
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of additives, ball milling and isotopic exchange in porous magnesium borohydride†
Michael Heere *ab,
Olena Zavorotynskaac,
Stefano Deleddaa,
Magnus H. Sørbya,
David Bookd,
Theodore Steriotise and
Bjørn C. Haubacka
*ab,
Olena Zavorotynskaac,
Stefano Deleddaa,
Magnus H. Sørbya,
David Bookd,
Theodore Steriotise and
Bjørn C. Haubacka
aDepartment for Neutron Materials Characterization, Institute for Energy Technology, NO-2027 Kjeller, Norway
bInstitute for Applied Materials – Energy Storage Systems (IAM – ESS), Karlsruhe Institute of Technology (KIT), D–76344, Eggenstein-Leopoldshafen, Germany. E-mail: michael.heere@kit.edu
cInstitute for Mathematics and Physics, University of Stavanger, PO Box 8600 Forus, NO-4036 Stavanger, Norway
dSchool of Metallurgy and Materials, University of Birmingham, Birmingham B15 2TT, UK
eInstitute of Nanoscience and Nanotechnology, NCSR “Demokritos”, Ag. Paraskevi Attikis, Athens 15341, Greece
First published on 2nd August 2018
Abstract
Magnesium borohydride (Mg(BH4)2) is a promising material for solid state hydrogen storage. However, the predicted reversible hydrogen sorption properties at moderate temperatures have not been reached due to sluggish hydrogen sorption kinetics. Hydrogen (H) → deuterium (D) exchange experiments can contribute to the understanding of the stability of the BH4− anion. Pure γ-Mg(BH4)2, ball milled Mg(BH4)2 and composites with the additives nickel triboride (Ni3B) and diniobium pentaoxide (Nb2O5) have been investigated. In situ Raman analysis demonstrated that in pure γ-Mg(BH4)2 the isotopic exchange reaction during continuous heating started at ∼80 °C, while the ball milled sample did not show any exchange at 3 bar D2. However, during ex situ exchange reactions investigated by infrared (IR) and thermogravimetric (TG) analyses a comparable H → D exchange during long exposures (23 h) to deuterium atmosphere was observed for as received, ball milled and γ-Mg(BH4)2 + Nb2O5, while the Ni3B additive hindered isotopic exchange. The specific surface areas (SSA) were shown to be very different for as received γ-Mg(BH4)2, BET area = 900 m2 g−1, and ball milled Mg(BH4)2, BET area = 30 m2 g−1, respectively, and this explains why no gas–solid H(D) diffusion was observed for the ball milled (amorphous) Mg(BH4)2 during the short time frames of in situ Raman measurements. The heat treated ball milled sample partially regained the porous γ-Mg(BH4)2 structure (BET area = 560 m2 g−1). This in combination with the long reaction times allowing for the reaction to approach equilibrium explains the observed gas–solid H(D) diffusion during long exposure. We have also demonstrated that a small amount of D can be substituted in both high surface area and low surface area samples at room temperature proving that the B–H bonds in Mg(BH4)2 can be challenged at these mild conditions.
Introduction
Research on solid state hydrogen storage materials has been enormously extended in the last 20 years, ever since Bogdanović and Schwickardi reported reversible hydrogenation in Ti-catalyzed sodium alanate, NaAlH4.1 Many compounds have been considered for hydrogen storage applications and magnesium borohydride, Mg(BH4)2, is still one of the most promising candidates, with a high hydrogen content (14.9 wt% H) and predicted reversible hydrogen sorption properties at moderate temperatures.2,3 The compound is potentially suitable for the operating temperature window of polymer electrolyte membrane (PEM) fuel cells (∼80 °C). The synthesis of Mg(BH4)2 from hydrogenation of MgB2 was first achieved at 400 °C and 900 bar hydrogen.4 Furthermore, decomposition of Mg(BH4)2 at moderate temperatures (∼200 °C) resulted in the formation of Mg(B3H8)2, which can almost completely be rehydrogenated to Mg(BH4)2 within 48 h at 250 °C and 120 bar H2,5 and thus reversibly cycling 2.5 wt% H. Nevertheless, Zavorotynska et al. reported similar results on a much shorter time scale.6 2.5 wt% hydrogen were reabsorbed at 260 °C under 120 bar H2 in 7–8 h, and the reaction was completed in less than 3 h at 280 °C.Efforts in tailoring the hydrogen storage properties of Mg(BH4)2 have been continuing. Addition of small amounts of various transition-metal compounds, with the goal to improve kinetics and reversibility of hydrogen desorption in Mg(BH4)2, has been a subject of several recent studies. Transition-metal (TM) oxides, chlorides or fluorides, with TM = Sc, Ti, V, Co, Ni, Zn, Nb, Mo, Ru and Pd7–17 have been employed and showed improvement of hydrogen release during the first desorption.18
Saldan et al.18 investigated the effect of various Ni-based additives in Mg(BH4)2 and reported that NiF2 and NiCl2 transformed into a phase “resembling Ni3B” after the first hydrogen desorption. Therefore, Ni3B appeared to be the most promising candidate to improve the hydrogen release properties of Mg(BH4)2. Mg(BH4)2 including 2 mol% Ni3B has shown a release of 2.7 wt% H during a 15 h desorption measurement at 220 °C under static vacuum,18 whereas it took 800 h at 200 °C to desorb the same amount from pure Mg(BH4)2.5
Mg(BH4)2 has a rather unique crystal chemistry.19 In total, there are currently seven known polymorphs: α-, β-, γ-, δ- and ζ-Mg(BH4)2 have been structurally characterized,20–23 two yet unsolved structures ε- and β′-Mg(BH4)2,24,25 and an unidentified structure was recently proposed.19 Furthermore, non-crystalline amorphous Mg(BH4)2 has been obtained from solvent-free synthesis,26 by ball milling of the crystalline α- and γ-phases22 and via pressure collapse of γ-Mg(BH4)2.27 γ-Mg(BH4)2 exhibits a characteristic porous structure, and its specific surface area (SSA) of 1160 m2 g−1 is the highest so far found in complex hydrides.22 It is higher than the SSA of high performing organosilicas28,29 and can be compared to the SSA of metal organic frameworks (MOFs). It has been shown that it is possible to reversibly adsorb N2, CH2Cl2 and even 3 wt% of hydrogen (at 83 K, 105 bar) in γ-Mg(BH4)2.22 Possible thermal decomposition pathways of different polymorphs of Mg(BH4)2 are reviewed by Zavorotynska et al.19
Isotopic experiments in group I and II metal borohydrides have been carried out by different research groups.30 Recently, Zavorotynska et al.31 demonstrated the H → D isotopic exchange reaction in porous γ-Mg(BH4)2 at much milder conditions than reported for isotopic exchange in the denser α-Mg(BH4)2 phase.32 In α-Mg(BH4)2, the isotopic exchange reaction was observed at 132 °C after prolonged treatment at 42 bar D2, whereas the exchange in porous γ-Mg(BH4)2 was recorded after ∼17 min at 100 °C under only 3 bar D2. The authors concluded that the porous nature and resulting high specific surface area of γ-Mg(BH4)2 was responsible for the rapid exchange reaction occurring at the surface at mild conditions.
In this work, isotopic exchange is used to obtain a deeper understanding of the effect of additives and/or ball milling, on the hydrogen sorption kinetics of γ-Mg(BH4)2. Firstly, we have compared the H2 release in as received and ball milled γ-Mg(BH4)2 as well as γ-Mg(BH4)2 milled with Nb- and Ni-based additives (2 mol%). Nb2O5, to the best of our knowledge, has not been explored for improving the properties of Mg(BH4)2, but has been reported to enhance kinetics in hydrogen desorption and absorption in MgH2.33 Secondly, we have shown the influence of additives and/or ball milling on the H → D isotopic exchange in γ-Mg(BH4)2. The isotopic exchange reactions were studied by thermogravimetric and differential scanning calorimetry (TG-DSC), infrared (IR) and in situ Raman measurements. Furthermore, synchrotron radiation powder X-ray diffraction (SR-PXD) measurements, including in situ SR-PXD, were carried out for all samples. Nitrogen adsorption isotherms were measured at 77 K for the as received, the ball milled Mg(BH4)2 as well as their heat treated analogues in order to determine their specific surface area.
Experimental details
γ-Mg(BH4)2 (95%) and Nb2O5 (99%) were purchased from Sigma Aldrich and used without further purification. The additive Ni3B was prepared as described in ref. 18.All sample manipulations were carried out under inert conditions. An MBraun glove box fitted with a recirculation system was used with oxygen and humidity levels kept below 1 ppm during all operations. Mechanochemical milling (ball milling, bm) with and without 2 mol% additives was carried out using a Fritsch Pulverisette 6 planetary mill. Steel vial and balls were used and a ball-to-powder ratio of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was employed. The powders were milled for 4 × 15 min at 300 rpm with breaks of 5 min to avoid overheating. The synthesis procedures of the four investigated samples, S1–S4, are summarized in Table 1.
