
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of nanostructured powders and thin films of iron sulfide from molecular precursors†
Laila Almanqura,
Inigo Vitorica-yrezabala,
George Whitehead a,
David J. Lewis*b and
Paul O'Brien*ab
a,
David J. Lewis*b and
Paul O'Brien*ab
aSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: paul.o'brien@manchester.ac.uk
bSchool of Materials, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: david.lewis-4@manchester.ac.uk
First published on 15th August 2018
Abstract
Iron(III) xanthate single-source precursors [Fe(S2COR)3] (R = methyl, ethyl, isopropyl and 1-propyl) were used to deposit iron sulfide thin films and nanostructures by two simple, efficient and low-cost methods (spin coating and solid state deposition). The single-crystal X-ray structures of the iron(III) n-propyl xanthate and iron(III) iso-propyl xanthate have been determined. Thermogravimetric analysis (TGA) studies of the complexes shows that decomposition of the complexes produces iron sulfide, pyrite or trolite. The crystallinity of iron sulfide thin films and powder samples was studied using X-ray diffraction (XRD), and their morphology was studied by scanning electron microscopy (SEM).
Introduction
Iron chalcogenides have attracted considerable attention over the last decade, due to their interesting optical, electrical and magnetic properties.1,2 Iron sulfide is the most earth-abundant chalcogenide mineral, and is used in several important applications, including batteries, catalytic processes and biomedical appliances and is also a potentially useful material for sustainable and inexpensive photovoltaics applications.3–5 It has many potential advantages over other materials, including its low cost, high abundance, negligible toxicity and unique magnetic, electric and optical properties.6–9 Iron sulfide crystallises in many different phases; these include pyrite (cubic FeS2), marcasite (orthorhombic FeS2), mackinawite (Fe1+xS), troilite (FeS), pyrrhotite (Fe1−xS), greigite (cubic spinel Fe3S4) and smythite (Fe3+xS4).10–12 The optical, magnetic and electrical properties of iron sulfides vary according to the stoichiometric ratio between the iron and sulfur atoms.13 Of these compounds, cubic pyrite (FeS2) is of interest for its potential use as an absorber material in photovoltaic and photo-electrochemical applications because of its suitable band gap (0.80–0.95 eV) and high absorption coefficient (∼105 M−1 cm−1) extending over the visible energy range.14–18 The favoured phases for Li-ion batteries are suggested to be troilite (FeS) and pyrrhotite (Fe1−xS). For supercapacitor applications, troilite (FeS) and greigite (Fe2S3) are potential candidate materials.19A variety of methods have been used to prepare iron sulfide nanoparticles including hot injection20–22 and colloidal methods,23 hydrothermal methods,24 and solvothermal methods.25,26 Nanoscale iron sulfide has been synthesized with various morphologies by the decomposition of several single source precursors such as iron alkyl dithiocarbamates,27,28 iron polysulfides, iron thiobiurets29 iron diethyldithiophosphates,30 FeS cubane clusters,31 iron thiosemicarbazones32 and iron alkyl xanthates.33 In addition, a great deal of attention has been paid to the deposition and characterization of iron sulfide thin films. Several deposition techniques have been employed to deposit iron sulfide thin films, including chemical bath deposition,34 chemical vapour deposition,35 chemical bath deposition,36 metal–organic chemical vapour deposition (MOCVD),37 and aerosol-assisted chemical vapour deposition (AACVD).38–40
Herein we report the synthesis of tris (O-alkylxanthato) iron(III) (alkyl Et, Me, iPr and nPr) complexes. These complexes have been used for the deposition of iron sulfide, troilite (FeS) and pyrrhotite (Fe1−xS) nanostructured materials, using a simple, cheap and low temperature synthesis methods – spin coating and solventless pyrolysis. These provide control over nanocrystal size and morphologies by variation of temperature and precursor structure.41,42
Experimental section
Materials
