
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTracking intramolecular energy redistribution dynamics in aryl halides: the effect of halide mass†
Xiaosong Liu a,
Yunfei Song‡
b,
Wei Zhanga,
Gangbei Zhub,
Zhe Lva,
Weilong Liu*a and
Yanqiang Yang*ab
a,
Yunfei Song‡
b,
Wei Zhanga,
Gangbei Zhub,
Zhe Lva,
Weilong Liu*a and
Yanqiang Yang*ab
aDepartment of Physics, Harbin Institute of Technology, Harbin, China
bNational Key Laboratory of Shock Wave and Detonation Physics, Institute of Fluid Physics, China Academy of Engineering Physics, Mianyang, China. E-mail: yqyang@caep.cn
First published on 23rd August 2018
Abstract
Selective excitation of C–H, C–C, CX1 and CX2 stretching vibrational modes in an orderly manner, detection of intramolecular energy redistribution and vibrational coupling in the electronic ground state of aryl halides are performed by time- and frequency-resolved Coherent Anti-Stokes Raman Scattering (CARS) spectroscopy. Intramolecular energy flow from parent modes to daughter modes is observed in the experiment. According to the experimental results, it is found that the up-hill vibrational energy flow from lower frequency modes to higher frequency ones is counterintuitive and energy redistribution efficiencies are controlled by the mass of the halide. The selectivity and directionality of energy flow are also discussed in view of vibrational symmetry.
Introduction
The substituent effect is one of the most important concepts in chemistry, biochemistry, and related fields.1–4 When substituted benzenes became the subject of a spectroscopic study,5–8 there was much confusion regarding the results, such as the influence of substituents on reactivity and stability of the very wide range of organic species containing aromatic moieties.9–12 Analysis of mono-substituted benzenes reveals that the influence of a single group will be essential.11,12Monosubstituted benzenes are chosen for this study because of our prior knowledge of benzene's intramolecular energy redistribution,13 and we can describe a series of molecules where the basic phenyl structure remains unchanged. According to our previous work,13 isolated-molecule energy flow of benzene was described, so the vibrational energy redistribution observed in liquid aryl halides X–Ph (X = F, Cl, Br, I; –Ph = C6H5) is entirely a consequence of intramolecular interactions. Using the narrow band CARS technique, it is easy to understand the effect of substituent mass on vibrational energy redistribution. We see two prominent ring stretching modes, νCH (∼3070 cm−1), νCC (∼1580 cm−1), whose frequencies in benzene and in aryl halides are quite similar, along with two substituent-dependent modes of νCX1 (∼1080 cm−1), and νCX2 (∼700 cm−1). In subsequent comparisons of intramolecular energy redistribution within the series of aryl halides F, Cl, Br and I, we will refer to increasing mass, that means halide substitution can affect the vibrational dynamic via mass effects.
In this work, vibrational modes of phenyl and substituent-dependent are selectively excited, and the normal intramolecular energy flow from high frequency phenyl modes to neighbouring bands and the counterintuitive up-hill energy flow from substituent-dependent modes to higher phenyl modes are detected. Quantum beats among these relevant modes in the vibrational ground state are observed, and the vibrational coupling information is extracted via Fourier analysis. The effects of halide mass on the efficiencies of intramolecular energy redistribution are described.
Experimental
The light source we used is a Ti:sapphire laser system (Spectra-Physics). The duration, repetitive frequency, single pulse energy, and center wavelength of the pulse generated from a regenerative amplifier (Spitfire, Spectra-Physics) are 110 fs, 1 kHz, 500 μJ and 800 nm, respectively. The output beam is split by a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 beam splitter, then the beam with 90% energy is focused into an optical parametric amplifier (OPA) and the output pulse is further split into two beams that are used as pump and probe pulses; the other beam with 10% energy is served as the Stokes pulse (800 nm, FHWM: 13.4 nm, 795–808 nm). Photon energy difference between the Stokes and pump pulses is tuned by changing the center wavelength of the pump pulse, which can achieve the selective excitation of a particular vibrational mode. Specifically, the center wavelengths of the pump and probe pulses are changed from 763 to 645 nm, the center wavelength of the Stokes pulse is fixed at 800 nm. The Raman active vibrational modes νCX2 ∼ 700 cm−1, νCX1 ∼ 1080 cm−1, νCC (in range from 1320 cm−1 to 1590 cm−1) and νCH ∼ 3070 cm−1 are selectively excited. Regarding the beam geometry, the folded BOXCAR configuration with properly chosen angles between the beams defined by the phase-matching condition is employed. Thus, the CARS signal is spatially separated from the other beams. The detection setup consists of a spectrometer (Bruker Optics 500 IS/SM) and an intensified charge coupled device (ICCD, Andor DU440-BU2).
:1 beam splitter, then the beam with 90% energy is focused into an optical parametric amplifier (OPA) and the output pulse is further split into two beams that are used as pump and probe pulses; the other beam with 10% energy is served as the Stokes pulse (800 nm, FHWM: 13.4 nm, 795–808 nm). Photon energy difference between the Stokes and pump pulses is tuned by changing the center wavelength of the pump pulse, which can achieve the selective excitation of a particular vibrational mode. Specifically, the center wavelengths of the pump and probe pulses are changed from 763 to 645 nm, the center wavelength of the Stokes pulse is fixed at 800 nm. The Raman active vibrational modes νCX2 ∼ 700 cm−1, νCX1 ∼ 1080 cm−1, νCC (in range from 1320 cm−1 to 1590 cm−1) and νCH ∼ 3070 cm−1 are selectively excited. Regarding the beam geometry, the folded BOXCAR configuration with properly chosen angles between the beams defined by the phase-matching condition is employed. Thus, the CARS signal is spatially separated from the other beams. The detection setup consists of a spectrometer (Bruker Optics 500 IS/SM) and an intensified charge coupled device (ICCD, Andor DU440-BU2).
