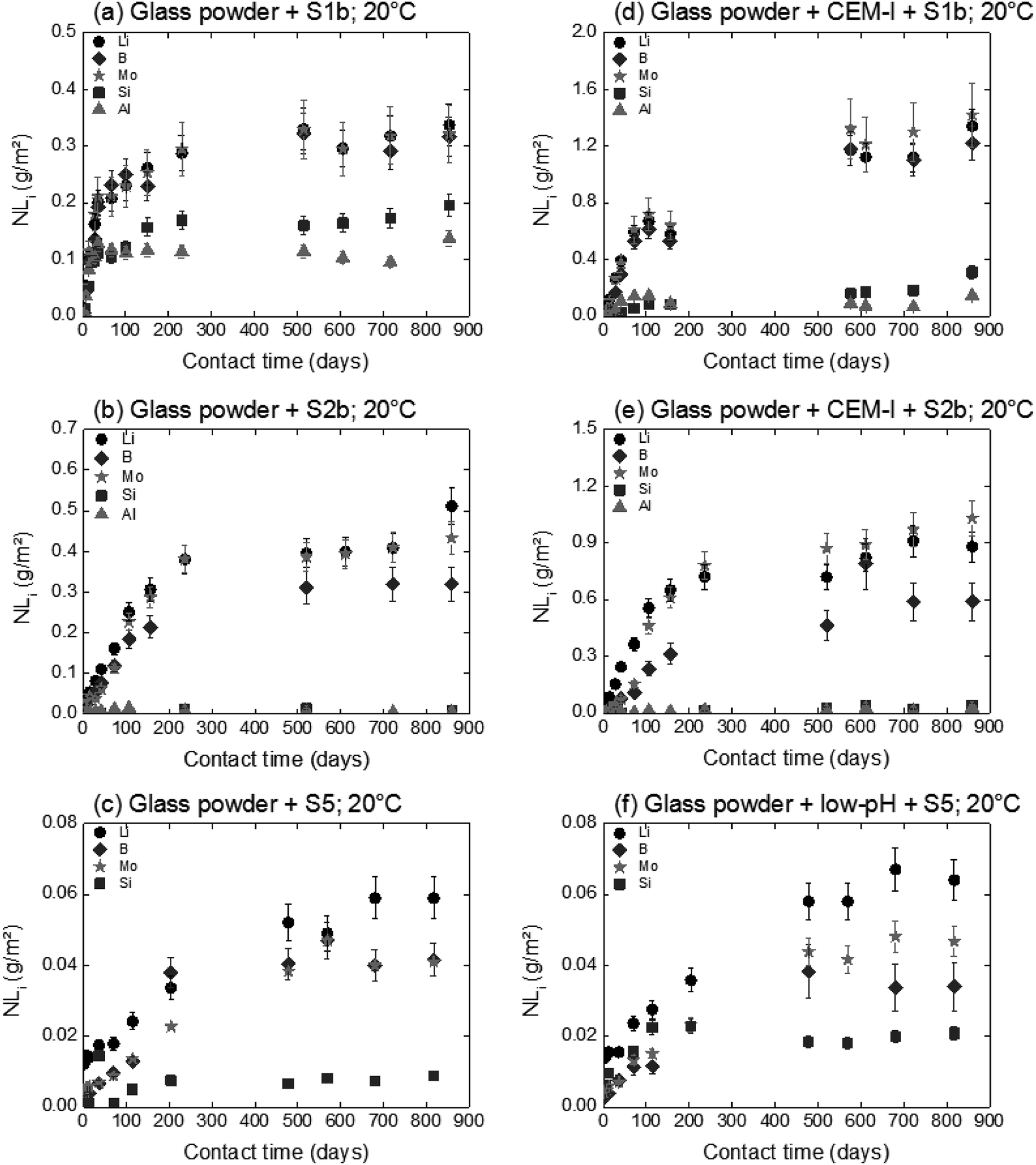 | ||
| Fig. 4 Normalized mass loss of selected elements during glass powder dissolution in S1b, S2b and S5 solution at 20 °C in the absence (Exp. 1) and presence of CEM-I, CEM-V and low-pH HCP (Exp. 6 and 7). | ||
In solutions S1b and S2b, B, Li and Mo are released congruently throughout the experiment duration. The short-term alteration rates of glass powder (NLRinit) between 0 and 50 days are 5.5 × 10−3 g m−2 d−1 in S1b solution and 2.9 × 10−3 g m−2 d−1 in S2b solution at 20 °C, and 3.5 × 10−2 g m−2 d−1 in S1b solution and 1.1 × 10−2 g m−2 d−1 in S2b solution at 50 °C (Table 6). The solution composition and the pH effects are key parameters relating to the dissolution process and they have been assessed in the literature.63 Typical initial alteration rates under batch conditions are in the range of ∼10−2 g m−2 d−1 between 25–90 °C near neutral pH, and they increase significantly with pH.6,45 However, these values evolve as a function of the composition of the leaching solution; in particular the initial alteration rates tend to be lower in highly concentrated alkali solutions. In the presence of Ca, K, and Na, representative of solution S1b, the surface of the glass develops resistance to corrosion via the direct adsorption of these elements at the surface. This mechanism avoids, therefore, the first reactions of inter-diffusion and hydrolysis, resulting in the formation of a protective layer.63