Methanol (99.8%), 2-propanol (99.5%), n-propanol (99.9%), potassium ethyl xanthogenate (96%), carbon disulfide (low benzene 99.9%), chloroform (99.8%), and iron(III) chloride (99%) were all acquired from Sigma-Aldrich, and used without further purification.Materials characterisation
Elemental analysis was conducted in the micro-analytical laboratory at the University of Manchester with a Carlo Erba EA 1108 elemental analyser. Thermogravimetric analysis (TGA) measurements were recorded from 0 °C to 600 °C at 10 °C min−1 heating rate under nitrogen, using a Seiko SSC/S200 TGA-DSC. Infrared spectra (IR) were obtained using a Specac single reflectance ATR instrument (4000–400 cm−1, resolution 4 cm−1). Melting points were obtained using a Barloworld SMP10 apparatus. Nuclear magnetic resonance (NMR) was recorded using a 400 MHz Bruker instrument. Mass spectra were recorded on Shimadzu Axima Confidence MALDI TOF mass spectrometer. Powder X-ray diffraction (p-XRD) measurements were carried out by using a Bruker D8 Advance and a Bruker Xpert diffractometer, utilising Cu-Kα radiation (1.54 Å). The samples were scanned between 20° and 80°, with a step size of 0.02° 2θ. Scanning electronic microscopy (SEM) images were obtained using a Philips XL30 FEG SEM. Inductively coupled plasma optical emission spectroscopy (ICP-OES) was carried out with a Perkin-Elmer Optima 5300 DV instrument. Raman spectra were recorded using a Renishaw 1000 microscope system equipped with laser excitation of 514 nm. Single crystal X-ray diffraction data for compounds were collected on a dual source Rigaku FR-X rotating anode diffractometer, using an MoKα wavelength at 150 K, and reduced using a CrysAlisPro 171.39.30c.43 The structure was solved and refined using Shelx-2016, implemented through Olex2 v1.2.9.44,45Synthesis of molecular precursors
Results and discussion
The four tris(O-alkylxanthato) iron(III) (alkyl = Me, Et, iPr and nPr) complexes, [Fe(S2COMe)3] (1), [Fe(S2COEt)3] (2), [Fe (S2COiPr)3] (3) and [Fe(S2COnPr)3] (4) were synthesised by metathesis of iron(III) salts in water. All complexes were soluble in common organic solvents such as chloroform, THF and toluene. These complexes were stored at −20 °C to avoid premature decomposition.Single-crystal X-ray structure
The single crystal X-ray structure of iron(III) isopropylxanthate, Fe[S2COiPr]3 (3) and iron(III) n-propylxanthate, Fe[S2COnPr]3 (4) are shown in Fig. 1. Complexes (3) and (4) crystallised in the monoclinic system with space group P21/c and P21/n respectively. The iron ion is coordinated to six sulfur atoms from the three bidentate xanthate ligands in a distorted octahedral environment. | ||
| Fig. 1 X-ray single crystal structure of (a) [Fe(S2COiPr)3] and (b) [Fe(S2COnPr)3]. Yellow spheres = sulfur, orange spheres = iron, red spheres = oxygen, grey spheres = carbon. | ||
The Fe–S bond length ranges in complex (3) are 2.29(8)–2.30(7) Å which is slightly longer than those for complex (4) which is in range of 2.2931–2.3162 Å. The Fe–S bond length obtained from both complexes is considerably closer to the distance of 2.3083–2.3263 Å reported previously for single crystal X-ray data of tris-(ethylxanthate) iron(III).47 The refinement data is given in ESI Table S1.† Bond lengths and angles are reported in ESI Table S2 and S3.†
Thermogravimetric analysis (TGA)
Thermal analysis of all complexes was performed up to 600 °C under a nitrogen atmosphere. The thermogram for complexes – [Fe(S2COEt)3] (2), [Fe(S2COiPr)3] (3) and [Fe(S2COnPr)3] (4) – show decomposition occurs through two steps, as shown in Fig. 2. These complexes exhibited approximately similar TGA profiles with a rapid mass loss within the temperature range of 120 to 300 °C, and gradual mass loss between 320 to 500 °C. [Fe(S2COMe)3] (1) decomposed in three steps, with the first step between 100 to 130 °C, and second and final steps similar to complexes (2–4). TGA results of the final residues are compiled in ESI Table S4.† All four complexes gave final solid residue amounts that matched with the calculated value for FeS2 or FeS. The complexes from (1) to (4) decomposed firstly to give FeS2; above 350 °C FeS is obtained from loss of sulfur. | ||
