
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile preparation of partially reduced graphite oxide nanosheets as a binder-free electrode for supercapacitors†
Juncai Zhanga,
Lingjuan Deng *a and
Zong-huai Liubc
*a and
Zong-huai Liubc
aSchool of Chemistry & Chemical Engineering, Xianyang Normal University, Xianyang 712000, P. R. China. E-mail: denglingjuan@163.com; Tel: +86-29-33720704
bKey Laboratory of Applied Surface and Colloid Chemistry, Shaanxi Normal University, Ministry of Education, Xi'an 710062, P. R. China
cSchool of Materials Science and Engineering, Shaanxi Normal University, Xi'an 710062, P. R. China
First published on 14th August 2018
Abstract
Preparation of graphene (GR) based electrode materials with excellent capacitive properties is of great importance to supercapacitors. Herein, we report a facile approach to prepare partially reduced graphite oxide (PRG) nanosheets by reducing graphite oxide (GO) using commercial Cu2O powder as a reduction agent, moreover, we demonstrate that the PRG nanosheets can act as building blocks for assembling hydrogels (PRGH) and flexible film (PRGF). The obtained PRGH and PRGF can be directly used as binder-free electrodes for supercapacitors and give high specific capacitance (292 and 273 F g−1 at a current density of 0.5 A g−1 in a three-electrode system, respectively) due to the existence of oxygen-containing functional groups in PRG nanosheets. PRG also gives excellent rate ability and cycle stability. This study suggests a facile pathway to produce GR-based materials with excellent capacitive properties and is meaningful for flexible supercapacitors.
1. Introduction
The ever-increasing energy demand has promoted the rapid development of various novel and high performance energy storage devices. Supercapacitors (also called electrochemical capacitors) have been demonstrated to be one of the most effective and practical technologies for electrochemical energy conversion and storage.1–3 Supercapacitors can be classified into two categories according to their mechanism. One is electrical double layer capacitors (EDLCs) which always use carbon-based materials with high surface area and suitable pore sizes as the electrode materials. During the charge/discharge process in EDLCs, there is pure physical charge accumulation rather than electrochemical reaction occurring at electrode/electrolyte interface.4 The other is pseudocapacitors, which employ electroactive conducting polymers or transition metal oxides for electrode materials. Pseudocapacitors store charges mainly through electrochemical reaction in the charge/discharge process.5 It has been proved that pseudocapacitors can supply much higher specific capacitance and energy density than those of EDLCs.6 Continuous development of wearable electronics and photonics devices in daily life, has resulted in huge demand for flexible supercapacitors.7,8 It is well known that the electrode material is a crucial factor which affects the capacitive properties of a supercapacitor,4 so preparing electrodes with intriguing capacitive properties and flexibility holds great promise to fulfill the need for flexible supercapacitors.As the most flexible one-atom-thick carbon nanosheets, graphene (GR) possess the most opportunity to be a flexible electrode for supercapacitors.9 When used as supercapacitors electrode, GR can give a theoretical specific capacitance of 550 F g−1 assuming all the surface area be fully used.10 Most reported GR based electrode materials, however, exhibit a moderate specific capacitances of about 150 F g−1 due to the irreversible agglomerates resulting from π–π stacking interaction and van der Waals force.11 In order to promote the capacitive property of GR, a number of approaches have been followed. The most classic method is incorporating electroactive materials to form GR-based composites.12–20 These composite can provide much higher capacitance than that of GR, moreover, an excellent rate ability and cycle stability than those of electroactive materials. Due to the lacking of chemical bond between GR and electroactive materials, the electroactive materials are easily fall off from the GR sheets during the cycle process, and thus produce unsatisfied cycle performance.16 Therefore, connecting the electroactive materials on the GR surface with chemical bond is of great importance to GR-based electrode materials for supercapacitors.
Among the various methods for preparation GR, chemical redox/reduction is supposing to be scalable and has been used widely. The most used precursor for chemical redox/reduction is graphite oxide (GO) produced by oxidation graphite.21 GO with abundant function groups, including hydroxyl, carboxyl and carbonyl can be easily exfoliated into individual sheet by ultrasonic treatment. The oxygen-functionalities could improves the wetting between GR and polar electrolytes, but also shut down the charge transport in GO nanosheets which corresponding to the poor conductivity of GO. Reducing GO by chemical process, fortunately, can reduce most oxygen-functionalities to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and provide satisfied electrical conductivity for GR. Hydrazine,22 NaOH,23 urea,24 Al powders,25 Fe powders,26 vitamin C27 and so on, have been employed to reduce GO. It is reported that the density of the oxygen-functionalities on GO can be adjusted by controlled the experiment condition, and the remained oxygen-containing functional groups can act as sources for pseudocapacitance.28 This result gives a meaningful signal to prepare excellent GR based electrode material for supercapacitors. Unfortunately, the reported GR are mostly in the form of graphite-like powders, which require additives (such as binders and conductive agent) to fabricate the ultimate electrode, and finally may degrade the overall capacitive performance.28 Therefore, the flexible GR based materials with satisfied capacitive property might give desired property for supercapacitors.
C and provide satisfied electrical conductivity for GR. Hydrazine,22 NaOH,23 urea,24 Al powders,25 Fe powders,26 vitamin C27 and so on, have been employed to reduce GO. It is reported that the density of the oxygen-functionalities on GO can be adjusted by controlled the experiment condition, and the remained oxygen-containing functional groups can act as sources for pseudocapacitance.28 This result gives a meaningful signal to prepare excellent GR based electrode material for supercapacitors. Unfortunately, the reported GR are mostly in the form of graphite-like powders, which require additives (such as binders and conductive agent) to fabricate the ultimate electrode, and finally may degrade the overall capacitive performance.28 Therefore, the flexible GR based materials with satisfied capacitive property might give desired property for supercapacitors.
Herein, partial reduced graphite oxide (PRG) nanosheets has been facilely prepared at room temperature using GO as precursor and commercial Cu2O powders as reduction agent, respectively. Oxygen-containing functional group on the PRG nanosheets can be adjusted by varying the amount of Cu2O powders. The PRG nanosheets can act as building block for assembling hydrogels (PRGH) in high ionic strength medium with three-dimensional architecture and flexible film (PRGF) with compact layered structure. The obtained PRGH and PRGF can be directly used as binder-free electrode for supercapacitors and give high specific capacitance (292 and 273 F g−1 at a 0.5 A g−1 in three-electrode system, respectively). This study suggests a facial pathway to produce GR-based materials with excellent capacitive property and is meaningful for flexible supercapacitors.