:1 was employed. The powders were milled for 4 × 15 min at 300 rpm with breaks of 5 min to avoid overheating. The synthesis procedures of the four investigated samples, S1–S4, are summarized in Table 1.
| Sample name | Composition | Procedure | M (g mol−1) | H-content (wt%) | Δm (wt%) | H loss (%) | Sample name after 23 h H → D exchange at 120 °C |
|---|---|---|---|---|---|---|---|
| S1 | γ-Mg(BH4)2 | As received | 53.99 | 14.94 | 13.5 ± 0.3 | 90 ± 2 | S1-D |
| S2 | γ-Mg(BH4)2 | Ball milling | 53.99 | 14.94 | 13.1 ± 0.3 | 87 ± 2 | S2-D |
| S3 | γ-Mg(BH4)2 + 2 mol% Ni3B | Ball milling | 57.73 | 13.97 | 12.3 ± 0.1 | 88 ± 1 | S3-D |
| S4 | γ-Mg(BH4)2 + 2 mol% Nb2O5 | Ball milling | 59.31 | 13.60 | 11.8 ± 0.5 | 86 ± 3 | S4-D |
Thermogravimetric and differential scanning calorimetry (TG-DSC) experiments were conducted using a Netzsch STA 449 F3 Jupiter analyser. Two sets of experiments were conducted. The first set was measured from room temperature (RT) to 285 °C at 5 °C min−1. The second set of experiments were carried out by heating from RT to 450 °C at heating rates of 1, 2, 5 and 7 °C min−1, respectively. All measurements were conducted within Al crucibles. The Ar flow (protective and purge gas) was 20 and 50 ml min−1, respectively. The peak search function in the Netzsch software (proteus thermal analysis) was used to determine peak temperatures in the DSC data. For overlapping events, the peak shape fitting function in the software Fityk34 was employed, with a Gaussian function, in which parameters such as peak height, position, half width at half maximum and shape were fitted one by one (ESI Fig. A1†). Temperature programmed desorption-mass spectrometry (TPD-MS) was performed in an in-house manufactured apparatus connected to a MKS Microvision-IP Residual Gas Analyser. The powder (∼10 mg) was loaded in a steel sample holder and heated from RT to 400 °C with a heating rate of 5 °C min−1 under dynamic vacuum.
The deuterium exchange experiments were executed in an in-house manufactured Sieverts type apparatus35 at 3 bar D2 and 120 °C. During all experiments the furnace was preheated to 120 °C before the steel autoclave with the sample was introduced. The D2 pressure was adjusted to 3 ± 0.05 bar at RT and did not rise significantly during heating due to the large volume of the Sieverts apparatus. The duration of the exchange reaction at 120 °C was chosen to 23 h. Samples were contained in sealed Al-crucibles with a pierced lid, which were placed inside an in-house manufactured steel autoclave. For H → D exchange at elevated temperature, the samples S1–S4 were treated individually. TG data were employed to calculate the deuterium content (ESI Fig. A2†).
Synchrotron radiation powder X-ray diffraction (SR-PXD) data were collected at the Swiss Norwegian Beam Line (SNBL), BM01 at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. For SR-PXD experiments at RT the samples were contained in 0.5 mm sealed boron glass capillaries. For in situ SR-PXD experiments, sapphire capillaries with 1.2 mm and 0.8 mm outer and inner diameter, respectively, were connected via Vespel ferrules to an in-house manufactured remote controlled gas rig and high pressure manifold cell. The sample-to-detector distances and the wavelength were calibrated from a NIST LaB6 standard. Data were collected using a Pilatus 2M detector. The exposure time was set to 30 s giving a temperature resolution of 2.5 °C per pattern and the sapphire capillary was rotated by 10° during exposure to improve powder averaging. Single crystal reflections from the sapphire tube were masked manually and the data were integrated to 1D diffraction patterns in Fit2D and Bubble.36,37
Attenuated total reflection IR (ATR-IR) measurements were performed using a Bruker Alpha-Platinum infrared spectrometer with a diamond crystal inside an Ar-filled glove box. The spectra were obtained in the wavenumber range of 4000–400 cm−1 with a resolution of 2 cm−1 at RT. Three measurements each including 32 scans were averaged for each spectrum and the background. IR spectra were ATR corrected and normalized using commercial spectroscopic software OPUS. The samples were measured without any dilution.
In situ Raman spectra were measured employing a Renishaw inVia Raman microscope with a 488 nm excitation laser (usually 2 mW on the sample). To focus the laser beam onto the sample a microscope objective with a spot diameter of 50 μm was used. An Instec sample HCS621 V cell with a fused silica window was employed for H → D exchange measurements at 3 bar D2 pressure while heating was conducted in this cell between RT and 175 °C at 2 °C min−1. The cell was flushed with argon before use. Each scan consists of 10 times 5 s data acquisition, with automatic refocusing after every fifth scan. The temperature resolution was 1.7 °C per pattern. Data processing with the software WiRe 4.0 included baseline correction and cosmic ray removal, as well as normalization for samples S1 and S2. Furthermore, the data was smoothed, with special emphasis that no peaks were removed during smoothing.
N2 adsorption isotherms were carried out at −196 °C (77 K) on an Autosorb-1MP volumetric analyzer equipped with a Gifford-McMahon cryocooler, which is a two-stage closed cycle refrigeration unit (CCR) working from −265 °C to +47 °C (8–320 K). The samples were outgassed at 35 °C under high vacuum (<10−6 mbar), usually for 8–10 h. Preliminary N2 adsorption measurements of the as received S1 at −196 °C (77 K) revealed extremely poor kinetics. For this reason very strict equilibration criteria (pressure change <0.00001 bar h−1) were used for S1 and S2, leading to equilibration times of several hours per point. Due to the ultra-microporous character of some samples low relative pressure measurements (p/p0 = 10−6 – 0.995) were employed. Similar measurements were carried out on the heat treated S1 and S2 samples. The heat treated samples were named S1-H and S2-H in analogy to S1-D and S2-D (Table 1) as the same conditions (120 °C and 3 bar H2 for 23 h) were used, but with hydrogen instead of deuterium. In all cases BET areas were calculated by taking into consideration the pertinent IUPAC consistency criteria for microporous materials. Alternative models for area determination such as Langmuir, Dubinin–Radushkevich (DR) and micropore analysis method (MP-method) were applied as well to validate the data.38–40 Total pore volumes were calculated from the plateau region by assuming liquid N2 density in the pores.