Results and discussion
Vibrational mode assignments
The vibrational modes of aryl halides in our work are essentially in agreement with the experimental assignments of the vibrational spectra,10,14–17 we group them into three categories, namely, the phenyl modes, the substituent modes, and the combination bands. The vibrational mods of theory calculation results by Gaussian 03 are listed in ESI (Tables 5–8†). The visible modes in the CARS experiments are sorted as follows:(1) Phenyl modes: the νCH (∼3070 cm−1, A1 or B1 symmetry); νCC (in range from 1320 cm−1 to 1590 cm−1, A1 or B1 symmetry); βCH (in range from 1000 cm−1 to 1290 cm−1, A1 or B1 symmetry); γCH (in range from 730 cm−1 to 980 cm−1, A2 or B2 symmetry); ϕCC (∼600 cm−1, B2, B1 or A2 symmetry).
(2) Substituent-dependent modes: νCX1 (∼1080 cm−1, A1 symmetry), νCX2 (∼700 cm−1, A1 symmetry).
(3) Combination bands: νCM1 ∼ 2230 cm−1 (B1 symmetry, νCX1 ∼ 1080 cm−1 + βCH ∼ 1150 cm−1); νCM2 ∼ 2500 cm−1 (A1 symmetry, νCC ∼ 1450 cm−1 + βCH ∼ 1066 cm−1).10,14
Selective excitation of the C–H stretching vibrational modes
Selective excitation of high frequency C–H stretching vibrational modes and detection of vibrational energy redistributes to lower frequency combination bands are shown in Fig. 1. The white lines are the experimental spontaneous Raman spectra of aryl halides. Vibrational modes in the frequency range from 2752 cm−1 to 3298 cm−1 are selectively excited by a pair of pump (642 nm, FHWM: 14 nm, 638–652 nm) and Stokes pulses (800 nm, FHWM: 13 nm, 795–808 nm), combination bands arising from vibrational energy redistribution are shown in Fig. 1(a)–(d), respectively. The high frequency C–H stretching vibrational modes around 3070 cm−1 of aryl halides are excited instantaneously. It is found that the intramolecular vibrational energy flow to the lower frequency substituent-dependent modes νCX1 ∼ 1080 cm−1, νCC (in range from 1320 cm−1 to 1590 cm−1) and βCH (in range from 1000 cm−1 to 1290 cm−1). Moreover, the signal at 2230 cm−1 is not fundamental modes, it emerges due to the Fermi resonance of νCX1 ∼ 1080 cm−1 and βCH ∼ 1150 cm−1 modes, another combination band at 2500 cm−1 emerges due to the Fermi resonance of νCC ∼ 1450 cm−1 and βCH ∼ 1066 cm−1 modes.5,10 Time-domain CARS spectra of fluorobenzene (F–Ph), chlorobenzene (Cl–Ph), bromobenzene (Br–Ph), and iodobenzene (I–Ph) are shown in Fig. 1(a)–(d), respectively. Quantum beats in CARS spectra are derived from vibrational coupling of C–H stretching modes and combination bands. Through Fourier analysis, the dynamic characteristics of the modes or combination bands involved in vibrational coupling can be extracted.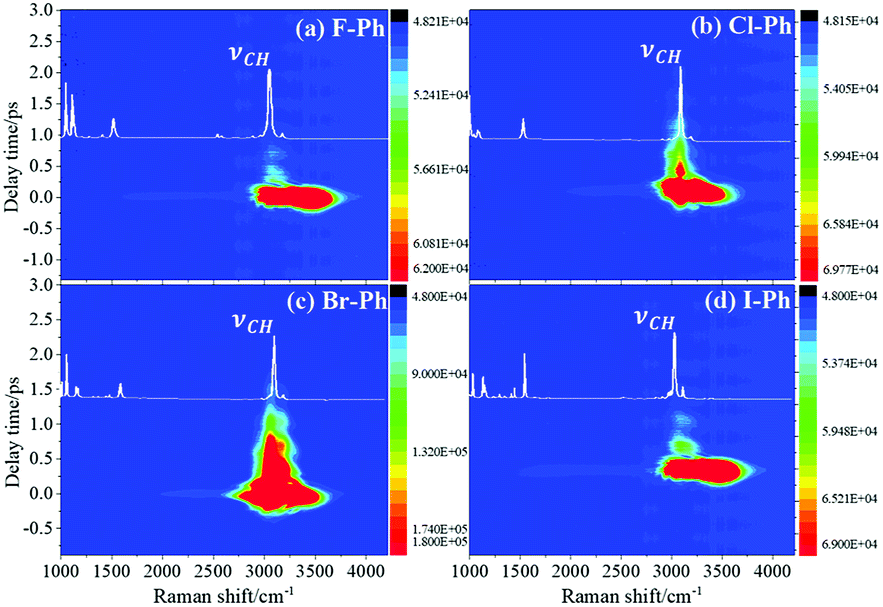 | ||
| Fig. 1 The C–H stretching vibrational modes of aryl halides are selectively excited, intramolecular energy flow to combination bands. Aryl halides: (a) F–Ph, (b) Cl–Ph, (c) Br–Ph, and (d) I–Ph. | ||
FT power spectra contain vibrational couplings information of high frequency C–H stretching vibrational modes and lower frequency combination bands which are located outside of the direct excitation region, as shown in Fig. 2. Peak positions in the FT power spectra indicate the frequency difference between the two neighbouring modes. Fig. 2(a)–(d) correspond to the Fourier analysis of CARS signals of F–Ph, Cl–Ph, Br–Ph and I–Ph, respectively. The peaks of Fourier transform (FT) power spectra that marked by Qi are corresponding to the vibrational coupling of high frequency C–H stretching vibrational modes and combination bands. Detailed information of coherent coupling among these relevant modes and their assignments are listed in ESI (Table 1†), frequency difference between the identified vibrational modes matches well with the results from the FT spectra of aryl halides.