2. Experimental
2.1. Material preparation
GO was fabricated from crude flake graphite (Qingdao Aoke Co.) using the classic Hummers method with some modifications.29 50 mg as-prepared GO powders were dispersed in 100 mL distilled water by ultrasonic treated using a KQ-5200kDE digital ultrasonic cleaning device (200 W, 100% amplitude) for 3 hours, and the GO homogeneous aqueous dispersion (0.5 mg mL−1) was obtained.PRG was prepared as follows: commercial Cu2O powder (0.125, 0.25, 0.5, 0.75 and 1.0 g) was mixed with 50 mL GO aqueous dispersion (0.5 mg mL−1) and then stirred violently at 25 °C for 1 h. Successively, HCl aqueous solution (6 mol L−1) was added to the above system to remove the unreacted Cu2O powder.
PRGF was made through vacuum filtration of the above aqueous dispersion through an membrane filter (47 mm in diameter, 0.2 mm pore size) followed by drying in air at room temperature, and were abbreviated as PRGF-0.125, PRGF-0.25, PRGF-0.5, PRGF-0.75 and PRGF-1.0, respectively.
GO homogeneous dispersion (100 mL, 0.25 mg mL−1) was mixed with hydrazine solution (35 μL) and ammonia solution (400 μL, 25%), followed by stirring for 10 min and refluxed at 95 °C for 1 h. The above mixture was washed and vacuum filtered to obtain GR film (GRF). GO film (GOF) was also prepared by filter the GO dispersion (50 mL, 0.5 mg mL−1) for comparison.
PRG hydrogel (PRGH) was synthesized as follows: let the obtained PRG mixture (reduced by 0.75 g commercial Cu2O powder) stand for 12 h, the PRG nanosheets aggregation and suspended in the system. The as-prepared PGRH were taken out of the vial using tweezer and medicinal ladle carefully, and then the PRGH was immersed in pure water to remove adsorbed Cu2+ for the following tests.
2.2. Characterization
X-ray diffraction (XRD) measurements were carried out using an Ultima IV diffractometer with Cu Kα (λ = 0.154 nm), using an operation voltage and current of 40 kV and 30 mA, respectively. A Quanta 600 FEG field emission scanning electron microscope (FSEM) and transmission electron microscope (TEM) (JEM2010-HR) were used to observe the morphology of the obtained materials. The surface electronic states about the obtained materials were investigated by X-ray photoelectron spectroscopy (XPS; AXIS ULTRA Kratos Analytical Ltd.). FT-IR spectra were collected using a Nicolet IR 200 FTIR spectrometer (KBr pellets). Thermogravimetric analysis (TGA) was carried out on a thermogravimetric analyzer (TA-60Ws, Shimadzu Co.). Raman spectra were taken at an excitation wavelength of 1064 nm on a Bruker RFS 100/S Raman spectroscopy system. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements of different electrodes were carried out on a CH Instruments, Model CHI660E electrochemical workstation. Galvanostatic charge/discharge and cycle stabilities measurements were measured using a battery testing system (LAND, Model CT2001A).2.3. Electrochemical measurement
PRGF electrodes used for electrochemical test were prepared as follows: The as-synthesized PRGF pieces with size of ∼1 cm2 and a mass about 2 mg were first cut from the pristine obtained flexible film, and then were sandwiched between two stainless steel net layers (500 mesh, one piece: 1.5 cm × 1.5 cm, and the other piece: 1.5 cm × 10 cm, see Fig. s1†) under a pressure of ∼10 MPa for 1 min.PGRH electrode was prepared as follows: slices of PRGH with a thickness of ∼1 mm was first cut from the purified PRGH, and then were sandwiched between two stainless steel net layers (500 mesh, one piece: 1.5 cm × 1.5 cm, and the other piece: 1.5 cm × 10 cm) under a pressure of ∼6 MPa for 1 min. The mass of PGRH in electrode was determined by calculating the weight difference before and after dried at 110 °C for 12 h.
