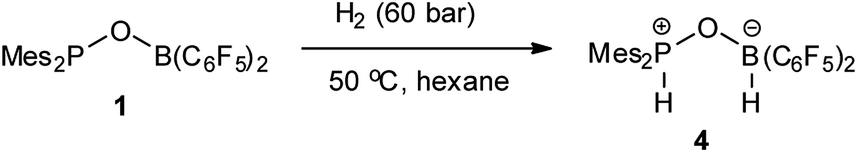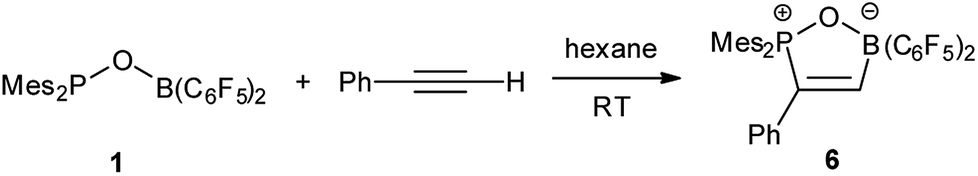DOI:
10.1039/C8RA05108A
(Paper)
RSC Adv., 2018,
8, 26271-26276
Synthesis of an oxygen-linked germinal frustrated Lewis pair and its application in small molecule activation†
Received
14th June 2018
, Accepted 17th July 2018
First published on 23rd July 2018
Abstract
Reaction of Mes2P(O)Li with (C6F5)2BCl gave access to an oxygen-linked germinal intramolecular frustrated Lewis pair Mes2P(O)B(C6F5)2 (1). Compound 1 is stable at room temperature and only decomposes when heated to 90 °C. NMR analysis and theoretical analysis revealed the frustrated nature between the boron and phosphorus centers. Compound 1 shows typical frustrated Lewis pair reactivity when treated with dihydrogen, carbon dioxide, alkyne and alkene. The reaction of 1 with isoprene resulted in selective formation of 3,4-phosphoryl/boryl addition product 8.
Introduction
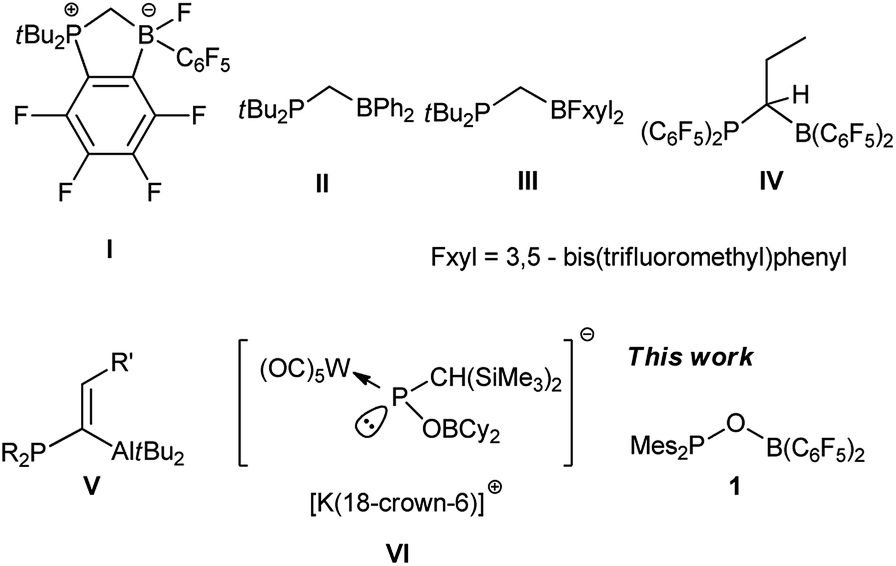
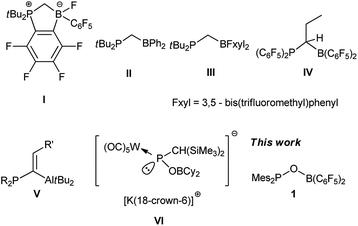
Ever since the groundbreaking discovery by Stephan et al. that an unquenched phosphine/borane pair can activate dihydrogen under ambient conditions, the concept of a “frustrated Lewis pair” (FLP) has been successfully applied in small-molecule activation, organic synthesis and catalysis.1 Mechanism investigations revealed that one of the most prominent features of FLP mediated reactions is that substrates are usually synergistically activated by both the Lewis acid and base centers.2 To enhance such a synergistic effect, the most often employed strategy is to pre-organize Lewis acid and base functionalities through a linker.3 Among a variety of intramolecular FLPs, germinal FLPs in which donor and acceptor sites are separated by one atom have attracted substantial attention because the proximity of their Lewis acid and base centers can facilitate their synergistic reactivities against small molecules. Furthermore, despite such proximity, intramolecular interaction between Lewis acid and base centers in germinal FLPs is largely excluded because of the high constrain of formation of a three-membered ring. Attempts to introduce frequently applied –B(C6F5)2 and –PtBu2 functionalities to C1-linked FLPs were thwarted by facile intramolecular nucleophilic substitution of fluoride on the C6F5 ring by the phosphine moiety (I).4 To circumvent this problem, Slootweg, Lammertsma and coworkers employed –BPh2 instead of –B(C6F5)2 functionality and synthesized a CH2-bridged tBu2PCH2BPh2 (II).5 It was discovered that this germinal FLP can activate H2, CO2 and other unsaturated substrates despite of the mild Lewis acidity of the boron center. The groups of Wagner6 and Erker7 also reported a number of methylene-bridged germinal FLPs employing –BFxyl2 (Fxyl = 3,5-bis(trifluoromethyl)phenyl) as the Lewis acid center (III) or less nucleophilic –P(C6F5)2 as the Lewis base center (IV). Besides of methylene linkers, alkylidene moiety was also employed as a linker for germinal FLPs. Uhl and coworkers prepared a series of alkylidene-bridged Al/P based FLPs (V) which can not only coordinate to a variety of substrates,8 but also function as phase transfer catalyst9 or B–N dehydrogenation catalyst.10 Additionally, a few alkylidene-bridged FLPs based on Ga/P,11 B/P12 and Zr/P13 functionalities were reported by Uhl and Erker groups. In spite of the progress in the study of germinal FLPs, the linker is almost exclusively based on carbon. Only very recently, Streubel and coworkers synthesized an anionic oxygen-linked germinal FLP (VI) and investigated its reactivity against CO2.14 Since the nucleophilicity of phosphinite is markedly lower than alkyl- or alkenyl-substituted phosphine, we reasoned that an oxygen-linked germinal FLP containing –B(C6F5)2 and –PMes2 functionalities (1) would be less susceptible to intramolecular nucleophilic substitution, which could render this FLP stable enough for isolation. Herein, we report the synthesis of oxygen-linked germinal FLP 1 and its reactivity against small molecules (Fig. 1).