| Fig. 2 Thermogravimetric analysis (TGA) profiles of complexes [Fe(S2COMe)3] (1), [Fe(S2COEt)3] (2), [Fe(S2COiPr)3] (3), [Fe(S2COnPr)3] (4). | ||
Deposition of thin films by spin coating
Iron sulfide precursors (1–4) were deposited on glass substrates by spin coating, using chloroform as solvent. Films of the precursors were then annealed at different growth temperatures, between 300 to 500 °C, in a furnace tube under N2 for 60 min. The resulting thin films of iron sulfides were uniform and black in colour and were characterised by p-XRD and SEM.The p-XRD patterns of thin films prepared from a chloroform solution of [Fe(S2COMe)3], (1) [Fe(S2COEt)3] (2), [Fe(S2COiPr)3] (3) and [Fe(S2COnPr)3] (4) and annealed at three different temperatures (300, 400 and 500 °C) were recorded. The XRD pattern of iron sulfide thin films annealed at 400 and 500 °C corresponded to hexagonal troilite (FeS, ICDD no: 01-075-2165) Fig. 3. The diffraction peaks could be assigned to the (110), (201), (114), (107) and (008) planes. The p-XRD pattern of thin film of [Fe(S2COMe)3] (1) annealed at 300 °C exhibited a pure phase hexagonal troilite (FeS) (ESI Fig. S1†) No peaks were observed at a growth temperature below 400 °C when precursor [Fe(S2COEt)3] (2) was used. The p-XRD patterns of thin films deposited at 300 and 350 °C from a chloroform solution of [Fe(S2COiPr)3] (3) [Fe (S2COnPr)3] (4) show a similar trend to that observed from thin films deposited at 300 °C from complex (1) (ESI Fig. S2 and S3†). No crystalline deposition products were observed below 350 °C. It is clear that as annealing temperature increases that the peaks grow sharper as evidenced by a reduction of the full width at half maximum (FWHM); this generally indicates that the material becomes more microcrystalline according to the Scherrer equation.
 | ||
| Fig. 3 Powder X-ray diffraction (p-XRD) patterns of the iron sulfide thin films obtained by spin coating [Fe(S2COMe)3] (a), [Fe(S2COEt)3] (b), [Fe(S2COiPr)3] (c) and [Fe(S2COnPr)3] (d) followed by annealing under nitrogen at 400–500 °C for 60 min. The black sticks represent hexagonal troilite phase (FeS). (ICDD: 01-075-2165). | ||
Energy dispersive X-ray (EDX) spectroscopy of the iron sulfide thin films showed an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of Fe to S, consistent with troilite. The results are presented in ESI Table S5.†
:1 ratio of Fe to S, consistent with troilite. The results are presented in ESI Table S5.†
The surface morphology of iron sulfide thin films derived from pyrolysis of precursors (1–4) was studied using SEM. The SEM images obtained from an iron sulfide thin film derived from precursor (1) and deposited at 300 °C exhibited pseudo spherical crystallites in the size range of 260–300 nm (ESI Fig. S4†). At 400 and 500 °C clusters of crystallites with an average size of 200–250 nm (Fig. 4(1a) and (1b)) were apparent. The SEM images of iron sulfide thin films from [Fe(S2COEt)3] (2) at 400 and 500 °C are shown in Fig. 4(2a) and (2b) respectively. The morphology of the sample growth at 400 °C exhibited fairly uniform cluster like crystallites, consisting of spherical nanoparticles with open pores. At 500 °C, cluster-like crystallites consisting of irregular nanoparticles were obtained. SEM images of iron sulfide thin films derived from (3) at 300 °C show uniform cluster-like crystallites, as in ESI Fig. S5.† In contrast, the films deposited at 400 °C consist of smaller individual spherical crystallites with a size range of 140–180 nm, as in (Fig. 4(3a)). The films deposited at 500 °C show plate-like crystallites with an average size of 250–300 nm Fig. 3(3b). SEM images of iron sulfide thin films from (4) displayed the growth of randomly shaped crystallites at 350 °C (ESI Fig. S6†). The film growth at 400 °C showed a cluster of densely packed crystallites Fig. 4(4a), whereas a flower-like cluster were observed when films were deposited at 500 °C, as shown in Fig. 4(4b). In summary, the obtained images of the prepared thin films exhibited different sizes and morphologies. The SEM images of films deposited from all precursors show that the morphology of iron sulfide thin films is dependent on the growth temperature.
