
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDecorating a single giant DNA with gold nanoparticles†
Jose M. Carnerero a,
Shinsuke Masuokab,
Hikari Babab,
Yuko Yoshikawab,
Rafael Prado-Gotor*a and
Kenichi Yoshikawa*b
a,
Shinsuke Masuokab,
Hikari Babab,
Yuko Yoshikawab,
Rafael Prado-Gotor*a and
Kenichi Yoshikawa*b
aDepartment of Physical Chemistry, Faculty of Chemistry, Universidad de Sevilla, c/Profesor García González, 1, 41012, Seville, Spain. E-mail: pradogotor@us.es
bFaculty of Life and Medical Sciences, Doshisha University, Kyotanabe 610-0394, Japan. E-mail: keyoshik@mail.doshisha.ac.jp
First published on 25th July 2018
Abstract
We decorated a single giant DNA (1.66 × 105 base pairs) with gold nanoparticles through the simple procedure of mild warming, without denaturation of the DNA molecule. Single-molecule observation with fluorescence microscopy revealed that individual decorated DNA molecules stay in the bulk solution by avoiding aggregation and precipitation, and exhibit translational and conformational fluctuation, i.e., Brownian motion. An analysis of the intra-chain fluctuation of single DNA molecules revealed that the apparent spring constant and damping coefficient of a DNA chain increased by ca. 13- and 5-fold, respectively, upon decoration with gold nanoparticles. Observation by transmission electron microscopy revealed that gold nanoparticles were stably attached to the DNA skeleton. UV-visible measurements revealed the absence of any detectable change in surface plasmon resonance, suggesting that the gold nanoparticles assemble without the formation of a densely packed aggregate. CD measurements showed that the secondary structure of decorated DNA is still essentially the B-form.
1. Introduction
Nanoparticles (NPs) have attracted considerable interest with regard to both their potential applications in medicine and basic understanding of biological systems. The evolution of nanoscience has led to the development of a wide variety of methods to fight cancer and image damaged organisms.1,2 In gene therapy, DNA is transferred into the living cells of patients. Nevertheless, the principal challenge in the design of gene delivery systems lies in the carrier's ability to overcome different biological barriers and reach the target in an efficient manner.1,3,4 Due to the limitations of a modified virus for this purpose, non-viral vectors (and particularly NPs) have become important.4 On the other hand, the easy control of NPs' size and charge allows mimicking proteins and other kinds of macromolecules and studying their effects in the DNA structure. For example, cationic nanoparticles have been used to study the DNA's wrapping or bending by electrostatic interaction with the purpose of simulating the chromatin-like structures at different levels of organization.5–7 Not only the effect of the charge in the DNA structure can be studied throughout this method, but also the effect of the occupied volume by macromolecules, making nanoparticles an excellent tool for studying the crowding effect in living cells.8Among the many types of NPs, those with noble metal cores are especially popular due to their amazing optic properties9 which gives rise to a whole new field involving the engineering and architecture of DNA–noble metal NPs conjugates.10–12 Oscillation of the electron cloud of nanoparticles upon interaction with the electric field of light causes the colloidal system to absorb electromagnetic radiation, and, for noble metal NPs, this absorption is in the visible range.13 This strong and broad band is called the Surface Plasmon Resonance (SPR) band and its characteristics depend on the physical properties of the NPs and the environment.13,14 Among all existing nanoparticle types, gold nanoparticles (AuNPs) have enjoyed the greatest rise in popularity, despite the fact that silver nanoparticles display a stronger and narrower band than colloidal gold.15 This is in no small part due to the inert nature and biocompatibility of gold,16 which allows for generating different structures with a wide range of properties according to the “construction block”.17–19 DNA–AuNPs systems have received a large boost over the past few years, and the number of publications on this topic has grown exponentially.20 This marked interest has been principally due to the versatile interactions between AuNPs and thiolated oligonucleotides, which allow for the easy and selective functionalization of these colloids.20,21 In addition, the nucleobases exhibit a strong tendency to interact with the AuNP surface, and each shows a different adsorption affinity.22 In this way, appropriate oligonucleotide design could allow for the anchoring of short DNA molecules to a nanoparticle surface without the need for a sulfur bond. These adsorption processes enable, among many other applications, the detection of oligonucleotide hybridization upon the addition of salt, since non-complementary molecules tend to stick to the nanoparticle surface, increasing the colloid's charge and its stability against aggregation processes.23,24 Consequently, studies about noble metal NPs and DNA have mainly focused on the interaction of single-stranded oligonucleotides with AuNPs, and much less is known about the interactions of these systems with long DNA molecules.25 As a consequence of their predominant use as sensors, there has been little interest in the physical effects of AuNPs on DNA, including oligonucleotides. As an example, a recent study examined the influence of cationic colloidal silver on the DNA structure; the structural changes of the polymer depended on the nanoparticle concentration and charge, and partial strand separation was achieved.26
In the present work, we report the formation of AuNP/DNA complexes by a novel and easy method involving giant DNA (T4-DNA; 166 kbp) and anionic AuNPs, without the need for any surface alteration. It should be noted that the procedure adopted in the present study is markedly different than those in studies using DNA as a template, where the nanoparticles' functionalization was required, either with cationic ligand to reach the interaction via electrostatic attraction with anionic phosphate groups27,28 or using oligonucleotides to make the assembly by DNA hybridization mechanism.11,29 Besides we have focused our attention on the interaction of AuNPs with genome-sized giant DNA molecules because it is well known that the persistence length of double-stranded DNA molecules is around 170 bp (or 50 nm), so this implies that DNA below the size of kbp behaves as a stiff rod whereas giant DNA molecules above the size of several tens of kbp are regarded as semi-flexible polymer chains. Actually, giant DNA molecules with lengths on the order of 100 kbp undergo a large discrete transition, contrary to short DNA molecules which tend to aggregate without a folding transition.30,31 In addition, changes in the higher-order structure of DNA are believed to be involved in the regulation of gene expression.32,33 Furthermore, studies on the basic properties of giant DNA molecules are expected to shed light on the underlying mechanism of epigenetic phenomena.34 Here we have evaluated and obtained the spring constant on genome sized DNA though the observation of fluctuating molecules. Although there have been so many studies on the measurements of the Brownian fluctuation of DNA molecules, these studies have failed to obtain the precise information on the intrachain fluctuation eliminating the influence of translational and rotational fluctuations.30,31 The procedure developed here to obtain the spring constant means the base for future studies in the higher order structure of genome sized DNA by the interaction with different kind of molecules, for example, anticancer agents.