 |
| | Fig. 1 Germinal intramolecular FLPs. | |
Results and discussion
Synthesis of 1 was achieved by treatment of Mes2P(O)H with nBuLi and subsequently (C6F5)2BCl in hexane and it can be isolated as a pale yellow oil with 83% yield after workup (Scheme 1). Compound 1 was characterized by multinuclear NMR analysis. The 31P NMR spectrum of 1 showed a resonance at δ 126.4 ppm, similar to that observed for phosphinite Ph2P(OMe) (117 ppm).15 In the 11B NMR spectrum of 1 a broad singlet was observed at δ 42.4 ppm, indicating the existence of a tri-coordinated boron center. This was corroborated by the appearance of signals at δ −131.4, −148.8, −161.2 ppm in the 19F NMR spectrum of 1. The identity of 1 was further confirmed by derivatization. Addition of equimolar of pyridine to 1 in hexane resulted in quantitative formation of adduct 2, which was characterized by X-ray crystal structure analysis. Compound 1 is stable at room temperature both in solution and neat form. Upon heating to 90 °C in a toluene solution, 1 undergoes intramolecular nucleophilic substitution to afford 3 (Scheme 1), which was isolated in 36% yield and fully characterized by NMR spectroscopy and X-ray analysis.
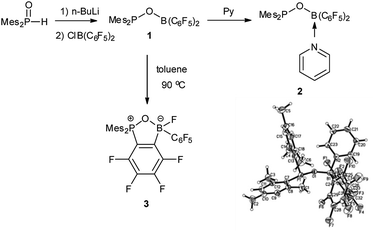 |
| | Scheme 1 Synthesis and thermolysis of 1 and molecular structure of 2 (thermal ellipsoids are shown with 30% probability). | |
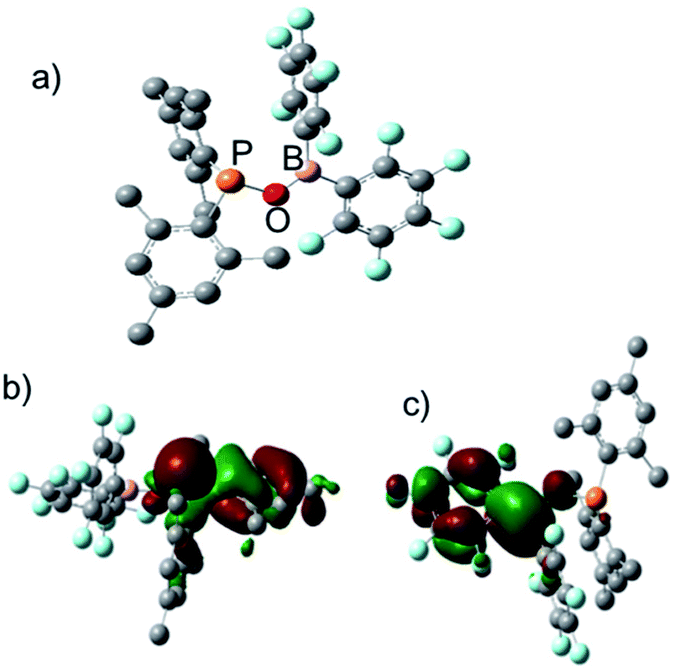
To understand the electronic structure of 1, DFT (M06-2X) calculation16 was carried out. In the calculated structure, the P–O–B bond angle is 114° and the distance between the P and B atoms is 2.57 Å (Fig. 2a), which is shorter than the observed intramolecular P⋯B distance in tBu2PCH2B(Fxyl)2 (2.90 Å).6 Population analysis suggested that the interaction between the P and B atoms is very weak and the covalent P–B Wiberg bond order is only 0.02. The HOMO of 1 mainly corresponds to the lone pair on phosphorus, with some contribution from π orbital of the mesityl substitutent (Fig. 2b). The LUMO is largely distributed over the boron center as well as one C6F5 ring and the oxygen linker, indicative of π electron donation from the oxygen atom to the empty p orbital of the boron center (Fig. 2c).