 | ||
| Fig. 4 SEM images of iron sulfide nanoparticles by spin coating method from [Fe(S2COMe)3] (1a) at 400 °C and (1b) at 500 °C, [Fe(S2COEt)3] (2a) at 400 °C and (2b) at 500 °C, Fe(S2COiPr)3] (3a) at 400 °C and (3b) at 500 °C and [Fe(S2COnPr)3] (4a) at 400 °C and (4b) at 500 °C. | ||
Solid state thermolysis of complexes
Solventless pyrolysis was employed for synthesis of iron sulfide crystallites using iron alkyl xanthates complexes (1–4). The solid complexes were decomposed under a nitrogen atmosphere in a tube furnace in the temperature range 300–500 °C for 60 min. The p-XRD patterns of the iron sulfide obtained from all four precursors [Fe(S2COMe)3] (1), [Fe(S2COEt)3] (2), [Fe(S2COiPr)3] (3) and [Fe(S2COnPr)3](4), at 400 and 500 °C as shown in Fig. 5(a)–(d). The results show the formation of a pure hexagonal pyrrhotite (Fe1−xS) phase (ICDD no: 00-022-1120). Reflections in the powder pattern corresponding to the (100), (101), (102), (110) and (201) planes of pyrrhotite (Fe1−xS) were dominant in the sample obtained from all precursors. The XRD patterns obtained from the complexes [Fe(S2COiPr)3] (3) and [Fe(S2COnPr)3] (4) gave a pure pyrrhotite (Fe1−xS) at all growth temperatures of 300, 400 and 500 °C, while [Fe(S2COMe)3] (1) and [Fe(S2COEt)3], (2) gave a pure phase at high temperatures of 400 °C and 500 °C. However, the XRD patterns obtained at a growth temperature of 300 and 350 °C from the complex [Fe(S2COMe)3] (1) and [Fe(S2COEt)3] (2), respectively show that some additional peaks corresponded to cubic pyrite (FeS2) (ICDD no: 01-071-0053) (denoted by the symbol*) (ESI Fig. S7 and S8†). The average crystallites size of iron sulfide is affected by growth temperature. The average crystallite size of iron sulfide from precursor (1) to (4) at two different temperatures 400 and 500 °C were calculated using the Scherer equation (ESI Table S6†). The average crystallite size of iron sulfide from complex (1) at growth temperature of 400 °C was 22.3 ± 2.6 nm. Whereas the crystalline obtained from growth