Results and discussion
Effect of additives vs. ball milling
The SR-PXD data at RT for samples S1–S4 are presented in Fig. 1. For S1 only Bragg peaks of γ-Mg(BH4)2 are observed on a low background.22 However, the ball milled S2 and S3 show strong diffuse scattering and only traces of Bragg peaks from γ-Mg(BH4)2, indicating almost complete amorphization during the milling process.22 Bragg peaks of the Ni3B additives could not be observed in S3, which is due to its amorphous nature and in agreement with previous work.18 In S4 diffuse scattering is present as well, but with more intense Bragg peaks of the γ-Mg(BH4)2 phase compared to S2.18 Bragg peaks of Nb2O5 are identified for S4. No contaminations have been found in S1–S4. The first order phase transitions of γ-Mg(BH4)2 to ε-Mg(BH4)2 (ref. 25) and from ε- into β′-Mg(BH4)2 (ref. 24) have previously been reported. In situ SR-PXD measurements were employed in order to study how additives influence the phase transitions in crystalline Mg(BH4)2. Fig. 2a shows in situ SR-PXD data of S4 heated from RT to 285 °C at 5 °C min−1 followed by a 1 h isotherm, while Fig. 2b is presenting the same SR-PXD data at selected temperatures for a better visualization. For clarity the data are only given up to 2θ = 10°, and thus excluding most Bragg peaks of Nb2O5. 285 °C was chosen as the end temperature because earlier reports suggested melting and/or formation of amorphous phases above 285 °C,14 followed by the formation of very stable B12H12 phases at higher temperatures.4 Bragg peaks of γ-Mg(BH4)2 and Nb2O5 are present at RT. Upon heating, Bragg peaks of γ-Mg(BH4)2 sharpen until 142 °C (red curve in Fig. 2b). A crystallization reaction is the probable cause for peak sharpening, corresponding to an increase in crystallinity. Upon further heating the Bragg peaks of γ-Mg(BH4)2 decrease in intensity and vanish at 173 °C. Bragg peaks corresponding to ε-Mg(BH4)2 (ref. 25) appear at 154 °C and have their highest intensity at 178 °C (Fig. 2b, blue curve). ε-Mg(BH4)2 is transformed directly into β′-Mg(BH4)2,24 appearing at 197 °C and the highest intensity is observed at 248 °C (magenta curve). The disappearance of Bragg peaks after 18 min in the 285 °C isotherm has been reported before and is due to formation of an amorphous phase and possibly a reversible product which is very likely Mg(B3H8)2.13 In the isothermal regime no further reactions occur. The in situ SR-PXD data of S3 (ESI Fig. A4†) show Bragg peaks of γ-Mg(BH4)2 which appear between RT and ∼150 °C. Bragg peaks of β′-Mg(BH4)2 are observed between 195 and 285 °C, while ε-Mg(BH4)2 is not observed.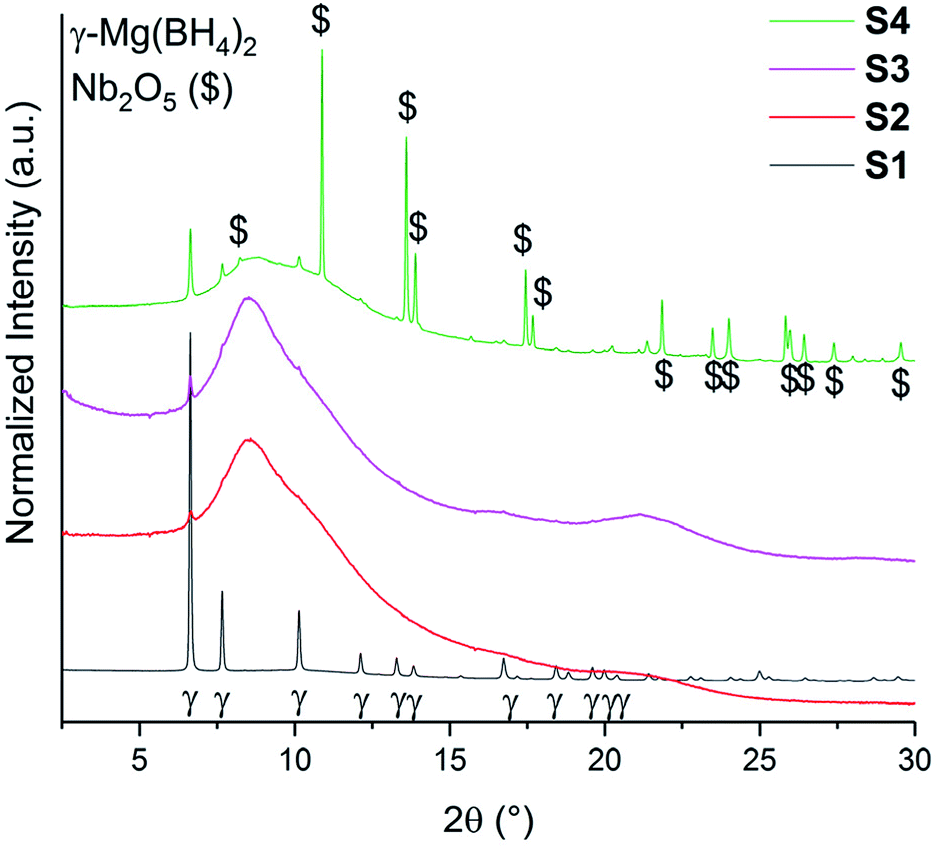 | ||
| Fig. 1 SR-PXD data of γ-Mg(BH4)2 samples (S1–S4) at RT. All samples have been normalized to its maximum intensity and are shifted on the y-axis for better visibility. λ = 0.7454 Å. | ||
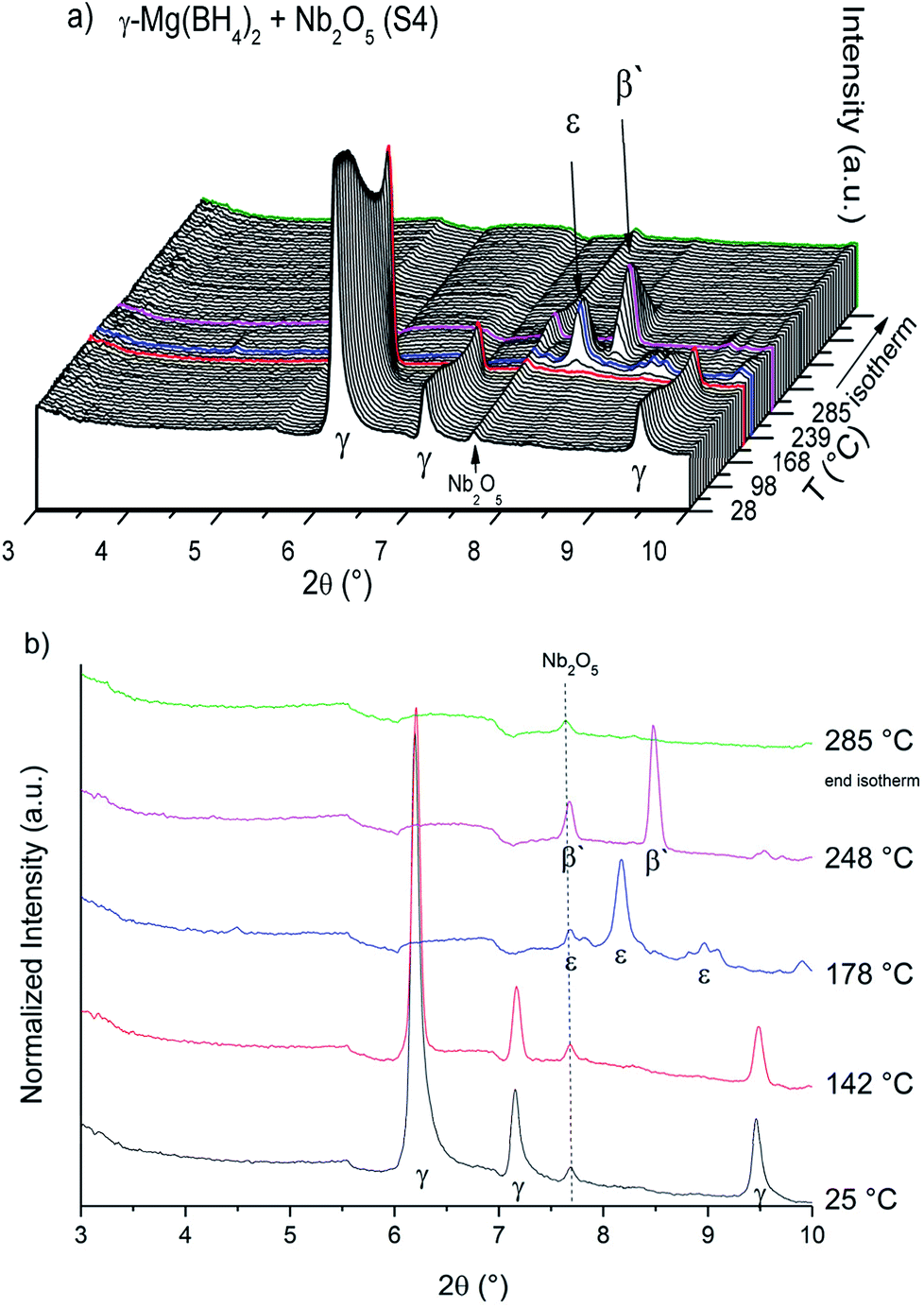 | ||
| Fig. 2 (a) In situ SR-PXD data of ball milled Mg(BH4)2 + Nb2O5 (S4) heated from RT to 285 °C under 2.3 bar H2 (temperature ramp of 5 °C min−1) and 1 h isotherm. (b) SR-PXD data of S4 at specific temperatures. Curves in different colours are used from (a) for better visualization. λ = 0.6973. Å. | ||
TG-DSC data of S1–S4 heated from RT to 285 °C (5 °C min−1) are presented in Fig. 3. The first exothermic peak in the DSC data of S2 and S4 at 100 °C and of S3 at 120 °C stems from the crystallization of amorphous into γ-Mg(BH4)2. For S3 an elevated baseline and increased temperature of crystallization was observed. However, for S1 the first order transitions from γ-phase into ε-Mg(BH4)2 (Tonset = 153 °C) and from ε- into β′-Mg(BH4)2 (Tonset = 185 °C) can be clearly observed. Notably, they are shifted to slightly lower temperatures for the other samples. The onset temperatures of phase transitions for S2–S4 are very similar, 150 °C and 177 °C, respectively. These observations suggest a decreased stability of γ-phase in S2–S4, which is likely to have been induced by ball milling. The TG data between RT and 285 °C show that all samples have fairly similar weight loss (3.7 wt%), when considering that TG data are not corrected for the amounts of the additives (raw data in Fig. 3).