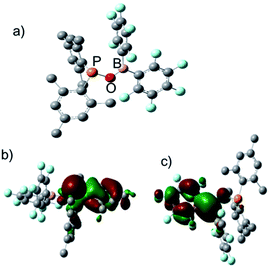 |
| | Fig. 2 (a) calculated structure of 1; (b) HOMO of 1; (c) LUMO of 1. | |
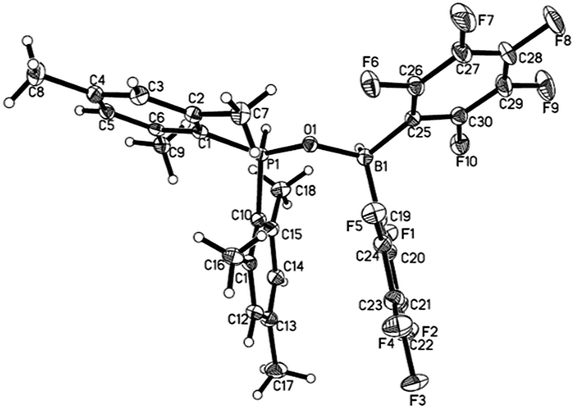
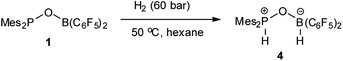
While 1 does not react with H2 under 6 bar at room temperature, it reacts with H2 under harsher conditions (60 bar, 50 °C) to afford compound 4 with 58% yield after workup (Scheme 2). Compound 4 can be also independently synthesized from Mes2P(O)H and HB(C6F5)2. Existence of a P–H moiety was confirmed by the appearance of a doublet at δ 31.5 ppm (1JP–H = 514 Hz) in the 31P NMR spectrum, with a corresponding doublet at δ 8.02 ppm (1JP–H = 514 Hz) observed in the 1H NMR spectrum. However, both the 11B and 11B{1H} NMR spectra displayed a broad signal at −4.79 ppm, excluding study of the coupling constant with the adjacent H atom. The 1H NMR spectrum displayed a broad singlet at δ 4.59 ppm with integration of 1, which was assigned to the BH moiety. The identity of 4 was further validated by X-ray crystal structure analysis (Fig. 3).
 |
| | Scheme 2 Reaction of 1 with H2. | |
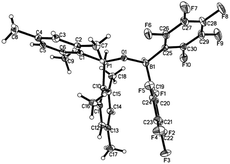 |
| | Fig. 3 Molecular structure of 4 (thermal ellipsoids are shown with 30% probability). | |
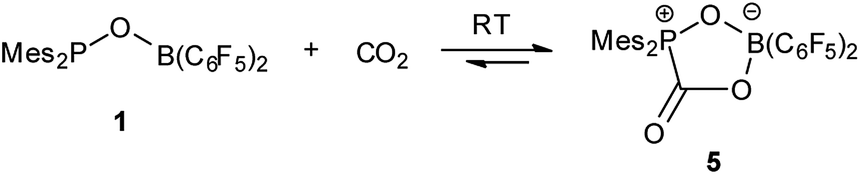
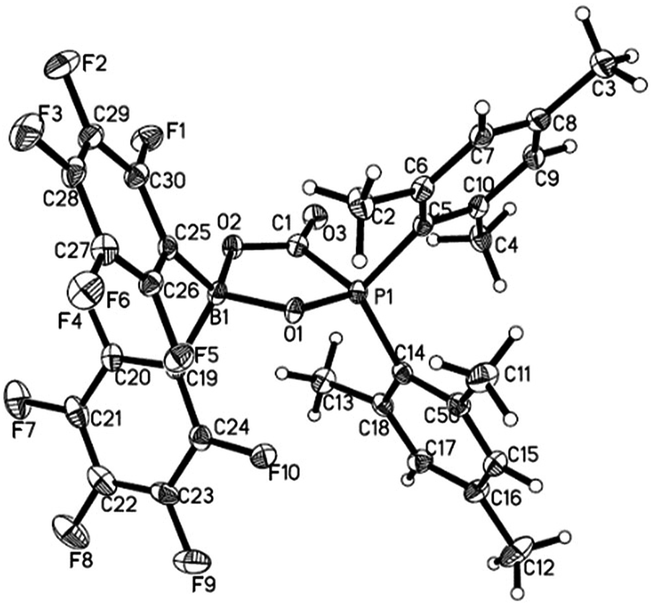
Treatment of 1 with CO2 (1.8 bar) at room temperature in hexane resulted in formation of a CO2 adduct 5, in an analogous way with other germinal FLPs (Scheme 3).5a,6,8a,11,13b,14 5 was characterized by both NMR spectroscopy and X-ray analysis (Fig. 4). 5 is stable infinitely as solid. But unlike germinal B/P FLPs II5a and III,6 the solution of 5 in C6D6 slowly released CO2 under N2 atmosphere at room temperature. In a sealed NMR tube at room temperature, equilibrium was reached after 24 hours and about 25% of 5 was converted to 1.