2. Experimental methods
2.1. Materials
Hydrogen tetrachloroaurate(III) trihydrate was purchased from Sigma-Aldrich (St. Louis, MO, USA), and trisodium citrate was from Alfa Aesar (Haverhill, MA, USA); they were used without further purification. All chemicals were of analytical reagent grade. Solutions were prepared with deionized water with a conductivity of less than 10−6 S m−1.T4 GT7 phage DNA (166 kbp with a contour length of 57 μm) was purchased from Nippon Gene Co., Ltd. LTP (Toyama, Japan). Calf thymus DNA (CT DNA: 8–15 kbp) was purchased from Sigma-Aldrich. The fluorescent dye GelGreen was obtained from Biotium (Fremont, CA, USA). The antioxidant 2-mercaptoethanol (2-ME) was purchased from Wako Pure Chemical Industries (Osaka, Japan).
2.2. Synthesis and characterization of gold nanoparticles
Citrate-capped AuNPs with an average size of around 15 nm were prepared according to a variation of Turkevich's method, by reduction of a gold complex in a liquid phase.35 97 ml of HAuCl4 (0.01%) was heated under reflux at 90 °C, and then, at this temperature, 3 ml of tri-sodium citrate (38.8 mM) was added. Heating under reflux was continued for an additional 10 minutes, during which time the color changed to deep red. The solution was then left stirring at room temperature for 24 hours.In the UV-Vis absorption spectra, the absorption maximum wavelength was located at 520 nm. This wavelength is similar to that obtained in previous studies on AuNPs with a size of ca. 15 nm, making it possible to estimate the extinction coefficient: ε = 3.9 × 108 M−1 cm−1.36 The concentration can be obtained by applying the Lambert–Beer law: [AuNPs] = 3.9 × 10−9 M.
2.3. Preparation of assemblies of single DNAs with AuNPs
10 μl of T4 DNA solution (10 μM) was added to 790 μl of Tris–HCl buffer solution (pH 7.5). This sample was placed in a water bath at 60 °C for 10 minutes. Next, without moving the bath container, 200 μl of a 15 nm AuNP dispersion was added to give final concentrations of [DNA] = 0.1 μM and [AuNP] = 0.8 nM. The sample was kept in the bath for an additional 5 minutes, and then left to cool to room temperature (24 °C). A DNA solution at the same concentration and treated in the same way as the sample was used as a control. Another sample with its respective control but without any heating was also prepared.2.4. Fluorescence microscopy (FM) measurements
2-ME and the dye GelGreen were added to 950 μl of the sample, with final concentrations of 4% (v/v) and 5 μM, respectively. 2-ME was used as a free-radical scavenger to reduce fluorescent fading and light-induced damage of DNA. GelGreen was added to visualize individual DNA molecules by FM. In a previous work, we confirmed that GelGreen has a negligible effect on the conformation of DNA molecules at the concentrations used in observations by fluorescence microscopy.37 Single-DNA observations were performed with an Axiovert 200 inverted fluorescence microscope (Carl Zeiss, Oberkochen, Germany) equipped with a 100× oil-immersion objective lens and fluorescent illumination from a mercury lamp (100 W) via a filter set (Zeiss-10, excitation BP 450–490; beam splitter FT 510; emission BP 515–565). Images were recorded onto a DVD at 30 frames per second with a high-sensitivity EBCCD camera (Hamamatsu Photonics, Shizuoka, Japan) and analyzed with an image-analysis software, Cosmos32 (Library Co., Ltd., Tokyo, Japan).2.5. Transmission electron microscopy (TEM) measurements
For TEM examinations, a single drop (10 μl) of the sample was placed on a carbon film-coated copper grid, which was then left to dry in air for several hours at room temperature. TEM observations were performed with an energy filtered-TEM JEM2100F electron microscope (JEOL, Tokyo, Japan) at 200 kV. The samples were analyzed at different magnifications (4k, 6k, 8k, 10k, 12k, 15k, 40k, 100k, 300k), and then magnified 30-fold with a CCD camera (Olympus, Tokyo, Japan).2.6. UV-visible spectra
AuNP spectra were recorded from 400 to 800 nm with a Cary 500 spectrophotometer (Agilent, Santa Clara, CA, USA). The path length of the quartz cuvette was 1 cm. Experiments were carried out under different temperature conditions at a fixed colloidal gold concentration ([AuNPs] = 8 × 10−10 M) in the absence and presence of CT DNA ([DNA] = 5 × 10−5 M).2.7. Circular dichroism (CD) spectra