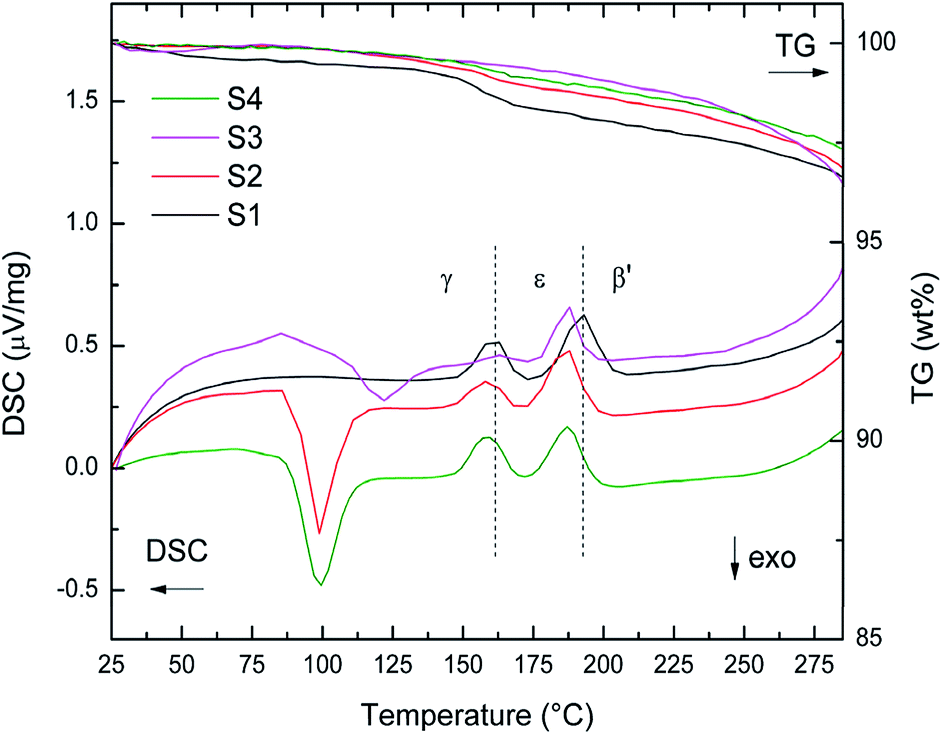 | ||
| Fig. 3 TG-DSC data of S1–S4 between RT and 285 °C heated at 5 °C min−1. Dashed lines at 153 °C and 185 °C indicate phase transition of as received γ-Mg(BH4)2 into ε-Mg(BH4)2 and β′-Mg(BH4)2. | ||
The decomposition of γ-Mg(BH4)2 can be divided into four steps, all taking place above 285 °C. TG-DSC data between RT and 450 °C are shown in ESI Fig. A3a–d.† A new fifth decomposition step is present in S2 and S4, and has not been assigned yet. It is an endothermic event and thus a decomposition of an intermediate Mg–B–H species is likely.41 Nevertheless, all decomposition events are possibly accompanied by the formation of intermediate Mg–B–H species including higher boranes.42 The decomposition of S1–S4 have been investigated by four TG-DSC measurements at different heating rates (1, 2, 5, 7 °C min−1) from RT to 450 °C. The average weight loss (Δm) from the TG measurements are given in Table 1 with standard deviations, and compared to the hydrogen contents of the samples. TPD-MS experiments in the range RT – 450 °C (ESI Fig. A5†) show that the mass losses are almost solely due to hydrogen release. Only traces of diborane were detected. The samples lose 80–90% of their hydrogen during the TG-DSC measurements and the relative losses (when accounting for the mass of the additive) are similar within the standard deviations. The TPD-MS experiments also show that the onset of hydrogen release is slightly reduced when comparing S1 to S2–S4, S4 showing the largest reduction.
Isotopic exchange
Isotopic exchange reactions were carried out to study the effect of additives and/or ball milling on the hydrogen sorption kinetics. These measurements were carried out both ex situ (long-term exposure) and in situ (short-term exposure) for each sample. The samples S1–S4 are re-named S1-D to S4-D after long-term exposure to 3 bar D2 atmosphere for 23 h at 120 °C. The chosen temperature (120 °C) is above the crystallization temperature of amorphous Mg(BH4)2 into γ-Mg(BH4)2 as it was shown earlier that storage or ball milling induces formation of amorphous Mg(BH4)2.27 The SR-PXD data at RT of S1-D to S4-D are presented in Fig. 4. The major phase in all samples is porous γ-Mg(BH4)2. A possible partial substitution of H with D cannot be determined by SR-PXD. Some diffuse scattering is observed, meaning that parts of the samples remain amorphous. In S1-D, a small amount of α-Mg(BH4)2 (ref. 20) is present (Fig. 4). Since S1 showed very little diffuse scattering, it appears that this α-phase is formed from γ-Mg(BH4)2. Larger amounts of α-Mg(BH4)2 are present present in S2-D to S4-D, as well as Bragg peaks of ε-Mg(BH4)2. The latter is unexpected at RT as the phase transition γ- → ε-Mg(BH4)2 is observed around 150 °C in TG-DSC and in situ SR-PXD measurements, which is well above the temperature used during the isotopic exchange. Thus, it appears that the transformation occurred at 120 °C, but at too slow a rate to be detected in the heating ramp (SR-PXD and TG-DSC) experiments.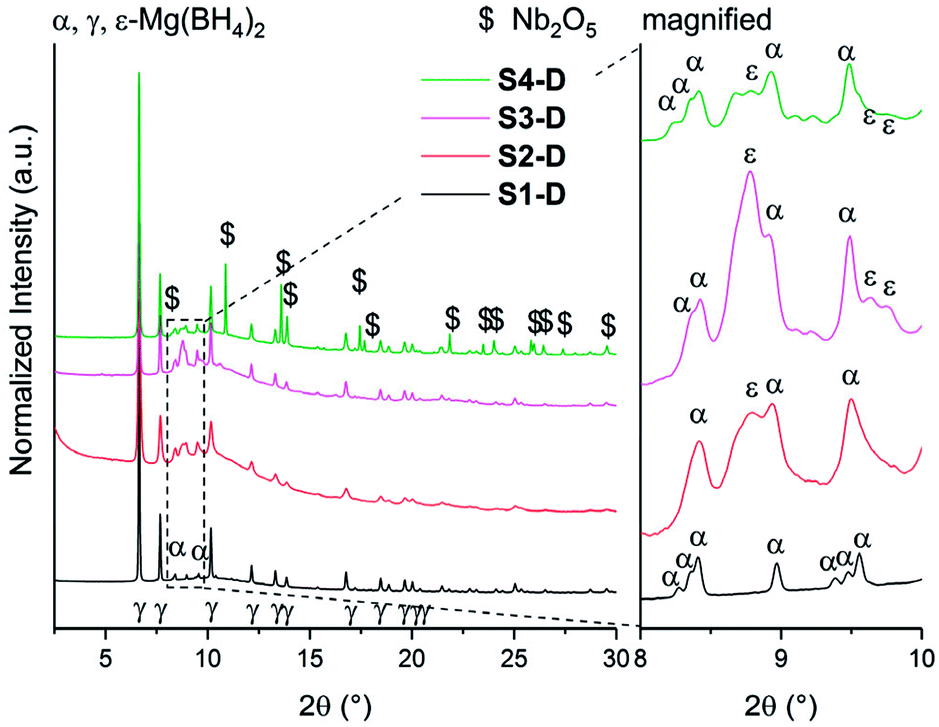 | ||
| Fig. 4 SR-PXD data of γ-Mg(BH4)2 samples (S1-D to S4-D) at RT. The samples S1 to S4 were exposed to 3 bar D2 at 120 °C for 23 h. For comparative reasons, all samples have been normalized to its maximum intensity and are shifted on the y-axis for better visibility. Inset showing the magnification of Bragg peaks between 2θ = 8–10°. λ = 0.7454 Å. | ||
The lack of Bragg peaks from ε-Mg(BH4)2 in S1-D is possibly due to the slightly higher onset temperature for the γ- → ε-phase transition in the as received sample. A control experiment was conducted (see details in ESI Fig. A6†) and confirms our assumption that the phase transition temperature was not reached. It is worth noting that a previous attempt to preserve ε-Mg(BH4)2 at RT was unsuccessful, even by rapid quenching.25 However once it is formed, it is stable on cooling to RT in all our investigated samples.