In solution S5, the normalized mass loss values of glass obtained are 10 times lower than those measured in solutions S1b and S2b: 9.8 × 10−5 g m−2 d−1 at 20 °C and 4.5 × 10−4 g m−2 d−1 at 50 °C. Fig. 5 shows the Li-concentration released from glass powder in the absence of HCP at 50 °C in solution S5. The results are compared with those obtained in solutions S1b and S2b and are represented as a function of the square root of the contact time in the binary system (glass/solution). In solution S5, there is a linear correlation between the Li concentration and the square root of time, indicating a diffusive type Li release regime during glass alteration. In general, the so-called interdiffusion phase occurs in the first hours of glass alteration, when the cation modifiers of the glass network, which are the least bound, are released in solution. The reaction is then governed by an exchange of a proton with an alkaline element, here lithium, based on the reaction:
| Si–O–Li + H+ ↔ Si–O–H + Li+ |
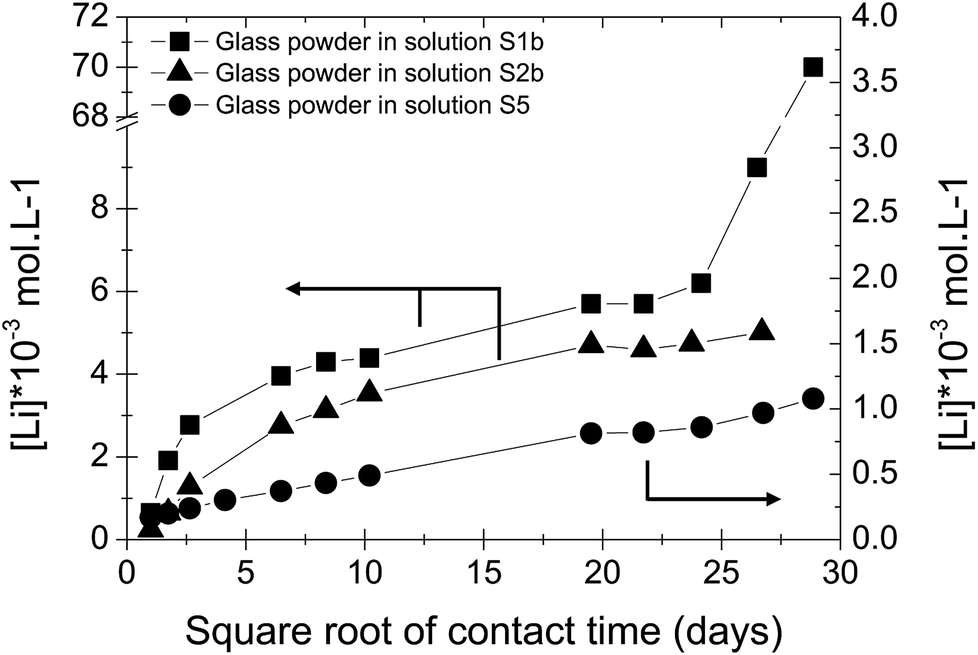 | ||
| Fig. 5 Concentration of Li released from glass powder as a function of the square root of contact time in S1b, S2b and S5 solution at 50 °C (Exp. 1). | ||
This reaction also shows that this is promoted in an acidic and neutral environment. However, the results of the alteration experiments in S5 solution with a pH of 9–10 show that a continuous diffusion mechanism can be expected throughout the duration of the experiment, i.e., over nearly 850 days.