Electronic CD spectra were recorded in a Mos-450 spectropolarimeter (Bio-Logic, Seyssinet-Pariset, France). A standard quartz cell with a path length of 1 cm was used. The spectra were expressed in terms of ellipticity. Scans were taken from 230 nm to 320 nm for the intrinsic region of the DNA. For each spectrum, 5 runs were averaged at different temperatures. All the spectra were obtained with a fixed concentration of CT DNA ([DNA] = 5 × 10−5 M), both in the absence of and at a fixed concentration of AuNPs ([AuNPs] = 8 × 10−10 M).3. Results and discussion
In several preliminary experiments on the interaction of AuNPs with giant DNA molecules, we noticed that most AuNPs tend to effectively bind DNA molecules under mild warming at around 60–70 °C. To avoid the effect of the melting transition on double-stranded DNA, we chose a warming temperature at 60 °C and observed the obtained assemblies by several different methods. Here, we present the results of observations of complexes between DNA and AuNPs, by comparing the results for samples after warming at 60 °C with control specimens prepared at 24 °C (room temperature).Fig. 1 exemplifies images of T4 DNA (166 kbp) observed by fluorescence microscopy (FM). Fig. 1A and B correspond to control DNA samples in the absence of AuNPs, before and after warming at 60 °C, respectively. In both cases, elongated DNA molecules exhibit translational and intrachain fluctuation as Brownian motion in the bulk aqueous solution. The observations at room temperature (24 °C) are similar to those reported in previous studies.7,38 With the addition of AuNPs to DNA solution at room temperature (Fig. 1C), DNA molecules remain in an elongated conformation. In the sample after warming (Fig. 1D), DNA molecules exhibit a rigid conformation, i.e., almost no intrachain fluctuation is observed except for fluctuating translational and tumbling motions. Additionally, bright dots are arranged along a single DNA molecule.
 | ||
| Fig. 1 Real-time monitoring of single T4 DNA molecules in bulk solution as observed by fluorescence microscopy (at room temperature, 24 °C). The time-interval between neighboring frames is 0.5 s. (A) In the absence of AuNPs without warming; (B) in the absence of AuNPs after warming at 60 °C for 15 min; (C) in the presence of AuNPs without warming; (D) in presence of AuNPs after warming at 60 °C for 15 min. Corresponding quasi-three-dimensional (3D) images indicating the profiles of the fluorescence intensity distribution are also shown. The scale bar is 5 μm. | ||
To evaluate the degree of intramolecular Brownian motion, we analyzed fluctuation on fluorescence images of single DNA molecules before and after decoration by AuNPs. The red line in Fig. 2 shows the time-dependent fluctuation of the long-axis length for a T4 DNA molecule after warming in the presence of AuNPs. The blue line shows the fluctuation for a natural DNA molecule in the absence of AuNPs. It is apparent that the single DNA in an assembly with AuNPs after gentle warming exhibits a rather stiff structure with a significant decrease in intrachain fluctuation. Interestingly, a single-DNA assembly is essentially the same size as that of native DNA without AuNPs. It has been well established30,39 that many chemical agents, such as polyamines, cationic surfactants, crowding solvable polymers, etc., cause the tight compaction of giant DNA molecules. Thus, the effect of AuNPs on the higher-order structure of DNA is rather specific, and leads to a marked change in fluctuation by maintaining the conformation without compaction/condensation.
 | ||
| Fig. 2 Time dependent change in the long-axis length of T4 DNA molecules without and with attachment to AuNPs as depicted by blue and red lines, respectively. The blue and red lines correspond to the samples in Fig. 1B and D, respectively. Both measurements were performed at room temperature after warming at 60 °C for 15 min. The blue line shows the time trace for DNA without AuNPs, and the red line shows that for the AuNP–DNA assembly. For convenience, schematic illustrations of the conformations are shown on the right side. | ||
Next, we evaluated the autocorrelation C(τ) from the time-dependent fluctuation of the long-axis length, L(τ):
C(τ) = 〈L(τ) − ![[L with combining macron]](https://www.rsc.org/images/entities/i_char_004c_0304.gif) 〉〈L(0) − 〉 〉〈L(0) − 〉
| (1) |
is the time-average of the long-axis length.