Spectroscopic studies
Raman and IR spectroscopy are used to identify deuterium substitution in the BH4 anion. BH4 has tetrahedral (Td) symmetry and four normal modes of vibrations. The modes in an isolated Td molecule are divided into the Raman-active and/or IR-active as follows: the symmetric stretching and bending modes, ν1 (A1) and ν2 (E) are only Raman-active, while the asymmetric stretching and bending modes, ν3 (F2) and ν4 (F2) are Raman-active and IR-active. The E mode is doubly degenerate, while the F mode is triply degenerate. It is, however, possible that the inactive mode become active in solids, while the degenerate modes can split due to various solid-state effects.19 Since D has twice the mass of H, IR and Raman spectroscopy can easily distinguish between stretching and bending modes of B–H and B–D bonds, as they are well separated in energy spectrum.Fig. 5a and b present IR data at RT for S1 to S4 and for S1-D to S4-D, respectively. In Fig. 5a S1 to S4 show the asymmetric stretching and bending modes ν3 and ν4. In good agreement with SR-PXD, our normalized IR data of the ball milled S2–S4 show a slight broadening of ν3 and ν4 modes compared to S1, which can be attributed to the amorphization observed in S2–S4. The integrated peak areas of the B–H stretching region are summarized in ESI Table A2† and support the visual inspection: S1 having the smallest area, followed by an increase between 26–30% for the ball milled samples, with tendency S2 < S4 < S3.
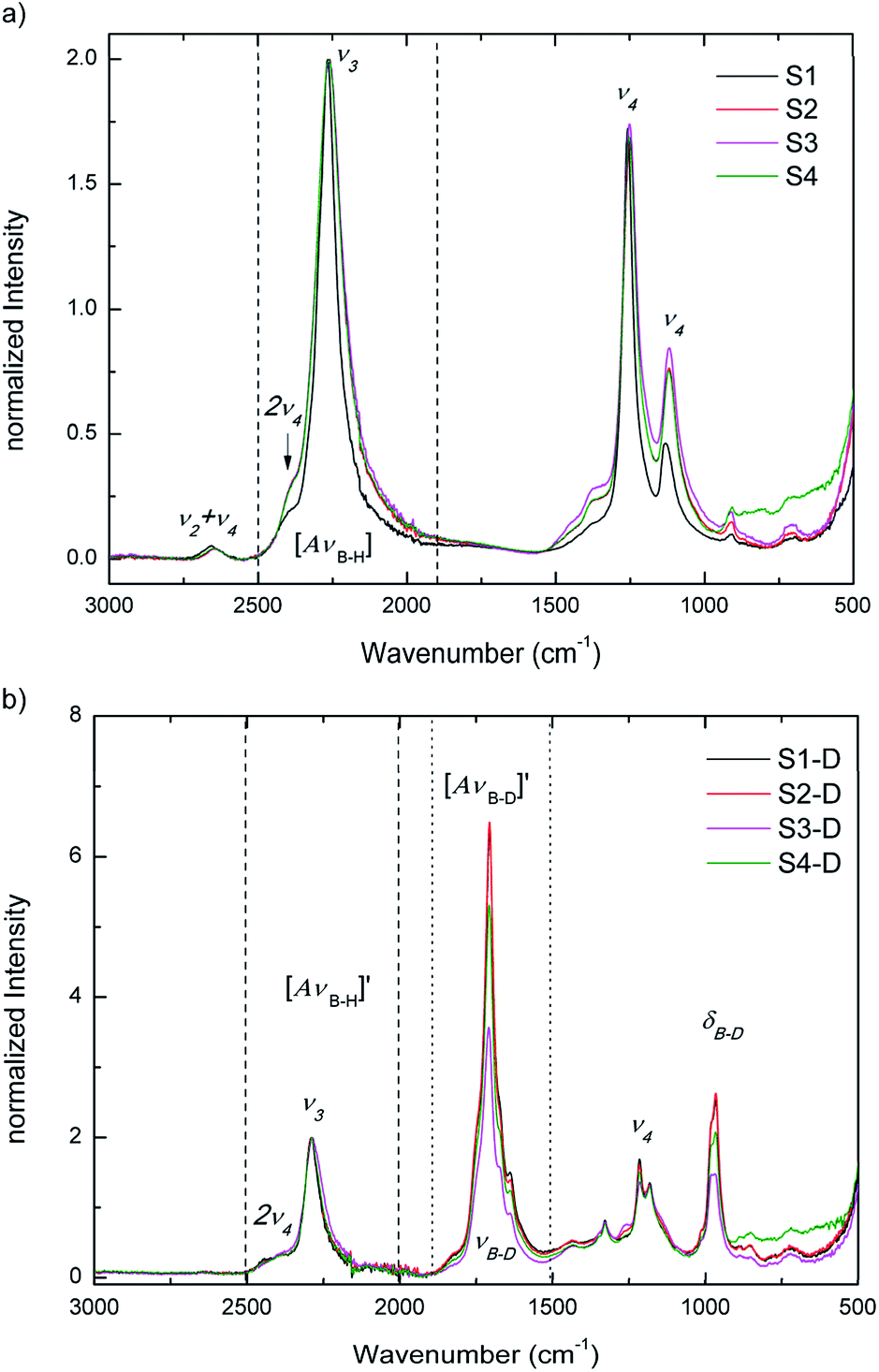 | ||
| Fig. 5 IR data at RT. (a) of S1–S4; and (b) of S1-D to S4-D after 23 h at 120 °C and 3 bar D2. | ||
After the treatment in deuterium atmosphere, the isotopic exchange is evident in S1-D to S4-D from the appearance of the B–D stretching mode νB–D at ∼1707 cm−1 and the B–D bending region δB–D at ∼965 cm−1 (νB–D and δB–D in Fig. 5b). The observed B–D modes are in good agreement with literature.31,32 S1-D and S2-D have gained the most in intensity in B–D stretching followed by S4-D and S3-D, when comparing the B–H stretching mode ν3 and combined B–D stretching modes νB–D after H → D exchange. These observations give a first indication on the extent of exchange after 23 h and imply that the pure materials may have higher exchange rates than those with additives.
It is worth noting, IR data only give a qualitative indication of the isotope exchange, and thus the weight losses measured from TG-DSC measurements (ESI Fig. A3a to A3d†) were used to draw quantitative conclusions. The procedure of data treatment is explained in the ESI,† and the results are presented in Table 2 with the actual weight loss compared to masses of D in Mg(BD4)2. When comparing S1-D, S2-D and S4-D, the deuterium exchanges are similar and within the standard deviations of the measurement, while S3-D has a fairly lower amount of deuterium exchanged. This is in agreement with the IR spectroscopy data after exchange, as the integrated B–D stretching mode νB–D – area is the lowest among all samples. Furthermore, S3–D shows the largest fraction of the dense polymorphs α- and ε-Mg(BH4)2 from SR-PXD (visual inspection) and also an increased crystallization temperature from the TG-DSC data. Thus, these data support the hypothesis of lower hydrogen–deuterium exchange in S3-D.
| # | D content (wt%) in Mg(BD4)2 | ΔTG data (wt%) | x in Mg(BH4−xDx)2 |
|---|---|---|---|
| S1-D | 26.0 | 19.0 ± 0.2 | 1.4 ± 0.1 |
| S2-D | 26.0 | 19.5 ± 0.6 | 1.5 ± 0.2 |
| S3-D | 24.5 | 16.7 ± 0.2 | 1.0 ± 0.1 |
| S4-D | 24.0 | 17.2 ± 0.4 | 1.3 ± 0.1 |
In situ Raman spectroscopy and adsorption studies
In situ Raman measurements were carried out in order to understand the reaction pathway during the isotopic exchange. In contrast to the isotope exchange investigated by IR, the timescale of these measurements is rather short, 75 min, and thus it is expected that the processes on the surface, dominate the exchange process.The first set of the in situ Raman experiments were carried out using samples S1 and S2. Fig. 6 shows the in situ Raman data of S1 heated from RT to 175 °C at 2 °C min−1 under 3 bar D2. Initially, the symmetric stretching (ν1) and bending (ν2) modes of the BH4 anion are observed at 2318 and 1400 cm−1, respectively. Furthermore, the asymmetric BH4 stretching and bending modes ν3 and 2ν4 are observed at 2280 and 2208 cm−1. The isotopic exchange seems to start at 81 °C as a new peak appears at 1718 cm−1 and corresponding to the reported region of symmetric B–D stretching (νB–D).43 This characteristic peak for B–D stretching is observed at a lower temperature than that reported by Zavorotynska et al.31 As previously reported,31 this observation is supported by a decrease in 2v4 and red shift in v1. The latter observation can be explained by the fact that the peak at ∼2318 cm−1 actually consists of several overlapping peaks, and is dominated by ν1 in pure Mg(BH4)2. Upon deuterium substitution new modes corresponding to the B–H stretching of various BH4−xDx ions appear causing the apparent broadening and the red shift.