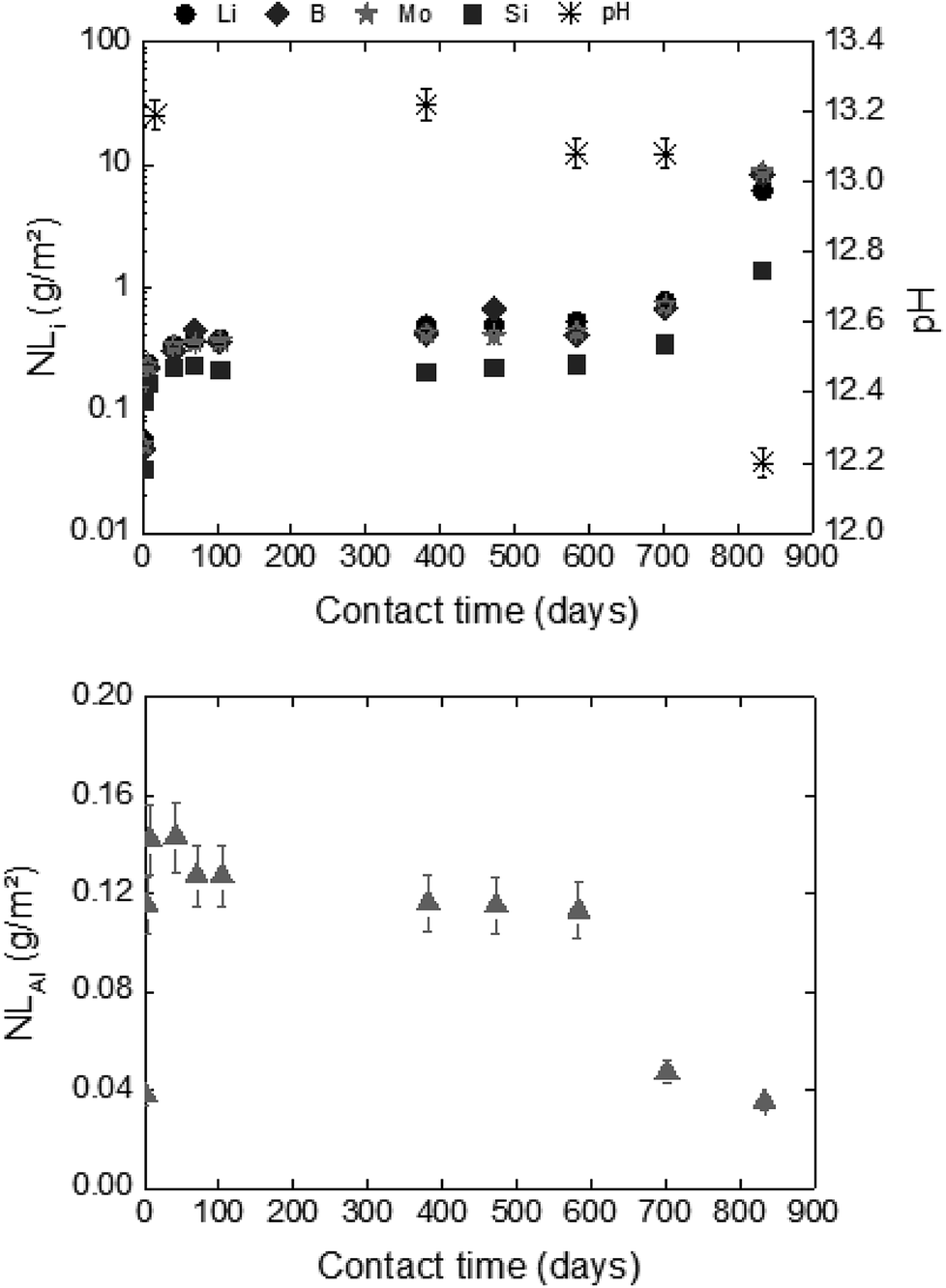 | ||
| Fig. 6 Normalized mass loss of glass powder dissolution in solution S1b at 50 °C (Exp. 1). | ||
For experiments in the presence of hydrated cement pastes (CEM-I, CEM-V and low-pH), the same trend is observed with short-term dissolution rates at 50 °C (4 to7 times higher than at 20 °C) with S1b and S5 solutions. For solution S2b, the temperature effect on glass dissolution seems to be counterbalanced by a slight decrease in the pH value from 12.1 at 20 °C to 11.8 at 50 °C; the latter is less aggressive with regard to glass dissolution. The short-term dissolution rates in S2b (CEM-I, CEM-V) at 50 °C are then only 2–3 times higher than at 20 °C.
For long-term dissolution rates at 50 °C, no steady-state in the ternary system (glass/HCP/S1b; Exp. 4) is reached, even after 776 days of experiments. The long-term dissolution rates are 26 to 75 times higher at 50 °C than at 20 °C for CEM I and CEM V. As an example, Fig. 7 presents the normalized mass loss of glass powder dissolution in S1b solution at 50 °C in the presence of CEM-I HCP.