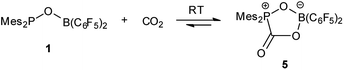 |
| | Scheme 3 Reaction of 1 with CO2. | |
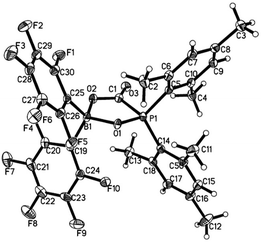 |
| | Fig. 4 Molecular structure of 5 (thermal ellipsoids are shown with 30% probability). | |
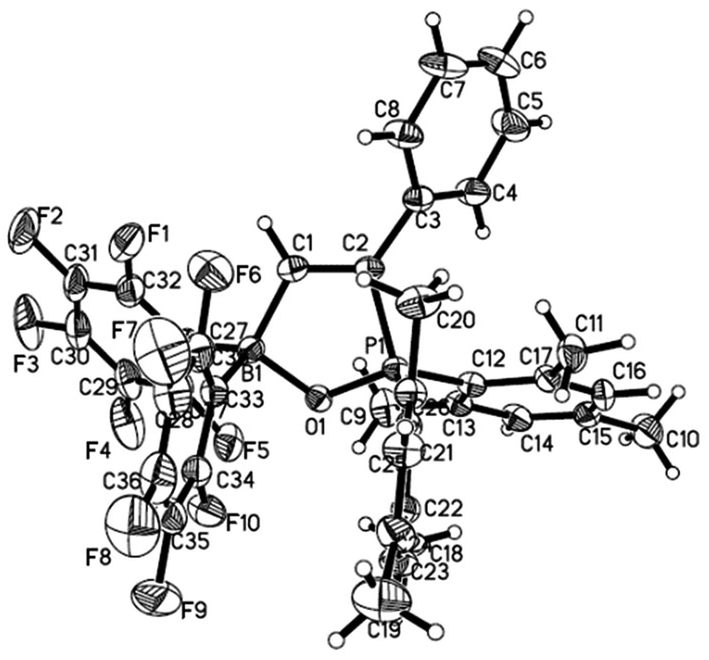
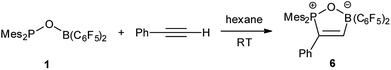
Reaction of 1 with phenylacetylene in hexane led to addition of both phosphoryl and boryl moieties to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond to give 6 in 73% yield (Scheme 4). This is in line with the discovery by Stephan17 and Erker7 that FLPs with phosphine moiety of low basicity tend to afford addition products. In the solid structure of 6 (Fig. 5), the distances of B–O and P–O are similar to those observed in tBu2P(μ-O)(μ-C6H4)B(C6F5)2 (P–O 1.546, B–O 1.550 Å) reported by Wagner.6
C bond to give 6 in 73% yield (Scheme 4). This is in line with the discovery by Stephan17 and Erker7 that FLPs with phosphine moiety of low basicity tend to afford addition products. In the solid structure of 6 (Fig. 5), the distances of B–O and P–O are similar to those observed in tBu2P(μ-O)(μ-C6H4)B(C6F5)2 (P–O 1.546, B–O 1.550 Å) reported by Wagner.6
 |
| | Scheme 4 Reaction of 1 with phenylacetylene. | |
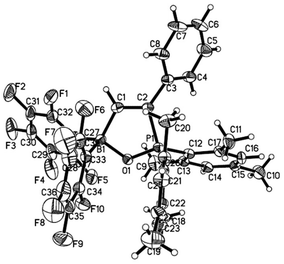 |
| | Fig. 5 Molecular structure of 6 (thermal ellipsoids are shown with 30% probability). | |
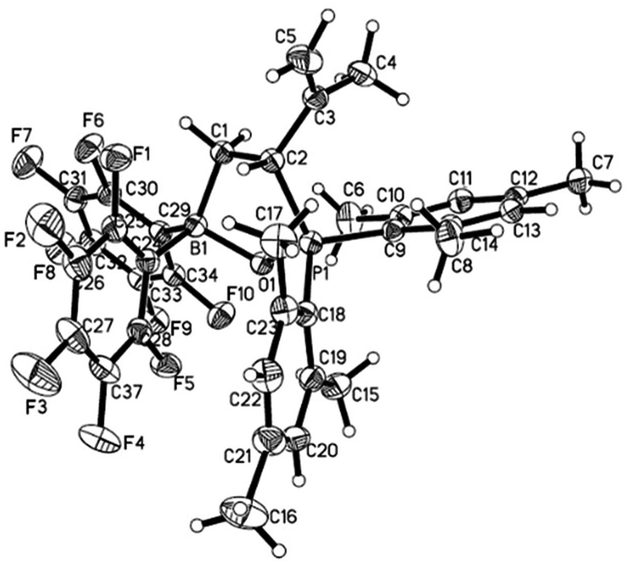
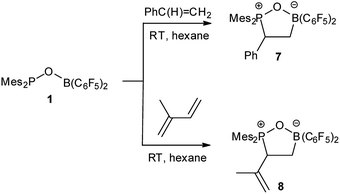
Compound 1 reacted with equimolar of styrene in hexane at room temperature to yield a white precipitate, which was identified as 1,2-boryl/phosphoryl addition product 7 via NMR spectroscopy and X-ray analysis.18 When 1 was treated with isoprene in hexane, 3,4-phosphoryl/boryl addition product 8 was formed as the only regioisomer, which can be isolated as a white solid in 79% yield (Scheme 5). 8 was characterized by both NMR spectroscopy and X-ray analysis (Fig. 6). This observed regioselectivity is in contrast to previous reports by Stephan19 and Lerner20 that activation of dienes with intermolecular FLP tBu3P/B(C6F5)3 or intramolecular FLP di-t-butyl-phosphaboradibenzofulvene affords predominately 1,4-addition products. It is likely that in the case of intramolecular FLP 1, the favored formation of a five-member ring dictates the observed 3,4-selectivity.
 |
| | Scheme 5 Reaction of 1 with styrene and isoprene. | |
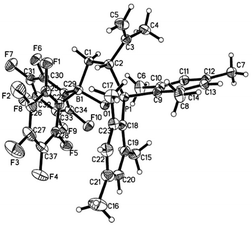 |
| | Fig. 6 Molecular structure of 8 (thermal ellipsoids are shown with 30% probability). | |
Conclusions
By employing an oxygen atom as the linker, a germinal FLP containing both electrophilic B(C6F5)2 and nucleophilic PMes2 moieties was synthesized for the first time. This intramolecular FLP readily reacts with a series of small molecules, such as H2, CO2, phenylacetylene, styrene and isoprene. Among them, regioselective 3,4-addition to a diene is unprecedented. Current research is focusing on deoxygenation of addition adducts, which might provide a novel way for the synthesis of intramolecular FLPs.
Experimental section
General experimental methods
Solvents were dried by reflux under N2 over sodium or CaH2 and freshly distilled prior to use. Air-sensitive compounds were handled under a N2 atmosphere using standard Schlenk and glovebox techniques. NMR spectra were recorded on Bruker SPECT NMR (400 MHz for 1H, 376 MHz for 19F, 100 MHz for 13C) and Bruker DMX500 NMR (500 MHz for 1H, 160 MHz for 11B) spectrometers. Most assignments were based on a series of 2D NMR experiments. HRMS analyses were performed at Bruker micrOTOF II. Crystallographic data for the structure reported in this paper have been deposited with the Cambridge Crystallographic Data Center: CCDC 1843771–1843777 (compound 2–8) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre at www.ccdc.cam.ac.uk/data_request/cif. Mes2P(O)H (Mes = 2,4,6-trimethylphenyl)21 and ClB(C6F5)2 (ref. 22) were prepared as reported.
