
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSensitive and selective detection of the highly toxic pesticide carbofuran in vegetable samples by a molecularly imprinted electrochemical sensor with signal enhancement by AuNPs†
Peipei Qiabcd,
Jiao Wangabcd,
Xiangyun Wangabcd,
Zhiwei Wangabcd,
Hao Xuabcd,
Shanshan Diabcd,
Qiang Wangabcd and
Xinquan Wang *abcd
*abcd
aInstitute of Quality and Standard of Agro-products, Zhejiang Academy of Agricultural Sciences, Hangzhou 310021, P. R. China. E-mail: wangxinquan212@163.com; Fax: +86 571 86419060; Tel: +86 571 86404355
bState Key Laboratory Breeding Base for Zhejiang Sustainable Pest and Disease Control, Hangzhou 310021, P. R. China
cAgricultural Ministry Key Laboratory for Pesticide Residue Detection, Hangzhou 310021, P. R. China
dKey Laboratory of Detection for Pesticide Residues and Control of Zhejiang, Hangzhou 310021, P. R. China
First published on 16th July 2018
Abstract
An imprinted electrochemical sensor was constructed for the detection of carbofuran with high sensitivity and selectivity. AuNPs were used as the electron wire for signal amplification, and molecularly imprinted polymer was used as the recognition element. The preparation process of the modified electrode was optimized, and the electrode was characterized using scanning electron microscopy, cyclic voltammetry and differential pulse voltammetry. The results proved that the prepared sensor can selectively detect carbofuran and the AuNPs can increase its sensitivity. The method validation included the systematic evaluation of the linearity, sensitivity, selectivity, reproducibility and stability. The optimized sensor showed a wide linear response to carbofuran in the range from 5.0 × 10−8 to 4.0 × 10−4 mol L−1 with a low detection limit of 2.4 × 10−8 mol L−1. The sensor also exhibited a high selectivity to carbofuran. The real sample analysis for vegetable samples suggested its potential application in the analysis of vegetable samples.
1. Introduction
Carbofuran is widely used in agriculture due to its high effectiveness and low cost.1 It can shorten the grain growth cycle and increase agricultural production. However, the accompanying environmental pollution and harm to human health by the frequent use of carbofuran have raised the level of concern. Therefore, developing an efficient detection method for carbofuran is necessary for further evaluation, monitoring and management.2 Until now, it has been reported that carbofuran can be determined by gas chromatography,3 high performance liquid chromatography,4 gas chromatography-mass spectrometry5 and immunosensors.6,7 Chromatographic methods, especially coupled to mass spectrometry, can give rise to accurate qualitative and quantitative determination of carbofuran. However, they require expensive instrumentation as well as tedious extraction and clean-up steps, which make them unsuitable for the on-site and rapid determination of carbofuran.An electrochemical sensor has a significant advantage in terms of rapid determination, especially for on-site detection due to its compact instrument and easy operation. It is thus extensively developed and coupled to many types of recognition elements, such as selective antibodies,8 DNA aptamers,9 recognizable polymers and so on. The recognition element plays an essential role in the selective and accurate determination of the target analyte. Therefore, the construction of the recognition element onto the electrode is the core of developing an efficient sensor. As is known to us, an antibody is specifically selective towards the antigen molecule, but it is difficult to prepare the antibody. Screening for a suitable DNA aptamer is also a time-consuming process. On the contrary, artificial antibody molecularly imprinted polymer (MIP) has provided a new possibility for the selective recognition of the target analyte.
MIP has been widely used as the chromatographic stationary phase, solid phase extraction adsorbent and recognition element of the sensor.10–13 During the preparation of the MIP, the template molecule was pre-synthesized with the functional monomer and cross-linker into the polymer. Removal of the template was then performed to give rise to lots of selective recognition sites, which are complementary to the size, shape and functional groups of the template.14–16 It is reported that the MIP-modified sensors show high selectivity for the template molecules over other structurally related compounds because of the sites formed in the MIPs.17–19 Until now, MIP sensors such as amperometric sensors,20,21 quartz crystal microbalance sensors22,23 and electrochemical sensors24 were fabricated for the detection of different analytes. However, the disadvantages of the low proton electron transfer rate and low sensitivity of the MIP sensor needed to be overcome.25,26 Nanomaterials, such as nanoparticles, nanotubes16,27 and nanocomposites, have been introduced for providing higher surface area and conductivity.19,22 AuNPs possess chemical stability and excellent conductivity. They can also be readily prepared in the electrode by electro-deposition. Hence, AuNPs were introduced to enhance the sensitivity of prepared sensor in the present work.
The purpose of the present work is to develop an electrochemical sensor for the detection of carbofuran using MIP as the recognition element and AuNPs as the signal enhancement material on a glassy carbon electrode (GCE). The introduction of AuNPs can effectively increase the electric conductivity and surface area of the electrode. The electrical aggregated functional monomers and template molecules can result in the formation of MIP on the surface of an AuNPs-modified GCE. After the removal of template molecules, the MIP-based electrochemical sensor was obtained. This sensor was proven to possess good stability, sensitivity and selective recognition ability toward carbofuran. The real sample analysis results also demonstrated its good prospective application in carbofuran residue analysis in vegetable samples.
2. Experimental
2.1. Chemicals
Standards, such as carbofuran (98.5%), isoprocarb (99.9%), methomyl (98.3%), pirimicarb (99.0%), aldicard (98.5%) and metholcarb (1000 mg L−1 in acetone) were purchased from the Shanghai Pesticide Research Institute (Shanghai, China). p-Aminobenzoic acid (99.0%) (ABA) was purchased from J&K Scientific Ltd. (Shanghai, China). HPLC-grade methanol was purchased from Merck (Darmstadt, Germany). Acetic acid, H2SO4, Na2HPO4, NaH2PO4, K3Fe(CN)6, K4Fe(CN)6 and KCl were of analytical purity. Water used was purified using a Milli-Q gradient A10 system (Millipore Corp., Billerica, MA, USA). A stock solution of ABA (100 mmol L−1) was prepared in methanol. A 0.05 mol L−1 phosphate buffered solution (PBS, pH = 7.0) was prepared with Na2HPO4 and NaH2PO4 solutions. The supporting electrolytewas1 mmol L−1 K3Fe (CN)6/K4Fe(CN)6 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) containing 0.1 mol L−1 KCl solution for the detection of the analyte.