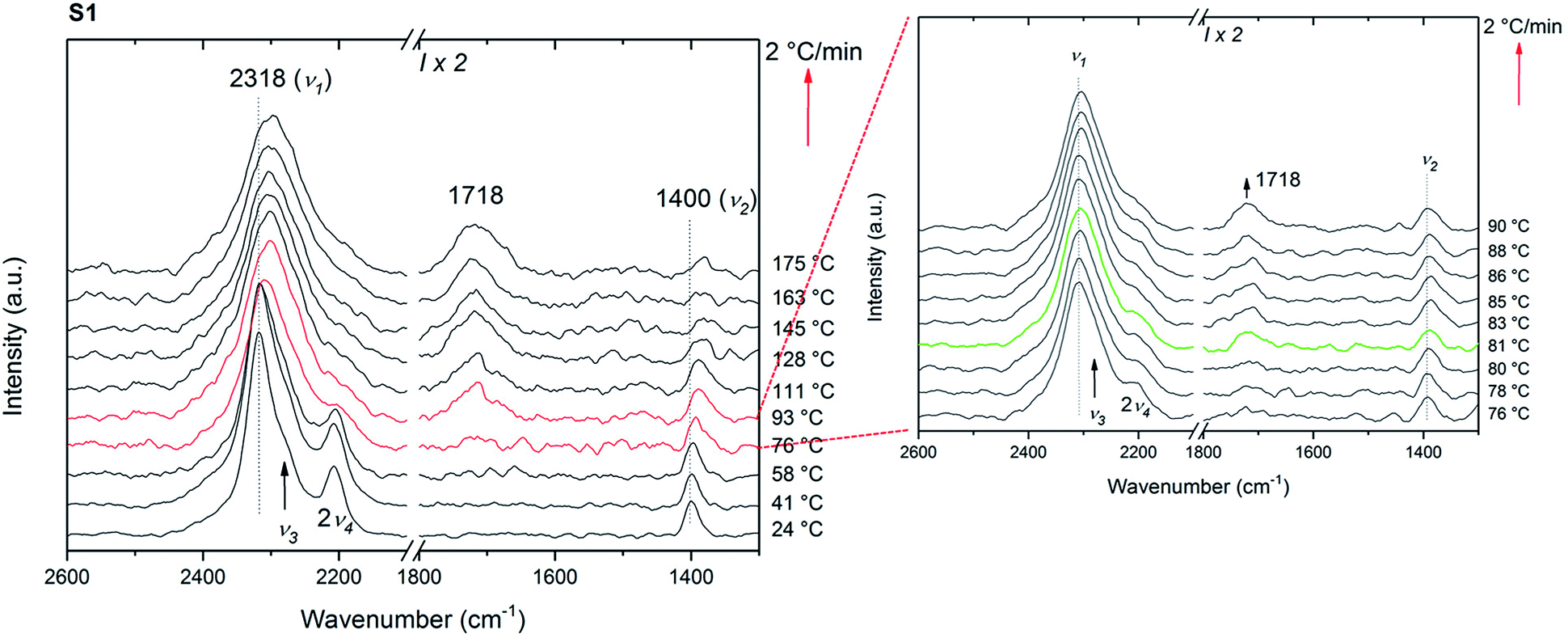 | ||
| Fig. 6 In situ Raman data of S1 including the heating from RT to 175 °C by 2 °C min−1 under 3 bar D2. Every 10th measurement is shown except for the inset, where every measurement is presented from 76 to 90 °C. Inset displays the B–D stretching at 81 °C (green curve). All data is translated in y axis for clarity. | ||
Under the same conditions, no new peak around 1718 cm−1, no decrease in 2v4 nor shift in v1 was observed for sample S2 (ESI Fig. A7a†). This clearly indicates that no H → D exchange takes place in the ball milled sample within the time scale of the measurement. ∼38% of the hydrogen was exchanged with deuterium for the sample S2-D during the 23 h exposure described above. However, during the in situ Raman measurements the time where the isotopic exchange can occur (between ∼80 and ∼150 °C) was only ∼35 min. Amorphous Mg(BH4)2 which is the major component in S2 is not porous as a BET area of only 30 m2 g−1 was measured (compared to 900 m2 g−1 for γ-Mg(BH4)2), see Fig. 7 and ESI† Part B – BET area. Therefore, it is predicted to behave like a dense polymorph, relying on slow, long-range bulk diffusion to achieve appreciable isotopic exchange.32
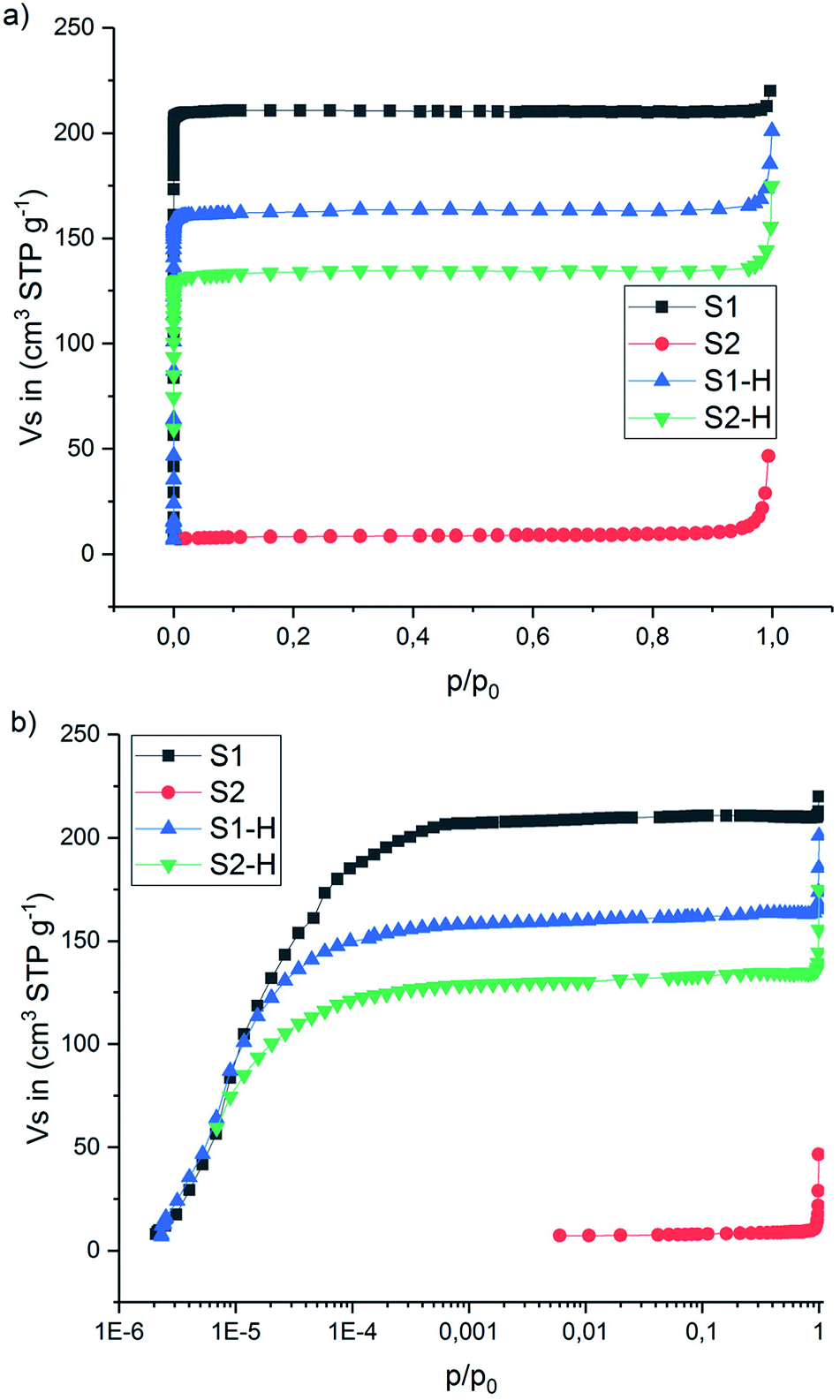 | ||
| Fig. 7 N2 adsorption–desorption isotherms (77 K) of the S1, S2 and thermally treated S1-H, S2-H. (a) x-axis in p/p0 and (b) x-axis in log scale p/p0. | ||
The N2 adsorption–desorption isotherms measured at −196 °C (77 K) are presented in Fig. 7 and Table 3 (more details can be found in the ESI Fig. B1–B4†). The isotherm for S1 is of type I(a) (IUPAC classification38), showing a clear plateau region without any signs of hysteresis and highlighting the ultra-microporous character of this material. The calculated BET area is 900 m2 g−1. Unlike S1, sample S2 (the ball-milled version of S1) revealed very small BET area (30 m2 g−1) and gave a type II isotherm.38 This is typical for adsorption on non-porous solids, and showing clearly that ball-milling leads to amorphization and to collapse of the porous network (compare SR-PXD in Fig. 1, red curve). However, for comparison the samples S1 and S2 have been heat treated under hydrogen (120 °C, 23 h, 3 bar H2, sample named SX-H in analogy to SX-D, X = 1, 2). N2 adsorption measurements gave again an ultra-microporous isotherm shape in S1-H and S2-H and a BET area value of 560 m2 g−1 for the latter which is 62% of the BET area of the as received S1 sample (900 m2 g−1). This suggests that at least part of the porous structure has been regenerated in S2-H. The same is proposed for S2-D and supported by the SR-PXD results shown in Fig. 4 (red curve).