The electrochemical test of the individual electrode was performed in a three-electrode cell at room temperature, in which platinum foil, saturated calomel electrodes (SCE) and 0.5 mol L−1 H2SO4 aqueous solution were used as counter reference electrodes, referenced electrode and electrolyte, respectively. The specific capacitance C (F g−1) of the electrode was determined by means of galvanostatic charge/discharge test as follow:
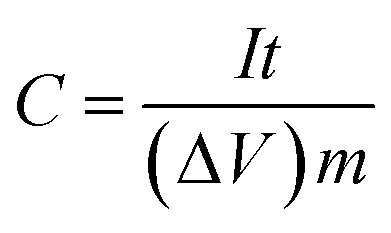 | (1) |
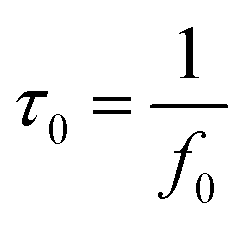 | (2) |
3. Results and discussion
Fig. 1 shows the schematic for preparation of PRG. The exfoliated GO dispersion was light brown due to the introduction of oxygen-containing functional group in the drastically oxidation process. After adding commercial Cu2O powders, the color of the mixture becomes brick-red, which should be ascribes to the adsorption of Cu2O powders on the surface of GO. After violently stirring for 60 min, and sequentially stand for 10 min, the colloid system coagulated on the bottom of the beaker. After putting same quantity of Cu2O powders in GO dispersion (0.5 mg mL−1) and stirring for 10 min, the upper clear liquid is still light brown while Cu2O deposition on the bottom of the beaker, indicating the GO dispersion was stable (see Fig. s2†). As well known, the stability of a colloidal dispersion can be destroyed easily by opposite charged electrolyte ions. Owing to the existence of the oxygen-containing functional group (such as –COOH, –OH and so on), the GO colloid dispersion could stand stably driven by the negative electrostatic repulsion. So, in the first mix stage, the GO colloid was coagulated by the Cu2+ which produced by the reaction between the Cu2O and GO. The existence of Cu2+ has been evidenced by slight blue Cu(OH)2 precipitate after adding NaOH solution to the upper clear liquid (see Fig. s3†). The reducing power in this period is too small to reduce the GO to GR, but the amount of Cu2+ is adequate to make GO colloid coagulation. Finally, the moderate HCl aqueous solution was added to the system, there is a disproportionation reaction between Cu2O and H+ as follow.| Cu2O + 2H+ = Cu2+ + Cu + H2O |
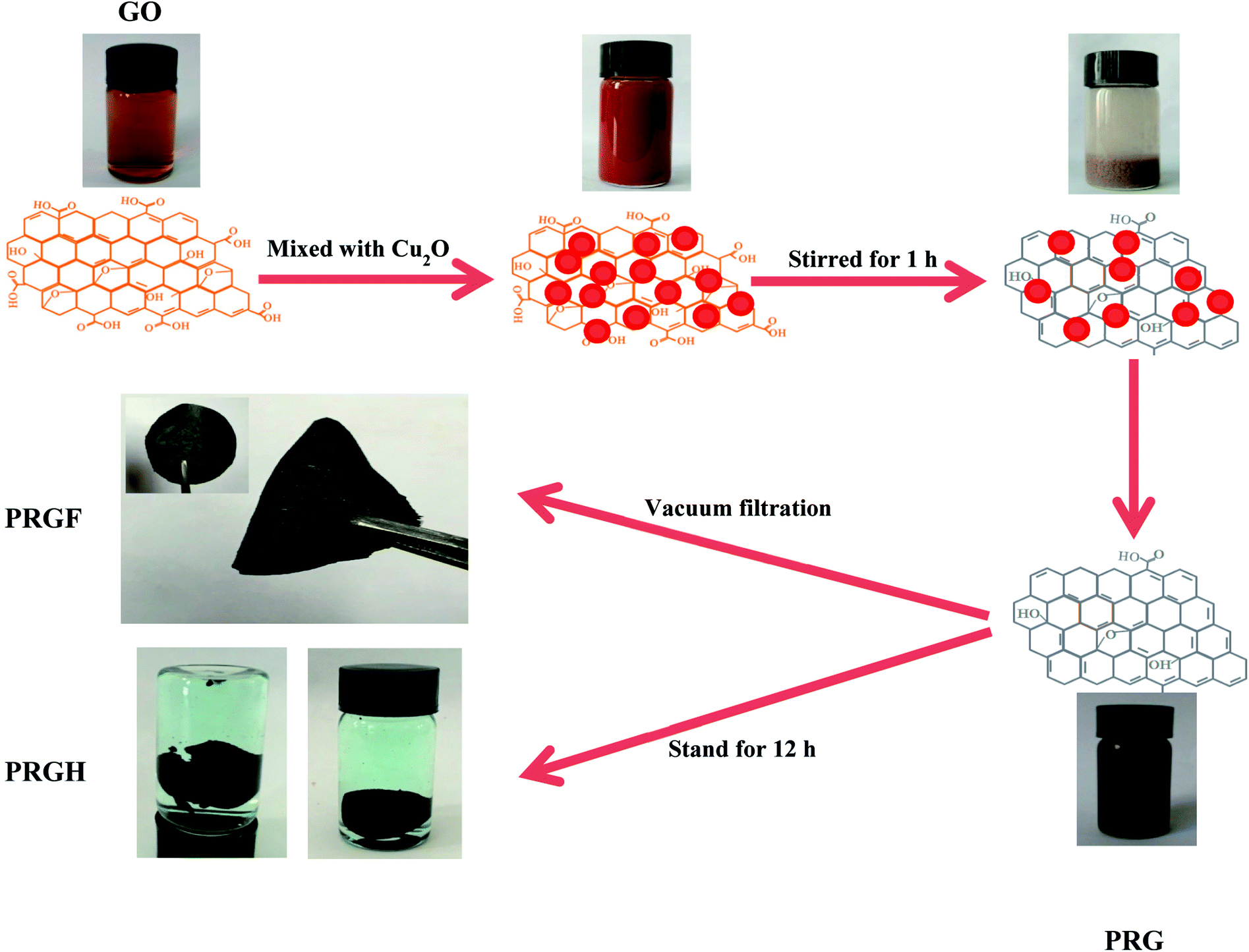 | ||
| Fig. 1 Schematic for preparation of PRGF and PRGH. | ||
The mass of the product would be high considering the water-insoluble elemental Cu if Cu2O powders were only react with HCl (even if the lowest addition mass of 0.125 g, the quality of the final product would be 47.95 mg). The mass of all the obtained PRGF, however, are about 27 mg. The introduction of HCl could not only dissolve Cu2O powders but also improve the reduction potential of Cu2O/Cu2+.26 Furthermore, the redox reaction is easy to be driven due to the close contact between GO and Cu2O particles by the electrostatic attraction, and the Cu2O plays an important role for reduction the GO. So, the dispersion becomes black thoroughly after adding HCl solution. The standard reduction potential of Cu2O/Cu2+ (−0.2065 V) is higher than that of the reported Al/Al3+ (−1.68 V), Fe/Fe2+ (−0.447 V), BH4−/BO2− (−1.24 V) and N2H4/N2 (−1.16 V), so the obtained materials are supposed to be partial reduced.
Fig. 2a shows the XRD patterns of GO, Cu2O, PRGF-0.125, PRGF-0.25, PRGF-0.5, PRGF-0.75, PRGF-1.0 and GR. Owing to the introduction of oxygenated functional groups (such as carbonyl, hydroxyl, epoxy, and so on) on carbon sheets, the basal distance of GO obtained from the (002) peak located at 2 theta equals to 10.3° is approximately 0.83 nm, which is larger than that of the graphite (0.34 nm).16 During reduction process, the intensity of the (002) diffraction peak (see the dotted frame in Fig. 2a) gradually weakened and finally disappeared with increasing the amount of the Cu2O. Simultaneously, the intensity of the diffraction peak at 23° (corresponding to a d-spacing of 0.37 nm) increased. Moreover, there is no characteristic diffraction peak of Cu2O and Cu in the obtained PRGF materials, suggesting the Cu2O powder are removed thoroughly by HCl, and the small amount of the Cu existed as amorphous state.