:1) containing 0.1 mol L−1 KCl solution for the detection of the analyte.
2.2. Preparation of the carbofuran MIP/AuNPs/GCE
An Autolab PGSTAT 204 electrochemistry workstation (Metrohm, Switzerland) was used for the preparation of the MIP- and AuNP-modified electrode. As demonstrated in Fig. 1, the preparation procedures contain the following steps: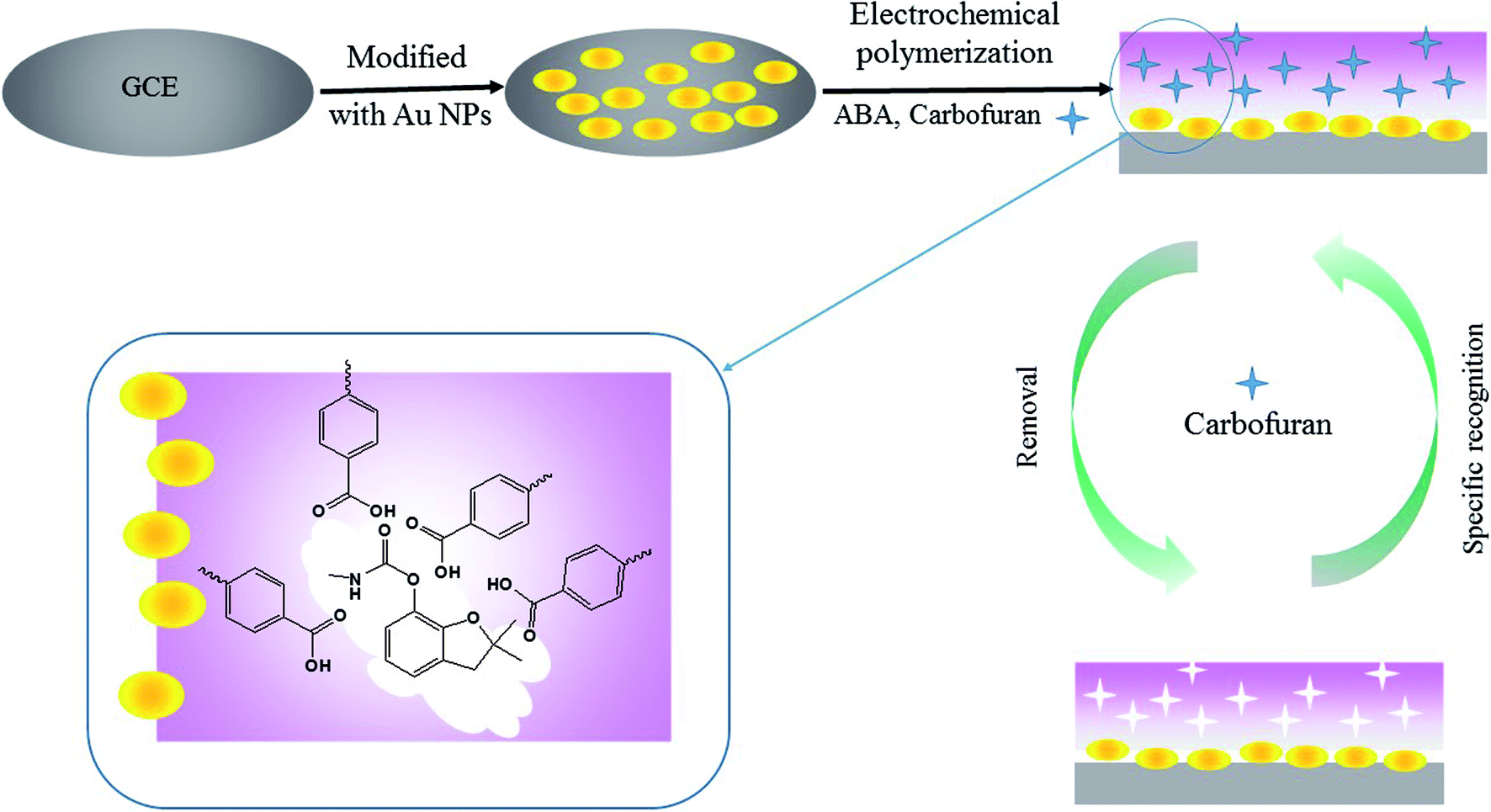 | ||
| Fig. 1 The schematic diagram of MIP/Au NPs/GCE preparation procedure. | ||
(1) Pre-treatment of the bare GCE. The GCE was polished with an alumina (particle size: 0.05 μm) slurry using a polishing cloth and subsequently ultra-sonicated in ethanol and water for 5 min, respectively. The electrode was scanned by cyclic voltammetry (CV) between −0.4 V and 1.6 V for 20 cycles at a scan rate of 0.1 V s−1 in 0.1 mol L−1 H2SO4 solution.
(2) Electro-deposition of AuNPs. The bare polished GCE was immersed in 3 mmol L−1 HAuCl4 solution and treated at a constant potential of −0.2 V for 200 s. The obtained electrode was named AuNPs/GCE.
(3) Electro-polymerization of MIP on the AuNPs/GCE. CV was employed for the electro-polymerization of the MIP membrane. The AuNPs/GCE was further immersed in the PBS solution containing 4.0 mmol L−1 ABA and 1.0 mmol L−1 carbofuran. The PBS solution was pre-treated with high-purity nitrogen for 10 min to remove the oxygen. The potential ranged from −0.4 V to 1.0 V for 7 consecutive cycles at a scan rate of 0.05 V s−1.
(4) Removal of the template from the MIP-modified electrode. The obtained electrode was placed into 10 mL of a 0.1 mol L−1 sodium hydroxide methanol solution under magnetic stirring for 15 min to remove the template molecule. The modified electrode was rinsed with water and dried at room temperature. This electrode was named MIP/AuNPs/GCE.