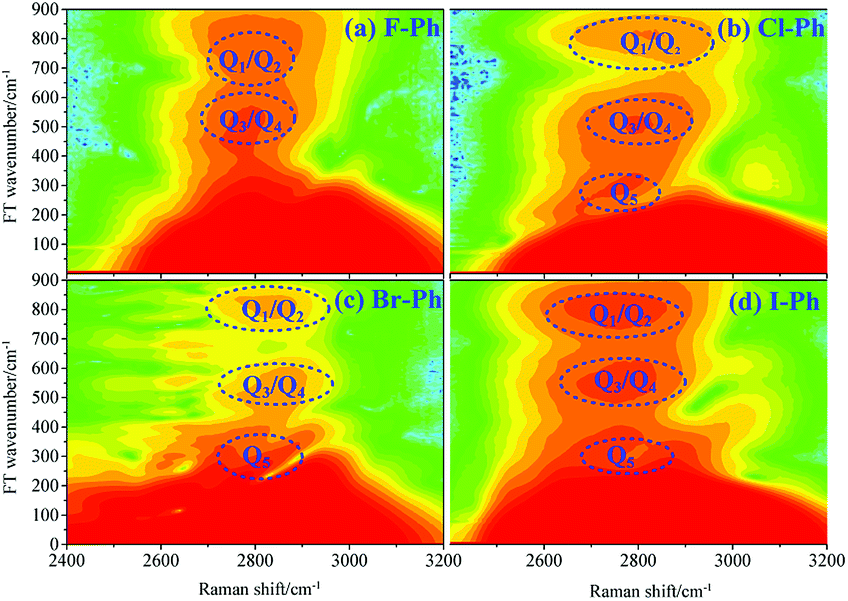 | ||
| Fig. 2 FT power spectra of the C–H stretching vibrational modes and combination bands. Aryl halides: (a) F–Ph, (b) Cl–Ph, (c) Br–Ph, and (d) I–Ph. | ||
Combining the time-domain CARS spectra and its FT power spectra, vibrational couplings between high frequency C–H stretching vibrational modes are confirmed. Meanwhile lower frequency combination bands νCM1 ∼ 2230 cm−1 and νCM2 ∼ 2500 cm−1 which are secondly excited by vibrational energy redistribution are also identified.
Selective excitation of the C–C stretching vibrational modes
Vibrational modes in the frequency range from 1362 to 1788 cm−1 are selectively excited by a pair of pump (711 nm, FHWM: 11 nm, 706–717 nm) and Stokes pulses (800 nm, FHWM: 13 nm, 795–808 nm), combination bands arising from intramolecular energy redistribution are shown in Fig. 3. There are five C–C stretching vibrational modes and all of them are excited simultaneously. Fig. 3(a)–(d) are the time-resolved CARS spectra of F–Ph, Cl–Ph, Br–Ph and I–Ph, respectively. The white-coded lines represent the experimental spontaneous Raman spectra of aryl halides. Combination bands (νCM1 ∼ 2230 cm−1 and νCM2 ∼ 2500 cm−1) and in-plane C–H deformations (βCH ∼ 1150 cm−1) modes emerge in the CARS spectra even though they are outside of direct excitation range. It denotes vibrational energy can flow from lower frequency modes to higher ones. Vibrational couplings will lead to quantum beats in the time-resolved CARS spectra, and the characteristic frequencies of beats can be extracted through Fourier analysis.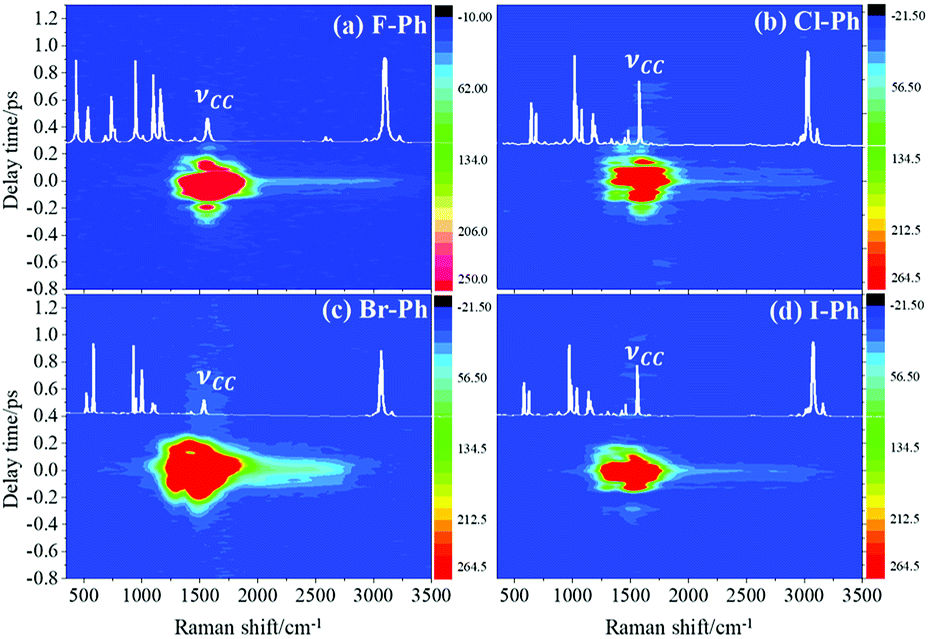 | ||