Based on a theoretical model of thermal fluctuation under a harmonic potential, the autocorrelation function is represented as in eqn (2) to a reasonable approximation:40
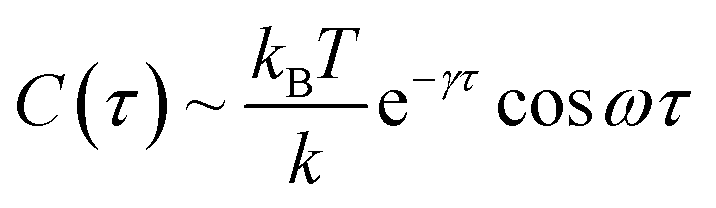 | (2) |
 From the vertical axis at τ = 0 in Fig. 3, C(0) is calculated as 0.46 ± 0.09 and 0.034 ± 0.007 μm2 for natural and Au-decorated DNA, respectively. Thus, the spring constants k of natural DNA and Au-decorated DNA are k0 = (8.9 ± 1.8) × 10−9 N m−1 and kAu = (1.2 ± 0.3) × 10−7 N m−1, respectively, by taking T ≈ 300 K. In other words, the spring constant of Au-decorated DNA is ca. 13 times larger than that of natural DNA. Next, we deduce physico-parameters from the fitting curves based on eqn (2), as in the broken lines in Fig. 3. The damping coefficients for natural and Au-decorated DNAs are thus evaluated to be γ0 = 1.1 ± 0.3 s−1 and γAu = 5 ± 2 s−1, respectively. As for the angular frequencies, we found that ω0 = 3.2 ± 0.7 rad s−1 and ωAu = 24 ± 6 rad s−1, respectively. From the fundamental characteristics of a harmonic oscillator with spring constant k, we assume the relationship to be k ∝ ω2. Although the experimental error for the angular frequency of Au-decorated DNAs is rather large, the relationship of k ∝ ω2 seems to be supported by the data obtained from the analysis of the autocorrelation function. Recently, there has been an increasing number of experimental trials to measure the elasticity of DNA by use of the experimental technique of laser tweezers. Unfortunately, most of the current studies reported the observed spring constant for short DNA molecules smaller than several kbp, which is due to the technical difficulty of measuring the elastic properties of giant DNA molecules. Instead, we can roughly estimate the Hooke spring constant for long DNA based on a simple theoretical prediction:41–43
From the vertical axis at τ = 0 in Fig. 3, C(0) is calculated as 0.46 ± 0.09 and 0.034 ± 0.007 μm2 for natural and Au-decorated DNA, respectively. Thus, the spring constants k of natural DNA and Au-decorated DNA are k0 = (8.9 ± 1.8) × 10−9 N m−1 and kAu = (1.2 ± 0.3) × 10−7 N m−1, respectively, by taking T ≈ 300 K. In other words, the spring constant of Au-decorated DNA is ca. 13 times larger than that of natural DNA. Next, we deduce physico-parameters from the fitting curves based on eqn (2), as in the broken lines in Fig. 3. The damping coefficients for natural and Au-decorated DNAs are thus evaluated to be γ0 = 1.1 ± 0.3 s−1 and γAu = 5 ± 2 s−1, respectively. As for the angular frequencies, we found that ω0 = 3.2 ± 0.7 rad s−1 and ωAu = 24 ± 6 rad s−1, respectively. From the fundamental characteristics of a harmonic oscillator with spring constant k, we assume the relationship to be k ∝ ω2. Although the experimental error for the angular frequency of Au-decorated DNAs is rather large, the relationship of k ∝ ω2 seems to be supported by the data obtained from the analysis of the autocorrelation function. Recently, there has been an increasing number of experimental trials to measure the elasticity of DNA by use of the experimental technique of laser tweezers. Unfortunately, most of the current studies reported the observed spring constant for short DNA molecules smaller than several kbp, which is due to the technical difficulty of measuring the elastic properties of giant DNA molecules. Instead, we can roughly estimate the Hooke spring constant for long DNA based on a simple theoretical prediction:41–43| k ∼ kBT/lpL | (3) |
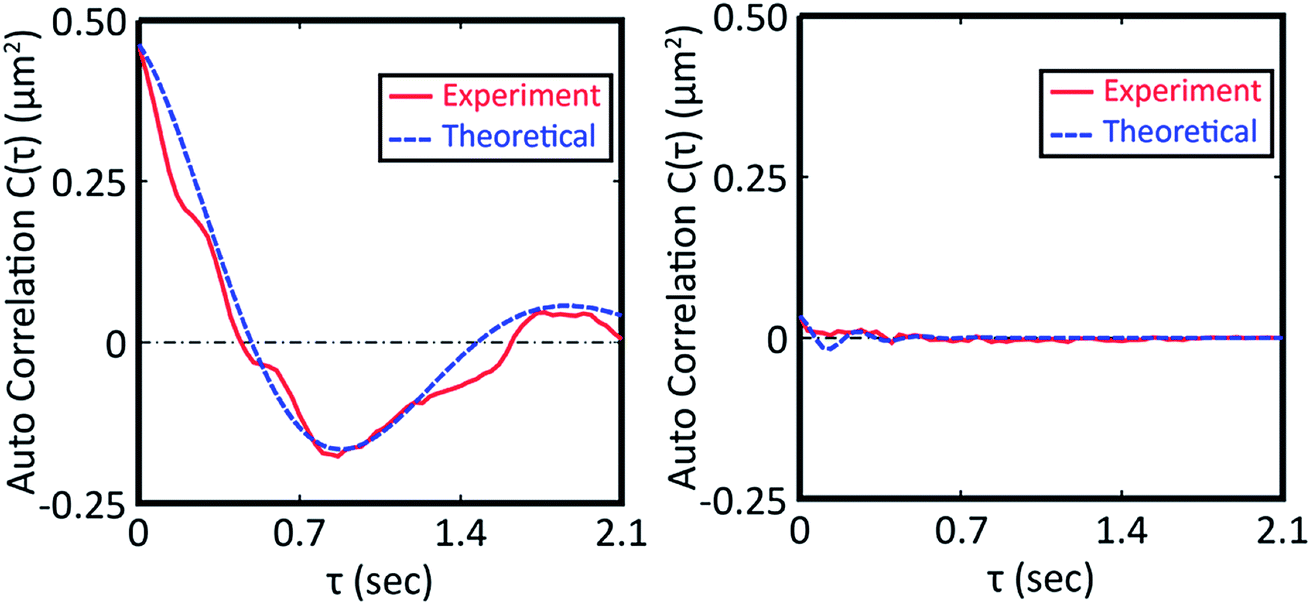 | ||