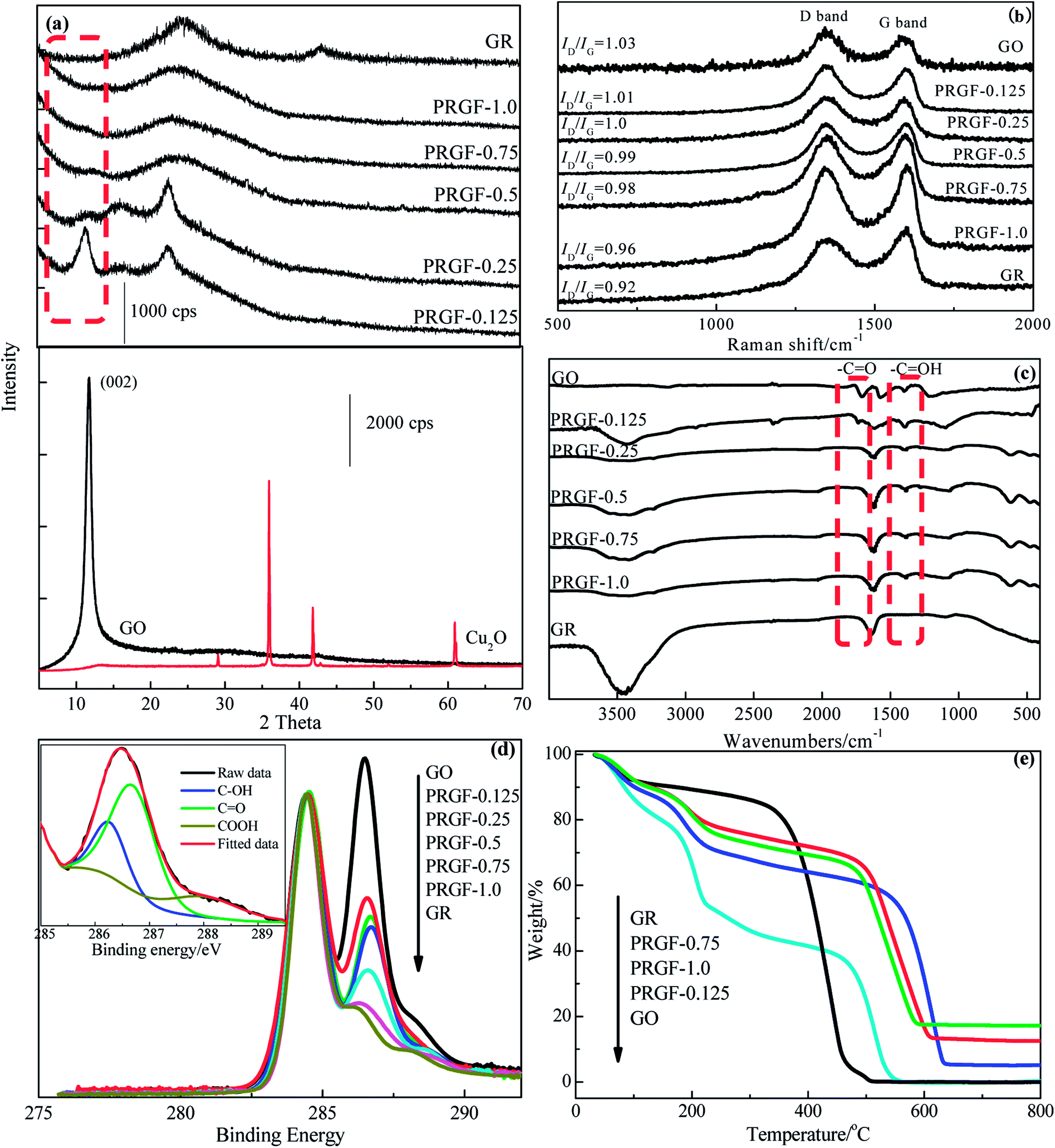 | ||
| Fig. 2 (a) XRD patterns of GR, PRGF-0.125, PRGF-0.25, PRGF-0.5, PRGF-0.75, PRGF-1.0, GO and Cu2O, respectively. (b) Raman spectra, (c) FTIR spectra, (d) C 1s XPS spectra normalized with respect to the C–C peak, of GO, PRGF-0.125, PRGF-0.25, PRGF-0.5, PRGF-0.75, PRGF-1.0 and GR, respectively. (e) TGA curves (in air) of GR, PRGF-0.125, PRGF-0.75, PRGF-1.0 and GO, respectively. | ||
Raman, FTIR and XPS were employed for monitor the microstructure of the PRGF. It is well known that the G band located at about 1600 cm−1 is associated with sp2-bonded carbon atoms in the 2D hexagonal carbon framework, while the D band at about 1350 cm−1 is attributed to sp3-bonded carbon atoms at the edges of the hexagonal carbon framework and indicative of the extent of defects of GR.30 As shown in Fig. 2b, the obtained PRGF materials show decreased ID/IG value (from 1.03 to 0.96) while increasing the amount of Cu2O powders (from 0.125 g to 1.0 g), indicating decreased oxygen functionalities content and fewer defects than that of GO. As exhibits in Fig. 2c, GO shows three remarkably bands at 1720, 1630 and 1394 cm−1 are ascribes to CO, CC and –C–OH stretching vibration, respectively, and which is totally different from that of GR that only exhibits one band at 1620 cm−1 attributed to the skeletal vibration.28 While increasing the amount of Cu2O, the intensity of the –CO decreasing sharply and nearly disappeared for that of PRGF, and also indicating that CO band is reduced easily than that of –C–OH band in this process.
Fig. 2d gives the C 1s XPS spectra normalized with respect to the C–C peak of GO, PRGF-0.125, PRGF-0.25, PRGF-0.5, PRGF-0.75, PRGF-1.0 and GR, respectively. Normally, the peak at 284.4 eV in C (1s) core-level spectrum is assigned to the sp2-hybridized carbon atoms (insert in Fig. 2d), and the peak located in the higher section is due to the oxygen functionalities such as –C–O (286.2 eV), –CO (287.7 eV) and –COOH (288.7 eV).31–33 As shown in Fig. 2d, GO shows the highest content of oxygen-containing functional group among the three materials caused by the violently oxidation during the preparation process. Accompanied by increasing the mass of the Cu2O, the obtained PRG materials show partial deoxygenation which is ascribed to the Cu2O-assisted reduction. After deconvolution the shoulder peak of C 1s XPS spectrum, the content of –CO decreased from 59.28% for GO to 11.56% for PRGF-1.0, suggesting most of the –CO has been reduced during this process, and this conclusions are accord with that of the FTIR results.