| Fig. 3 The C–C stretching vibrational modes are selectively excited, intramolecular energy flow to combination bands and in-plane C–H deformations modes (βCH ∼ 1150 cm−1). Aryl halides: (a) F–Ph, (b) Cl–Ph, (c) Br–Ph, and (d) I–Ph. | ||
Fourier analysis is performed to identify the vibrational modes involved in coherent coupling in the direct excitation region around 1500 cm−1, as shown in Fig. 4. The peaks of FT power spectra that marked by Qi (i = 1–6) are corresponding to the vibrational coupling of C–C stretching vibrational modes and in-plane C–H deformations (βCH ∼ 1150 cm−1) modes of aryl halides, as described in Fig. 4(a)–(d). Detailed assignments of coherent couplings of C–C stretching vibrational modes and in-plane C–H deformations modes are listed in ESI (Table 2†). The vibrational couplings of C–C stretching νCC (in range from 1320 cm−1 to 1590 cm−1) and βCH (in range from 1000 cm−1 to 1290 cm−1) can be identified easily.
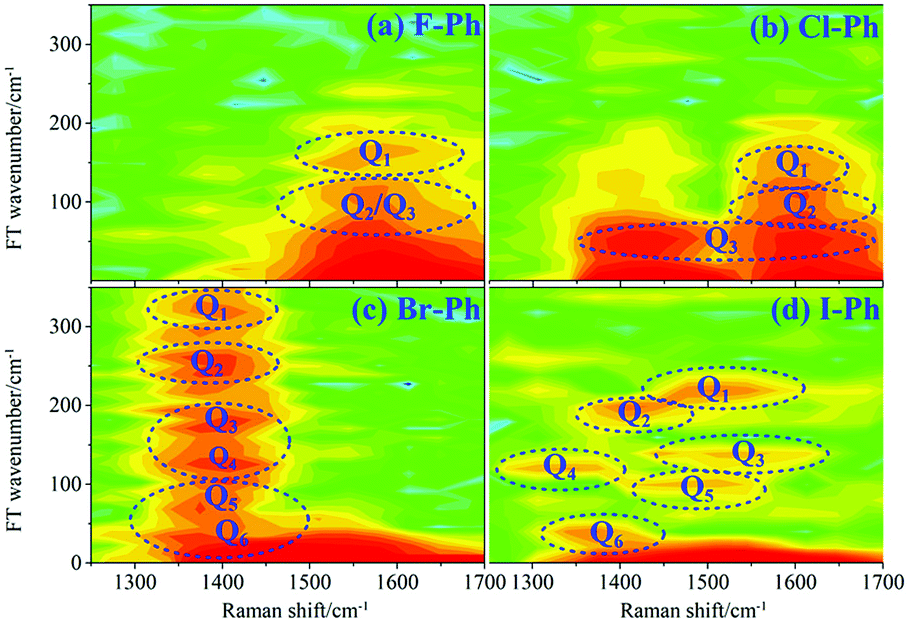 | ||
| Fig. 4 FT power spectra of C–C stretching vibrational modes νCC (∼1580 cm−1) and in-plane C–H deformations modes (βCH ∼ 1150 cm−1). Aryl halides (a) F–Ph, (b) Cl–Ph, (c) Br–Ph, and (d) I–Ph. | ||
Combining the results of time-domain CARS spectra of aryl halides and their FT power spectra, it can be confirmed that the vibrational energy can flow from C–C stretching vibrational modes to combination bands and the in-plane C–H deformation modes. Vibrational modes of C–C stretching νCC (in range from 1320 cm−1 to 1590 cm−1, A1 or B1 symmetry) involved in couplings are confirmed.