Meanwhile, the non-imprinted polymer membrane modified electrode (NIP/AuNPs/GCE) was prepared by the same procedure without the addition of the template in step (3). The MIP/GCE was also fabricated without the electro-deposition of AuNPs in step (2). These electrodes assisted in evaluating the selectivity and sensitivity of the MIP/AuNPs/GCE.
2.3. Morphological characterization of the electrodes
Field-emission scanning electron microscopy (FE-SEM) was performed on an Utral 55 instrument (Corl-zeisD, Germany) for the morphological characterization of the modified electrodes.2.4. Electrochemical measurements of the electrodes
Electrochemical measurements were based on the standard three-electrode configuration with a saturated calomel electrode and platinum wire as the reference electrode and counter electrode, respectively. Both CV and differential pulse voltammetry (DPV) were employed to evaluate the preparation procedure of the MIP/AuNPs/GCE.Before the electrochemical measurements, the extraction of the target analytes was carried out. Briefly, the modified GCEs were immersed into solutions spiked with different concentrations of analyte or the real sample solution under continuous stirring for 10 min. After extraction, the modified GCE was then washed with water and dried using nitrogen gas. The modified GCE was transferred to an electrolytic cell containing 1 mmol L−1 K3Fe(CN)6, 1 mmol L−1 K4Fe(CN)6 and 0.1 mol L−1 KCl as the supporting electrolyte. The CV was recorded from −0.2 V to 0.6 V at a scan rate of 0.05 V s−1. The DPV was performed by scanning from 0.6 V to −0.2 V with a pulse width of 50 ms, interval time of 0.5 s, step potential of 0.005 V, and modulation amplitude of 0.05 V. All of the electrochemical measurements were conducted at room temperature.
2.5. Real sample analysis
Vegetable samples were purchased from a local supermarket in Hangzhou, China. The samples were determined after acetonitrile extraction and dilution by ultra-pure water. Briefly, 25 g of vegetable sample was weighed, followed by the addition of 25 mL of acetonitrile. After high-speed homogenization for 2 min, the sample was filtered, and the filtrate was transferred to a measuring cylinder with 5 g of NaCl for the salt-outing process. One millilitre of supernatant was transferred into a weighing bottle containing 3 mL of ultrapure water. The MIP/AuNPs/GCE was immersed into the above solution for extraction, followed by DPV measurement according to the method stated in 2.4.3. Results and discussion
3.1. Construction of the MIP/AuNPs/GCE sensor
For the preparation of the MIP/AuNPs/GCE sensor, electro-deposition was used for the formation of AuNPs on the surface of the GCE according to the literature.19 Electro-polymerization was further utilized for the preparation of the MIP membrane on the AuNPs/GCE. During the electro-polymerization, several factors play important roles in the performance of the MIP membrane, such as the ratio of template to functional monomer, scan cycles and so on. The removal of the template from the MIP membrane and the recognition of the analyte also influenced the sensitivity of the MIP sensor. Therefore, these factors were optimized to explore the suitable conditions for the preparation of the MIP membrane and the recognition of the target analyte.During the electro-synthesis process, the potential range was set from −0.4 to 1.0 V in order to avoid over-oxidation of the synthesized polymer. As shown in Fig. 2, an oxidation peak at 0.86 V was clearly discerned on the first scan, and the peak currents declined with an increasing number of scan cycles. This could be attributed to the formation of amino cation radicals. A pair of redox peaks was observed at 0.11 and 0.17 V, which may be ascribed to the reduction and oxidation of the poly-ABA film.28 As demonstrated in the inset of Fig. 2, the recognized mechanism was speculated to include the formation of a radical cation from ABA, the further generation of a dimeric species by meta-coupling between the radical cation with either an unoxidized monomer or another radical cation, and finally facilitating the propagation of the polymerization.29 Before electro-polymerization, the template carbofuran can form non-covalent interactions with the ABA. Consequently, the template was embedded into the polymer membrane. The continual formation of the MIP membrane would hinder the charge transfer, leading to a decrease of the peak current. These results indicated that electro-polymerization occurred and the MIP membrane was formed on the electrode surface.