Thermal stability of the PRGF is also studied and the results are showed in Fig. 2e. GO gives nearly a 60% weight loss at ∼200 °C, arising from the decomposition of oxygen-containing functional group,28 and completely decomposes at ∼520 °C. In contrast, PRGF show about 40% weight loss centering round 200 °C, and complete decomposition at ∼600 °C, suggesting that the remaining oxygen-containing functional group in favor of its thermal stability. PRGF-1.0 maintains 16% weight retention at 800 °C, and which should ascribe to the oxidation of Cu powders in air. This results also confirm our previous speculation.
In order to choose the optimum PRGF for supercapacitors, cycle voltammetry (CV), galvanostatic charge/discharge measurements were employed in 0.5 mol L−1 H2SO4 aqueous solution electrolyte. As shown in Fig. 3a, GOF electrode exhibits luffa-like CV curve indicating the unsatisfied capacitive performance resulting from the awful electrical conductivity. PRGF-0.125 and PRGF-0.25 electrodes show moderate capacitive property which are evidenced by the distorted rectangle CV curves. PRGF-0.5 electrode also shows distorted rectangle CV curve with several obvious redox peaks, indicating a complex supercapacitive, namely pseudocapacitance and EDLC. According to the literature, the oxygenated groups, such as quinone and carbonyl/carboxyl groups, remained on the surface of GR are mainly responsible for the pseudocapacitance. The unconspicuous redox peaks (Fig. 3a) in the distorted CV curves correspond to reduction or oxidization of the electrochemically active oxygenated groups in the electrolyte: e.g. –CO + H+ + e− → –C–O–H, similar to other works proposed previously.34–36 The redox peaks in the CV curves of PGRF-0.75 and PRGF-1.0 electrodes are more evident than other electrodes, suggesting the percentage of the pseudocapacitance is more dominate for these materials. At the same scan rate, it is clear that the PRGF-0.75 electrode exhibits a larger current density than those of the other electrodes (Fig. 3a), indicating a larger ion accessible specific surface area and faster ion diffusion rate than the other electrodes. So, PRGF-0.75 is more suitable to be an electrode material for supercapacitors.
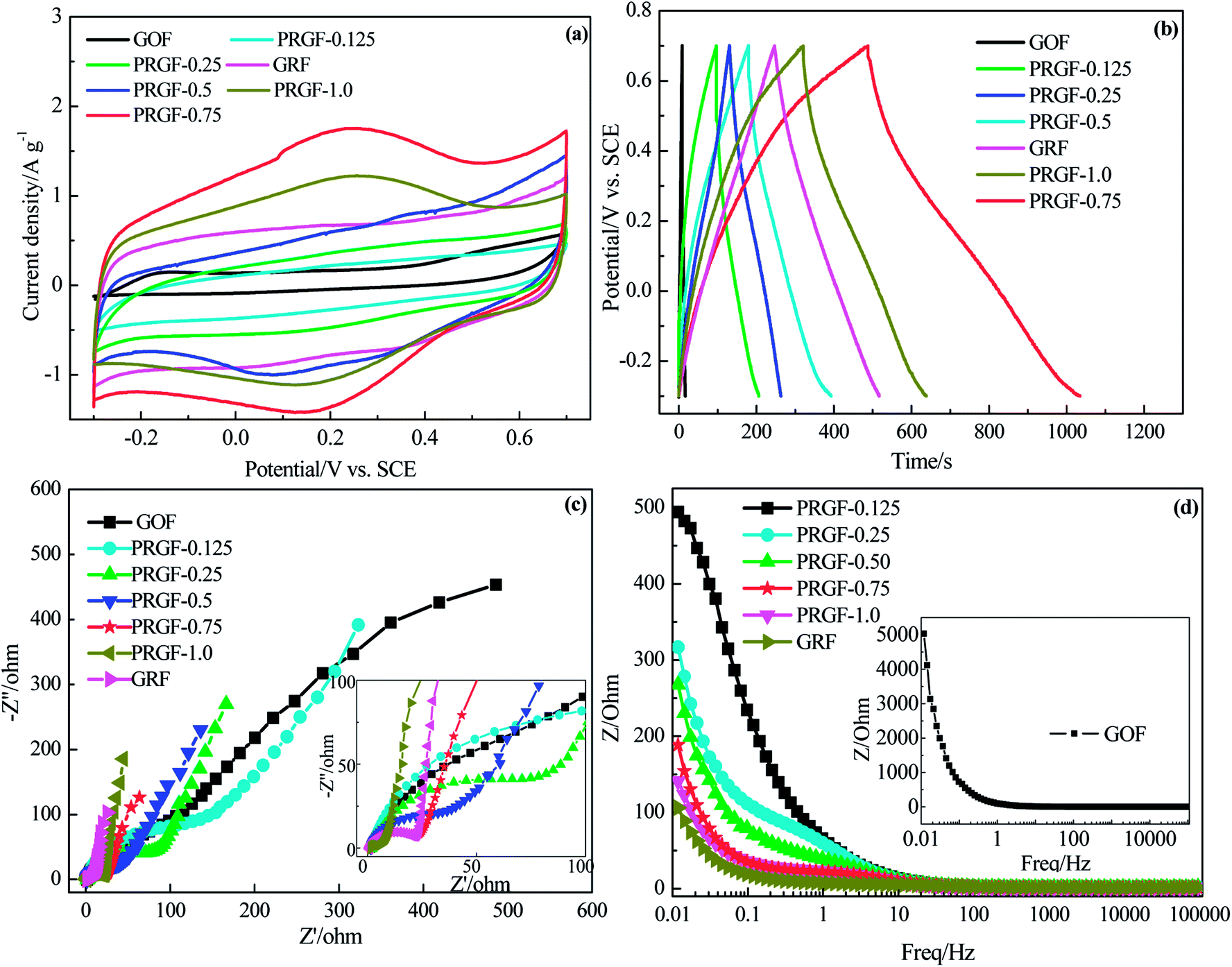 | ||
| Fig. 3 Electrochemical capacitive properties of GRF, PRGF-0.125, PRGF-0.25, PRGF-0.5, PRGF-0.75, PRGF-1.0, and GOF electrodes measured using a three-electrode system in 0.5 mol L−1 H2SO4 electrolyte: (a) CV curves at scan rate of 5 mV s−1. (b) Galvanostatic charge/discharge profiles at a current density of 0.5 A g−1. (c) Nyquist plots measured at frequent range of 100 kHz to 0.1 Hz. Inset: expanded high frequency region of Nyquist plots. (d) Plot of total impedance versus frequency. | ||
Fig. 3b shows the charge/discharge curves of GOF, GRF and PRGF electrodes at a current density of 0.5 A g−1. From a rough view of Fig. 3b, GOF displays nearly a line-like charge/discharge curve, indicating the poor capacitive property. The specific capacitances of the tested electrodes at 0.5 A g−1 are 4, 55, 66, 106, 273, 206 and 135 F g−1 for GOF, PRGF-0.125, PRGF-0.25, PRGF-0.5, PRGF-0.75, PRGF-1.0 and GRF electrode, respectively. PRGF-0.75 electrode shows the biggest specific capacitance among all the electrodes, which is accord with the results of the CV test. Although the specific capacitance of PRGF-0.75 electrode is smaller than that of the theoretical value for GRF, it is still superior to those of similar electrodes, such as holey GR paper (201 F g−1 at 1 A g−1),37 GO/CNT sandwich paper (151 F g−1 at 0.5 A g−1),34 GR/mesoporous carbon nanosphere film electrode (211 F g−1 at 0.2 A g−1),38 graphene/multiwalled carbon nanotube film electrode (265 F g−1 at 0.1 A g−1),31 and so on.