Selective excitation of the substituent-dependent mode νCX1
Vibrational modes in the frequency range from 880 cm−1 to 1303 cm−1 are directly excited by pump (745 nm, FHWM: 12 nm, 739–751 nm) and Stokes pulses (800 nm, FHWM: 13.4 nm, 795–808 nm), the excited modes including the substituent-dependent mode νCX1 (∼1080 cm−1), βCH (in range from 1000 cm−1 to 1290 cm−1) and γCH (in range from 880 cm−1 to 980 cm−1). Intramolecular energy flow from parent modes to daughter modes of C–C stretching in liquid aryl halides are observed clearly in Fig. 5. Parent modes: vibrational modes which are directly excited by the pump and Stokes pulses; daughter modes: vibrational modes accepting the vibrational energy through intramolecular vibrational energy redistribution. The white-coded lines represent the experimental Raman spectra of aryl halides. The contour plots of Fig. 5(a)–(d) are the time-resolved CARS spectra of F–Ph, Cl–Ph, Br–Ph and I–Ph, respectively. Signals outside of excitation range emerge around 1580 cm−1 are indirectly excited through intramolecular energy redistribution, and they correspond to the C–C stretching vibrational modes. Even though the lower frequency modes cannot be distinguished directly due to the low resolution in frequency domain in ultrafast CARS spectra, they can be extracted through Fourier transform of the time-domain CARS spectra.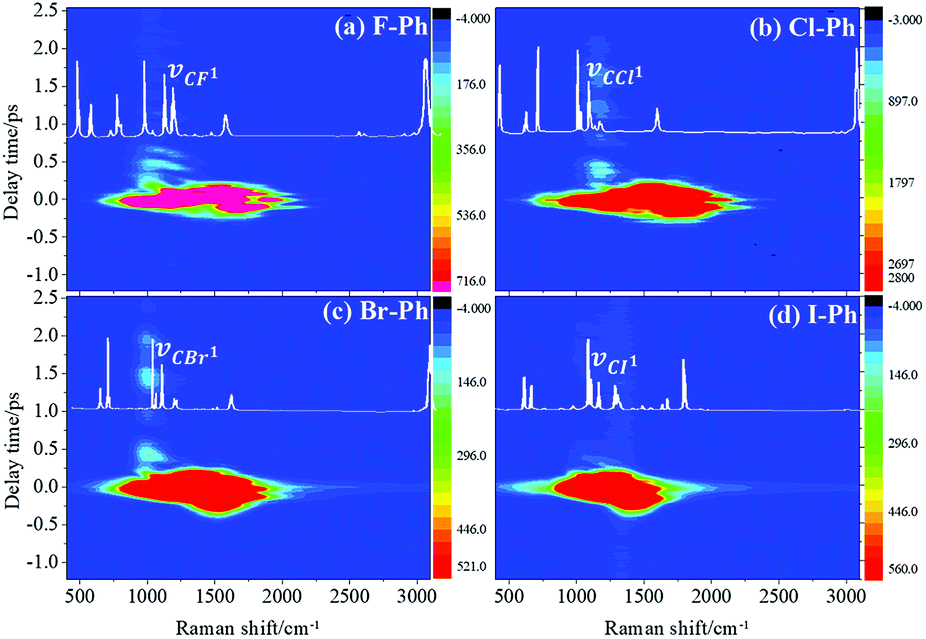 | ||
| Fig. 5 Substituent-dependent mode νCX1 (∼1080 cm−1) are selectively excited, the vibrational energy flow to the C–C stretching vibrational modes νCC (in range from 1320 cm−1 to 1590 cm−1). Aryl halides: (a) F–Ph, (b) Cl–Ph, (c) Br–Ph, and (d) I–Ph. | ||
FT power spectra contain vibrational couplings information of vibrational modes that range from 900 to 1300 cm−1 and C–C stretching modes which are located outside of the directly excitation region, as shown in Fig. 6. The peaks position in the FT power spectra indicate the frequency difference between two neighbouring modes and mark by Qi (i = 1–11), Fig. 6(a) represent the FT power spectra of F–Ph, Cl–Ph, Br–Ph and I–Ph, respectively. Detailed information of coherent couplings of νCX1 (∼1080 cm−1), βCH (in range from 1000 cm−1 to 1290 cm−1), γCH (in range from 730 cm−1 to 980 cm−1) and νCC (in range from 1320 cm−1 to 1590 cm−1) are listed in ESI (Table 3†).
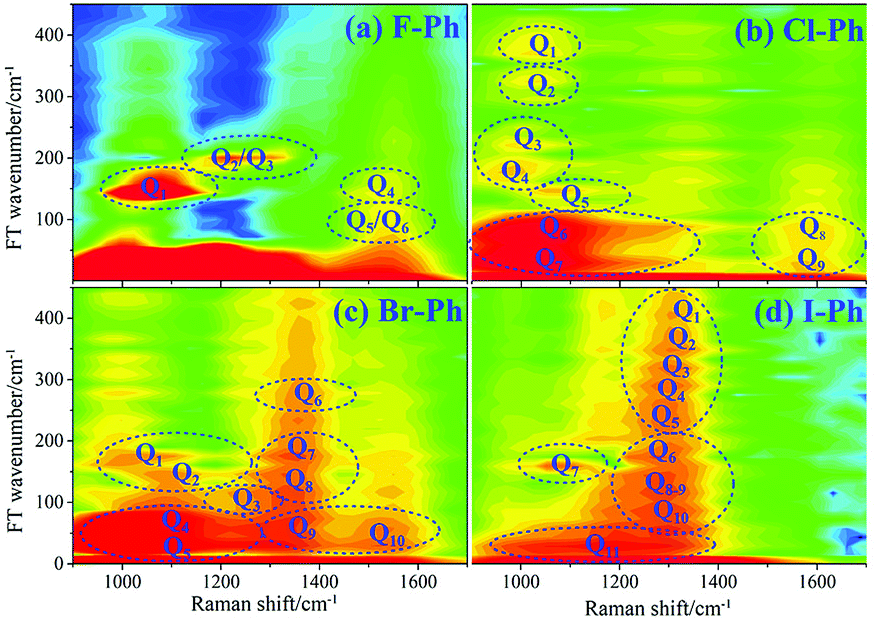 | ||
| Fig. 6 FT power spectra of the modes in range from 900 to 1600 cm−1. Aryl halides (a) F–Ph, (b) Cl–Ph, (c) Br–Ph, and (d) I–Ph. | ||
According to the results of the time-domain CARS spectra and its FT power spectra, the vibrational couplings of νCX1 (∼1080 cm−1), βCH (in range from 1000 cm−1 to 1290 cm−1) and γCH (in range from 730 cm−1 to 980 cm−1) are confirmed, meanwhile C–C stretching modes νCC (in range from 1320 cm−1 to 1590 cm−1) which are excited secondly by vibrational energy redistribution are also identified.