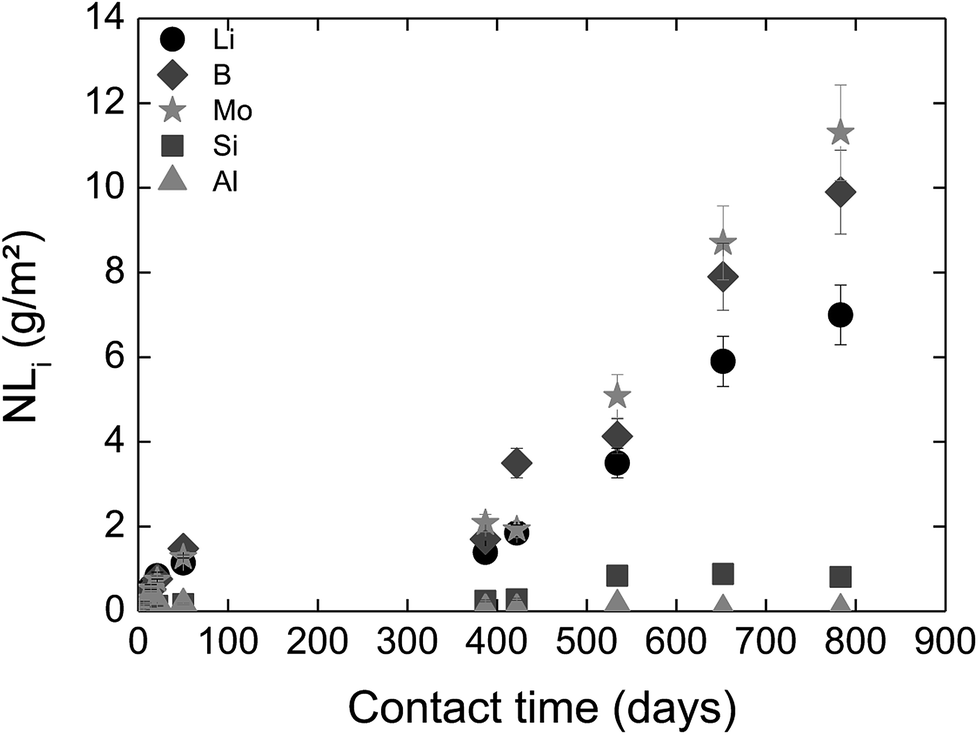 | ||
| Fig. 7 Normalized mass loss of glass powder dissolution in S1b solution at 50 °C in the presence of CEM-I HCP powder (Exp. 6). | ||
| CEM-I | S1b | S2b | ||
|---|---|---|---|---|
| 20 °C | 50 °C | 20 °C | 50 °C | |
| Li | (1.1 ± 0.1) × 10−5 | (1.1 ± 0.1) × 10−5 | (1.1 ± 0.3) × 10−5 | (1.2 ± 0.2) × 10−5 |
| B | — | (5.7 ± 0.1) × 10−5 | — | (5 ± 2) × 10−5 |
| Mo | — | — | — | (1.3 ± 0.1) × 10−7 |
| Si | — | (5 ± 2) × 10−5 | — | (6 ± 1) × 10−5 |
| Al | (3.5 ± 0.5) × 10−5 | (5.7 ± 0.4) × 10−5 | (1.2 ± 0.7) × 10−5 | (1.2 ± 0.5) × 10−5 |
| CEM-V | S1b | S2b | ||
|---|---|---|---|---|
| 20 °C | 50 °C | 20 °C | 50 °C | |
| Li | (6.6 ± 0.6) × 10−5 | (5.9 ± 0.5) × 10−5 | (6.5 ± 0.7) × 10−5 | (6.1 ± 0.4) × 10−5 |
| B | — | (7 ± 1) × 10−5 | — | (4.5 ± 0.1) × 10−5 |
| Mo | — | — | (1.2 ± 0.3) × 10−7 | |
| Si | (1.4 ± 0.5) × 10−4 | (2.3 ± 0.4) × 10−4 | (5.0 ± 0.3) × 10−5 | (3.2 ± 0.3) × 10−5 |
| Al | (4.3 ± 0.5) × 10−4 | (1.5 ± 0.4) × 10−3 | (3.6 ± 0.5) × 10−5 | (4.2 ± 0.4) × 10−6 |
| Low-pH | S5 | |||
|---|---|---|---|---|
| 20 °C | 50 °C | |||
| Li | (3.1 ± 0.9) × 10−5 | (3.1 ± 0.2) × 10−5 | ||
| B | — | (2 ± 1) × 10−5 | ||
| Mo | (1.4 ± 0.1) × 10−7 | (1.6 ± 0.5) × 10−7 | ||
| Si | (1.3 ± 0.3) × 10−3 | (1.9 ± 0.7) × 10−3 | ||
| Al | — | — | ||
- Lithium is released instantaneously into the solutions at 20 °C and 50 °C at about 10−5 mol L−1;
- The boron concentration is only measured at 50 °C to have an average concentration of 10−5 mol L−1;
- The molybdenum concentration is very low, with a mean value between 10−7 and 10−8 mol L−1;
- The aluminum concentration varies with solution type, HCP type and temperature (10−6 to 10−3 mol L−1).
In the same way, the silicon concentration directly depends on the HCP type. In the presence of CEM-I HCP, the total concentration measured in solutions S1b and S2b at 50 °C is 6 × 10−5 mol L−1. This value is an order of magnitude higher than the expected value calculated via geochemical models.62 For low-pH HCP, the concentration value is high, 1 × 10−3 mol L−1, which corresponds to the solubility value of amorphous silica.62 This is consistent with the low-pH HCP composition (30% silica fume), in which hydration leads to the formation of amorphous silica. Amorphous silica represents a third of the total mass of the low-pH cement paste.