Synthesis of compound 1
A solution of n-BuLi (2.5 M, 0.80 mL, 2.0 mmol) was added to a suspension of Mes2P(O)H (0.57 g, 2.0 mmol) in hexane (8 mL) at 0 °C, affording a pale yellow solution. After stirring for 4 h at room temperature, a solution of ClB(C6F5)2 (0.76 g, 2.0 mmol) in hexane (6 mL) was added at 0 °C. The reaction mixture was warmed up to room temperature and stirred for 12 h. The resulting white slurry was filtered and the filtrate was dried under vacuum affording 1 as a pale yellow oil (1.05 g, 81%). 1H NMR (400 MHz, C6D6): δ [ppm] = 6.62 (d, 4H, 4JP–H = 2 Hz, meta-Mes), 2.28 (s, 12H, ortho-CH3), 2.02 (s, 6H, para-CH3). 13C NMR (101 MHz, C6D6): δ [ppm] = 147.9 (dm, 1JC–F = 248 Hz, ortho-C6F5), 143.2 (dm, 1JC–F = 257 Hz, meta-C6F5), 137.6 (dm, 1JC–F = 253 Hz, para-C6F5), 141.3 (d, 2JP–C = 17 Hz, ortho-Mes), 140.2 (para-Mes), 133.3 (d, 1JP–C = 28 Hz, ipso-Mes), 130.6 (d, 3JP–C = 3 Hz, meta-Mes), 22.2 (d, 3JP–C = 4 Hz, ortho-CH3), 20.9 (para-CH3). 19F NMR (376 MHz, C6D6): δ [ppm] = −131.4 (m, ortho-C6F5), −148.8 (t, para-C6F5), −161.2 (m, meta-C6F5). 31P NMR (162 MHz, C6D6): δ [ppm] = 126.4. 11B NMR (160 MHz, C6D6): δ [ppm] = 42.4. HRMS (m/z): calcd for C30H23OPBF10 [M + H]+: 631.1420, found: 631.1434.
Synthesis of compound 2
A solution of pyridine (25 mg, 0.32 mmol) in hexane (1 mL) was added to a solution of 1 (130 mg, 0.206 mmol) in hexane (2 mL), affording a white precipitate. The reaction mixture was stirred for 30 min and stored at −30 °C for 10 h. Then the supernatant was removed by filtration and the residue was washed with hexane (1 mL) and dried under vacuum, affording 2 as a white powder (118 mg, 81%). Crystals suitable for X-ray diffraction were grown by slow diffusion of hexane into a solution of 2 in CH2Cl2. 1H NMR (400 MHz, C6D6): δ [ppm] = 8.12 (br s, 2H, ortho-Py), 6.66 (s, 4H, meta-Mes), 6.54 (br s, 1H, para-Py), 6.21 (br s, 2H, meta-Py), 2.47 (s, 12H, ortho-CH3), 2.10 (s, 6H, para-CH3). 13C NMR (101 MHz, C6D6): δ [ppm] = 148.6 (dm, 1JC–F = 240 Hz, ortho-C6F5), 140.8 (dm, 1JC–F = 251 Hz, meta-C6F5), 137.8 (dm, 1JC–F = 251 Hz, para-C6F5), 144.3 (ortho-Py), 141.0 (para-Py), 125.0 (meta-Py), 141.5 (d, 2JP–C = 17 Hz, ortho-Mes), 138.3 (para-Mes), 136.9 (d, 1JP–C = 32 Hz, ipso-Mes), 130.5 (d, 3JP–C = 2 Hz, meta-Mes), 21.5 (d, 3JP–C = 16 Hz, ortho-CH3), 20.9 (para-CH3). 19F NMR (376 MHz, C6D6): δ [ppm] = −131.6 (m, ortho-C6F5), −155.8 (t, para-C6F5), −163.3 (m, meta-C6F5). 31P NMR (162 MHz, C6D6): δ [ppm] = 102.9. 11B NMR (160 MHz, C6D6): δ [ppm] = 4.37.
Synthesis of compound 3
A solution of 1 (132 mg, 0.206 mmol) in toluene (1 mL) was heated to 90 °C for 15 h. Then, all volatiles were removed under vacuum. The resulting oily residue was extracted with hexane (2 mL). Afterwards hexane was removed under vacuum affording an oily solid. Another portion of hexane (1 mL) was introduced and an insoluble oil was formed which became crystals after standing at room temperature for 30 min. The supernatant was removed and the crystals were dried under vacuum affording 3 as a white crystalline solid (47 mg, 36%). 1H NMR (400 MHz, C6D6): δ [ppm] = 6.43, 6.27 (each d, each 2H, 4JP–H = 5 Hz, meta-Mes), 2.19, 2.05 (each s, each 6H, ortho-CH3), 1.84, 1.71 (each s, each 3H, para-CH3). 13C NMR (101 MHz, C6D6): δ [ppm] = 145.2, 144.6 (d, 4JP–C = 3 Hz, para-Mes), 143.5, 141.8 (d, 2JP–C = 12 Hz, ortho-Mes), 132.4, 131.5 (d, 3JP–C = 13 Hz, meta-Mes), 23.1, 21.8 (d, 3JP–C = 5 Hz, ortho-CH3), 20.9, 20.7 (d, 5JP–C = 1 Hz, p-CH3). 19F NMR (376 MHz, C6D6): δ [ppm] = −129.5 (m, 2F, C6F4), −134.5 (m, 2F, ortho-C6F5), −144.1 (m, 1F, C6F4), −149.5 (br s, 1F, BF), −153.6 (m, 1F, C6F4), −158.0 (t, 1F, para-C6F5), −164.6 (m, 2F, meta-C6F5). 31P NMR (162 MHz, C6D6): δ [ppm] = 58.3. 11B NMR (160 MHz, C6D6): δ [ppm] = 8.72. HRMS (m/z): calcd for C30H22OPBF10Na [M + Na]+: 653.1239, found: 653.1258.