Selective excitation of the substituent-dependent mode νCX2
Vibrational modes in the frequency range from 418 to 833 cm−1 are selectively excited by a pair of pump (763 nm, FHWM: 12 nm, 757–769 nm) and Stokes pulses (800 nm, FHWM: 13 nm, 795–808 nm). The lower frequency substituent-dependent mode νCX2 ∼ 700 cm−1, γCH (in range from 730 cm−1 to 833 cm−1) and ϕCC ∼ 600 cm−1 are excited coherently, as shown in Fig. 7. The CARS spectra of F–Ph, Cl–Ph, Br–Ph, and I–Ph are presented in Fig. 7(a)–(d), respectively. The vibrational modes of βCH (in range from 1000 cm−1 to 1290 cm−1) and νCX1 ∼ 1080 cm−1 appear in the CARS spectra even though they are outside of the direct excitation region. This means the vibrational energy can flow from lower frequency modes to higher frequency ones.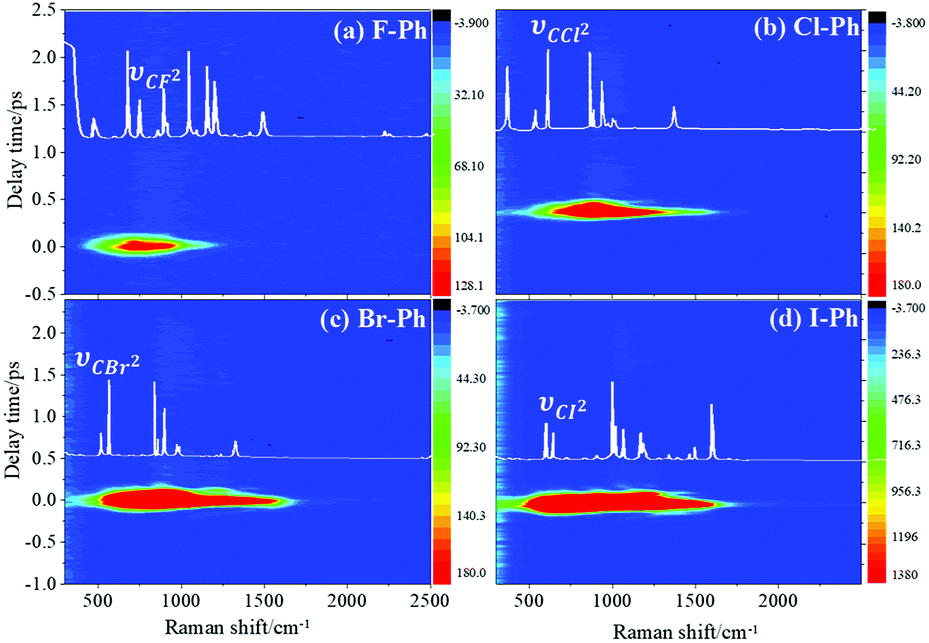 | ||
| Fig. 7 Vibrational modes of νCX2 ∼ 700 cm−1 is selectively excited, intramolecular energy flow to neighbouring modes βCH ∼ 1150 cm−1 and νCX1 ∼ 1080 cm−1. Aryl halides: (a) F–Ph, (b) Cl–Ph, (c) Br–Ph, and (d) I–Ph. | ||
FT power spectra is performed to identify the vibrational modes involved in their coherent couplings in the direct excitation region around 700 cm−1, and in the second excitation region from 900 cm−1 to 1200 cm−1, as shown in Fig. 8. The peaks of FT power spectra are marked by Qi (i = 1–10) and correspond to the vibrational couplings of substitution modes νCX2 ∼ 700 cm−1, βCH (in range from 1000 cm−1 to 1290 cm−1) and νCX1 ∼ 1080 cm−1 of aryl halides, as described in Fig. 8(a)–(d). Detailed assignments of coherent couplings of νCX2 ∼ 700 cm−1, γCH (in range from 730 cm−1 to 980 cm−1) and ϕCC ∼ 600 cm−1 are listed in ESI (Table 4†).