A comparison of glass dissolution rates obtained for binary (glass/solution) and ternary (glass/HCP/solution) systems shows two trends (Fig. 4, Table 6): (1) short-term rates are almost similar, with the maximum ratio of NLRinit(with HCP cement) to NLRinit(no HCP) not exceeding 1.8, and (2) long-term rates are higher for ternary systems with S1b and S2b solution. As an example, in the system with solution S1b and CEM-I or CEM-V, dissolution rates at 50 °C are higher than those in solution alone by factors of 115 and 162, respectively. Furthermore, the difference in dissolution rates between S1b (+HCP) and S2b (+HCP) can be explained by the difference in pH, which is higher for S1b (pH = 12.6) than for S2b (pH 11.6, see Table 2). Another parameter to take into account is that the Ca concentration is 10 times higher in S2b than in S1b solution. Calcium in cementitious systems is known to counterbalance the effects of high pH on glass dissolution by structuring the gel layer formed on the glass surface, thus slowing down water diffusion into the glass matrix.28,33 The S5 solution/low-pH cement system had no effect on glass long-term dissolution. This could be attributed to both the low pH value (pH = 9.8) and high silica content in the low-pH HCP, leading to high silica concentrations in solution (2 × 10−3 mol L−1).
To summarize, one can state that C–S–H phases are expected to precipitate in all systems with a higher Ca/Si ratio at 50 °C than at 20 °C, which could contribute to a glass dissolution increase at 50 °C. In the S5-low-pH system, the higher silica concentration is controlled by crystalline silica phases rather than by C–S–H phases, which partially explains the difference in glass dissolution kinetics.
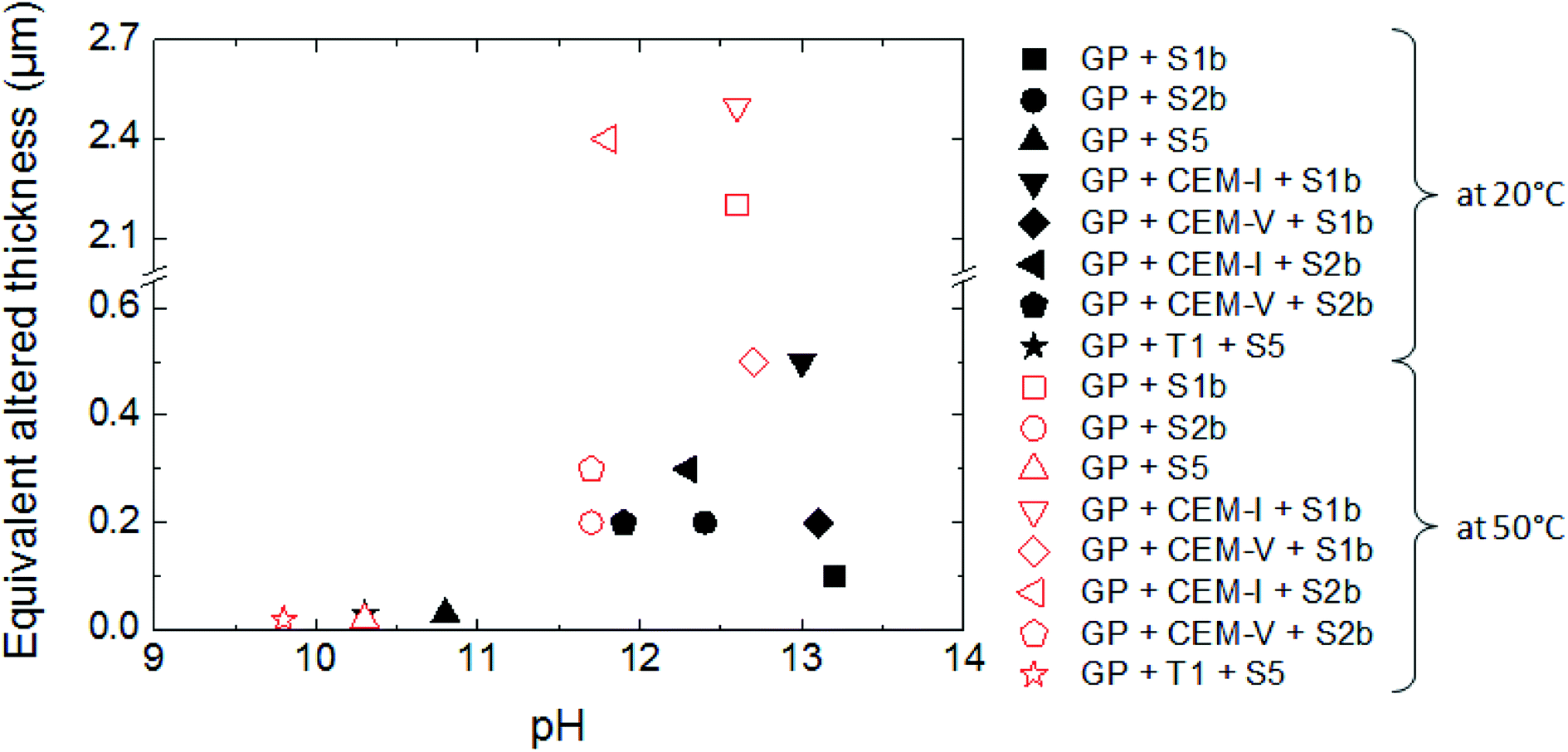 | ||
| Fig. 8 The equivalent altered thickness of glass powder (GP) as a function of pH: the effects of S1b, S2b and S5 solution compositions, the presence of CEM-I, CEM-V and low-pH HCP powder, and temperature (Exp. 1, 5, 6 and 7). | ||
4. Conclusions
Intermediate level waste (ILW) glass dissolution in cementitious systems has been studied under different conditions that could be encountered during deep geological disposal, with relation to temperature, solution chemistry, and cement composition.In the scenario of a repository of intermediate level vitrified waste, water re-saturation of the site starts with the degradation of cement, leading to highly alkaline pore water (solution S1b), followed by the control of the system by portlandite, with a lower pH of 12.4 (solution S2b). During long-term alteration, the pH is controlled by C–S–H phases and decreases to 9. This work shows that the dissolution of glass powder is influenced by the composition and the pH of solutions S1b (pH 13), S2b (pH 12.4) and S5 (pH 9): the dissolution rates and the equivalent altered thicknesses increase with pH.