The ion diffusion kinetic within the prepared electrode was monitored by electrochemical impedance spectra (EIS). Fig. 3c shows the Nyquist plot of GOF, GRF and PRGF electrodes within the frequency range from 10 mHz to 100 kHz at a signal voltage of 5 mV. A close-up view of the Nyquist plots reveals a semicircle and a short 45° Warburg region for all the electrodes. At very high frequency, the real part of resistance (Z′) of the intercept of plot with real axis, represents the equivalent series resistance (Rs) provided by the ionic resistance of the electrolyte, the intrinsic resistance of the activate materials and contact resistance with the current collector.35 All the electrodes show nearly the same Rs value (about 1.5 Ω) because the same test device is used. At medium-high frequencies, a distinct semicircle loop is observed, which stands for charge transfer resistance (Rct) at the interface between electrolytes and electrode. After fitting EIS spectra through the equivalent circuit diagram, the Rct are182.6, 152.9, 80.0, 47.9, 22.5, 21.7 and 4.1 Ω for GOF, PRGF-0.125, PRGF-0.25, PRGF-0.5, PRGF-0.75, PRGF-1.0, and GRF electrodes, respectively. GOF electrode shows the biggest Rct value resulting from the poor electrical conductivity. The PRGF electrode, however, exhibit smaller Rct value than that of GOF electrode, indicating the electrical conductivity of PRGF electrodes can be improved through reducing by Cu2O.
The relationship of the total impedance to the frequency for GOF, GRF and PRGF electrodes is shown in Fig. 3d. At low frequency, electrolyte ions can migrate from bottom of the porthole to all the electrode materials pores, and therefore the alternating current signal must penetrate through different depth to exhibit a big resistance. However, the electrolyte ions only migrate near the porthole of the pores to give a small resistance value at high frequency. The penetration degree is inversely proportional to the frequency.39 As exhibits in Fig. 3d, GOF electrode shows tremendous resistance below 1 Hz, suggesting an appalling electrical conductivity. The obtained PRGF electrodes locating between GOF and GRF electrodes are illustrative of improvement of electrical conductivity. Based on the discussion above, PRGF-0.75 is the optimum one for supercapacitors.
FSEM and TEM images of PRGF-0.75 are shown in Fig. 4. PRGF-0.75 is composed of flake-like PGR nanosheets (Fig. 4a) which can be further assembled into large-area flexible paper with layered structure (Fig. 4b) by vacuum filtration process. Interestingly, the cross-sectional FSEM image of PRGF shows loose, wavy, and some pores which are beneficial to fast charge transportation due to adsorption of Cu2O before adding HCl. The thicknesses of PRG films were estimated to be about 10 μm according to the SEM images. From Fig. 4c, we can see large PRG sheets exhibit a very stable nature under the electron beam, and the sheets appear transparent under the electron microscope, indicating their similarity to GR sheets prepared by other approaches.
 | ||
| Fig. 4 Planar (a) and cross-sectional (b) FSEM images of PRGF-0.75. TEM image (c) of PRGF-0.75, respectively. | ||
CV curves of PRGF-0.75 electrode at various scan rates are shown in Fig. 5a. All of the CV curves exhibit approximately rectangle-like shapes with a mirror-image feature, and the shapes of these CV curves do not significantly change as increasing the scan rate from 5 to 200 mV s−1, it reveals the ideal capacitive behaviors and good high-rate capabilities of PRGF-0.75. Rate capability is an important determining factor for the ECs especially in power applications. An ideal electrochemical energy storage device is required to provide high energy density at a high charge/discharge rate. The galvanostatic charge/discharge investigation of PRGF-0.75 exhibits excellent rate performance (Fig. 5b). PGRF-0.75 electrode not only shows high specific capacitance values from 273 to 183 F g−1 as the current densities increased from 0.25 to 20 A g−1, but also maintains 67% of its initial specific capacitance at a high current density. After 5000 consecutive charge/discharge cycles at a current density of 5 A g−1, the specific capacitances of PRGF-0.75 electrode is 205 F g−1, and giving capacitance retention of 97% (Fig. 5c). The obvious capacitance grow in the initial 500 cycles could be ascribed to the electrode/electrolyte interface thorough wetting owing to the existence of a few oxygen-containing groups on PRGF-0.75.