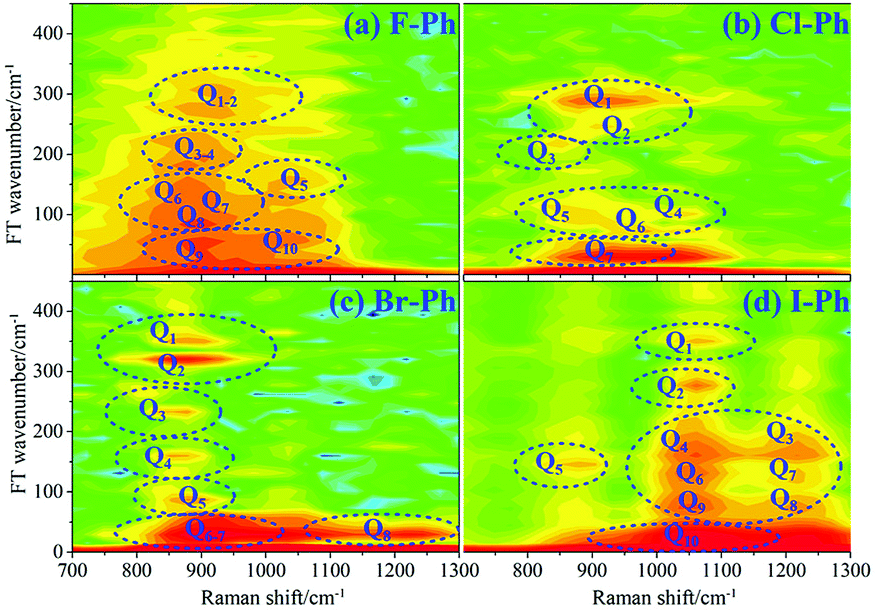 | ||
| Fig. 8 FT power spectra of dominant excitation modes centred around 700 cm−1 and 1080 cm−1. Aryl halides: (a) F–Ph, (b) Cl–Ph, (c) Br–Ph, and (d) I–Ph. | ||
According to the analysis of the time-domain CARS spectra and its FT power spectra of aryl halides, it can be found that substitution modes νCX2 ∼ 700 cm−1, out-plane C–H deformation modes γCH (in range from 730 cm−1 to 980 cm−1) and out-plane ring deformation modes ϕCC ∼ 600 cm−1 are selectively excited, neighbouring modes βCH (in range from 1000 cm−1 to 1290 cm−1) and νCX1 ∼ 1080 cm−1 outside of excitation range are emerged via vibrational energy redistribution.
Intramolecular energy redistribution processes of the liquid aryl halides
The above findings have implications for ultrafast physicochemical behaviours of the aryl halides and the characteristics of intramolecular energy redistribution can be summarized as follows: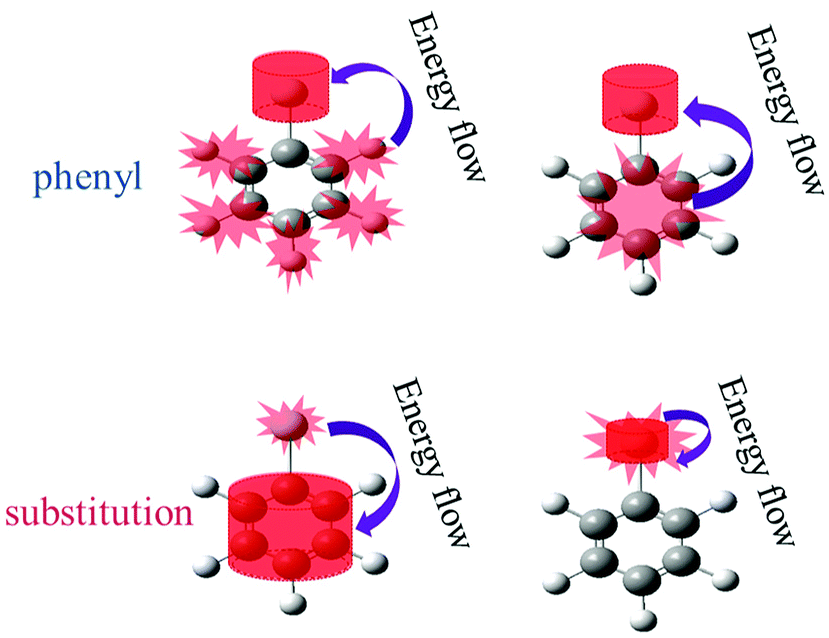 | ||
| Fig. 9 The parent modes are selective excited, vibrational energy flow to the daughter mode. The upper two plots displayed the excitation modes of the phenyl (νCH and νCC); the lower ones displayed the excitation modes of the substituent-dependent modes (νCX1 and νCX2), the purple arrows represented the direction of intramolecular energy redistribution. | ||
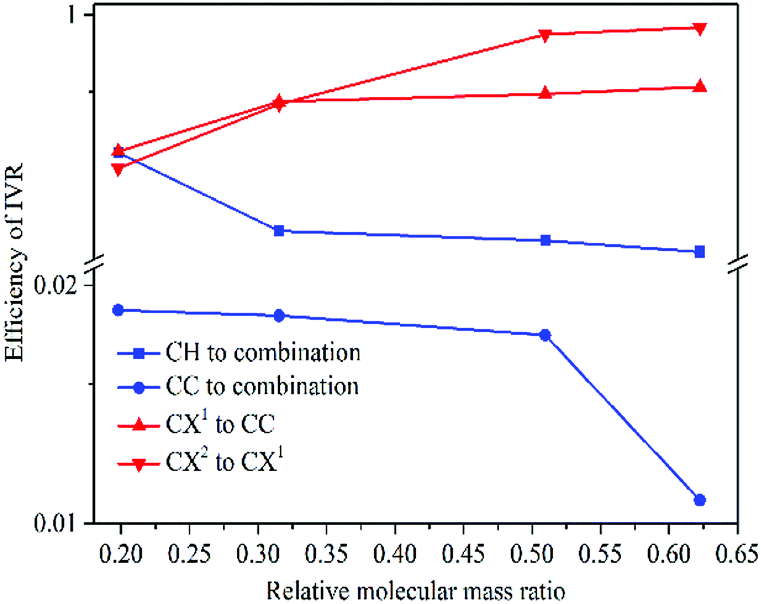 | ||
| Fig. 10 Scatterplot of vibrational energy redistribution efficiencies. | ||
(1) While substituent-dependent modes (νCX1 and νCX2) are selectively excited, vibrational energy flow from substituent-dependent modes to phenyl modes (CH stretching and CC stretching), the efficiencies locate on the upper of the 2D scattering plot (red-coded symbol lines). While phenyl modes (νCH and νCC) of the aryl halides are selectively excited, vibrational energy flow from phenyl mode to substituent-dependent modes (νCX1 and νCX2), the efficiencies locate on the lower of the 2D scattering plot (blue-coded symbol lines). It is found that the efficiencies of vibrational energy transfer from substituent-dependent modes to phenyl modes are bigger than that of the inverse.