Synthesis of compound 4
H2 (60 bar) was introduced to a solution of 1 (324 mg, 0.515 mmol) in hexane (2.5 mL) in an autoclave. The reaction mixture was stirred for 40 h at 50 °C, affording a white precipitate. The supernatant was removed by filtration and the solid was dried under vacuum affording 4 as a white powder (188 mg, 58%). Crystals suitable for X-ray diffraction were grown by slow diffusion of hexane into a solution of 4 in toluene. 1H NMR (400 MHz, C6D6): δ [ppm] = 8.02 (d, 1H, 1JP–H = 514 Hz, PH), 6.34 (d, 4H, 4JP–H = 5 Hz, meta-Mes), 4.59 (br s, 1H, BH), 2.07 (s, 12H, ortho-CH3), 1.86 (s, 6H, para-CH3). 13C NMR (101 MHz, C6D6): δ [ppm] = 148.4 (dm, 1JC–F = 240 Hz, ortho-C6F5), 139.9 (dm, 1JC–F = 247 Hz, meta-C6F5), 137.4 (dm, 1JC–F = 256 Hz, para-C6F5), 145.0 (d, 4JP–C = 3 Hz, para-Mes), 143.1 (d, 2JP–C = 11 Hz, ortho-Mes), 131.2 (d, 3JP–C = 12 Hz, meta-Mes), 117.8 (d, 1JP–C = 107 Hz, ipso-Mes), 21.0 (d, 3JP–C = 6 Hz, ortho-CH3), 20.9 (d, 5JP–C = 2 Hz, para-CH3). 19F NMR (376 MHz, C6D6): δ [ppm] = −133.9 (m, ortho-C6F5), −159.8 (t, para-C6F5), −165.1 (m, meta-C6F5). 31P NMR (162 MHz, C6D6): δ [ppm] = 31.5 (d, 1JP–H = 514 Hz). 11B NMR (160 MHz, C6D6): δ [ppm] = −4.79.
Synthesis of compound 5
CO2 (1.8 bar) was introduced to a degassed solution of 1 (551 mg, 0.875 mmol) in hexane (2.5 mL) at room temperature and the reaction mixture was stirred for 24 h, affording a white precipitate. The supernatant was removed by filtration and the residue was dried under vacuum affording 5 as a white powder (430 mg, 73%). Crystals suitable for X-ray diffraction were grown from toluene at −30 °C. 1H NMR (400 MHz, C6D6): δ [ppm] = 6.30 (s, 4H, meta-Mes), 2.19 (s, 12H, ortho-CH3), 1.79 (s, 6H, para-CH3). 13C NMR (101 MHz, C6D6): δ [ppm] = 165.9 (d, 1JP–C = 108 Hz, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 148.4 (dm, 1JC–F = 243 Hz, ortho-C6F5), 141.0 (dm, 1JC–F = 253 Hz, meta-C6F5), 137.6 (dm, 1JC–F = 251 Hz, para-C6F5), 145.7 (para-Mes), 143.3 (d, 2JP–C = 12 Hz, ortho-Mes), 131.7 (d, 3JP–C = 13 Hz, meta-Mes), 118.0 (d, 1JP–C = 92 Hz, ipso-Mes), 21.8 (d, 3JP–C = 6 Hz, ortho-CH3), 20.9 (para-CH3). 19F NMR (376 MHz, C6D6): δ [ppm] = −133.9 (ortho-C6F5), −155.9 (para-C6F5), −163.5 (meta-C6F5). 31P NMR (162 MHz, C6D6): δ [ppm] = 35.0. 11B NMR (160 MHz, C6D6): δ [ppm] = 7.94. HRMS (m/z): calcd for C31H22O3PBF10Na [M + Na]+: 697.1138, found: 697.1123.
O), 148.4 (dm, 1JC–F = 243 Hz, ortho-C6F5), 141.0 (dm, 1JC–F = 253 Hz, meta-C6F5), 137.6 (dm, 1JC–F = 251 Hz, para-C6F5), 145.7 (para-Mes), 143.3 (d, 2JP–C = 12 Hz, ortho-Mes), 131.7 (d, 3JP–C = 13 Hz, meta-Mes), 118.0 (d, 1JP–C = 92 Hz, ipso-Mes), 21.8 (d, 3JP–C = 6 Hz, ortho-CH3), 20.9 (para-CH3). 19F NMR (376 MHz, C6D6): δ [ppm] = −133.9 (ortho-C6F5), −155.9 (para-C6F5), −163.5 (meta-C6F5). 31P NMR (162 MHz, C6D6): δ [ppm] = 35.0. 11B NMR (160 MHz, C6D6): δ [ppm] = 7.94. HRMS (m/z): calcd for C31H22O3PBF10Na [M + Na]+: 697.1138, found: 697.1123.