temperature of 500 °C has an estimated diameter of 28.6 ± 1.6 nm the observed decrement in crystalline size with temperature indicated the influence of temperature. ICP-OES and EDX spectroscopy of iron sulfide samples confirmed the phase obtained from samples at high temperature of 500 °C is pyrrhotite Fe1−xS summary of results are shown in (ESI Table S7 and S8†). In addition, Raman spectroscopy confirmed the formation of pyrrhotite Fe1−xS with three vibration mode observed at wavenumbers of 214, 284 cm−1 and 399 cm−1 for all complexes (1–4).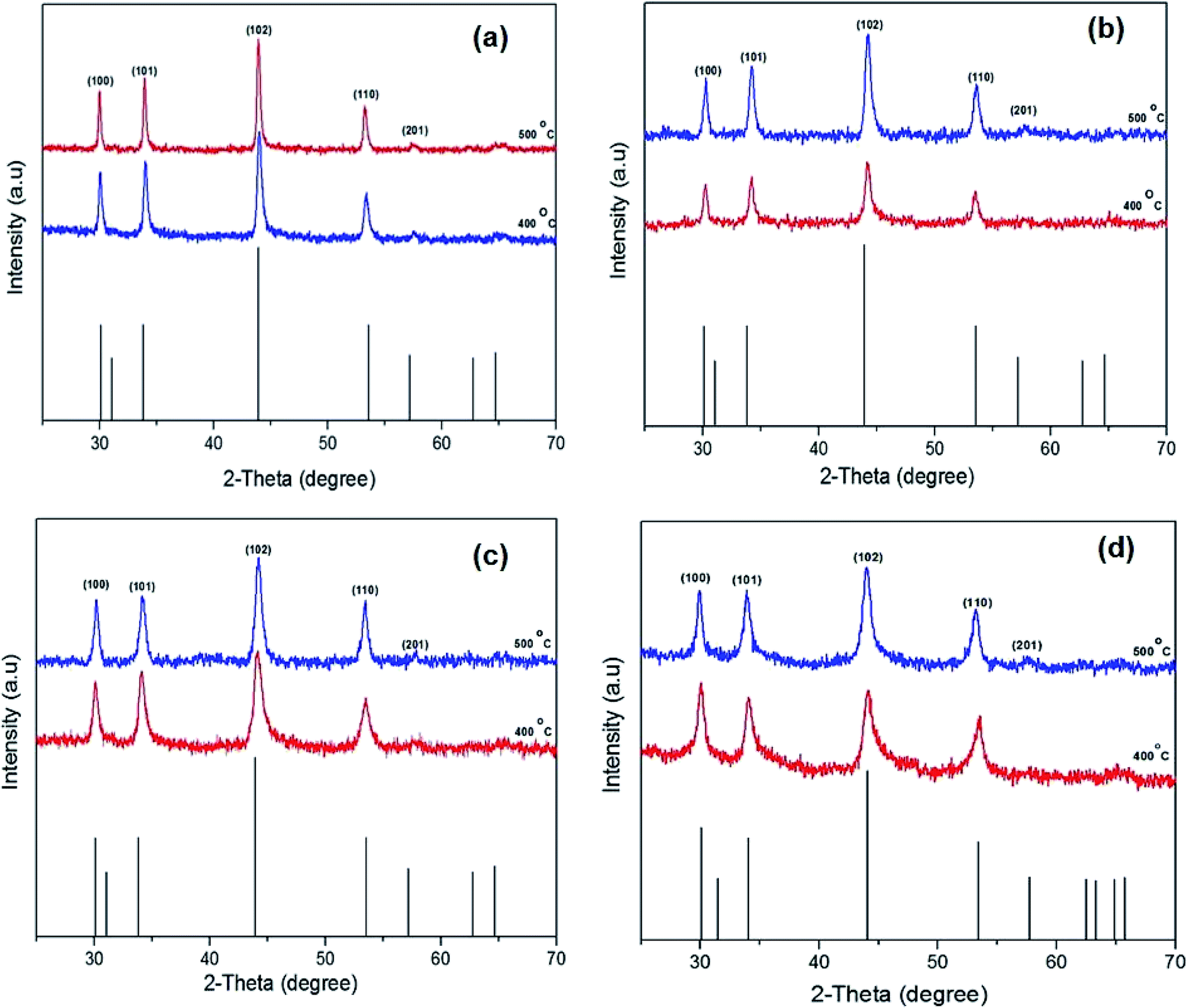 | ||
| Fig. 5 The powder X-ray diffraction (p-XRD) patterns of the iron sulfide nanostructure obtained by pyrolysis method [Fe(S2COMe)3] (a), [Fe(S2COEt)3] (b), [Fe(S2COiPr)3] (c) and [Fe(S2COnPr)3] (d) at temperature between 400 and 500 °C for 60 min. The black sticks represent hexagonal pyrrhotite (Fe1−xS) phase (ICDD no: 00-022-1120). | ||
Our results are in good agreement with that reported previously for iron sulfide pyrrhotite.48–50 The Raman spectra of pyrrhotite obtained from the pyrolysis of complexes (1–4) are presented in Fig. 6 and ESI Fig. S11.†
 | ||
| Fig. 6 Raman spectra of hexagonal pyrrhotite phase (Fe1−xS) from complexes [Fe(S2COMe)3] (a), [Fe(S2COEt)3] (b), [Fe(S2COiPr)3] (c) and[Fe(S2COnPr)3] (d) at 400 and 500 °C. | ||
SEM images of the iron sulfide crystallites growth at 400 °C and 500 °C derived from the thermolysis of complex (1) are presented in Fig. 7. The morphology of crystallites at growth temperatures of 300 °C (ESI Fig. S12†) and 400 °C showed clusters of densely packed crystallites, whilst hexagonal crystallites were observed at a growth temperature of 500 °C. SEM images from the pyrolysis of precursor [Fe(S2COEt)3] (2) at 300 °C (ESI Fig. S13†) and 400 and 500 °C are shown in Fig. 7. At all growth temperatures of 300–500 °C, regular spherical-like crystallites were observed. SEM images of iron sulfide crystals grown from the pyrolysis of [Fe(S2COiPr)3] (3) and [Fe(S2COnPr)3] (4) exhibited similar morphologies with spherical-like crystals observed and with considerable variation in particle size as shown in Fig. 7. The morphology of crystallites obtained from complexes (1–4) was clearly dependent on the temperature and the type of precursor.