| Fig. 3 Autocorrelation function for the time-dependent fluctuation given in Fig. 2. The fitting curve was depicted based on eqn (2). The left picture is the auto correlation function for DNA molecules in the absence of colloidal gold while that on the right is for DNA molecules decorated with gold nanoparticles. | ||
As for the DNA–AuNP assemblies generated by mild warming, by visual inspection there was no apparent color change for the sample solution before and after warming, suggesting that the AuNPs exhibit negligible aggregation. To check the solution conditions including aggregation in a quantitative manner, we carried out UV-Vis absorption measurements. Fig. 4 shows the SPR band of AuNPs in the presence and absence of DNA at different stages of the process, which reveals essentially the same absorption spectrum before and after warming. Here, we show the spectrum for the DNA–AuNP assembly after warming at 70 °C, to show the minimum effect of warming on plasmon absorption. (There was no detectable difference between samples after warming at 60 °C or 70 °C.) It is well known that the SPR band shifts to a higher wavelength when the size of AuNPs becomes larger due to electric field coupling. In addition, it has been reported that symmetry-breaking of spherical Au particles as the result of tight contact between them causes a quadrupolar absorption band at around 600 nm.44
 | ||
| Fig. 4 UV-Vis absorption spectra of AuNPs in the absence (A) and presence (B) of calf thymus DNA under different conditions: room temperature, 70 °C and room temperature after the warming procedure (AH, after heating). | ||
As mentioned above regarding the measurements by fluorescence microscopy and UV-Vis spectroscopy, we have shown that decorated individual giant DNA molecules are soluble in solution by avoiding aggregation and precipitation. However, the resolution of fluorescence microscopy is relatively low and is on the order of 0.5 μm. To obtain information on the detailed morphology of individual giant DNA decorated with AuNPs, we performed TEM observations. Fig. 5A shows AuNPs prepared in the absence of DNA, indicating the presence of randomly dispersed particles absorbed on a carbon film. Fig. 5B shows the results for a mixture of T4 DNA and AuNPs without warming, revealing that, while nanoparticles tend to attach to the DNA chain, a certain proportion remain unbound and are dispersed on the solid substrate. Interestingly, all of the particles bound to the DNA skeleton and resulted in greater density along the DNA chain (see Fig. 5C), in contrast to the sample prepared at room temperature. In accordance with the experimental results by fluorescence microscopy, the fully decorated sample prepared by mild warming exhibits only slight shrinkage compared to that prepared without warming. A more detailed comparison of the warmed and un-warmed samples is presented in the ESI (see Fig. S1–3†). Interestingly, the obtained structures are different than what would be obtained if the biopolymer acted as a mere template (DNA metallization), where the assembly of metallic nanoparticles would be more branched and ordered, or could even behave as a nanowire.45,46 In addition, the morphology observed by FM is similar to that observed by TEM, despite the large difference in spatial resolution in addition to the difference in the observation conditions, i.e., in solution or dry on a solid substrate (see Fig. S4†). On the other hand, as mentioned above, there was no apparent difference in the UV-visible spectrum or the SPR band, which implies that aggregation among AuNPs is almost negligible or that the interaction between neighboring colloids along the DNA molecule is relatively weak.