Synthesis of compound 6
PhCCH (120 mg, 1.18 mmol) was added to a solution of 1 (567 mg, 0.900 mmol) in hexane (3 mL) at room temperature, affording a white precipitate. After the reaction mixture was stirred for 17 h, the supernatant was removed by filtration and the solid was dried under vacuum affording 6 as a white powder (480 mg, 73%). Crystals suitable for X-ray diffraction were grown by slow diffusion of hexane into a solution of 6 in toluene. 1H NMR (400 MHz, C6D6): δ [ppm] = 9.01 (d, 1H, 3JP–H = 46 Hz, CCH), 7.17 (m, 2H, PhH), 6.94 (m, 3H, PhH), 6.40 (d, 4H, 4JP–H = 4 Hz, meta-Mes), 2.10 (s, 12H, ortho-CH3), 1.85 (s, 6H, para-CH3). 13C NMR (101 MHz, C6D6): δ [ppm] = 176.7 (br s, BCH), 147.8 (dm, 1JC–F = 241 Hz, ortho-C6F5), 139.9 (dm, 1JC–F = 250 Hz, meta-C6F5), 137.1 (dm, 1JC–F = 254 Hz, para-C6F5), 144.0 (d, 4JP–C = 3 Hz, para-Mes), 142.7 (d, 2JP–C = 11 Hz, ortho-Mes), 136.7 (d, 2JP–C = 21 Hz, ipso-Ph), 135.2 (d, 1JP–C = 81 Hz, PCC), 131.5 (d, 3JP–C = 12 Hz, meta-Mes), 129.2 (Ph), 127.5 (d, 3JP–C = 5 Hz, ortho-Ph), 124.9 (d, 1JP–C = 97 Hz, ipso-Mes), 23.0 (d, 3JP–C = 5 Hz, ortho-CH3), 20.8 (d, 5JP–C = 1 Hz, para-CH3). 19F NMR (376 MHz, C6D6): δ [ppm] = −132.0 (m, ortho-C6F5), −159.2 (t, para-C6F5), −164.5 (m, meta-C6F5). 31P NMR (162 MHz, C6D6): δ [ppm] = 68.4 (d, 3JP–H = 46 Hz). 11B NMR (160 MHz, C6D6): δ [ppm] = 2.65. HRMS (m/z): calcd for C38H28OPBF10Na [M + Na]+: 755.1709, found: 755.1706.
Synthesis of compound 7
PhCHCH2 (220 mg, 2.12 mmol) was added to a solution of 1 (1.00 g, 1.59 mmol) in hexane (6 mL) at room temperature, affording a white precipitate. After the reaction mixture was stirred for 15 h, the supernatant was removed by filtration and the solid was dried under vacuum affording 7 as a white powder (890 mg, 76%). Crystals suitable for X-ray diffraction were grown by slow diffusion of hexane into a solution of 7 in CH2Cl2. 1H NMR (400 MHz, C6D6): δ [ppm] = 6.92–6.74 (5H, Ph), 6.42 (d, 2H, 4JP–H = 3 Hz, meta-Mes), 6.32 (br s, 2H, meta-Mes), 4.76 (dt, 1H, 3JH–H = 13 Hz, 2JP–H = 6 Hz, CH), 2.96, 1.95 (each m, each 1H, BCH2), 2.38–1.84 (18H, Mes-CH3). 13C NMR (101 MHz, C6D6): δ [ppm] = 148.1 (dm, 1JC–F = 241 Hz, ortho-C6F5), 139.7 (dm, 1JC–F = 248 Hz, meta-C6F5), 137.5 (dm, 1JC–F = 249 Hz, para-C6F5), 143.8, 143.4, 138.2 (ortho-Mes and para-Mes), 131.7, 131.4 (meta-Mes), 129.1, 128.5, 127.6 (Ph), 51.3 (d, 1JP–C = 46 Hz, PCH), 35.6 (BCH2), 23.5, 22.4, 20.7 (Mes-CH3). 19F NMR (376 MHz, C6D6): δ [ppm] = −133.0 (dm, ortho-C6F5), −159.4 (t, para-C6F5), −164.4 (meta, m-C6F5). 31P NMR (162 MHz, C6D6): δ [ppm] = 82.4 (d, 2JP–H = 39 Hz). 11B NMR (160 MHz, C6D6): δ [ppm] = 3.16. HRMS (m/z): calcd for C38H30OPBF10Na [M + Na]+: 757.1865, found: 757.1865.
Synthesis of compound 8
Isoprene (50 mg, 0.74 mmol) was added to a solution of 1 (331 mg, 0.525 mmol) in hexane (3 mL) at room temperature, affording a white precipitate after stirring for about 24 h. The supernatant was removed by filtration and the solid was dried under vacuum affording 8 as a white powder (290 mg, 79%). Crystals suitable for X-ray diffraction were grown by slow diffusion of hexane into a solution of 8 in CH2Cl2. 1H NMR (400 MHz, C6D5Br): δ [ppm] = 6.79 (s, 2H, meta-Mes), 6.76 (d, 2H, 4JP–H = 4 Hz, meta-Mes), 5.16 (d, 1H, 2JH–H = 3 Hz, CCH2), 5.03 (s, 1H, CCH2), 4.58 (dt, 1H, 3JH–H = 12 Hz, 2JP–H = 7 Hz, CH), 2.66, 2.02 (each m, each 1H, BCH2), 2.57–2.01 (18H, Mes-CH3), 1.49 (s, 3H, CH3). 13C NMR (101 MHz, C6D5Br): δ [ppm] = 147.6 (dm, 1JC–F = 241 Hz, ortho-C6F5), 139.3 (dm, 1JC–F = 248 Hz, meta-C6F5), 137.0 (dm, 1JC–F = 249 Hz, para-C6F5), 143.9, 143.1, (ortho-Mes and para-Mes), 142.3 (CCH2), 131.8, 131.3 (meta-Mes), 116.9 (d, 3JP–C = 12 Hz, CCH2), 52.1 (d, 1JP–C = 46 Hz, PCH), 29.9 (BCH2), 23.4, 23.0, 21.1, 20.9 (Mes-CH3), 19.9 (CH3). 19F NMR (376 MHz, C6D5Br): δ −132.3 (dm, ortho-C6F5), −158.9 (dt, para-C6F5), −163.6 (m, meta-C6F5). 31P NMR (162 MHz, C6D5Br): δ [ppm] = 79.8 (d, 2JP–H = 37 Hz). 11B NMR (160 MHz, C6D5Br): δ [ppm] = 2.59. HRMS (m/z): calcd for C35H30OPBF10Na [M + Na]+: 721.1865, found: 721.1857.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the National Natural Science Foundation of China (21372048, 21573044, 21672039) and Shanghai Science and Technology Committee (16DZ2270100) is gratefully acknowledged. We also thank the High Performance Computer Center of Fudan University for the allocation of computing time. We thank Dr Yue-Jian Lin and Ms Haiwen Tian for help on crystallography study.