 | ||
| Fig. 7 SEM images of iron sulfide nanoparticles from solventless pyrolysis of [Fe(S2COMe)3] (1a) at 400 °C and (1b) at 500 °C, [Fe(S2COEt)3] (2a) at 400 °C and (2b) at 500. [Fe(S2COiPr)3] (3a) at 400 °C and (3b) at 500 °C and [Fe(S2COnPr)3] (4a) at 400 °C and (4b) at 500 °C. | ||
Conclusion
In summary, a series of iron alkyl xanthate complexes, including [Fe(S2COMe)3] (1) [Fe(S2COEt)3] (2) [Fe(S2COiPr)3] (3) and [Fe(S2COnPr)3] (4) have been successfully synthesised, using a single-source route. The X-ray crystal structures of [Fe(S2COiPr)3] (3) and [Fe(S2COnPr)3] (4) have been determined. These four complexes were used for the deposition of iron sulfide crystallites. Two simple methods have been described for the growth of iron sulfide nanocrystals which are spin coating and solventless pyrolysis methods. Different deposition parameters such as deposition method, deposition temperature and precursor type were investigated in this study. The deposited iron sulfide phases and the morphology of iron sulfide crystallites were significantly influenced by the deposition method used. p-XRD results revealed the formation of troilite when a spin coat-annealing method was used while iron sulfide pyrrhotite Fe1−xS was mainly formed when the solventless pyrolysis method was used. Both methods are promising for the low temperature production of iron sulfide materials with control of the crystalline phase produced.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
LA acknowledges Magmmah Univeristy in Saudi Arabia for financial support. DJL and POB thank EPSRC for support (EP/R022518/1).References
- A. K. Dutta, S. K. Maji, D. N. Srivastava, A. Mondal, P. Biswas, P. Paul and B. Adhikary, ACS Appl. Mater. Interfaces, 2012, 4, 1919–1927 CrossRef PubMed.