 | ||
| Fig. 5 (A) TEM images of dispersed gold nanoparticles at 4 × 10−9 M. (B) TEM images of nanoparticles in the presence of T4 DNA; [AuNPs] = 0.8 nM; [DNA] = 1 μM. (C) TEM images of nanostructures formed by heating a solution of [AuNP] = 0.8 nM and [DNA] = 1 μM. | ||
In any case, the differences between Fig. 5B and C show extremely different behaviors depending on the heating process. These differences in structure are due to enhanced DNA–AuNP interactions in the complexes formed upon warming. A possible explanation is that there is a partial separation of T4 DNA during the heating process which causes a loss of DNA stiffness and further exposes the bases to the gold surface; for this reason, the aforementioned complex structures are not observed in the unheated sample. The interaction way of DNA molecules with citrate capped AuNPs has been extensively studied, being established through the base fragments.22,47–50 In fact this interaction is so strong that it can break the hydrogen bond of base pairs in oligonucleotides, although that was observed with 5 nm AuNPs and not for 15 nm.48 On the other hand, the phosphate groups of DNA molecules only interact when the colloidal gold has positive charge, due to electrostatic attractive forces, and it is noteworthy that the interaction is with the cationic functional group and not with the gold surface.5,51,52 It has also be considered that at room temperature the AuNPs and DNA molecules are inclined to be distant (due to they present the same charge) and therefore the binding is rather difficult.24,53 As the temperature increases the thermal fluctuation too, so that the chances to collide of the AuNPs with DNA molecules are higher, being possible surpassing the energy barrier which avoids their approximation. In addition the partial denaturation of DNA can happen at higher temperatures, so the exposition of DNA's bases to the outside is viable and the interaction between bases and AuNPs does not need to overlap the repulsion electrostatic energy. Anyway, more studies are necessary to understand the mechanism that provokes an increase in the interaction between T4 DNA and anionic AuNPs at higher temperature.
Finally, to check if these interactions provoke changes in the secondary structure of DNA, the effects of temperature and the presence of NPs on the circular dichroism (CD) spectra of DNA were studied. Fig. 6A shows CD spectra for the samples at room temperature. The addition of AuNPs induces a slight decrease in the intensity of the positive band, although the basic character of the B-structure is preserved. An isosbestic point also appears over 258 nm, as has been previously observed for anionic nanoparticles and large DNA chains.53 This small conformational change does not involve an opening of the double helix due to the nanoparticles; the negative band does not decrease in intensity, which would correspond to a loss of helicity of the DNA, and the crossover point does not change, while Dias and coworkers found that for melting processes, a 3 nm shift could be expected.54 Even at 70 °C, the essential feature characteristic to the B-form remains (see Fig. 6B), accompanied by a small decrease in the band height, suggesting the induction of a helix-coil transition at some portion of DNA.
 | ||
| Fig. 6 CD spectra of calf thymus DNA in the absence and presence of AuNPs at different temperature conditions: (A) room temperature (without any heating); (B) 70 °C; (C) room temperature (after heating). | ||
Although no significant changes in DNA structure are observed at room temperature or adding AuNPs at 70 °C, this is not the case at room temperature after heating (see Fig. 6C): an important decrease of intensity of the positive band (around 20%) consisting in the DNA compaction appears. Some authors have attributed these changes to bending phenomena,55 which are associated with small losses of π-stacking interactions and helicity. The same phenomenon has been described working with functionalized cationic gold nanoparticles and 37-residue oligonucleotides (see Fig. 2 in the ref. 5). However in this case the DNA compaction took place by electrostatic interaction between negative charge of phosphate groups with the positive charge of the nanoparticle surface. This fact implies that not only a marked decrease intensity at 280 nm is present in the circular dichroism of DNA: it is coupled with a shift in the maximum wavelength. This point is not observed in the present study and cannot be concluded a total conversion of denatured double strand unlike the study carried out by Goodman and collaborators.5 However, at no stage of the process do the DNA molecules show a conformation being largely and remarkably different from the B-form, suggesting that the secondary structure of DNA remains almost same as that before the decoration with AuNPs.