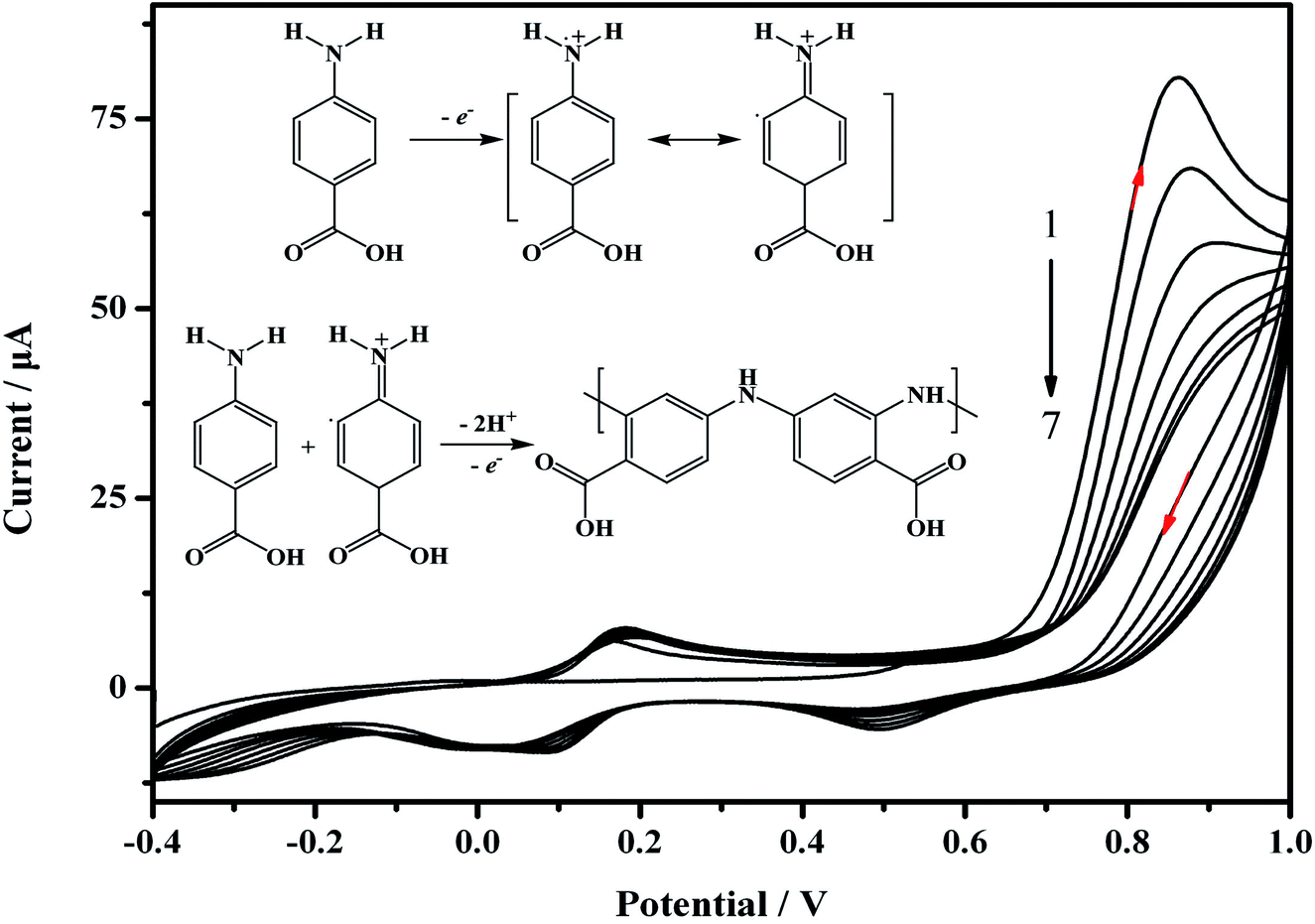 | ||
| Fig. 2 Cyclic voltammograms for the electro-polymerization of the MIP/AuNPs/GCE at the AuNPs/GCE surface and the polymerization mechanism (inset figure). The polymerization solution contained 4.0 mmol L−1 ABA and 1.0 mmol L−1 carbofuran in 0.05 mol L−1 PBS at pH 7.0. Scan rate: 0.05 V s−1, scan cycles: 7. | ||
As is well-known to us, the thickness of the polymer membrane dictated the amount of active sites, which influenced the sensitivity of the MIP-based sensor. A thin film would lead to a low adhesive ability and low sensitivity. Although the number of active sites increased with an increase in the number of polymerization laps, it was not easy to remove the target molecules in the deep part of membranes. Moreover, the target molecules had difficulty accessing active sites in the deep positions because of the mass-transfer resistance. Therefore, the number of scan cycles was investigated to control the thickness of the polymer membrane. In Fig. S1A,† the current response to carbofuran reached a maximum at seven cycles, and then, it decreased beyond seven cycles. Thus, seven cycles were used for the electro-polymerization of the MIP sensor.
The ratio of template to functional monomer during electro-polymerization had a great influence on the performance of the MIP membrane.10,14 The concentration of carbofuran was fixed at a constant value of 1.0 mmol L−1. The concentration of monomers was optimized in the present work by varying the monomer to carbofuran ratio to include the following: 1:1, 2:1, 4:1, 6:1 and 8:1. As shown in Fig. S1B,† at low concentrations of monomer (from 1.0 to 4.0 mmol L−1), ΔI increased with an increase of monomer due to the formation of more reaction sites. However, when the concentration of monomer exceeded 4.0 mmol L−1, a considerable decrease in the current signal of the MIP/AuNPs/GCE was observed. This may be the result of generating a large degree of cross-linker and a small amount of active sites in the matrix. In view of the best performance, the ratio of monomer to carbofuran was set at 4:1 for the next step in construction of the sensor.
The removal of template molecules from the MIP membrane is essential for the recognition of the target analyte. Many methods can be used to remove the template molecules, such as over-oxidation,30 supercritical fluid desorption, microwave-assisted extraction,31 redox extraction17 and solvent extraction.11 The solvent extraction is widely used and based on the principle of competitive desorption. Thus, the choice of the solvent would be dependent on the interaction of the template and the functional monomer. Methanol–acetic acid solution (9:1, v/v), 0.1 mol L−1 sulphuric acid solution and 0.1 mol L−1 sodium hydroxide methanol solution were used in attempts to remove the template molecules. The CV curve displayed that sodium hydroxide methanol solution was effective for removing the template molecules, while neither methanol–acetic acid nor sulphuric acid can give rise to the active sites for recognition (Fig. S2†). The removal time was also tested from 5 min to 30 min. Current responses of the MIP/AuNPs/GCE were recorded after being pre-treated with sodium hydroxide solution. The results revealed that the peak current increased with increasing the removal time as a result of the release of the active cavities (Fig. S1C†). When the removal time was longer than 15 min, the current response was not obviously enhanced any more. Thus, 15 min was used for the further experiments.
During the recognition process, the modified GCE was immersed into the sample solution for grabbing the target analyte, carbofuran. The extraction time is a critical parameter. As shown in Fig. S1D,† the current responses of the MIP/AuNPs/GCE in the analyte solution increased from 2 min to 20 min and then reached equilibrium. Therefore, 10 min was used for the further experiments.