This study gives some information about the potential influence of the different solutions on glass alteration. In contact with HCP (CEM-I and CEM-V) in solution S1b, a steady state is not reached, even after 776 days of alteration. In solution S2b, classical tracers of glass dissolution (Li, B and Mo) are strongly influenced by the compositions of solutions enriched in calcium. Boron may precipitate to form calcium borate. In low pH solution (S5), the initial dissolution rate is decreased by a factor of ∼18, suggesting that Si enrichment in solution, released by silica fume from low-pH HCP, controls the dissolution of ILW glass.
A mechanism of diffusion type has been identified over the whole duration of the experiments (850 days). The study of low-pH HCP dissolution in contact with glass gives promising results in terms of dissolution rates and the chemical durability of glass. Monolith glass alteration in the absence/presence of HCP and glass powder shows the formation of an altered surface layer, mainly constituted of calcium silicate hydrates with low Ca/Si ratios. These phases consume aqueous silica and thus sustain glass dissolution.
Finally, in the context of ILW glass disposal in a cement environment, developing low-pH cements as an alternative to high-pH cements could be a promising alternative. Nevertheless, long-term dissolution experiments and a thorough characterization of the alteration products are required for the understanding and modelling of long-term glass dissolution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Andra for the financial support, and CEA and Orano Cycle for supplying ILW glass samples. We would like to express our sincere gratitude to Véronique Baty (Subatech laboratory) for ion chromatography analyses, Damien Salou (Subatech laboratory) for performing batch experiments, and Nicolas Stephant (Institut des Matériaux – Jean Rouxel de Nantes, IMN) for SEM/EDX analyses.References
- ASN, in Les activités contrôlées par l’ASN, 2010 Search PubMed.
- ANDRA, Dossier 2005: Référentiel des matériaux d’un stockage de déchets à haute activité et à vie longue, 2005 Search PubMed.
- E. Y. Vernaz and N. Godon, MRS Online Proc. Libr., 1991, 257, 37 CrossRef.
- S. Gin, N. Godon, I. Ribet, P. Jollivet, Y. Minet, P. Frugier, E. Vernaz, J. M. Cavedon, B. Bonin and R. D. Quang, Mater. Res. Soc. Symp. Proc., 2004, 824, 327–332 CrossRef CAS.
- S. Gin, P. Jollivet, J. P. Mestre, M. Jullien and C. Pozo, Appl. Geochem., 2001, 16, 861–881 CrossRef CAS.
- S. Gin and J. P. Mestre, J. Nucl. Mater., 2001, 295, 83–96 CrossRef CAS.
- T. Advocat, P. Jollivet, J. L. Crovisier and M. Del Nero, J. Nucl. Mater., 2001, 298, 55–62 CrossRef CAS.
- S. Peuget, J.-N. Cachia, C. Jegou, X. Deschanels, D. Roudil, V. Broudic, J. M. Delaye and J.-M. Bart, J. Nucl. Mater., 2006, 354, 1–13 CrossRef CAS.