| S1 | S1-H | S2 | S2-H | |
|---|---|---|---|---|
| BET (m2 g−1) | 900 | 690 | 30 | 560 |
| Langmuir (m2 g−1) | 915 | 700 | 36 | 570 |
| DR (m2 g−1) | 915 | 700 | 38 | 570 |
| MP (m2 g−1) | 880 | 670 | 29 | 560 |
| Total pore volume (cm3 g−1) | 0.33 | 0.25 | — | 0.20 |
| % Surface area (N2) | 100 | 76 | 4 | 62 |
| % Pore volume (N2) | 100 | 76 | — | 61 |
It is shown that the adsorption studies reveal that it is possible to partially regain the porous structure from amorphous Mg(BH4)2 by annealing under hydrogen or deuterium. In combination with the long reaction times allowing for the reaction to approach equilibrium, this explains why the isotope exchange during long term D2 exposure was possible in initially partially amorphous ball-milled samples. Furthermore, part of the porous structure in S1-D collapses under heat treatment (BET area = 690 m2 g−1), resulting in a BET similar to the other samples and thus to a comparable extent of isotopic exchange in S1-D and S2-D.
In situ Raman data of S3 and S4 are not presented, as the signal-to-noise ratio over the whole spectrum was too low.
Room temperature isotopic exchange
Fig. 8 presents the IR data of S1–S4 after 6 days under 3 bar D2 at RT. Even though with low intensities, B–D stretching is observed for all samples (S1–S4). The normalized IR data of the ball milled S2–S4 show broadening of the stretching and bending modes compared to S1 due to the increased disorder in the local structure of BH4− in these largely amorphous samples. To the best of our knowledge, this is the first time that the isotopic exchange reaction was observed at RT under this low pressure of D2 (3 bar) in stable (alkali- and alkaline-earth) borohydrides.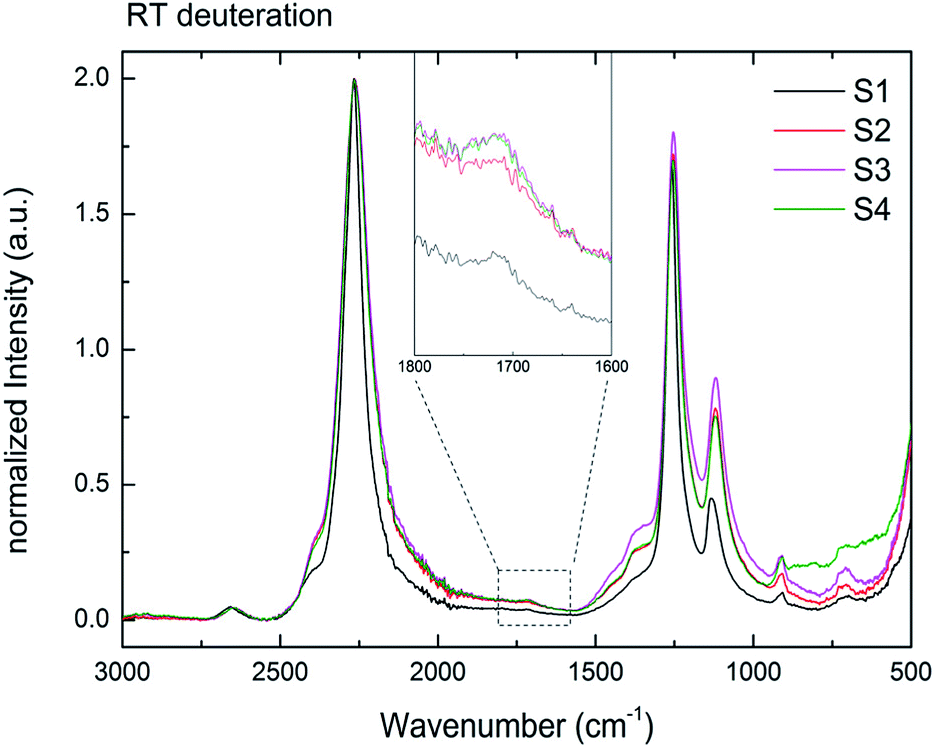 | ||
| Fig. 8 RT H → D exchange of samples S1–S4 for six days under 3 bar D2. Inset showing a magnification of 1800–1600 cm−1. The B–D stretching mode formed in all samples at RT. | ||
The isotopic exchange at RT was observed for both amorphous (S2–S4) and crystalline (S1) samples. Furthermore, the similar intensities of the B–D stretching in all samples suggest also the similar amounts of the exchanged hydrogen. As shown by the BET measurements for S2 (Table 3), the amorphous Mg(BH4)2 has a very low surface area and a non-porous structure. Therefore, the H–D exchange observed should be explained mostly by the long-range diffusion of deuterium into the bulk. It can be suggested then that at the long reaction times the equilibrium (at a given temperature) H/D ratio was achieved in both high and low surface Mg(BH4)2 samples. Indeed, it was shown before for LiBH4 that the amount of the substituted D and the D distribution between the five possible BH4−nDn units is comparable with the Boltzmann statistical distribution at a given temperature calculated from the zero-point vibrational energy difference between the BH4−nDn units.44
Conclusions
The effect of additives, such as Ni3B and Nb2O5, and the effect of ball milling porous γ-Mg(BH4)2 have been investigated with respect to both, dehydrogenation and H → D exchange. The additives and the milling process seem to influence the porous γ-phase and slightly lower the phase transition temperature. From TPD-MS it was observed that the hydrogen release temperature decreased most for the Nb2O5 containing sample and the decomposition pathway is altered, yet not completely understood.The isotopic exchange reactions have shown that neither the milling process nor the additives enhance the solid-state deuterium diffusion reaction in 23 h treated samples. The TG data show that as-received, ball milled and Mg(BH4)2 with Nb2O5 have an H → D exchange between 32 and 37% during the extended treatment in 3 bar deuterium atmosphere, while ball milled Mg(BH4)2 with Ni3B only shows 25% isotope exchange.
The in situ Raman measurements have shown that the isotopic exchange reaction in as-received γ-Mg(BH4)2 started at temperatures of ∼80 °C under 3 bar of D2, and thus almost 20 °C lower than reported before. Furthermore, the comparison of the results of in situ deuteration observed by the Raman measurements and the ex situ deuteration analyzed by the FTIR spectroscopy indicated that the gas–solid H(D) exchange is not the rate-limiting step in the H–D exchange reactions. The concentration of the BD species on the surface of the dense phases was simply too small to be detected by the short Raman measurements. On the other hand, an appreciable exchange in as-received Mg(BH4)2 with a high BET area (900 m2 g−1) was observable. Similar amount of exchanged D was observed after the long exchange reactions (23 h & 6 days) due to the fact that the long reaction times allowed for the solid-state D diffusion in the bulk of the (partially) amorphous phases.
Finally, IR results show that if all samples are kept for 6 days at 3 bar D2 the H → D exchange reaction takes place even at RT. To the best of our knowledge, this is the first time that an isotopic exchange reaction has been observed in stable (alkali- and alkaline-earth) boron-based complex metal hydrides at room temperature, indicating that the B–H bonds in Mg(BH4)2 can be challenged at mild conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results has received funding from the People Program (Marie Curie Actions) of the European Union's Seventh Framework Program FP7/2007–2013/under REA grant agreement no. 607040 (Marie Curie ITN ECOSTORE). The authors acknowledge the skilful assistance from the staff of the Swiss–Norwegian Beamline (SNBL), at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. The help and expertise of Dr Georgios N. Kalantzopoulos is acknowledged by the authors as well Prof. Stian Svelle from University of Oslo for providing beam-time and performing the in situ SR-PXD experiments resulted in the findings presented in ESI Fig. A6.† The contributions by Dr Daniel Reed in providing help for the Raman data acquisition and analysis are gratefully acknowledged. M. H. receives funding from the project 05K16VK2 “Energy research with Neutrons (ErwiN)” by the German Federal Ministry of Education and Research (BMBF).References
- B. Bogdanović and M. Schwickardi, J. Alloys Compd., 1997, 253, 1 CrossRef.