3.2. Characterizations of the MIP-based sensor
 | ||
| Fig. 3 FE-SEM images of bare GCE (A), AuNPs/GCE (B), MIP/AuNPs/GCE before template removal (C) and MIP/AuNPs/GCE after template removal (D). | ||
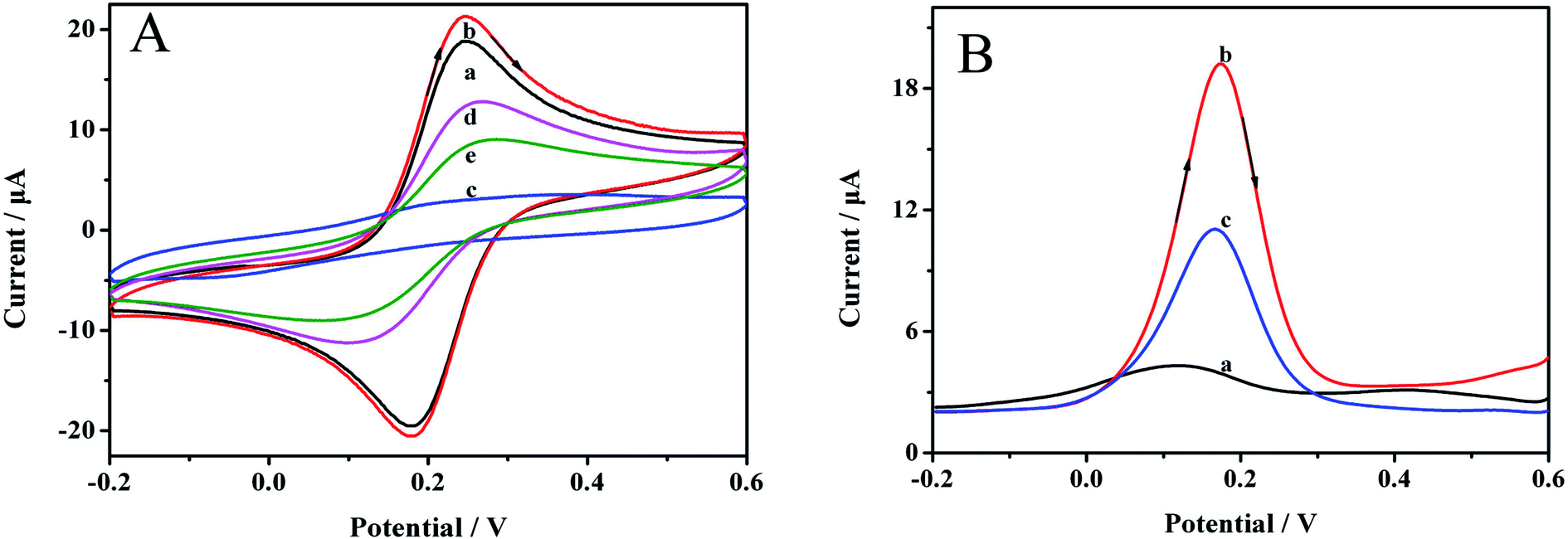 | ||
| Fig. 4 (A) CV curves of (a) bare GCE, (b) Au NPs/GCE, (c) MIP/Au NPs/GCE before the template removal, (d) MIP/Au NPs/GCE after template removal, (e) MIP/Au NPs/GCE after rebinding with 0.1 mmol L−1 carbofuran for 10 min. Scan rate: 0.05 V s−1. (B) The DPV curves of (a) MIP/Au NPs/GCE before the template removal, (b) MIP/Au NPs/GCE after the template removal, (c) MIP/Au NPs/GCE after being incubated in 0.1 mmol L−1 carbofuran for 10 min. Scan rate: 0.05 V s−1. | ||
DPV can also clearly reveal the preparation process. The results are demonstrated in Fig. 4B. Before removing the imprinting molecules, almost no signal was observed at 0.18 V (curve a), which was ascribed to the dense polymer membrane on the electrode surface. After the template was removed, an obvious reductive signal was considerably enhanced (curve b), indicating that the target molecule could be effectively removed. The signal was reduced when the MIP/AuNPs/GCE was immersed in 0.1 mmol L−1 carbofuran solution for 10 min (curve c), manifesting the affinity of the sensor for carbofuran.
Simultaneously, the anodic and the cathodic peak currents of the electrode were nearly symmetric, and both the peak currents and peak-to-peak separation increased when scan rates increased from 0.04 to 0.2 V s−1 (Fig. S3†). It can obviously be observed that both the anodic and the cathodic peak currents linearly scaled with the scan rate and could be expressed as the equations I = 124.92V + 11.18 (R2 = 0.988) and I = −101.46V − 10.69 (R2 = 0.987), respectively. These results indicated that the surface reaction was a reversible and surface-controlled electrochemical process.
3.3. Method validation of the MIP/AuNPs/GCE sensor
To evaluate the performance of the developed sensor, standard solutions of carbofuran at 5.0 × 10−8, 2.0 × 10−7, 5.0 × 10−7, 1.0 × 10−6, 6.0 × 10−6, 3.0 × 10−5, 1.0 × 10−4, 2.0 × 10−4 and 4.0 × 10−4 mol L−1 were analysed by DPV due to its higher sensitivity. The results in Fig. 5 show that the peak current decreased with increasing carbofuran concentrations. It was indicated that when the target analyte was seized by the MIP membrane, the channel for electron mass transfer was blocked, and the current signal was thus suppressed. The more active sites that were occupied, the lower the peak current was. Further analysis was carried out to explore the relationship between the concentration of the target analyte and the peak current. It was proven that the difference of the peak current (ΔI, μA) at the predetermined concentration (c, mmol L−1) compared to the initial current value was linearly correlated to the logarithm of the concentration (logc). The linear calibration equation was ΔI = 4.95logc + 26.64 (R2 = 0.971), while the linear range was from 5.0 × 10−8 mol L−1 to 4.0 × 10−4 mol L−1. The detection limit was 2.4 × 10−8 mol L−1 based on 3σ values of blank signals.