- E. Vernaz, S. Gin, C. Jegou and I. Ribet, J. Nucl. Mater., 2001, 298, 27–36 CrossRef CAS.
- F. Delage, D. Ghaleb, J. L. Dussossoy, O. Chevallier and E. Vernaz, J. Nucl. Mater., 1992, 190, 191–197 CrossRef CAS.
- P. Frugier, S. Gin, J. E. Lartigue and E. Deloule, Mat. Res. Soc. Symp. Proc., 2006, 932, 305–312 CrossRef CAS.
- P. Frugier, T. Chave, S. Gin and J. E. Lartigue, J. Nucl. Mater., 2009, 392, 552–567 CrossRef CAS.
- B. Grambow, Mater. Res. Soc. Symp. Proc., 1985, 44, 15–27 CrossRef CAS.
- B. Grambow and R. Müller, J. Nucl. Mater., 2001, 298, 112–124 CrossRef CAS.
- P. Frugier, S. Gin, Y. Minet, T. Chave, B. Bonin, N. Godon, J. E. Lartigue, P. Jollivet, A. Ayral, L. De Windt and G. Santarini, J. Nucl. Mater., 2008, 380, 8–21 CrossRef CAS.
- S. Gin, I. Ribet and M. Couillard, J. Nucl. Mater., 2001, 298, 1–10 CrossRef CAS.
- E. Y. Vernaz and J. L. Dussossoy, Appl. Geochem., 1992, 7, 13–22 CrossRef.
- T. Geisler, A. Janssen, D. Scheiter, T. Stephan, J. Berndt and A. Putnis, J. Non-Cryst. Solids, 2010, 356, 1458–1465 CrossRef CAS.
- R. Hellmann, S. Cotte, E. Cadel, S. Malladi, L. S. Karlsson, S. Lozano-Perez, M. Cabié and A. Seyeux, Nat. Mater., 2015, 14, 307 CrossRef CAS PubMed.
- Ministère De L’écologie/De L’énergie/Du Développement Durable Et De La Mer, J. Off. la République Française, Décret no 2009-961 du 31 juillet 2009.
- AREVA, AREVA – BG AVAL, Dir. Matières Déchets Radioact. NOTE Tech. DMDR NT 15-0010.
- IRSN, Avis de l’IRSN relative à la spécification référencée 300-AQ-061 pour le conditionnement par vitrification d’effluents de moyenne activité, 2009 Search PubMed.
- ANDRA, Catalogue descriptif des familles, 2015 Search PubMed.
- ANDRA, Référentiel des matériaux d’un stockage de déchets de haute activité et de déchets de moyenne activité et à vie longue, Tome 2: les matériaux cimentaires, 2012 Search PubMed.
- ANDRA, Sites en exploitation: La Hague, Basse Normandie, 2016 Search PubMed.
- A. Ait Chaou, A. Abdelouas, Y. El Mendili, R. Bouakkaz, S. Utsunomiya, C. Martin and X. Bourbon, R. Soc. Chem. Adv., 2015, 5, 64538–64549 CAS.
- S. Depierre, Etude Des Mécanismes d'altération Du Verre Par Des Eaux Cimentaires, Université de Montpellier II, France, 2012 Search PubMed.
- S. Mercado-Depierre, F. Angeli, F. Frizon and S. Gin, J. Nucl. Mater., 2013, 441, 402–410 CrossRef CAS.
- D. G. Bennett and R. Gens, J. Nucl. Mater., 2008, 379, 1–8 CrossRef CAS.
- P. A. Bingham, N. C. Hyatt and R. J. Hand, Glass Technol.: Eur. J. Glass Sci. Technol., Part A, 2012, 53, 83–100 CAS.
- C. A. Utton, R. J. Hand, P. A. Bingham, N. C. Hyatt, S. W. Swanton and S. J. Williams, J. Nucl. Mater., 2013, 435, 112–122 CrossRef CAS.
- S. M. Wickham, M. B. Crawford and D. G. Bennett, Eval. Ref. Des. Galson Sci. LTD, Oakham, 2005 Search PubMed.
- T. Chave, P. Frugier, S. Gin and A. Ayral, Geochim. Cosmochim. Acta, 2011, 75, 4125–4139 CrossRef CAS.
- P. Jollivet, P. Frugier, G. Parisot, J. P. Mestre, E. Brackx, S. Gin and S. Schumacher, J. Nucl. Mater., 2012, 420, 508–518 CrossRef CAS.
- G. J. Stockmann, D. Wolff-Boenisch, S. R. Gislason and E. H. Oelkers, Chem. Geol., 2011, 284, 306–316 CrossRef CAS.
- C. Cau Dit Coumes, S. Courtois, D. Nectoux, S. Leclercq and X. Bourbon, Cem. Concr. Res., 2006, 36, 2152–2163 CrossRef CAS.