4. Conclusions
In the present article, successful decoration of a giant DNA molecule with AuNPs through the simple procedure of mild warning was described. Upon such decoration, the DNA molecule becomes stiffer while retaining an elongated conformation. On the other hand, there is no apparent change in the surface plasmon resonance of AuNPs, suggesting that they are dispersed, rather than aggregated. Electron microscopy observations support such dispersed positioning. In addition, it was confirmed that the secondary structure of DNA stays mostly as in the B-form after AuNP decoration. Further studies will be needed to gain a basic understanding of the structure and properties of AuNP-decorated giant DNA, and also to develop and optimize DNA–AuNPs complexes for practical applications in medicine.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Miwako Toda for her assistance with obtaining TEM images, and Aila Jimenez-Ruiz (E-mail: ailjimrui@alum.us.es) for help in revising English usage throughout this manuscript. This work was financed by the Spanish Ministerio de Economía y Competitividad (MinECo), CTQ2016-78703-P (AEI/FEDER, UE), by the V Plan Propio Grupos Emergentes (V Own Gran Plan for Emerging Groups) of the Universidad de Sevilla, and by the OTRI (2010/00000762). We also thank Universidad de Sevilla for a predoctoral grant for the V-Plan Propio de Investigación (VPPI-US). This work was partially supported by JSPS KAKENHI Grant Numbers 15H02121 and 25103012.References
- B. Pelaz, C. Alexiou, R. A. Alvarez-Puebla, F. Alves, A. M. Andrews, S. Ashraf, L. P. Balogh, L. Ballerini, A. Bestetti, C. Brendel, S. Bosi, M. Carril, W. C. W. Chan, C. Chen, X. Chen, X. Chen, Z. Cheng, D. Cui, J. Du, C. Dullin, A. Escudero, N. Feliu, M. Gao, M. George, Y. Gogotsi, A. Grünweller, Z. Gu, N. J. Halas, N. Hampp, R. K. Hartmann, M. C. Hersam, P. Hunziker, J. Jian, X. Jiang, P. Jungebluth, P. Kadhiresan, K. Kataoka, A. Khademhosseini, J. Kopeček, N. A. Kotov, H. F. Krug, D. S. Lee, C. M. Lehr, K. W. Leong, X. J. Liang, M. Ling Lim, L. M. Liz-Marzán, X. Ma, P. Macchiarini, H. Meng, H. Möhwald, P. Mulvaney, A. E. Nel, S. Nie, P. Nordlander, T. Okano, J. Oliveira, T. H. Park, R. M. Penner, M. Prato, V. Puntes, V. M. Rotello, A. Samarakoon, R. E. Schaak, Y. Shen, S. Sjöqvist, A. G. Skirtach, M. G. Soliman, M. M. Stevens, H. W. Sung, B. Z. Tang, R. Tietze, B. N. Udugama, J. S. VanEpps, T. Weil, P. S. Weiss, I. Willner, Y. Wu, L. Yang, Z. Yue, Q. Zhang, Q. Zhang, X. E. Zhang, Y. Zhao, X. Zhou and W. J. Parak, ACS Nano, 2017, 11, 2313–2381 CrossRef PubMed.
- O. S. Wolfbeis, Chem. Soc. Rev., 2015, 44, 4743–4768 RSC.
- M. Riley and W. Vermerris, Nanomaterials, 2017, 7, 94 CrossRef PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef PubMed.
- C. M. Goodman, N. S. Chari, G. Han, R. Hong, P. Ghosh and V. M. Rotello, Chem. Biol. Drug Des., 2006, 67, 297–304 CrossRef PubMed.
- J. A. Nash, A. Singh, N. K. Li and Y. G. Yingling, ACS Nano, 2015, 9, 12374–12382 CrossRef PubMed.
- A. A. Zinchenko, T. Sakaue, S. Araki, K. Yoshikawa and D. Baigl, J. Phys. Chem. B, 2007, 111, 3019–3031 CrossRef PubMed.
- A. Zinchenko, Adv. Colloid Interface Sci., 2016, 232, 70–79 CrossRef PubMed.
- M. Quinten, Optical Properties of Nanoparticle Systems: Mie and Beyond, Wiley-VCH, Weinheim, 2011 Search PubMed.
- I. Capek, DNA Engineered Noble Metal Nanoparticles, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2015 Search PubMed.
- J. J. Storhoff and C. A. Mirkin, Chem. Rev., 1999, 99, 1849–1862 CrossRef PubMed.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553–556 CrossRef PubMed.
- A. Moores and F. Goettmann, New J. Chem., 2006, 30, 1121–1132 RSC.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef.
- E. Petryayeva and U. J. Krull, Anal. Chim. Acta, 2011, 706, 8–24 CrossRef PubMed.
- H. An and B. Jin, Biotechnol. Adv., 2012, 30, 1721–1732 CrossRef PubMed.
- B. Kemper, L. Zengerling, D. Spitzer, R. Otter, T. Bauer and P. Besenius, J. Am. Chem. Soc., 2018, 140, 534–537 CrossRef PubMed.
- J. M. Dixon and S. Egusa, J. Am. Chem. Soc., 2018, 140, 2265–2271 CrossRef PubMed.
- R. Ye, A. V. Zhukhovitskiy, R. V. Kazantsev, S. C. Fakra, B. B. Wickemeyer, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2018, 140, 4144–4149 CrossRef PubMed.
- J. Liu, Phys. Chem. Chem. Phys., 2012, 14, 10485–10496 RSC.
- K. M. Koo, A. A. I. Sina, L. G. Carrascosa, M. J. A. Shiddiky and M. Trau, Anal. Methods, 2015, 7, 7042–7054 RSC.
- J. J. Storhoff, R. Elghanian, C. A. Mirkin and R. L. Letsinger, Langmuir, 2002, 18, 6666–6670 CrossRef.
- J. Yang, J. Y. Lee, H.-P. Too, G.-M. Chow and L. M. Gan, Chem. Phys., 2006, 323, 304–312 CrossRef.
- H. Li and L. Rothberg, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14036–14039 CrossRef PubMed.
- J. M. Carnerero, A. Jimenez-Ruiz, P. M. Castillo and R. Prado-Gotor, ChemPhysChem, 2016, 18, 17–33 CrossRef PubMed.