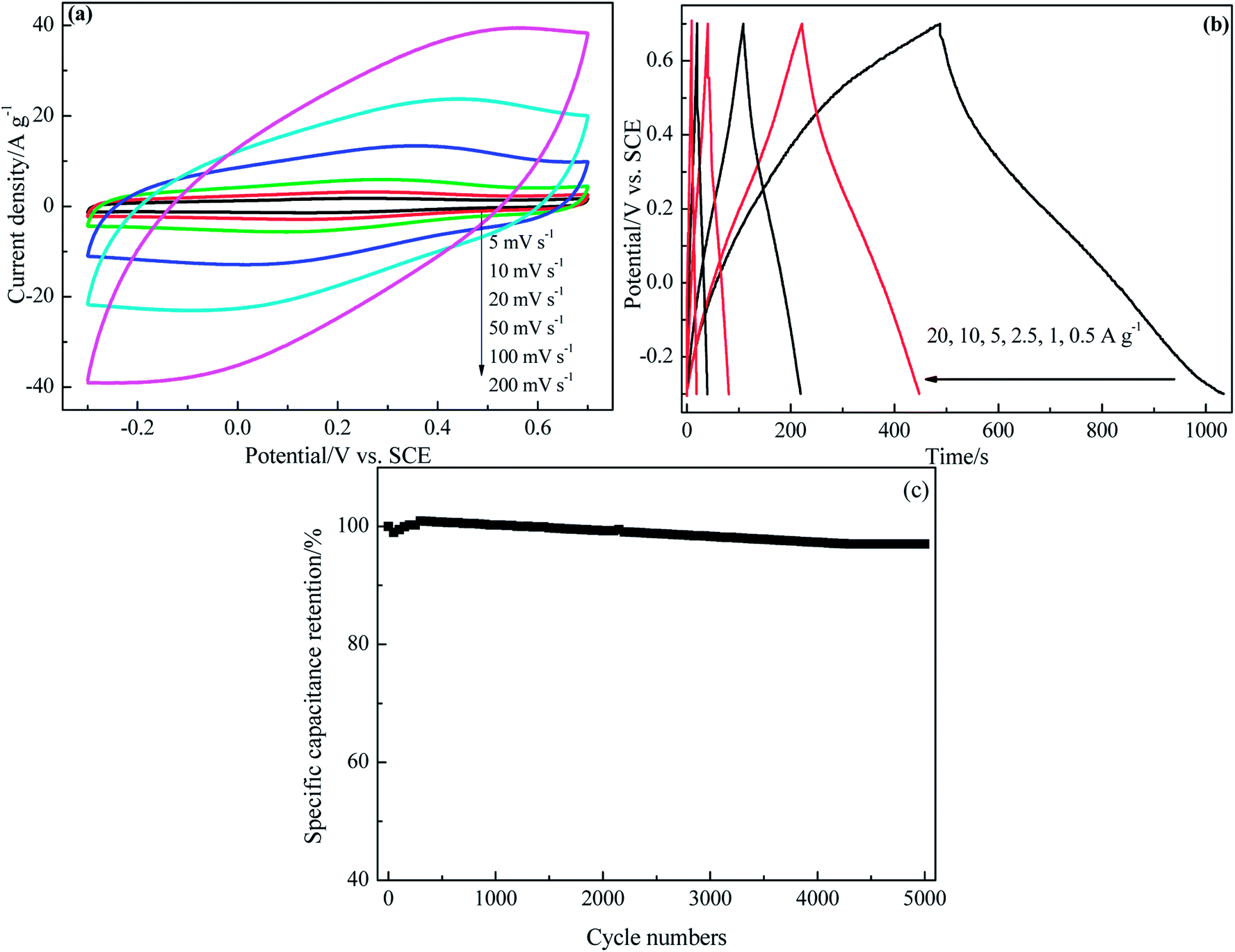 | ||
| Fig. 5 Electrochemical capacitive properties of PRGF-0.75 electrode measured using a three-electrode system in 0.5 mol L−1 H2SO4 electrolyte: (a) CV curves at different scan rates. (b) Galvanostatic charge/discharge profiles at different current densities. (c) Cycle stability at a current density of 5 A g−1. | ||
The superior electrochemical capacitive performance of PRGF-0.75 can be attributed to the following factors: (1) the remained oxygenated groups (–OH and –CO) on the surface of PRGF-0.75 provide pseudocapacitance, and therefore, a hybrid capacitance named pseudocapacitance and EDLC is obtained. (2) The moderate electrical conductivity of PRGF-0.75 is responsible for its excellent rate ability.
Similar to GO, the obtained PRG nanosheets can also be used as building block for supramolecular assembly. In this work, the PRG nanosheets can loosely self-assemble to form PRGH in strong ionic strength medium (Fig. 6a), and the sizes and shapes of PRGH can be easily tailored by changing the reaction vessels. PRGH shows an interconnected 3D porous network and the pore walls consisting of thin layers of stacked PRG sheets, as revealed by SEM images (Fig. 6b–d). Methylene blue (MB) dye adsorption method was employed to determine the specific surface areas (SSA) of PRGH.40 The PRGH showed a very high accessible SSA of ∼1330 m2 g−1, indicating a higher the accessible surface area.
 | ||
| Fig. 6 (a) Photo image of PRGH. SEM images of PRGH at different magnification (b–d). | ||
The unique 3D hierarchical porosity of PRGH arising from the self-assembly of PRG sheets makes it a highly promising material for high-performance supercapacitor electrodes. We have next studied the electrochemical performances of PRGH as binder-free electrodes. CV studies show a less oblique loop and larger current density for PGRH compared to PRGF at a high scan rate of 5 mV s−1 (Fig. 7a), suggesting that the PRGH has a higher ion accessible SSA and faster ion diffusion rate than that of PRGF. The distorted rectangular CV curve of PRGH also indicate a complex supercapacitive similar to that of PRGF. The PRGH electrode exhibits a specific capacitance of 285 F g−1 at a current density of 1 A g−1, 8% higher than that of PRGF electrode (260 F g−1) (Fig. 7b). When the current density was increased up to 20 A g−1, the PGRH could retain as high as ∼84% of its initial value (228 F g−1), while the PRGF give ∼70% capacitance retention (191 F g−1) (Fig. 7d). Furthermore, the PRGH showed a smaller voltage (IR) drop (0.21 V) than that of PRGF (0.37 V) at the current density of 20 A g−1 (Fig. 7c), implying a lower equivalent series resistance for PRGH.