- D. Jasion, J. M. Barforoush, Q. Qiao, Y. Zhu, S. Ren and K. C. Leonard, ACS Catal., 2015, 5, 6653–6657 CrossRef.
- H. Geng, L. Zhu, W. Li, H. Liu, L. Quan, F. Xi and X. Su, J. Power Sources, 2015, 281, 204–210 CrossRef.
- M. R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- H. Chen, L. Zhu, H. Liu and W. Li, J. Power Sources, 2014, 112(245), 406 Search PubMed.
- X. Chen, Z. Wang, X. Wang, J. Wan, J. Liu and Y. Qian, Inorg. Chem., 2005, 44, 951–954 CrossRef PubMed.
- H. Wang and I. Salveson, Phase Transform., 2005, 78, 547 CrossRef.
- S. Mlowe, D. J. Lewis, M. A. Malik, J. Raftery, E. B. Mubofu, P. O'Brien and N. Revaprasadu, Dalton Trans., 2016, 45, 2647–2655 RSC.
- J. Puthussery, S. Seefeld, N. Berry, M. Gibbs and M. Law, J. Am. Chem. Soc., 2011, 133, 716–719 CrossRef PubMed.
- K. Ramasamy, M. A. Malik, N. Revaprasadu and P. O'Brien, Chem. Mater., 2013, 25, 3551–3569 CrossRef.
- S. A. Kissin and S. D. Scott, Econ. Geol., 1982, 77, 1739 CrossRef.
- D. Rickard and G. W. Luther, Chem. Rev., 2007, 107, 514–562 CrossRef PubMed.
- P. Bai, S. Zheng, C. Chen and H. Zhao, Cryst. Growth Des., 2014, 14, 4295–4302 CrossRef.
- K. Ramasamy, M. A. Malik, M. Helliwell, F. Tuna and P. O'Brien, Inorg. Chem., 2010, 49, 8495–8503 CrossRef PubMed.
- R. Schieck, A. Hartmann, S. Fiechter, R. Könenkamp and H. Wetzel, J. Mater. Res., 1990, 5, 1567–1572 CrossRef.
- I. Dodony, M. Pósfai and A. R. Buseck, Am. Mineral., 1996, 81, 119–125 Search PubMed.
- D. Fan, M. Afzaal, M. A. Mallik, C. Q. Nguyen, P. O'Brien and P. J. Thomas, Coord. Chem. Rev., 2007, 251, 1878–1888 CrossRef.
- P. D. Matthews, P. D. McNaughter, D. J. Lewis and P. O'Brien, Chem. Sci., 2017, 8, 4177–4187 RSC.
- P. Matthews, M. Akhtar, M. Malik, N. Revaprasadu and P. O'Brien, Dalton Trans., 2016, 45, 18803–18812 RSC.
- J. Puthussery, S. Seefeld, N. Berry, M. Gibbs and M. Law, J. Am. Chem. Soc., 2010, 133, 716–719 CrossRef PubMed.
- C. Steinhagen, T. B. Harvey, C. J. Stolle, J. Harris and B. A. Korgel, J. Phys. Chem. Lett., 2012, 3, 2352–2356 CrossRef PubMed.
- C. Xu, Y. Zeng, X. Rui, N. Xiao, J. Zhu, W. Zhang, J. Chen, W. Liu, H. Tan, H. H. Hng and Q. Yan, ACS Nano, 2012, 6, 4713–4721 CrossRef PubMed.
- F. Cao, W. Hu, L. Zhou, W. Shi, S. Song, Y. Lei, S. Wang and H. Zhang, Dalton Trans., 2009, 42, 9246–9252 RSC.
- S. Kar and S. Chaudhuri, Chem. Phys. Lett., 2004, 398, 22–26 CrossRef.
- Q. Xuefeng, X. Yi and Q. Yitai, Mater. Lett., 2001, 48, 109–111 CrossRef.
- K. Anuar, W. Tan, N. Saravanan, S. Ho and S. Gwee, J. Sci. Technol., 2009, 10, 801–805 Search PubMed.
- Y. Zhang, Y. Du, H. Xu and Q. Wang, CrystEngComm, 2010, 12, 3658–3663 RSC.
- W. Han and M. Gao, Cryst. Growth Des., 2008, 8, 1023–1030 CrossRef.
- A. L. Abdelhady, M. A. Malik, P. O'Brien and F. Tuna, J. Phys. Chem. C, 2012, 116, 2253–2259 CrossRef.
- C. Wadia, Y. Wu, S. Gul, S. K. Volkman, J. Guo and A. P. Alivisatos, Chem. Mater., 2009, 21, 2568–2570 CrossRef.
- P. Vanitha and P. O'Brien, J. Am. Chem. Soc., 2008, 130, 17256–17257 CrossRef PubMed.
- S. D. Disale and S. S. Garje, Adv. Sci. Lett., 2010, 3, 80–86 CrossRef.
- M. Akhtar, M. A. Malik, F. Tuna and P. O'Brien, J. Mater. Chem., 2013, 1, 8766–8774 RSC.
- J. Oertel, K. Ellmer, W. Bohne, J. Röhrich and H. Tributsch, J. Cryst. Growth, 1999, 198, 1205–1210 CrossRef.
- M. Akhtar, A. L. Abdelhady, M. A. Malik and P. O'Brien, J. Cryst. Growth, 2012, 346, 106–112 CrossRef.
- G. Smestad, A. Da Silva, H. Tributsch, S. Fiechter, M. Kunst, N. Meziani and M. Birkholz, Sol. Energy Mater., 1989, 18, 299–313 CrossRef.
- M. R. Lazell, P. O'Brien, D. J. Otway and J.-H. Park, Chem. Mater., 1999, 11, 3430–3432 CrossRef.
- S. Saeed, R. Hussain and R. J. Butcher, J. Coord. Chem., 2014, 67, 1693–1701 CrossRef.
- S. Khalid, E. Ahmed, M. A. Malik, D. J. Lewis, S. A. Bakar, Y. Khan and P. O'Brien, New J. Chem., 2015, 39, 1013–1021 RSC.
- K. Ramasamy, M. A. Malik, M. Helliwell, F. Tuna and P. O'Brien, Inorg. Chem., 2010, 49, 8495–8503 CrossRef PubMed.
- E. Lewis, P. McNaughter, Z. Yin, Y. Chen, J. Brent, S. Saah, J. Raftery, J. Awudza, M. Malik, P. O'Brien and S. Haigh, Chem. Mater., 2015, 27, 2127–2136 CrossRef.
- M. Sigman, A. Ghezelbash, T. Hanrath, A. Saunders, F. Lee and B. Korgel, J. Am. Chem. Soc., 2003, 125, 16050–16057 CrossRef PubMed.
- Rigaku Oxford Diffraction, CrysAlisPro Software system, version 1.171.39.30c, Rigaku Corporation, Oxford, UK, 2017 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. J. Puschmann, Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- E. A. Lewis, P. D. McNaughter, Z. Yin, Y. Chen, J. R. Brent, S. A. Saah, J. Raftery, J. A. Awudza, M. A. Malik, P. O'Brien and S. J. Haigh, Chem. Mater., 2015, 27, 2127–2136 CrossRef.
- B. F. Hoskins and B. P. Kelly, J. Chem. Soc. D, 1970, 45–46 RSC.
- Y. El Mendili, A. Abdelouas, H. El Hajj and J. F. Bardeau, RSC Adv., 2013, 3, 26343–26351 RSC.
- W. L. Liu, X. H. Rui, H. T. Tan, C. Xu, Q. Y. Yan and H. H. Hng, RSC Adv., 2014, 4, 48770–48776 RSC.
- Y. El Mendili, A. Abdelouas and J. F. Bardeau, RSC Adv., 2013, 3, 15148–15156 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal data for CCDC 1846996 and 1846997. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra04917c |
| This journal is © The Royal Society of Chemistry 2018 |