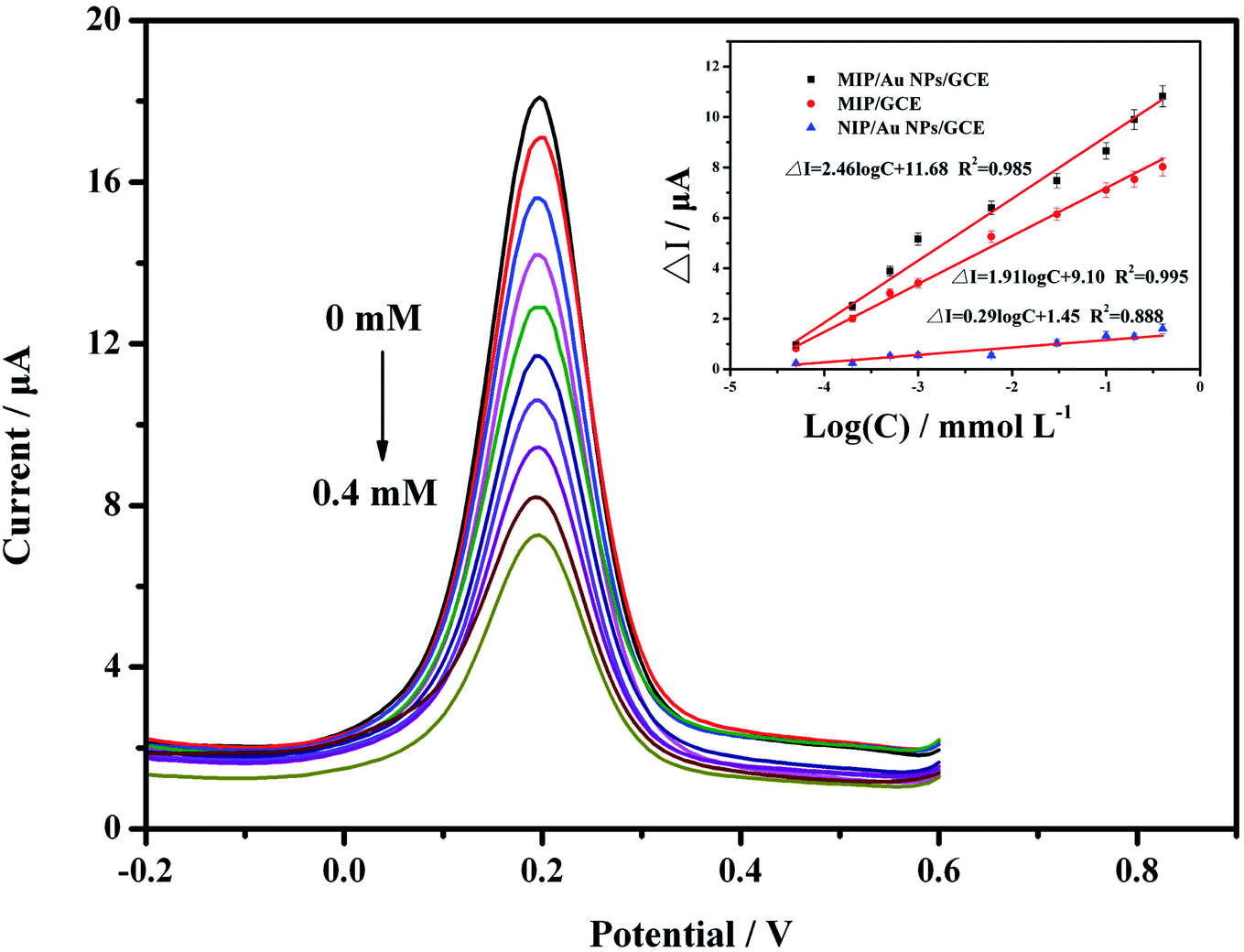 | ||
| Fig. 5 The DPV responses of the MIP/AuNPs/GCE in detection solution containing 1 mmol L−1 K3Fe (CN)6/K4Fe(CN)6 (1:1), 0.1 mol L−1 KCl solution and different concentrations of carbofuran. (Inset) The calibration curves of carbofuran detected by the MIP/AuNPs/GCE, MIP/GCE and NIP/AuNPs/GCE, respectively. | ||
For comparison, the MIP/GCE and NIP/AuNPs/GCE sensors were prepared and validated using the same conditions as for the MIP/AuNPs/GCE. Both sensors had lower responses than the MIP/AuNPs/GCE. The difference between the MIP/GCE and MIP/AuNPs/GCE sensors demonstrated that AuNPs could enhance the peak current signal by providing a higher surface area and electron transfer efficiency. The difference between the NIP/AuNPs/GCE and MIP/AuNPs/GCE sensors could display the specific selectivity of the MIP technology. Besides, the extremely low current response of the NIP/AuNPs/GCE could be attributed to that the electrode was mostly covered by the polymer film. Unlike the MIP/AuNPs/GCE, there were still many channels for the redox probe to connect with the electrode after template removal.
The selectivity of the MIP/AuNPs/GCE sensor was also investigated with the pesticides isoprocarb, methomyl, pirimicarb, aldicard and metholcarb as possible interfering compounds. These pesticides and carbofuran all belong to the carbamate pesticides. They are similar in structure and physical characteristics. As shown in Fig. 6, the carbofuran gave rise to the highest response (ΔI) on the MIP/AuNPs/GCE sensor among all the compounds. However, the responses of all of the pesticides were equivalent on the NIP/AuNPs/GCE sensor, further validating the absence of the recognizable sites and channels on the NIP/AuNPs/GCE sensor. The selectivity of the sensor can be quantitatively assessed by the imprinting factor (IF), which was usually defined as the ratio of the ΔI on the MIP-based sensor to the value on the NIP-based sensor. The higher the IF value was, the larger the selectivity of the prepared MIP sensor to the template molecule. In other words, the obtained highly selective MIP sensor would display a good anti-interference ability and accurate qualitative analysis ability. In the present work, the IF value of the carbofuran was close to 30, largely higher than those of the other pesticides. This result fully exhibited the selectivity of the prepared MIP/AuNPs/GCE sensor.