- P. Gisbert-Quilis, M. Masetti, J. Morla-Folch, J. M. Fitzgerald, N. Pazos-Perez, E. Garcia-Rico, V. Giannini, R. A. Alvarez-Puebla and L. Guerrini, Adv. Mater. Interfaces, 2017, 4, 1–10 Search PubMed.
- H. J. Kim, Y. Roh and B. Hong, J. Vac. Sci. Technol., A, 2006, 24, 1327–1331 CrossRef.
- M. G. Warner and J. E. Hutchison, Nat. Mater., 2003, 2, 272–277 CrossRef PubMed.
- J. Chao, Y. Zhang, D. Zhu, B. Liu, C. Cui, S. Su, C. Fan and L. Wang, Sci. China: Chem., 2016, 59, 730–734 CrossRef.
- K. Yoshikawa and Y. Yoshikawa, in Pharmaceutical Perspectives of Nucleic Acid-Based Therapeutics, ed. R. I. Mahato and S. W. Kim, Taylor & Francis, Abingdon, UK, 2002, pp. 137–163 Search PubMed.
- T. Iwataki, K. Yoshikawa, S. Kidoaki, D. Umeno, M. Kiji and M. Maeda, J. Am. Chem. Soc., 2000, 122, 9891–9896 CrossRef.
- H. Schiessel, J. Phys.: Condens. Matter, 2003, 15, R699–R774 CrossRef.
- T. Nozaki, R. Imai, M. Tanbo, R. Nagashima, S. Tamura, T. Tani, Y. Joti, M. Tomita, K. Hibino, M. T. Kanemaki, K. S. Wendt, Y. Okada, T. Nagai and K. Maeshima, Mol. Cell, 2017, 67, 282–293.e7 CrossRef PubMed.
- R. Cortini, M. Barbi, B. R. Caré, C. Lavelle, A. Lesne, J. Mozziconacci and J. M. Victor, Rev. Mod. Phys., 2016, 88, 1–29 CrossRef.
- C. P. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef.
- X. Liu, M. Atwater, J. Wang and Q. Huo, Colloids Surf., B, 2007, 58, 3–7 CrossRef PubMed.
- C. Tongu, T. Kenmotsu, Y. Yoshikawa, A. Zinchenko, N. Chen and K. Yoshikawa, J. Chem. Phys., 2016, 144, 205101 CrossRef PubMed.
- A. A. Zinchenko, F. Luckel and K. Yoshikawa, Biophys. J., 2007, 92, 1318–1325 CrossRef PubMed.
- V. A. Bloomfield, Curr. Opin. Struct. Biol., 1996, 6, 334–341 CrossRef PubMed.
- M. C. Wang and G. E. Uhlenbeck, Rev. Mod. Phys., 1945, 17, 323–342 CrossRef.
- J. F. Marko and E. D. Siggia, Macromolecules, 1995, 28, 8759–8770 CrossRef.
- M. D. Wang, H. Yin, R. Landick, J. Gelles and S. M. Block, Biophys. J., 1997, 72, 1335–1346 CrossRef PubMed.
- D. C. Appleyard, K. Y. Vandermeulen, H. Lee and M. J. Lang, Am. J. Phys., 2007, 75, 5–14 CrossRef.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef PubMed.
- L. I. Lopatina, E. A. Karpushkin, A. Zinchenko and V. G. Sergeyev, Mendeleev Commun., 2016, 26, 291–292 CrossRef.
- Z. Chen, C. Liu, F. Cao, J. Ren and X. Qu, Chem. Soc. Rev., 2018, 47, 4017–4072 RSC.
- H. Li and L. J. Rothberg, J. Am. Chem. Soc., 2004, 126, 10958–10961 CrossRef PubMed.
- J. Yang, B.-K. Pong, J. Y. Lee and H.-P. Too, J. Inorg. Biochem., 2007, 101, 824–830 CrossRef PubMed.
- L. Sun, Z. Zhang, S. Wang, J. Zhang, H. Li, L. Ren, J. Weng and Q. Zhang, Nanoscale Res. Lett., 2009, 4, 216–220 CrossRef PubMed.
- X. Zhang, M. R. Servos and J. Liu, Langmuir, 2012, 28, 3896–3902 CrossRef PubMed.
- G. Han, C. T. Martin and V. M. Rotello, Chem. Biol. Drug Des., 2006, 67, 78–82 CrossRef PubMed.
- R. Fu, C. Wang, J. Zhuang and W. Yang, Colloids Surf., A, 2014, 444, 326–329 CrossRef.
- J. M. Carnerero, A. Jimenez-Ruiz, E. M. Grueso and R. Prado-Gotor, Phys. Chem. Chem. Phys., 2017, 19, 16113–16123 RSC.
- M. Rosa, R. Dias, M. da Graça Miguel and B. Lindman, Biomacromolecules, 2005, 6, 2164–2171 CrossRef PubMed.
- P. J. Heath, J. B. Clendenning, B. S. Fujimoto and M. J. Schurr, J. Mol. Biol., 1996, 260, 718–730 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05088k |
| This journal is © The Royal Society of Chemistry 2018 |