(2) Efficiency can be controlled by varying the mass ratio of the halide in the aryl halide molecules. On one hand, while the substituent-dependent modes (νCX1 and νCX2) are selectively excited, efficiencies of vibrational energy redistribution are increased with increasing relative molecular mass ratio (kI–Ph > kBr–Ph > kCl–Ph > kF–Ph). On the other hand, while the phenyl modes (νCH and νCC) of the aryl halides are selectively excited, efficiencies of vibrational energy redistribution are decreased with the increasing relative molecular mass ratio (kI–Ph < kBr–Ph < kCl–Ph < kF–Ph).
The mass of phenyl is smaller than halides, the anharmonic couplings among these relevant modes are different and the efficiencies of vibrational energy redistribution are likely to be affected. While the heavier substitution-dependent modes are selectively excited, vibrational energy flow to lighter phenyl modes with larger amplitude oscillations, anharmonic coupling is more likely to happen and the efficiencies show a monotonic increase tendency with increasing mass of substituent of aryl halides. While the lighter phenyl modes are selectively excited, vibrational energy flow to substitution-dependent modes with the smaller amplitude oscillations, the anharmonic coupling is less likely to happen and the efficiencies show a monotonic decrease tendency with increasing mass of substituents.
Conclusions
Time-resolved CARS technique is performed to selectively excite parent modes and detect the daughter modes of aryl halides. With the help of Fourier analysis, vibrational modes that involved in the ultrafast coherent coupling are determined. Moreover, it is found that intramolecular energy flow is counterintuitive, especially the efficiencies of up-hill vibrational energy redistribution from lower frequency modes to higher ones are always larger than that of the opposite direction. Vibrational excitation on phenyl modes (νCH and νCC), intramolecular energy redistribution efficiencies decrease with the increasing halide mass; on the contrary, vibrational excitation on the substituent-dependent modes (νCX1 and νCX2), efficiencies increase with the increasing halide mass. It is noting that halide mass can control intramolecular energy redistribution efficiencies, and these results provide implications of substituent mass effects on photochemical reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Science Challenging Program (grant number TZ2016001) and the National Natural Science Foundation of China (grant number 21673211).Notes and references
- T. M. Krygowski and B. T. Stepień, ChemInform, 2005, 105, 3482 Search PubMed.
- L. Guan, M. G. Holl, C. R. Pitts, M. D. Struble, M. A. Siegler and T. Lectka, J. Am. Chem. Soc., 2017, 139, 14913 CrossRef PubMed.
- G. Szabó and L. Nyulászi, Struct. Chem., 2017, 28, 1–11 CrossRef.
- M. Huang, Z. Luo, T. Zhu, J. Chen, J. Z. Zhang and F. Xia, RSC Adv., 2017, 7, 51521–51527 RSC.
- B. C. Pein, Y. Sun and D. D. Dlott, J. Phys. Chem. B, 2013, 117, 10898–10904 CrossRef PubMed.
- B. C. Pein, Y. Sun and D. D. Dlott, J. Phys. Chem. A, 2013, 117, 6066–6072 CrossRef PubMed.
- K. Okamoto, N. Nomura, R. Fujiyoshi, K. Umegaki, H. Yamamoto, K. Kobayashi and T. Kozawa, J. Phys. Chem. A, 2017, 121, 9458–9465 CrossRef PubMed.
- R. V. Viesser, L. C. Ducati, C. F. Tormena and J. Autschbach, Phys. Chem. Chem. Phys., 2018, 20, 11247–11259 RSC.
- I. V. Omelchenko, O. V. Shishkin, L. Gorb, F. C. Hill and J. Leszczynski, Struct. Chem., 2012, 23, 1585–1597 CrossRef.
- A. M. Gardner and T. G. Wright, J. Chem. Phys., 2011, 135, 114305 CrossRef PubMed.
- N. Levy, J. S. Shpilman, H. C. Honig, D. T. Major and L. Elbaz, Chem. Commun., 2017, 53, 12942 RSC.
- W. Shen, W. Zhang and C. Zhu, Phys. Chem. Chem. Phys., 2017, 19, 23532 RSC.
- X. Liu, W. Zhang, Y. Song, G. Yu, Z. Zheng, Y. Zeng, Z. Lv, H. Song and Y. Yang, J. Phys. Chem. A, 2017, 121, 4948–4952 CrossRef PubMed.
- D. H. Whiffen, J. Chem. Soc., 1956, 4, 8 Search PubMed.
- B. C. Pein, N. H. Seong and D. D. Dlott, J. Phys. Chem. A, 2010, 114, 10500–10507 CrossRef PubMed.
- S. Singh, D. K. Singh, S. K. Srivastava and B. P. Asthana, Vib. Spectrosc., 2011, 56, 26–33 CrossRef.
- J. R. Scherer and J. C. Evans, Spectrochim. Acta, 1963, 19, 1739–1775 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04979c |
| ‡ This author contributed equally to this work and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2018 |