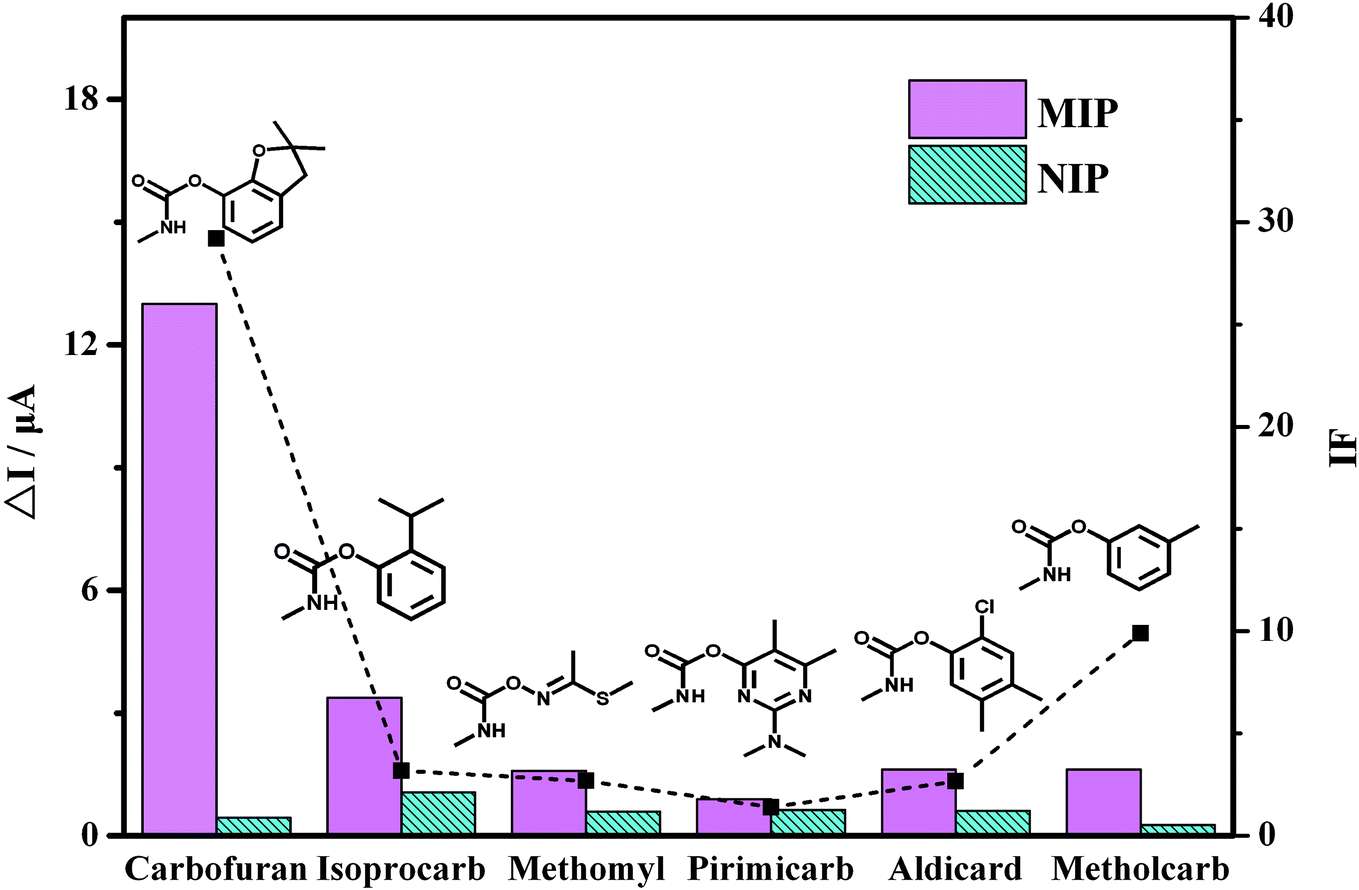 | ||
| Fig. 6 The current response of the pesticides by the MIP/AuNPs/GCE and NIP/AuNPs/GCE as well as the corresponding IF value of each pesticide. | ||
The reproducibility of one sensor was assessed in a 0.1 mmol L−1 carbofuran solution five times, and the calculated RSD was approximately 4.63%, indicating a good reproducibility. Five MIP/AuNPs/GCE electrodes were prepared in the same conditions and assessed in carbofuran solution at a constant concentration of 0.1 mmol L−1. The measured RSD was approximately 8.50%. The long-term stability of the electrode was evaluated by storage in air at room temperature for one week. The response of carbofuran by the MIP/AuNPs/GCE sensor decreased approximately 12.3% after storage for 7 days. These results indicated that the sensor was suitable for detecting carbofuran.
The proposed method was applied for determining carbofuran in vegetable samples to verify its feasibility in analysing real samples. The cowpea and pakchoi samples were spiked with carbofuran at concentrations of 0.02, 0.100 and 0.500 mg kg−1. As stated in Section 2.5, the sample was extracted by acetonitrile and then measured by DPV. As shown in Table 1, the recoveries of the carbofuran ranged from 71.0% to 91.0% with RSDs from 3.2% to 5.3%. Those results suggested that the developed sensor could be used for detecting carbofuran in real samples, displaying a potential application.
| Sample | Added (mg kg−1) | Found (mg kg−1) | Recovery (%) | RSD (%, n = 3) |
|---|---|---|---|---|
| Cowpea | 0.020 | 0.017 | 85.0 | 5.3 |
| 0.100 | 0.071 | 71.0 | 4.6 | |
| 0.500 | 0.406 | 81.2 | 3.2 | |
| Pakchoi | 0.020 | 0.018 | 90.0 | 3.3 |
| 0.100 | 0.091 | 91.0 | 2.8 | |
| 0.500 | 0.379 | 75.8 | 4.4 |
4. Conclusions
The present work described the fabrication of an electrochemical sensor for the selective determination of a highly toxic pesticide, carbofuran, based on MIP technology. AuNPs were introduced for signal amplification to improve the sensitivity of the obtained MIP sensor. The factors influencing the performance of the MIP/AuNPs/GCE were optimized to explore the suitable conditions. The morphological and electrochemical characterization could clearly illustrate the preparation process of the MIP/AuNPs/GCE and recognition of the target analyte. The comparison with the MIP/GCE sensor could prove the signal enhancement effect of the AuNPs. The method comparison with the NIP/AuNPs/GCE sensor could fully demonstrate the specific selectivity of the obtained MIP sensor to the template molecule, carbofuran. Systematic method validation showed that the MIP/AuNPs/GCE had good reproducibility, good stability and high selectivity toward carbofuran. Its application to a vegetable sample suggested its potential prospective application.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Special Fund for Agro-scientific Research in the Public Interest (Grant No. 201503107-12), the National Natural Science Foundation of China (Grant No. 31501556) and the Zhejiang Provincial Natural Science Foundation of China (Grant No. LQ15B050001).References
- R. A. Soomro, K. R. Hallam, Z. H. Ibupoto, A. Tahira, S. T. H. Sherazi, S. Juddin, S. Jawaid and M. Willander, Electroanalysis, 2016, 28, 1634–1640 CrossRef.