- M. J. v. Setten, G. A. d. Wijs, M. Fichtner and G. Brocks, Chem. Mater., 2008, 20, 4952 CrossRef.
- V. Ozolins, E. Majzoub and C. Wolverton, Phys. Rev. Lett., 2008, 100, 135501 CrossRef PubMed.
- G. Severa, E. Rönnebro and C. M. Jensen, Chem. Commun., 2010, 46, 421 RSC.
- M. Chong, A. Karkamkar, T. Autrey, S.-i. Orimo, S. Jalisatgi and C. M. Jensen, Chem. Commun., 2011, 47, 1330 RSC.
- O. Zavorotynska, S. Deledda and B. C. Hauback, Int. J. Hydrogen Energy, 2016, 41, 9885 CrossRef.
- E. G. Bardají, N. Hanada, O. Zabara and M. Fichtner, Int. J. Hydrogen Energy, 2011, 36, 12313 CrossRef.
- A. Al–Kukhun, H. T. Hwang and A. Varma, Int. J. Hydrogen Energy, 2012, 37, 17671 CrossRef.
- H.-W. Li, K. Kikuchi, Y. Nakamori, K. Miwa, S. Towata and S. Orimo, Scr. Mater., 2007, 57, 679 CrossRef.
- I. Saldan, C. Frommen, I. Llamas-Jansa, G. Kalantzopoulos, S. Hino, B. Arstad, R. Heyn, O. Zavorotynska, S. Deledda, M. Sørby, H. Fjellvåg and B. C. Hauback, Int. J. Hydrogen Energy, 2015, 40, 12286 CrossRef.
- D. Matsumura, T. Ohyama, Y. Okajima, Y. Nishihata, H.-W. Li and S.-i. Orimo, Mater. Trans., 2011, 52, 635 CrossRef.
- Z. Zhang, H. Wang, J.-W. Liu and M. Zhu, Thermochim. Acta, 2013, 560, 82 CrossRef.
- O. Zavorotynska, I. Saldan, S. Hino, T. Humphries, S. Deledda and B. Hauback, J. Mater. Chem. A, 2015, 3, 6592 RSC.
- O. Zavorotynska, S. Deledda, J. G. Vitillo, I. Saldan, M. N. Guzik, M. Baricco, J. C. Walmsley, J. Muller and B. C. Hauback, Energies, 2015, 8, 9173 CrossRef.
- J. Chen, Y. Zhang, Z. Xiong, G. Wu, H. Chu, T. He and P. Chen, Int. J. Hydrogen Energy, 2012, 37, 12425 CrossRef.
- E. Albanese, G. N. Kalantzopoulos, J. Vitillo, E. Pinatel, B. Civalleri, S. Deledda, S. Bordiga, B. Hauback and M. Baricco, J. Alloys Compd., 2013, 580, S282 CrossRef.
- G. Kalantzopoulos, J. Vitillo, E. Albanese, E. Pinatel, B. Civalleri, S. Deledda, S. Bordiga, M. Baricco and B. Hauback, J. Alloys Compd., 2014, 615, S702 CrossRef.
- I. Saldan, S. Hino, T. D. Humphries, O. Zavorotynska, M. Chong, C. M. Jensen, S. Deledda and B. C. Hauback, J. Phys. Chem. C, 2014, 118, 23376 CrossRef.
- O. Zavorotynska, A. El-Kharbachi, S. Deledda and B. C. Hauback, Int. J. Hydrogen Energy, 2016, 41, 14387 CrossRef.
- Y. Filinchuk, R. Černý and H. Hagemann, Chem. Mat., 2009, 21, 925 CrossRef.
- J.-H. Her, P. W. Stephens, Y. Gao, G. L. Soloveichik, J. Rijssenbeek, M. Andrus and J.-C. Zhao, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 561 CrossRef PubMed.
- Y. Filinchuk, B. Richter, T. R. Jensen, V. Dmitriev, D. Chernyshov and H. Hagemann, Angew. Chem., Int. Ed., 2011, 50, 11162 CrossRef PubMed.
- B. Richter, D. B. Ravnsbæk, N. Tumanov, Y. Filinchuk and T. R. Jensen, Dalton Trans., 2015, 44, 3988 RSC.
- W. David, S. Callear, M. Jones, P. Aeberhard, S. Culligan, A. Pohl, S. Johnson, K. Ryan, J. Parker and P. Edwards, Phys. Chem. Chem. Phys., 2012, 14, 11800 RSC.
- M. Paskevicius, M. P. Pitt, C. J. Webb, D. A. Sheppard, U. Filsø, E. M. Gray and C. E. Buckley, J. Phys. Chem. C, 2012, 116, 15231 CrossRef.
- C. Pistidda, S. Garroni, F. Dolci, E. G. Bardaji, A. Khandelwal, P. Nolis, M. Dornheim, R. Gosalawit, T. Jensen, Y. Cerenius, S. Surinach, M. D. Baro, W. Lohstroh and M. Fichtner, J. Alloys Compd., 2010, 508, 212 CrossRef.
- V. Ban, A. V. Soloninin, A. V. Skripov, J. Hadermann, A. Abakumov and Y. Filinchuk, J. Phys. Chem. C, 2014, 118, 23402 CrossRef.
- G. N. Kalantzopoulos, M. K. Antoniou, A. Enotiadis, K. Dimos, E. Maccallini, A. Policicchio, E. Colavita and R. G. Agostino, J. Mater. Chem. A, 2016, 4, 9275 RSC.
- G. N. Kalantzopoulos, A. Enotiadis, E. Maccallini, M. Antoniou, K. Dimos, A. Policicchio, E. Klontzas, E. Tylianakis, V. Binas, P. N. Trikalitis, R. G. Agostino, D. Gournis and G. E. Froudakis, Int. J. Hydrogen Energy, 2014, 39, 2104 CrossRef.
- M. Sharma, D. Sethio, V. D'Anna, J. C. Fallas, P. Schouwink, R. Černý and H. Hagemann, J. Phys. Chem. C, 2015, 119, 29 CrossRef.
- O. Zavorotynska, S. Deledda, G. Li, M. Matsuo, S. i. Orimo and B. C. Hauback, Angew. Chem., 2015, 127, 10738 CrossRef.
- H. Hagemann, V. D'Anna, J.-P. Rapin and K. Yvon, J. Phys. Chem. C, 2010, 114, 10045 CrossRef.
- G. Barkhordarian, T. Klassen and R. Bormann, Scr. Mater., 2003, 49, 213 CrossRef.
- M. Wojdyr, J. Appl. Crystallogr., 2010, 43, 1126 CrossRef.
- H. Brinks, A. Fossdal, R. Bowman and B. C. Hauback, J. Alloys Compd., 2006, 417, 92 CrossRef.
- A. Hammersley, FIT2D: An introduction and overview, ESRF Int. Rep., ESRF97HA02T, 1997 Search PubMed.
- V. Dyadkin, P. Pattison, V. Dmitriev and D. Chernyshov, J. Synchrotron Radiat., 2016, 23, 825 CrossRef PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. Sing, Pure Appl. Chem., 2015, 87, 1051 Search PubMed.
- M. M. Dubinin, J. Colloid Interface Sci., 1974, 46, 351 CrossRef.
- M. M. Dubinin and L. Radushkevich, Chem. Zentralbl., 1947, 1, 875 Search PubMed.
- W. I. F. David, S. K. Callear, M. O. Jones, P. C. Aeberhard, S. D. Culligan, A. H. Pohl, S. R. Johnson, K. R. Ryan, J. E. Parker, P. P. Edwards, C. J. Nuttall and A. Amieiro-Fonseca, Phys. Chem. Chem. Phys., 2012, 14, 11800 RSC.
- S. Guo, H. Y. L. Chan, D. Reed and D. Book, J. Alloys Compd., 2013, 580, S296 CrossRef.
- G. Renaudin, S. Gomes, H. Hagemann, L. Keller and K. Yvon, J. Alloys Compd., 2004, 375, 98 CrossRef.
- R. Gremaud, Z. Łodziana, P. Hug, B. Willenberg, A. M. Racu, J. Schoenes, A. J. Ramirez-Cuesta, S. J. Clark, K. Refson, A. Züttel and A. Borgschulte, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 100301 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05146a |
| This journal is © The Royal Society of Chemistry 2018 |