- D. Rothstein, J. J. Thomas, B. J. Christensen and H. M. Jennings, Cem. Concr. Res., 2002, 32, 1663–1671 CrossRef CAS.
- K. De Weerdt, M. Ben Haha, G. Le Saout, K. O. Kjellsen, H. Justnes and B. Lothenbach, Cem. Concr. Res., 2011, 41, 279–291 CrossRef CAS.
- B. Lothenbach, G. Le Saout, E. Gallucci and K. Scrivener, Cem. Concr. Res., 2008, 38, 848–860 CrossRef CAS.
- B. Lothenbach, T. Matschei, G. Möschner and F. P. Glasser, Cem. Concr. Res., 2008, 38, 1–18 CrossRef CAS.
- M. C. Alonso, L. Fernandez-Luco, J. L. Garcia, A. Hidalgo and F. Huertas, in Proceedings of the 12th International Congress on the Chemistry of Cement, Montreal, Canada, Citeseer, 2007, pp. 8–13 Search PubMed.
- A. Dauzères, P. Le Bescop, C. Cau-Dit-Coumes, F. Brunet, X. Bourbon, J. Timonen, M. Voutilainen, L. Chomat and P. Sardini, Cem. Concr. Res., 2014, 58, 76–88 CrossRef.
- A. Jenni, U. Mäder, C. Lerouge, S. Gaboreau and B. Schwyn, Physics and Chemistry of the Earth, Parts A/B/C, 2014, 70, 71–83 CrossRef.
- A. Dauzères, G. Achiedo, D. Nied, E. Bernard, S. Alahrache and B. Lothenbach, Cem. Concr. Res., 2016, 79, 137–150 CrossRef.
- K. Ferrand, S. Liu and K. Lemmens, Procedia Mater. Sci., 2014, 7, 223–229 CrossRef CAS.
- M. Codina, Les Bétons Bas PH. Formulation, Caractérisation et Étude à Long Terme, Institut National des Sciences Appliquées, Toulouse, France, 2007 Search PubMed.
- D. L. Parkhurst and C. A. J. Appelo, User's guide to Phreeqc (version 2) _ A computer program for speciation, batch reaction, one-dimensional transport, and inverse geochemical calculations, Water-resources investigations Report, Denver, CO USA, 1999 Search PubMed.
- L. Duro, M. Grivé and E. Giffaut, in MRS Proceedings, Cambridge Univ Press, 2012, vol. 1475, pp. imrc11-1475-nw35-o71 Search PubMed.
- L. Trotignon, H. Peycelon and X. Bourbon, Procedia Mater. Sci., 2006, 31, 610–617 Search PubMed.
- V. Gabis, Bull. Soc. Fr. Mineral. Cristallogr., 1958, 81, 183–185 CAS.
- D. Guimaraes, V. de A. Oliveira and V. A. Leao, J. Therm. Anal. Calorim., 2016, 124, 1679–1689 CrossRef CAS.
- R. B. Perkins and C. D. Palmer, Geochim. Cosmochim. Acta, 1999, 63, 1969–1980 CrossRef CAS.
- B. A. Clark and P. W. Brown, Cem. Concr. Res., 2000, 30, 233–240 CrossRef CAS.
- D. Damidot and F. P. Glasser, Cem. Concr. Res., 1992, 22, 1179–1191 CrossRef CAS.
- M. Fournier, P. Frugier and S. p Gin, Procedia Mater. Sci., 2014, 7, 202–208 CrossRef CAS.
- R. J. Kirkpatrick, J. L. Yarger, P. F. McMillan, Y. Ping and X. Cong, Adv. Cem. Based Mater., 1997, 5, 93–99 CrossRef CAS.
- G. Renaudin, R. Segni, D. Mentel, J.-M. Nedelec, F. Leroux and C. Taviot-Gueho, J. Adv. Concr. Technol., 2007, 5, 299–312 CrossRef CAS.
- J. Dweck, P. F. Ferreira da Silva, P. M. Büchler and F. K. Cartledge, J. Therm. Anal. Calorim., 2002, 69, 179–186 CrossRef CAS.
- J. Dweck, P. M. Buchler, A. C. V. Coelho and F. K. Cartledge, Thermochim. Acta, 2000, 346, 105–113 CrossRef CAS.
- K. Matsui, J. Kikuma, M. Tsunashima, T. Ishikawa, S. Matsuno, A. Ogawa and M. Sato, Cem. Concr. Res., 2011, 41, 510–519 CrossRef CAS.
- T. Sugama, L. E. Kukacka and W. Horn, J. Mater. Sci., 1981, 16, 345–354 CrossRef CAS.
- ANDRA, Référentiel des matériaux d’un stockage de déchets de haute activité et de déchets de moyenne activité et à vie longue, Tome 2: les matériaux cimentaires, 2012 Search PubMed.
- B. Grambow, in Uhlig's Corrosion Handbook: Third Edition, 2011 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |