
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInactivation of an urban wastewater indigenous Escherichia coli strain by cerium doped zinc oxide photocatalysis†
Ian Zammit a,
Vincenzo Vaianob,
Giuseppina Iervolinob and
Luigi Rizzo*a
a,
Vincenzo Vaianob,
Giuseppina Iervolinob and
Luigi Rizzo*a
aDepartment of Civil Engineering, University of Salerno, Via Giovanni Paolo II 132, 84084 Fisciano, SA, Italy. E-mail: l.rizzo@unisa.it; Fax: +39089969620; Tel: +39089969334
bDepartment of Industrial Engineering, University of Salerno, Via Giovanni Paolo II 132, 84084 Fisciano, SA, Italy
First published on 20th July 2018
Abstract
Heterogeneous photocatalysis (HPC) is a subset of Advanced Oxidation Processes (AOPs) with potential future applications in water disinfection. Herein, a zinc oxide photocatalyst was doped with cerium at various atomic ratios ranging from 0 to 0.1 Ce![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn. Keeping in mind that the application of HPC is often limited by its cost of use, a simple and easy to upscale method, that is the hydroxide induced hydrolysis of zinc nitrate in the presence of Ce3+ followed by calcination at 300 °C, was used to synthesise the catalysts. The catalysts have been characterized by different techniques such as X-ray diffraction (XRD), UV-vis diffuse reflectance (UV-vis DRS) and Raman spectroscopy. XRD results showed that Ce3+ ions were successfully incorporated into the ZnO lattice. UV-vis DRS spectra evidenced that Ce–ZnO samples present band-gap values of about 2.97 eV, lower than those of undoped ZnO (3.21 eV). These various photocatalysts, at 0.1 g L−1 in saline 0.85%, were used to inactivate Escherichia coli previously isolated from an urban wastewater treatment plant. Higher atomic ratios of Ce in the ZnO lattice, as confirmed by XRD and Raman spectroscopy, showed significant improvements to the inactivation rate; the resulting recommended optimum cerium loading of 0.04:1 Ce:Zn gave multiple orders of magnitude higher rate of inactivation after 60 min of treatment when compared to un-doped ZnO. This optimum loading of cerium was faster than the de facto literature standard TiO2-P25 tested under identical conditions.
:Zn. Keeping in mind that the application of HPC is often limited by its cost of use, a simple and easy to upscale method, that is the hydroxide induced hydrolysis of zinc nitrate in the presence of Ce3+ followed by calcination at 300 °C, was used to synthesise the catalysts. The catalysts have been characterized by different techniques such as X-ray diffraction (XRD), UV-vis diffuse reflectance (UV-vis DRS) and Raman spectroscopy. XRD results showed that Ce3+ ions were successfully incorporated into the ZnO lattice. UV-vis DRS spectra evidenced that Ce–ZnO samples present band-gap values of about 2.97 eV, lower than those of undoped ZnO (3.21 eV). These various photocatalysts, at 0.1 g L−1 in saline 0.85%, were used to inactivate Escherichia coli previously isolated from an urban wastewater treatment plant. Higher atomic ratios of Ce in the ZnO lattice, as confirmed by XRD and Raman spectroscopy, showed significant improvements to the inactivation rate; the resulting recommended optimum cerium loading of 0.04:1 Ce:Zn gave multiple orders of magnitude higher rate of inactivation after 60 min of treatment when compared to un-doped ZnO. This optimum loading of cerium was faster than the de facto literature standard TiO2-P25 tested under identical conditions.
1. Introduction
Due to the ever increasing (a) global human population, (b) proportion of irrigated farmland over rain fed and (c) water stress caused by the climate,1–3 conventional water sources are not sufficient to satisfy the demand for clean water. Consequently, alternative water sources, in particular water reuse practices, are expected to take a more central role in global water management. In developed countries with high water stress such as Italy, Spain, Australia, Cyprus, Malta and Israel the portion of reused treated wastewater is higher, reaching almost complete reuse in Cyprus and Israel.4,5 One of the main challenges in rendering wastewater safe for reuse, in particular for agricultural irrigation, is the removal of pathogens. While a European Union regulation is under discussion,6 some member countries have their own national regulations and corresponding limits of bacterial load for treated wastewater intended for agricultural reuse (e.g. ≤10 CFU of Escherichia coli per 100 mL in Italy). Conventional disinfection processes, such as chlorination and ozonation, which are typically used in urban wastewater treatment plants (UWTPs),7 increase effluent toxicity and this results in the formation of regulated and unregulated disinfection by-products.8,9 In order to overcome the problems related to conventional disinfection, alternative technologies, such as Advanced Oxidation Processes (AOPs), have been investigated in the last years. AOPs are based on highly oxidising reactive oxygen species (ROS), such as hydroxyl radicals (˙OH). AOPs were found to be highly effective in the inactivation of a wide range of waterborne pathogens.10–12 Among AOPs, heterogeneous photocatalysis (HPC) is an emerging treatment technology which can successfully address the challenge of pathogen removal from UWTPs effluents. Since the first application of semiconductor photocatalysis for bacterial inactivation in 1993,13 much research has been devoted to inactivation of bacteria (see a recent review14) and recently specifically antibiotic resistant bacteria.15–17 HPC may have potentially substantial advantages over chlorination and ozonation when it comes to disinfection by-products. Chlorination produces toxic chlorinated by-products18 that are regulated in some countries, HPC doesn't involve chlorine radicals and hence doesn't produce these chlorinated by-products. Ozonation on the other hand is well known to oxidise naturally occurring bromide ions to highly toxic bromates as well as low-molecular-weight aldehydes.18 It is thought that HPC has a lower tendency to generate disinfection by-products19 however the complexity and diversity of different wastewaters and applications makes a definite conclusion hard to reach. Thus potentially HPC can fill a niche in application where water quality is of the utmost importance and chlorination and ozonation cannot be used.In HPC, ROS are generated by the light induced generation of electron–hole pairs within the photocatalyst in the presence of water. Electron–hole pairs are only generated when the energy of the electromagnetic radiation absorbed by the semiconductor is of higher or equal energy than its band gap – the gap between the valence and conduction bands. Titanium dioxide is the most investigated photocatalyst partially due to its commercial availability and abundance but recently zinc oxide (ZnO) has gained traction over titanium dioxide due to the fact that it has the same advantages but is cheaper.20–25
Metal doping of semiconductors is a technique employed to change the photocatalytic properties of the material, such as the band gap, the recombination rate of electron–hole pairs and adsorption properties of the material, these in turn affect the generation of ROS and reaction kinetics.26,27
Doping of ZnO with cerium has been previously investigated in the development of sensors28–30 and coating fibers.31 More relevantly to the work herein, Ce doped ZnO was also investigated in the removal of chemical pollutants,32–36 showing promising results, at times better than titanium dioxide. However, to the best of our knowledge Ce doped ZnO has not been used for the inactivation of bacteria so far, and the promising results on chemical pollutants warrant research on bacterial inactivation. Additionally we test a wider range of cerium atom ratios in doping to evaluate possible improvements over the optimum of 0.01 previously tested against chemical compounds.37 Herein photocatalytic inactivation of an indigenous E. coli strain selected from the secondary effluent of an UWTP was investigated using Ce doped ZnO as the photocatalyst. The loading of cerium within ZnO was optimised with respect to its activity in inactivating the selected E. coli strain with the aim of improving the activity of the photocatalyst while maintaining low costs of production and a simple and easy to upscale method of synthesis. This work is part of an effort to develop a reactor using an easy to upscale optimised photocatalyst for the disinfection of wastewater allowing safe and economically feasible agricultural reuse.
2. Materials and methods
2.1 Chemicals
Zinc nitrate hexahydrate (Sigma-Aldrich: Purum, >99.0%) was used as the source of Zn2+ while cerium(III) nitrate hexahydrate (Sigma-Aldrich, ≥99%) for Ce3+. Sodium hydroxide (Carlo Erba, RPE) was used to form the hydroxide precipitate in the hydroxide induced hydrolysis synthesis. Bacteria were enumerated on TBX agar (Sigma-Aldrich) after appropriate serial dilution. The isolated strain of E. coli was grown overnight in LB Broth (Miller) (Sigma) before spiking in an isotonic solution of sodium chloride (Sigma-Aldrich, BioReagent). The isolated E. coli was also grown on Coliforms Chromogenic Agar (Conda) to confirm putative identification.2.2 Synthesis of zinc oxide based catalysts
Eight doped catalysts as well as undoped ZnO were synthesised via the hydroxide induced hydrolysis of zinc nitrate modified from.38 In brief, 5.0 g (16.8 millimoles) of zinc nitrate hexahydrate were dissolved in 75 mL of ultrapure water. Separately 2.2 g of NaOH were dissolved in 25 mL of ultrapure water. The NaOH solution was added dropwise to the dissolved zinc nitrate under vigorous stirring. A white precipitate (eqn 1) gradually formed with the addition of the base. The suspension was stirred for an additional 1 hour, centrifuged at 5000 rpm to remove water and dissolved impurities, resuspended in 100 mL of ultrapure water and centrifuged again at the same speed. After discarding the water the second time, the resulting paste was dried at 110 °C for 30 min and calcinated at 300 °C for 1 hour to form ZnO (eqn 2).| Zn(NO3)2·6H2O(aq.) + 2OH(aq.)− → Zn(OH)2(s) + 2NO3− | (1) |
| Zn(OH)2(s) + heat → ZnO(s) | (2) |
Seven doping levels of cerium in ZnO were also prepared using the same method. The only difference being that cerium(III) nitrate hexahydrate was dissolved and well homogenised with the zinc salt prior to co-precipitation on the addition of the base. The amount of cerium(III) nitrate hexahydrate used in the synthesis was in the range of 0.042 millimoles to 1.681 millimoles obtaining samples at different Ce:Zn molar ratio. In particular, the seven molar ratios of Ce:Zn prepared are 0.0025:1, 0.0050:1, 0.0100:1, 0.0150:1, 0.0400:1, 0.0700:1, 0.1000:1. Thus the range of Ce to Zn atom ratio ranged from nil, in the undoped, to 10%.
2.3 Photocatalysts characterisation
All synthesised catalysts were characterised from a chemical-physical point of view by different techniques. XRD measurements were realized using an X-ray micro diffractometer Rigaku D-max-RAPID, using Cu-Kα radiation. Raman spectra were obtained at room temperature with a Dispersive Micro Raman (Invia, Renishaw) equipped with 514 nm laser in the range of 200–2000 cm−1 Raman shift. The ultraviolet-visible diffuse reflectance spectra (UV-vis DRS) were recorded using a Perkin Elmer Lambda 35 spectrophotometer using a RSA-PE-20 reflectance spectroscopy accessory (Labsphere Inc., North Sutton, NH). The band gap values of photocatalysts were determined through the corresponding Kubelka–Munk function (KM) (which is proportional to the absorption of radiation) and by plotting (KM × hν)2 against hν. The specific surface area (SSA) analysis was performed by BET method using N2 adsorption with a Costech Sorptometer 1042 after a pre-treatment at 150 °C for 30 min in He flow (99.999%). The Ce/Zn molar ratio of the samples were determined by X-ray fluorescence spectrometry (XRF) in a Thermo Fischer ARL QUANT’X EDXRF spectrometer equipped with a rhodium standard tube as the source of radiation and with Si–Li drifted crystal detector.2.4 Isolation of E. coli from UWTP
Wastewater sample was taken from the effluent of the secondary treatment in a UWTP in Salerno province, Italy (40.6305700°, 014.8448900°). Blue colonies were isolated on selective chromogenic agar (TBX). X-Gluc in TBX agar identifies β-glucuronidase activity in E. coli and results in blue coloured colonies. One of these colonies was isolated, cultivated in LB broth and a mother stock preserved in glycerol. The strain was also tested on a second chromogenic agar, the expected dark blue colouration was also observed here. The strain is hence presumed to be E. coli.
2.5 Photocatalytic experiments
Seven doped zinc oxide catalysts, 1 undoped zinc oxide and commercial titanium dioxide Degussa P25, as the established reference photocatalyst in the literature, were used in our measurements. The following conditions were used for all measurements, the light source was an Osaka 125 W UVA lamp (1.26 mW cm−2 at 365 nm peak) placed 32 cm from the base of an open cylindrical vessel of 13 cm diameter and 4 cm water height, with a total volume of 500 mL of isotonic water (0.85% NaCl) containing 0.1 g L−1 of catalyst in powder form. The reactor was placed in a water bath to maintain the suspension at room temperature.The selected E. coli strain was grown overnight in LB broth, centrifuged at 3000 rpm for 10 min and a suitable volume to reach 106 CFU mL−1 was suspended in 250 mL of sterile isotonic water. Separately 0.05 g of photocatalyst was sonicated for 15 min in 250 mL of isotonic water. The volumes were mixed under stirring and the initial bacterial density sampled. The stirred mixture was left for 60 min in the dark to establish an equilibrium of adsorption between bacteria and the catalyst. After 60 min of dark phase the reactor was exposed to the UVA radiation from the 125 W lamp pre-warmed for 20 min. The stirred mixture was then sampled after 15 min, 30 min, 60 min, 120 min, and 135 min. Additionally two control measurements were also carried out, these being bacterial inactivation due to UVA radiation damage only, labelled as UVA control and the inactivation of bacteria with the optimum catalyst loading in the dark, labelled as Dark Ce-400.