- L. Liu, D. Xu, Y. Hu, S. Liu, H. Wei, J. Zheng, G. Wang, X. Hu and C. Wang, Food Control, 2015, 53, 72–80 CrossRef.
- B. Cavaliere, M. Monteleone, A. Naccarato, G. Sindona and A. Tagarelli, J. Chromatogr. A, 2012, 1257, 149–157 CrossRef PubMed.
- X.-Y. Song, Y.-P. Shi and J. Chen, Food Chem., 2013, 139, 246–252 CrossRef PubMed.
- S. E. Petropoulou, E. Gikas, A. Tsarbopoulos and P. A. Siskos, J. Chromatogr. A, 2006, 1108, 99–110 CrossRef PubMed.
- J. Yang, H. Wang, Y. Jiang, Y. Sun, K. Pan, H. Lei, Q. Wu, Y. Shen, Z. Xiao and Z. Xu, Molecules, 2008, 13, 871–881 CrossRef PubMed.
- X. Sun, S. Du and X. Wang, Eur. Food Res. Technol., 2012, 235, 469–477 CrossRef.
- J. Liu, J. Wang, T. Wang, D. Li, F. Xi, J. Wang and E. Wang, Biosens. Bioelectron., 2015, 65, 281–286 CrossRef PubMed.
- S. Singh, A. Kaushal, H. Gautam, S. Gupta and A. Kumar, Sens. Actuators, B, 2017, 246, 300–304 CrossRef.
- C. Baggiani, L. Anfossi, C. Giovannoli and C. Tozzi, Talanta, 2004, 62, 1029–1034 CrossRef PubMed.
- A. Gomezcaballero, M. A. Goicolea and R. J. Barrio, Analyst, 2005, 130, 1012–1018 RSC.
- G. Xu, L. Yang, M. Zhong, C. Li, X. Lu and X. Kan, Microchim. Acta, 2013, 180, 1461–1469 CrossRef.
- B. Rezaei, O. Rahmanian and A. A. Ensafi, Microchim. Acta, 2013, 180, 33–39 CrossRef.
- H. Yan and K. Row, Int. J. Mol. Sci., 2006, 7, 155–178 CrossRef.
- J. O. Mahony, A. Molinelli, K. Nolan, M. R. Smyth and B. Mizaikoff, Biosens. Bioelectron., 2006, 21, 1383–1392 CrossRef PubMed.
- H. Li, C. Xie, S. Li and K. Xu, Colloids Surf., B, 2012, 89, 175–181 CrossRef PubMed.
- L. Ozcan and Y. Şahin, Sens. Actuators, B, 2007, 172, 362–369 CrossRef.
- X. Yan, J. Deng, J. Xu, H. Li, L. Wang, D. Chen and J. Xie, Sens. Actuators, B, 2012, 171, 1084–1094 Search PubMed.
- X. Zhang, Y. Peng, J. Bai, B. Ning, S. Sun, X. Hong, Y. Liu, Y. Liu and Z. Gao, Sens. Actuators, B, 2014, 200, 69–75 CrossRef.
- C. Apetrei, M. L. Rodríguez-Méndezb and J. A. D. Sajac, Electrochim. Acta, 2011, 56, 8919–8925 CrossRef.
- L. Kong, X. Jiang, Y. Zeng, T. Zhou and G. Shi, Sens. Actuators, B, 2013, 185, 424–431 CrossRef.
- M. Stoytcheva, R. Zlatev, S. Cosnier, M. Arredondo and B. Valdez, Biosens. Bioelectron., 2013, 41, 862–866 CrossRef PubMed.
- G. Fang, H. Wang, Y. Yang, G. Liu and S. Wang, Sens. Actuators, B, 2016, 237, 239–246 CrossRef.
- J. Bai, X. Zhang, Y. Peng, X. Hong, Y. Liu, S. Jiang, B. Ning and Z. Gao, Sens. Actuators, B, 2017, 238, 420–426 CrossRef.
- A.-E. Radi, A.-E. El-Naggar and H. M. Nassef, J. Electroanal. Chem., 2014, 729, 135–141 CrossRef.
- X. Tan, Q. Hu, J. Wu, X. Li, P. Li, H. Yu, X. Li and F. Lei, Sens. Actuators, B, 2015, 220, 216–221 CrossRef.
- N. Karimian, M. B. Gholivand and G. Malekzadeh, J. Electroanal. Chem., 2016, 771, 64–72 CrossRef.
- A. Benyoucef, F. Huerta, M. I. Ferrahi and E. Morallon, J. Electroanal. Chem., 2008, 624, 245–250 CrossRef.
- Y. Liu, J. Deng, X. Xiao, L. Ding, Y. l. Yuan, H. Li, X. Li, X. Yan and L. Wang, Electrochim. Acta, 2011, 56, 4595–4602 CrossRef.
- S. P. Ozkorucuklu, Y. Sahin and G. Alsancak, Sensors, 2008, 8, 8463–8478 CrossRef PubMed.
- A. Ellwanger, C. Berggren, S. Bayoudh, C. Crecenzi, L. Karlsson, P. K. Owens, K. Ensing, P. A. G. Cormack, D. Sherrington and B. Sellergren, Analyst, 2001, 126, 784–792 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05022h |
| This journal is © The Royal Society of Chemistry 2018 |