Notes and references
-
(a) G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef PubMed;
(b) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef PubMed;
(c) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef PubMed;
(d) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef PubMed;
(e) D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
-
(a) T. A. Rokob, A. Hamza, A. Stirling, T. Soós and I. Pápai, Angew. Chem., Int. Ed., 2008, 47, 2435–2438 CrossRef PubMed;
(b) Y. Guo and S. Li, Inorg. Chem., 2008, 47, 6212–6219 CrossRef PubMed;
(c) T. A. Rokob, A. Hamza and I. Pápai, J. Am. Chem. Soc., 2009, 131, 10701–10710 CrossRef PubMed;
(d) S. Grimme, H. Kruse, L. Goerigk and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 1402–1405 CrossRef PubMed;
(e) T. A. Rokob, I. Bakó, A. Stirling, A. Hamza and I. Pápai, J. Am. Chem. Soc., 2013, 135, 4425–4437 CrossRef PubMed.
- G. Kehr, S. Schwendemann and G. Erker, Top. Curr. Chem., 2012, 332, 45–84 CrossRef PubMed.
- A. Schnurr, H. Vitze, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2010, 29, 6012–6019 CrossRef.
-
(a) F. Bertini, V. Lyaskovskyy, B. J. J. Timmer, F. J. J. Kanter, M. Lutz, A. W. Ehlers, J. C. Slootweg and K. Lammertsma, J. Am. Chem. Soc., 2012, 134, 201–204 CrossRef PubMed;
(b) E. R. M. Habraken, L. C. Mens, M. Nieger, M. Lutz, A. W. Ehlers and J. C. Slootweg, Dalton Trans., 2017, 46, 12284–12292 RSC.
- K. Samigullin, I. Georg, M. Bolte, H.-W. Lerner and M. Wagner, Chem.–Eur. J., 2016, 22, 3478–3484 CrossRef PubMed.
- A. Stute, G. Kehr, R. Fröhlich and G. Erker, Chem. Commun., 2011, 47, 4288–4290 RSC.
-
(a) C. Appelt, H. Westenberg, F. Bertini, A. W. Ehlers, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2011, 50, 3925–3928 CrossRef PubMed;
(b) W. Uhl and C. Appelt, Organometallics, 2013, 32, 5008–5014 CrossRef;
(c) W. Uhl, C. Appelt, A. Wollschläger, A. Hepp and E.-U. Würthwein, Inorg. Chem., 2014, 53, 8991–8999 CrossRef PubMed;
(d) M. Devillard, E. Nicolas, A. W. Ehlers, J. Backs, S. Mallet-Ladeira, G. Bouhadir, J. C. Slootweg, W. Uhl and D. Bourissou, Chem. Commun., 2014, 50, 14805–14808 RSC;
(e) M. Devillard, R. Declercq, E. Nicolas, A. W. Ehlers, J. Backs, N. Saffon-Merceron, G. Bouhadir, J. C. Slootweg, W. Uhl and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 4917–4926 CrossRef PubMed;
(f) L. Keweloh, H. Klöcker, E.-U. Würthwein and W. Uhl, Angew. Chem., Int. Ed., 2016, 55, 3212–3215 CrossRef PubMed.
- C. Appelt, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2012, 51, 5911–5914 CrossRef PubMed.
- C. Appelt, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2013, 52, 4256–4259 CrossRef PubMed.
- J. Possart and W. Uhl, Organometallics, 2018, 37, 1314–1323 CrossRef.
-
(a) C. Rosorius, C. G. Daniliuc, R. Fröhlich, G. Kehr and G. Erker, J. Organomet. Chem., 2013, 744, 149–155 CrossRef;
(b) Z. Jian, G. Kehr, C. G. Daniliuc, B. Wibbeling and G. Erker, Dalton Trans., 2017, 46, 11715–11721 RSC.
-
(a) X. Xu, R. Fröhlich, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Commun., 2012, 48, 6109–6111 RSC;
(b) X. Xu, G. Kehr, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2013, 135, 6465–6476 CrossRef PubMed.
- A. W. Kyri, R. Kunzmann, G. Schnakenburg, Z.-W. Qu, S. Grimme and R. Streubel, Chem. Commun., 2016, 52, 13361–13364 RSC.
- C. U. Grünanger and B. Breit, Angew. Chem., Int. Ed., 2008, 47, 7346–7349 CrossRef PubMed.
-
(a) Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed;
(b) Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2006, 110, 5121–5129 CrossRef PubMed.
- M. A. Dureen, C. C. Brown and D. W. Stephan, Organometallics, 2010, 29, 6594–6607 CrossRef.
-
(a) J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 4968–4971 CrossRef PubMed;
(b) C. M. Mömming, S. Frömel, G. Kehr, R. Fröhlich, S. Grimme and G. Erker, J. Am. Chem. Soc., 2009, 131, 12280–12289 CrossRef PubMed.
- M. Ullrich, K. S.-H. Seto, A. J. Lough and D. W. Stephan, Chem. Commun., 2009, 2335–2337 RSC.
- J. M. Breunig, A. Hübner, M. Bolte, M. Wagner and H.-W. Lerner, Organometallics, 2013, 32, 6792–6799 CrossRef.
- J. Ke, Y. Tang, H. Yi, Y. Li, Y. Cheng, C. Liu and A. Lei, Angew. Chem., Int. Ed., 2015, 54, 6604–6607 CrossRef PubMed.
- D. J. Parks, W. E. Piers and G. P. A. Yap, Organometallics, 1998, 17, 5492–5503 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: [DETAILS]. CCDC 1843771–1843777 (2–8). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra05108a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a and
Huadong Wang
a and
Huadong Wang