2.6 Bacterial enumeration
Spread plate method was used for bacterial enumeration using TBX Agar. All samples were appropriately serially diluted and 100 μL (200 μL for sampling times 120 min and 135 min) were plated in triplicates and incubated at 44 °C for 20–24 h. Plots of means of triplicate measurements together with the standard error of the mean are presented herein.3. Results and discussion
3.1 Catalysts characterisation
XRD patterns of the undoped ZnO and Ce-doped ZnO samples are shown in Fig. 1. XRD of undoped ZnO showed five main patterns located at 2θ 32.06°, 34.74°, 36.53°, 47.85° and 56.9°, respectively associated to the (1 0 0), (0 0 2), (1 0 1), (1 0 2) and (1 1 0) planes of hexagonal wurtzite ZnO crystal structure.39 The main diffraction patterns of ZnO crystalline structure were still observed after the doping with Ce, underlining that no change in the ZnO structure occurred upon Ce doping. For Ce-700 and Ce-1000 samples, an additional wide diffraction peak located at about 28.5° starts to be visible. This last signal could be due to cerium oxide formed on the photocatalysts surface.40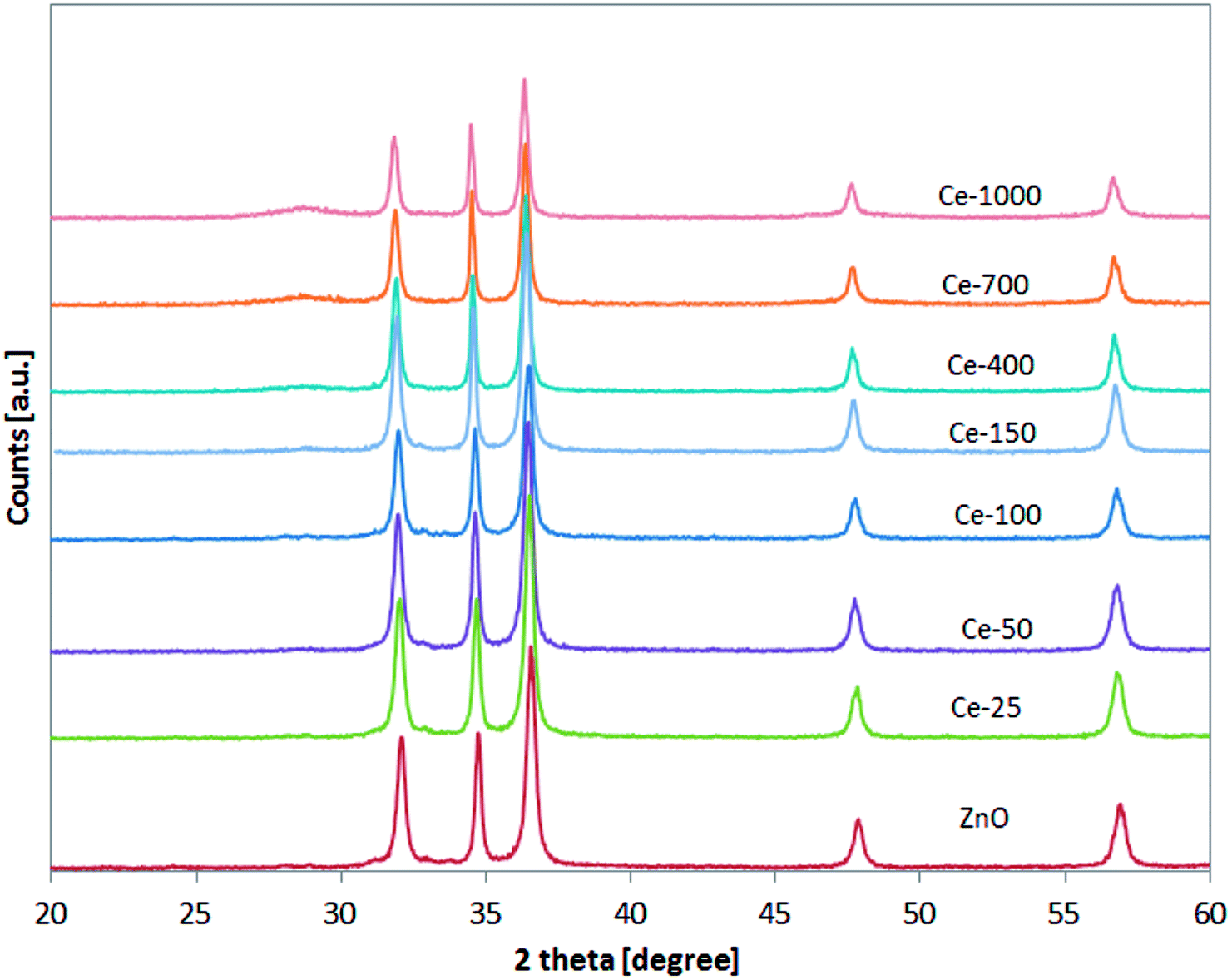 | ||
| Fig. 1 XRD analysis of all the synthesised photocatalysts in the range 20–60°. | ||
XRD spectra of the samples were analysed more accurately in the range 31–33° (Fig. 2). It is possible to observe that the position of the diffraction pattern associated to (1 0 0) plane shifted towards lower angle with increasing the Ce content, indicating that the lattice parameters of ZnO increased.41 This result can be explained considering that the radius of Ce3+ ion is much larger than that of Zn2+ (0.074 nm), so inducing a shift of diffraction peak and indicating that the Ce ions have been successfully incorporated into the ZnO lattice and substituted the Zn2+ sites.42
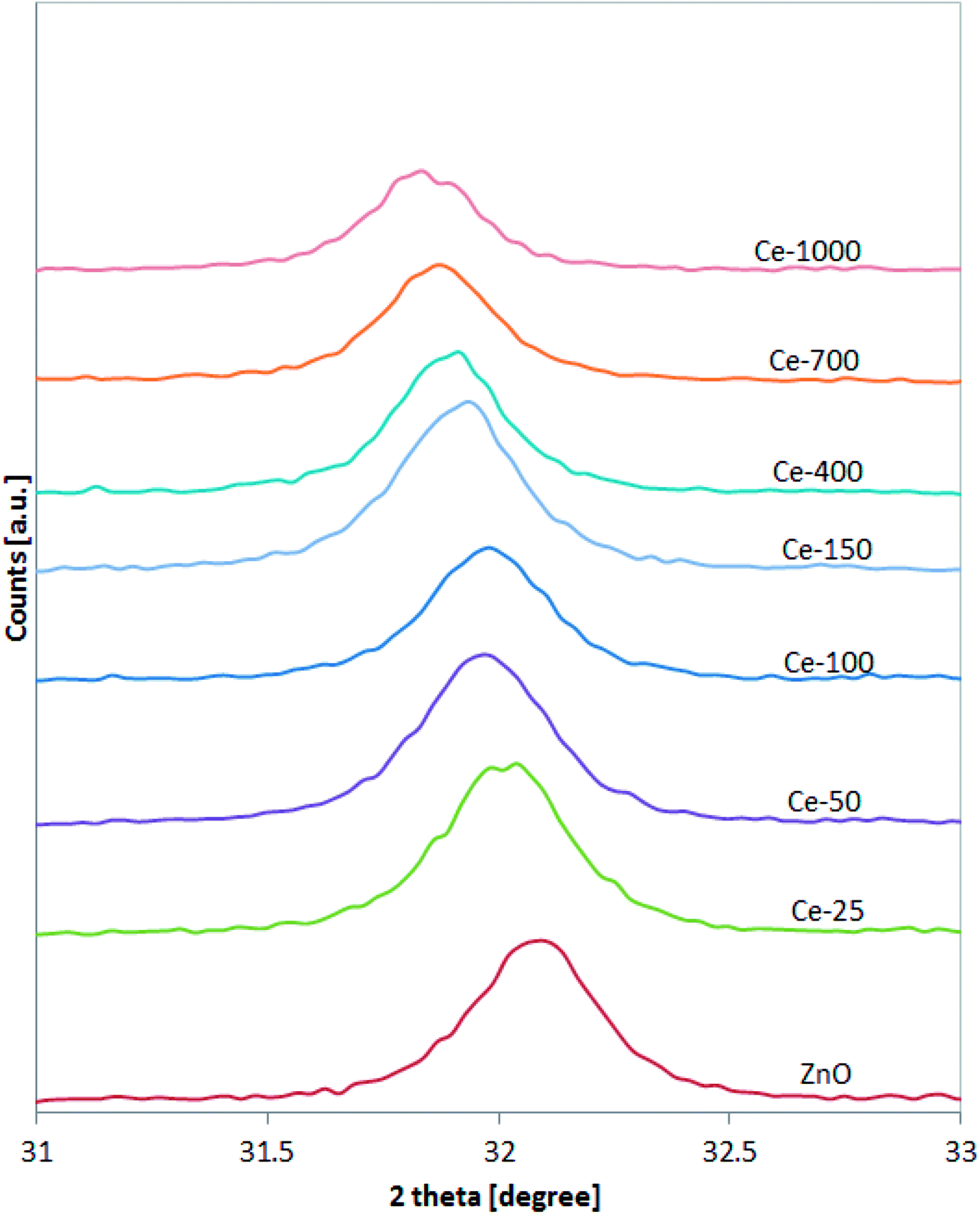 | ||
| Fig. 2 XRD analysis of all the synthesised photocatalysts in the range 31–33°. | ||
The successful doping with Ce and the formation of CeO2 was further confirmed by Raman spectroscopy (Fig. 3).
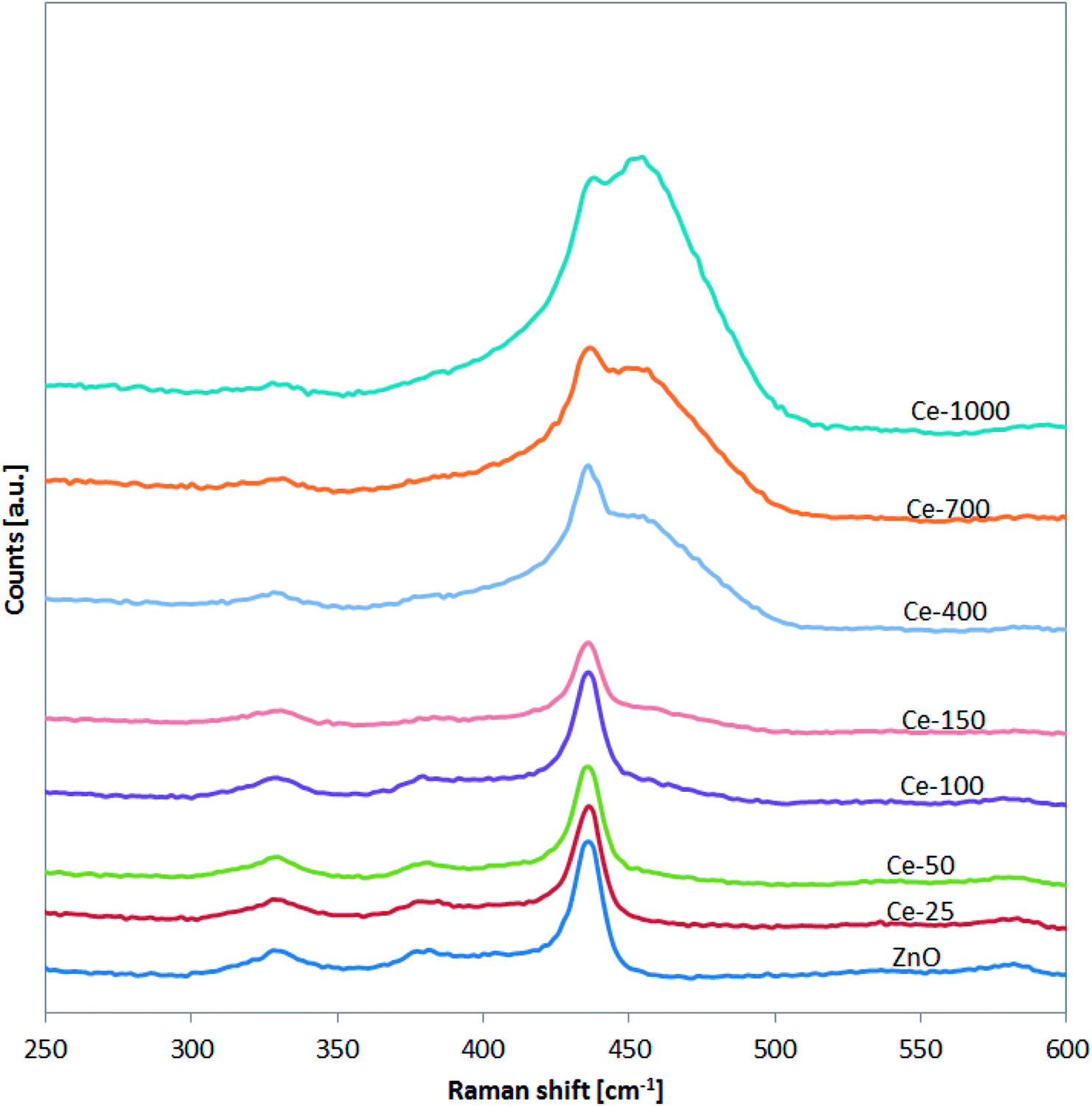 | ||
| Fig. 3 Raman spectra of undoped ZnO and Ce-doped ZnO photocatalysts in the range 250–600 cm−1. | ||
In particular, for the undoped ZnO and Ce-25, Ce-50, Ce-100 and Ce-150, it is possible to note the presence of the signals located at about 332, 379, and 437 cm−1 assigned to vibration modes of wurtzite phase of ZnO.43 Compared to undoped ZnO, the main Raman signal of the ZnO structure for Ce-400, Ce-700 and Ce-1000 samples was shifted from 437 to about 433 cm−1. This shift is due to stress phenomena induced by the lattice distortion of ZnO crystalline structure, in agreement with literature concerning Ce-doped ZnO.44,45 Moreover, in the Raman spectra of the same samples (Ce-400, Ce-700 and Ce-1000), the signal observed at about 456 cm−1 is originated from the Raman active mode characteristic of CeO2 fluorite-structured materials corresponding to the ceria Ce–O8 vibrational unit.46
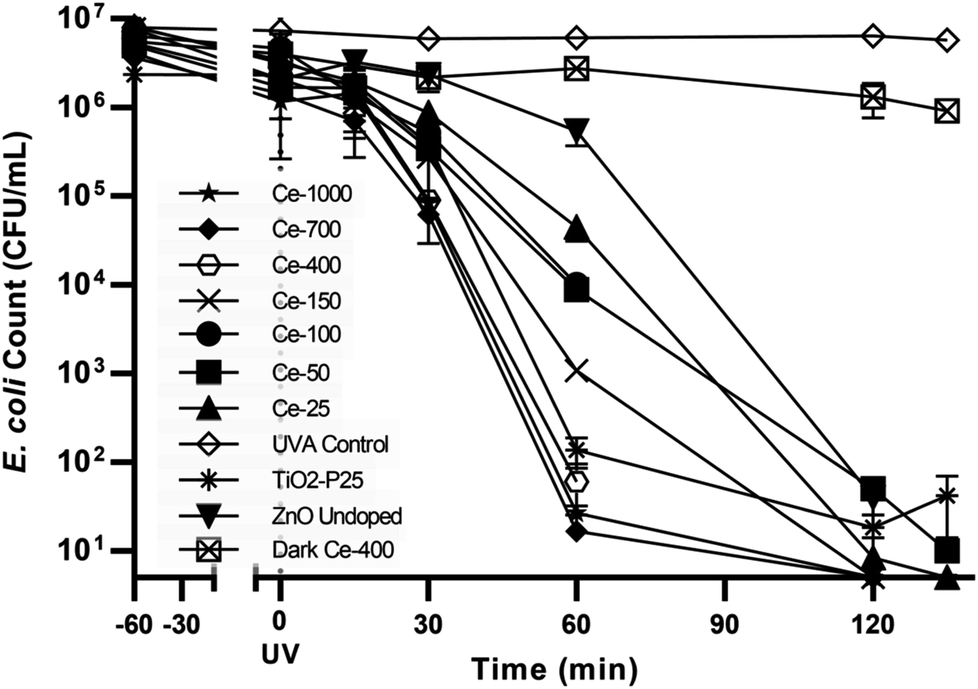 | ||
| Fig. 4 Control tests and E. coli photocatalytic inactivation for all the synthesised photocatalysts. | ||
UV-vis DRS spectra of undoped ZnO and Ce doped ZnO samples were used to calculate the energy band gap (Ebg) of the samples and the obtained values are reported in Table 1.
| Labelled as | Cerium content Ce:Zn at/at |
Cerium content as measured by XRF Ce:Zn at/at |
SSA (BET method), m2 g−1 | Ebg, eV |
|---|---|---|---|---|
| ZnO | No cerium | No cerium | 14 | 3.21 |
| Ce-25 | 0.0025:1 |
0.002:1 |
19 | 3.19 |
| Ce-50 | 0.0050:1 |
0.005:1 |
23 | 3.18 |
| Ce-100 | 0.0100:1 |
0.011:1 |
19 | 3.17 |
| Ce-150 | 0.0150:1 |
0.017:1 |
23 | 3.16 |
| Ce-400 | 0.0400:1 |
0.038:1 |
22 | 3.15 |
| Ce-700 | 0.0700:1 |
0.074:1 |
27 | 3.00 |
| Ce-1000 | 0.1000:1 |
0.140:1 |
28 | 2.97 |
| TiO2-P25 | N/A | N/A | 47 | 3.30 |
The decrease of Ebg from 3.21 eV of undoped ZnO to 2.97 eV of Ce-1000 sample can be ascribed to the formation of a shallow level inside the band gap, because of impurity atoms (Ce3+) introduced into the wurtzite ZnO crystalline structure.47 These results are in agreement with UV-vis absorption results previously described and with recent reports in literature.45
The specific surface areas (SSA) of all the investigated samples are also reported in Table 1. All the doped samples show a higher surface area than undoped ZnO (14 m2 g−1 cf. 28 m2 g−1 for ZnO and Ce-1000, respectively), in agreement with the scientific literature.41 The synthetic method used herein also produced higher SSA than that reported by Wang et al. using a similar method but involving temperature shifts.36 As for SSA with the Ce:Zn ratio, there is not a clear cut trend. The ZnO Ce-700 and ZnO Ce-1000 did produce a higher SSA but the lower atom ratios did not gradually give an increase. In all atom ratio doesn't seem to greatly impact the surface area.
The Ce:Zn molar ratio in the samples was determined by XRF analysis (Table 1). It is possible to observe that, in all cases, the measured Ce:Zn atomic ratio well fits the nominal values indicating a good yield of the doping process.
3.2 Inactivation of E. coli
The UVA only blank (UVA control) i.e. without any catalyst showed negligible bacterial inactivation excluding bacterial death due to UVA damage (Fig. 4). This is in agreement with literature48 since it is know that UVA does not have sufficient energy to ionize DNA directly as UVC has. A dark control for the activity of the optimal catalyst, ZnO Ce-400, was also carried out. The activity in the dark was rather limited with 0.6log10 decrease measured after 135 min. This excludes substantial bacterial cell inactivation due to any potential toxicity of the catalyst to the cells implying inactivation being due to photocatalytic oxidative damage. The reduction of culturable bacteria in the dark is attributed to interactions between the cells and the powdered photocatalyst and not photocatalysis (i.e. the action of ROS).49 For this reason, 60 min of dark phase are included prior to each photocatalytic experiment to specifically measure the rate of removal due to photocatalysis.
The optimisation of cerium load in zinc oxide yielded drastic improvements in inactivation efficiencies when compared to the undoped ZnO. This is most noticeable after 60 min of treatment, the removal efficiency of all the doped ZnO is 1 to 3 orders of magnitude higher then ZnO undoped (Fig. 5). A gradual increase in efficiency was observed with cerium load. E. coli inactivation was higher than 99.995% for Ce-400, Ce-700, Ce-1000 within 60 min treatment time. It is worthy to mention that they even performed better than the standard TiO2 Degussa P25. By the end of the treatment time the percentage of inactivation reach comparable levels for all catalysts tested. Calza et al. obtained similar results with 1% cerium doped ZnO,32 however the fastest of the doped catalysts therein was in the same range of TiO2 under UVA light, and only under UV-visible light did the cerium doped ZnO overtake TiO2. This is expected since the doped catalyst has a lower band gap energy and thus can harness a larger part of the visible light spectrum.
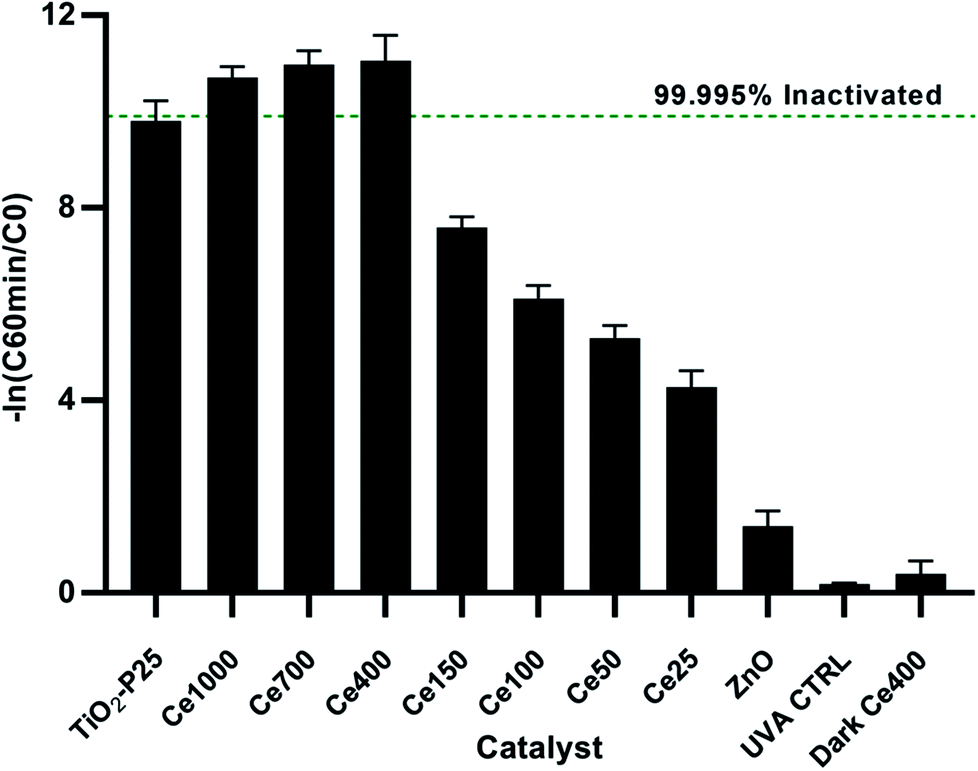 | ||
| Fig. 5 Relative inactivation after 60 min of treatment. | ||
3.3 Kinetics of E. coli inactivation
To better visualise the photocatalyst with highest activity, linear regression was fitted for the normalised inactivation in natural log form (ln[Ct/C60 min dark]) and time. This gives the pseudo first order equations of the inactivation kinetics of each photocatalyst. The slope of each linear fit represents the rate of inactivation efficiency, k in min−1 in this case, for its respective cerium doped photocatalyst. Ce-400 shows the largest slope i.e. highest activity due to having the highest rate constant. To better visualise the relationship between cerium load and inactivation efficiency, the rate constants are plotted against cerium load as measured by XRF (Table 1 and Fig. 6).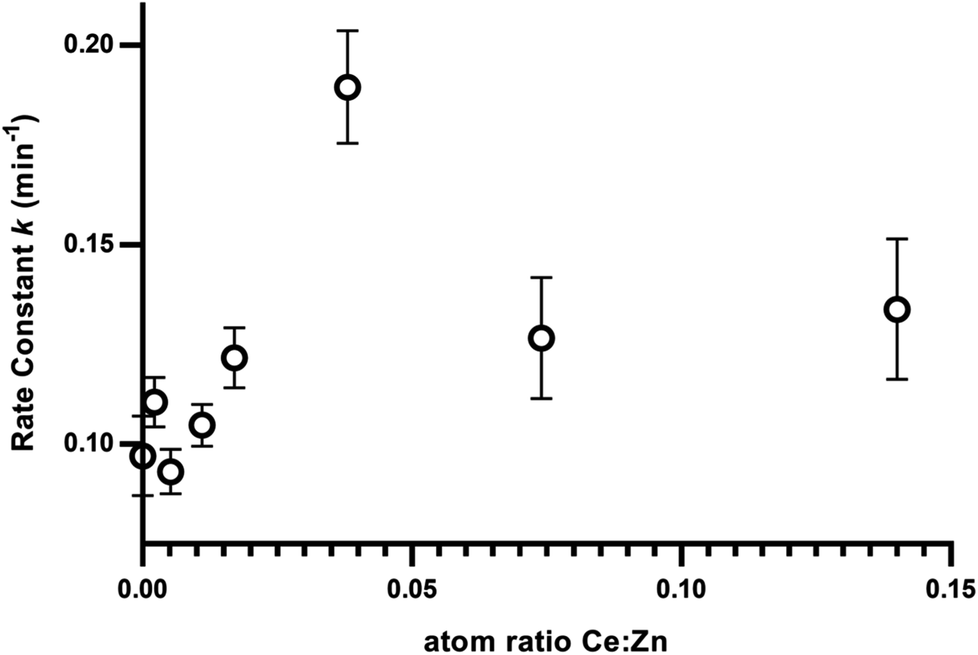 | ||
| Fig. 6 Rate constant from the linear regression of inactivation rates with the load of Ce. | ||
There seems to be a positive correlation between cerium load and efficiency of inactivation up to a peak at ZnO–Ce400 (i.e. 0.038:1 Ce:Zn as measured by XRF). The two higher doping levels of cerium doped catalysis (namely Ce-700 and Ce-1000) did not produce further improvement but maintained a high level of efficiency. The optimised quantity of cerium for bacterial inactivation is thus determined to be 0.04 atoms of cerium per atom of zinc.
To the best of our knowledge, cerium doped ZnO has not been used for photocatalytic bacterial inactivation, it has however been used in photocatalytic reactions. Paganini et al. investigated the doping of ZnO with 1% of cerium.37 Their results show a decisive improvement in the removal of phenol (>95% degraded for the doped ZnO vs. 50% for undoped ZnO) and up to four times faster for the three contrast dyes investigated using Ce doped ZnO over TiO2 P-25 and undoped ZnO for the same duration. Jiang et al. showed optimum performance of cerium doped ZnO against methyl orange at 0.3% Ce loading and slower removal rates at the two other Ce loadings of 0.1% and 0.5%.33 Calza et al. observed higher efficiencies in removal of acesulfame K at 1% Ce loading over 0.5%,32 The differences between optimums for different substrates indicates that the optimisation should be targeted towards the substrate of interest. Since there is a clear correlation between ˙OH radical generation and bacterial inactivation50 and since the removal of organic compounds by photocatalysis is based on the same radical chemistry, there might be room for additional improvement in the removal of chemicals by further increasing the cerium load to the optimum of 4% identified herein. However, the efficiency of removal of both chemical compounds and biological matter does not exclusively depend on quantity of ROS generation. Physicochemical conditions such as adsorption are affected by doping.26,27 This in turn affects reaction rates since a better adsorbed species is in closer proximity to the site of generation of the very short-lived radicals increasing the probability that the radicals take part in a reaction with the compound of interest.
A number of novel photocatalysts that show higher bacterial inactivation activity than Degussa TiO2 P-25 are reported in literature. Zhu et al. synthesised a composite film of P/Ag/Ag2O/Ag3PO4/TiO2 catalyst reporting fast disinfection of E. coli under high intensity solar light (50 mW cm−2) in a tubular reactor.51 Karaolia et al. used reduced graphene oxide–TiO2 composites for the disinfection of bacteria and antibiotic resistant bacteria showing increased activity in some cases over commercial TiO2.15 Higher activity was observed in co-deposited TiO2 and WO3 on reduced graphene oxide to suppress electron hole-pair recombination.52 Regrettably, photocatalysts involving carbon materials such as graphene oxide, reduced graphene oxide and graphene have the disadvantage of involving high cost in the synthesis and usually employing the Hummer's method which leave much to desire in terms of green synthetic practices. Ibrahim and Asal, used a similarly facile method as the one used herein to dope ZnO with 3 lanthanides giving improvements in bacterial inactivation using UV-A lamps at 0.9 mW cm−2 over undoped ZnO but the results were not compared against TiO2 in the same conditions.53 The three lanthanides used gave very similar results at 0.025 mole ratio of the dopant. However, the highest activity in inactivation efficiency for all three cases was relatively low with only approximately 80% of the initial 2.3 × 104 CFU mL−1 inactivated within 120 min of treatment time. The authors attributed the higher activity of the lanthanide doped compared to un-doped ZnO to modified surface properties that led to higher adsorption and bacterial inactivation rates. Their results however show only marginally improved activity of the optimal doped catalyst over their un-doped ZnO. In contrast the cerium doped catalysts used herein showed up to log3 higher efficiency of inactivation relative to un-doped ZnO after 60 min of treatment and at comparable UV irradiance of 1.26 mW cm−2. Li et al. synthesised ZnO/ZnFe2O4 coupled photocatalysts using high temperature treatment of sphalerite.54 At 1 g L−1 catalyst loading and 3.3 mW cm−2 of visible light with the UV region filtered out, the catalysts reported therein showed improvement over ZnO in terms of bacterial inactivation with the best performing catalyst achieving complete inactivation after 180 min of treatment while ZnO produced less than 1log after 180 min and less than 2log after 300 min. Dark controls for bacterial inactivation and activity relative to the established standard TiO2-P25 are not reported. The main limitation, together with long treatment times, is the high temperature (1200 °C) needed for the synthesis of the optimised catalysts. Kubacka et al. used zinc and silver modified titania at 0.6 g L−1 to improve the activity over unmodified titania against E. coli.55 However, the experimental setup reported therein (spectrophotometer used as light source) limits the comparability of results. Venieri et al. doped TiO2 using a similar method to the one used herein to produce photocatalysts with higher activity under simulated solar light at an intensity of 13.1 mW cm−2. Complete inactivation was recorded after 10–15 min for optimised catalysts and 30 min for TiO2-P25.56 Gupta et al. demonstrated an improvement in activity over unmodified ZnO by the interfacial coupling of copper and silver on iron doped ZnO. 6log10 inactivation was recorded for 3 wt% Cu coupled iron doped ZnO under visible light (>400 nm 68 klx) and 0.25 g L−1 catalyst load. For chemical pollutant degradation an enhanced removal rate over unmodified ZnO was observed and this improvement was attributed to decreased electron hole-pair recombination.57 It is also worth noting that the improvement in the rate of disinfection demonstrated with ZnO–Ce400 is not due to a simple shift in the band gap resulting in a higher portion of the electromagnetic spectrum being utilised. This suggests that the higher activity is potentially due to modified surface activity resulting in higher affinity to the bacterial cells and/or reduced recombination rates of electron–hole pairs.
4. Conclusions
There are two main limitations hindering the application of photocatalysis as tertiary treatment method in UWTPs: (1) the lack of regulations to control the release of micropollutants (also known as contaminants of emerging concern) and (2) difficult to upscale production of photocatalysts from literature to industrial methods. These issues make photocatalysis not competitive with alternative water treatment options available on the market. In the attempt to contribute to overcome limitation (2), a new photocatalyst was optimised in this work to effectively inactivate indigenous E. coli selected from the secondary treatment effluent of a UWTP. The catalyst was synthesised via the hydroxide induced hydrolysis of zinc nitrate demonstrating that a non time consuming, easy to upscale method which utilises inexpensive precursors produces substantial improvements to rate of inactivation of E. coli. ZnO doped with cerium at 0.04:1 at/at Ce:Zn at 0.1 g L−1 concentration inactivated 99.995% of the initial bacteria after 60 min and below the limits of quantification after 135 min of treatment and 1.26 mW cm−2 at 365 nm. The results achieved are highly encouraging, even exceeding the efficiency of the standard TiO2 Degussa P25 in identical conditions.
Conflicts of interest
There are no conflicts of interest.Acknowledgements
This work is part of a project that has received funding from the European Union's Horizon 2020, under the Innovative Training Networks (ITN-ETN) programme Marie Skłodowska-Curie grant (ANtibioticS and mobile resistance elements in WastEwater Reuse applications: risks and innovative solutions) agreement No. 675530. The content of this publication reflects only the authors' views and the Research Executive Agency is not responsible for any use that may be made of the information it contains.References
- B. Bates, Z. Kundzewicz, S. Wu and J. Palutikof, Climate Change, 2008, vol. 95, p. 96 Search PubMed.
- Food and Agriculture Organization of the United Nations, The State of the World's Land and Water Resources for Food and Agriculture (SOLAW) – Managing Systems at Risk, Rome and London, 2011 Search PubMed.
- N. V. Paranychianakis, M. Salgot, S. A. Snyder and A. N. Angelakis, Crit. Rev. Environ. Sci. Technol., 2015, 45, 1409–1468 CrossRef.
- BIO by Deloitte, Optimising water reuse in the EU – Final report prepared for the European Commission (DG ENV) Part 1, Report 07.0307/2013/658572/ENV.C1, 2015 Search PubMed.
- I. K. Kalavrouziotis, P. Kokkinos, G. Oron, F. Fatone, D. Bolzonella, M. Vatyliotou, D. Fatta-Kassinos, P. H. Koukoulakis and S. P. Varnavas, Desalin. Water Treat., 2015, 53, 2015–2030 CrossRef.
- L. Rizzo, R. Krätke, J. Linders, M. Scott, M. Vighi and P. de Voogt, Current Opinion in Environmental Science & Health, 2018, 2, 7–11 Search PubMed.
- A. di Cesare, E. M. Eckert, S. d'Urso, R. Bertoni, D. C. Gillan, R. Wattiez and G. Corno, Water Res., 2016, 94, 208–214 CrossRef PubMed.
- W. Lee and P. Westerhoff, Environ. Sci. Technol., 2005, 39, 879–884 CrossRef PubMed.
- J. Hollender, S. G. Zimmermann, S. Koepke, M. Krauss, C. S. McArdell, C. Ort, H. Singer, U. von Gunten and H. Siegrist, Environ. Sci. Technol., 2009, 43, 7862–7869 CrossRef PubMed.
- I. García-Fernández, M. I. Polo-López, I. Oller and P. Fernández-Ibáñez, Appl. Catal., B, 2012, 121–122, 20–29 CrossRef.
- G. Ferro, F. Guarino, A. Cicatelli and L. Rizzo, J. Hazard. Mater., 2017, 323, 426–433 CrossRef PubMed.
- A. Fiorentino, G. De Luca, L. Rizzo, G. Viccione, G. Lofrano and M. Carotenuto, Environ. Sci., 2018, 69, 95–104 CrossRef PubMed.
- J. C. Ireland, P. Klostermann, E. W. Rice and R. M. Clark, Appl. Environ. Microbiol., 1993, 59, 1668–1670 Search PubMed.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef PubMed.
- P. Karaolia, I. Michael-Kordatou, E. Hapeshi, C. Drosou, Y. Bertakis, D. Christofilos, G. S. Armatas, L. Sygellou, T. Schwartz, N. P. Xekoukoulotakis and D. Fatta-Kassinos, Appl. Catal., B, 2018, 224, 810–824 CrossRef.
- L. Rizzo, A. Della Sala, A. Fiorentino and G. Li Puma, Water Res., 2014, 53, 145–152 CrossRef PubMed.
- L. Rizzo, D. Sannino, V. Vaiano, O. Sacco, A. Scarpa and D. Pietrogiacomi, Appl. Catal., B, 2014, 144, 369–378 CrossRef.
- D. L. Sedlak and U. von Gunten, Science, 2011, 331, 42–43 CrossRef PubMed.
- X. Qu, P. J. J. Alvarez and Q. Li, Water Res., 2013, 47, 3931–3946 CrossRef PubMed.
- K. M. Lee, C. W. Lai, K. S. Ngai and J. C. Juan, Water Res., 2016, 88, 428–448 CrossRef PubMed.
- S. G. Kumar and K. S. R. K. Rao, RSC Adv., 2015, 5, 3306–3351 RSC.
- C. Sushma and S. Girish Kumar, Chem. Pap., 2017, 71, 2023–2042 CrossRef.
- W. Raza, S. M. Faisal, M. Owais, D. Bahnemann and M. Muneer, RSC Adv., 2016, 6, 78335–78350 RSC.
- S. Adhikari, R. Gupta, A. Surin, T. S. Kumar, S. Chakraborty, D. Sarkar and G. Madras, RSC Adv., 2016, 6, 80086–80098 RSC.
- K. Qi, B. Cheng, J. Yu and W. Ho, J. Alloys Compd., 2017, 727, 792–820 CrossRef.
- S.-m. Chang and W.-s. Liu, Appl. Catal., B, 2014, 156–157, 466–475 CrossRef.
- K. Wilke and H. D. Breuer, J. Photochem. Photobiol., A, 1999, 121, 49–53 CrossRef.
- H. Chen, H. Yu, S. Cui, J. Xu, Y. Zhang and C. Liu, Phys. B, 2017, 516, 36–40 CrossRef.
- F. M. Li, X. B. Li, S. Y. Ma, L. Chen, W. Q. Li, C. T. Zhu, X. L. Xu, Y. Chen, Y. F. Li and G. Lawson, J. Alloys Compd., 2015, 649, 1136–1144 CrossRef.
- A. J. Kulandaisamy, V. Elavalagan, P. Shankar, G. K. Mani, K. J. Babu and J. B. B. Rayappan, Ceram. Int., 2016, 42, 18289–18295 CrossRef.
- D. Gao, L. Lyu, B. Lyu, J. Ma, L. Yang and J. Zhang, Mater. Res. Bull., 2017, 89, 102–107 CrossRef.
- P. Calza, C. Gionco, M. Giletta, M. Kalaboka, V. A. Sakkas, T. Albanis and M. C. Paganini, J. Hazard. Mater., 2017, 323, 471–477 CrossRef PubMed.
- J. Jiang, K. Zhang, X. Chen, F. Zhao, T. Xie, D. Wang and Y. Lin, J. Alloys Compd., 2017, 699, 907–913 CrossRef.
- N. C. Birben, M. C. Paganini, P. Calza and M. Bekbolet, Photochem. Photobiol. Sci., 2017, 16, 24–30 RSC.
- C.-J. Chang, C.-Y. Lin and M.-H. Hsu, J. Taiwan Inst. Chem. Eng., 2014, 45, 1954–1963 CrossRef.
- L. Wang, Z. Ji, J. Lin and P. Li, Mater. Sci. Semicond. Process., 2017, 71, 401–408 CrossRef.
- M. C. Paganini, D. Dalmasso, C. Gionco, V. Polliotto, L. Mantilleri and P. Calza, ChemistrySelect, 2016, 1, 3377–3383 CrossRef.
- R. A. McBride, J. M. Kelly and D. E. McCormack, J. Mater. Chem., 2003, 13, 1196–1201 RSC.
- V. Vaiano, M. Matarangolo, O. Sacco and D. Sannino, Appl. Catal., B, 2017, 209, 621–630 CrossRef.
- H. Guo and Y. Qiao, Appl. Surf. Sci., 2008, 254, 1961–1965 CrossRef.
- M. Ahmad, E. Ahmed, F. Zafar, N. R. Khalid, N. A. Niaz, A. Hafeez, M. Ikram, M. A. Khan and Z. Hong, J. Rare Earths, 2015, 33, 255–262 CrossRef.
- C.-J. Chang, C.-Y. Lin, J.-K. Chen and M.-H. Hsu, Ceram. Int., 2014, 40, 10867–10875 CrossRef.
- K. A. Alim, V. A. Fonoberov, M. Shamsa and A. A. Balandin, J. Appl. Phys., 2005, 97, 124313 CrossRef.
- S. Maensiri, P. Laokul and V. Promarak, J. Cryst. Growth, 2006, 289, 102–106 CrossRef.
- R. Bomila, S. Srinivasan, A. Venkatesan, B. Bharath and K. Perinbam, Mater. Res. Innovations, 2017, 1–8, DOI:10.1080/14328917.2017.1324379.
- B. Chouchene, T. B. Chaabane, L. Balan, E. Girot, K. Mozet, G. Medjahdi and R. Schneider, Beilstein J. Nanotechnol., 2016, 7, 1338 CrossRef PubMed.
- T. B. Ivetić, M. R. Dimitrievska, I. O. Gúth, R. Đ. Lj and S. R. Lukić-Petrović, Journal of Research in Physics, 2012, 36, 43–51 Search PubMed.
- K. P. Kühn, I. F. Chaberny, K. Massholder, M. Stickler, V. W. Benz, H.-G. Sonntag and L. Erdinger, Chemosphere, 2003, 53, 71–77 CrossRef.
- O. Yamamoto, K. Nakakoshi, T. Sasamoto, H. Nakagawa and K. Miura, Carbon, 2001, 39, 1643–1651 CrossRef.
- M. Cho, H. Chung, W. Choi and J. Yoon, Water Res., 2004, 38, 1069–1077 CrossRef PubMed.
- Q. Zhu, X. Hu, M. S. Stanislaus, N. Zhang, R. Xiao, N. Liu and Y. Yang, Sci. Total Environ., 2017, 577, 236–244 CrossRef PubMed.
- X. Zeng, Z. Wang, G. Wang, T. R. Gengenbach, D. T. McCarthy, A. Deletic, J. Yu and X. Zhang, Appl. Catal., B, 2017, 218, 163–173 CrossRef.
- M. M. Ibrahim and S. Asal, J. Mol. Struct., 2017, 1149, 404–413 CrossRef.
- Y. Li, Y. Li, Y. Yin, D. Xia, H. Ding, C. Ding, J. Wu, Y. Yan, Y. Liu, N. Chen, P. K. Wong and A. Lu, Appl. Catal., B, 2018, 226, 324–336 CrossRef.
- A. Kubacka, M. J. Muñoz-Batista, M. Ferrer and M. Fernández-García, Appl. Catal., B, 2013, 140–141, 680–690 CrossRef.
- D. Venieri, A. Fraggedaki, M. Kostadima, E. Chatzisymeon, V. Binas, A. Zachopoulos, G. Kiriakidis and D. Mantzavinos, Appl. Catal., B, 2014, 154–155, 93–101 CrossRef.
- R. Gupta, N. K. Eswar, J. M. Modak and G. Madras, Catal. Today, 2018, 300, 71–80 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05020a |
| This journal is © The Royal Society of Chemistry 2018 |