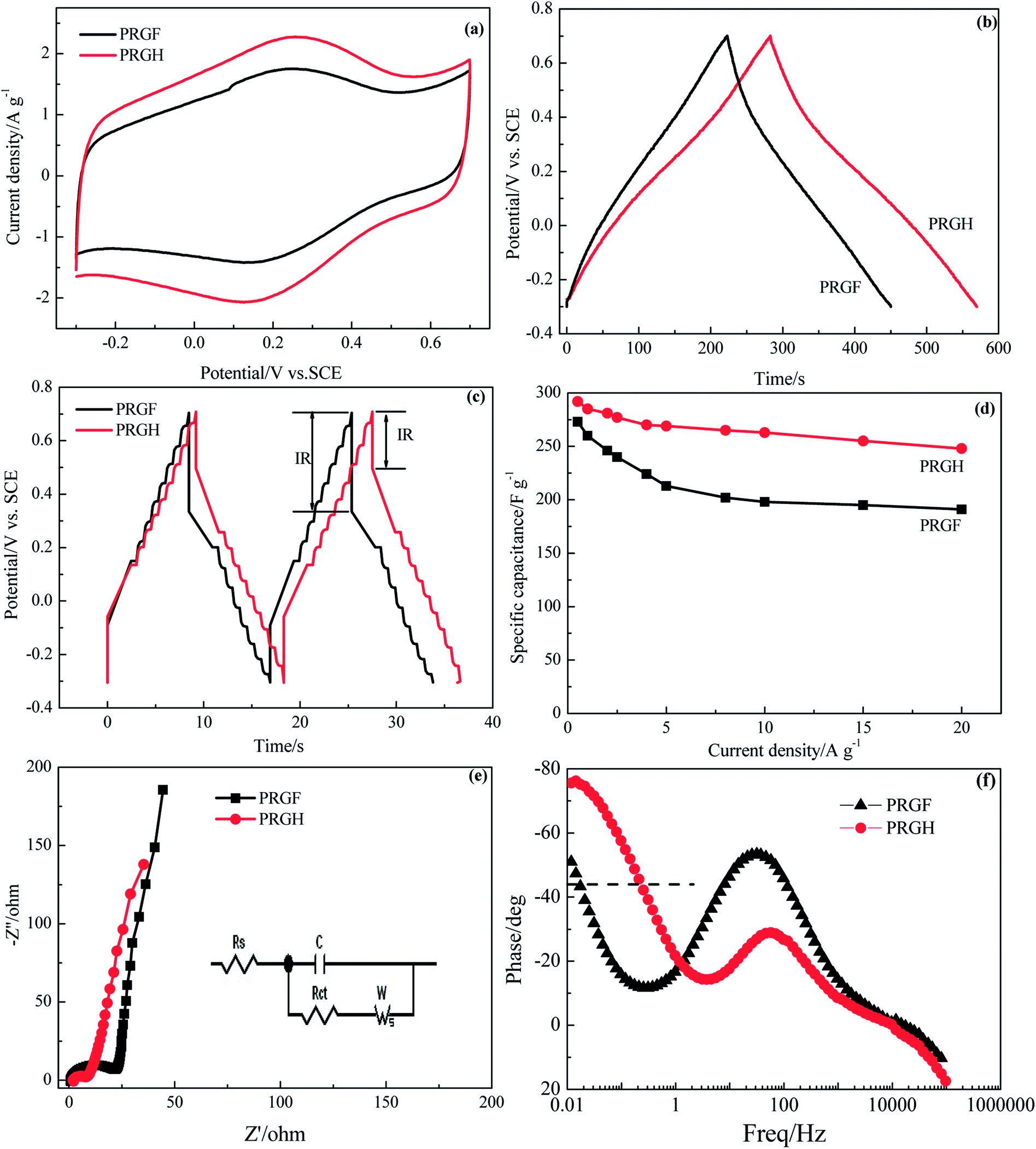 | ||
| Fig. 7 Electrochemical capacitive properties of PRGH and PRGF electrodes measured using a three-electrode system in 0.5 mol L−1 H2SO4 solution: (a) CV curves at scan rate of 5 mV s−1. Galvanostatic charge/discharge profiles at current densities of 1.0 A g−1 (b) and 20 A g−1 (c). (d) Specific capacitance at different current densities. (e) Nyquist plots. Inset: A simplified equivalent circuit. (f) Impedance phase angle versus frequency. | ||
The ion diffusion dynamics within the PRGH was further probed by EIS (Fig. 7e). The Nyquist plots obtained over a frequency range from 100 kHz to 10 mHz showed a vertical line in the low-frequency regime, indicating a nearly ideal capacitive property for PRGH and PRGF. A close-up view of the high-frequency regime of the Nyquist plots revealed a semicircle with smaller diameter and a short 45° Warburg region for PGRH, confirming a lower charge transfer resistance and more rapid ion diffusion within the PRGH due to its hierarchical porosity. By extrapolating the vertical portion of the plot to the real axis, an Rct could be derived to be 5.97 Ω for PRGH, almost one third of that of PRGF (20.93 Ω), which is consistent with the results of galvanostatic charge/discharge studies.
Fig. 7f shows the Bode plot (phase angle versus frequency) of PRGH and PRGF electrodes in three-electrode system. The relaxation time constant τ0 of supercapacitor, which is defined as the 1/f0 at a phase angle of −45°, represents the minimum time needed to discharge all the energy from the device with an efficiency of more than 50%.40,41 The characteristic frequencies f0 at the phase angle of −45° was measured to be 0.2105 and 0.01608 Hz in aqueous H2SO4, corresponding to a time constants τ0 of 4.75 and 62.18 s for PRGH and PRGF electrodes, respectively. The smaller relaxation time (4.75 s) of PRGH also indicates the bigger accessibility of the electrolyte ions to the outer surface and the improved electrical conductivity compared with that of PRGF. Owing to the unique structure, PRGH demonstrates excellent cycling stability with a nearly 99% capacitance retention over 5000 cycles at a current density of 5 A g−1.
4. Conclusions
In summary, we have reported a facile strategy to prepare PRG nanosheets by reducing GO using commercial Cu2O powder as a reduction agent at room temperature. The quantity of oxygen functionalities can be adjusted by controlling the usage of commercial Cu2O powder. Owing to the existence of some oxygen functionalities, the obtained PRG nanosheets can function as building blocks for assembling PRGH easily in high ion strength medium and PRGF by vacuum filtration technology. The existed oxygen functionalities of PRG act as sources for pseudocapacitance, and therefore present excellent capacitive properties when used as binder-free electrode for supercapacitor. This work will greatly impact the GR based materials for supercapacitors and flexible supercapacitors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Shaanxi province (2018JM5037) and the Key Laboratory of Applied Surface and Colloid Chemistry (Shaanxi Normal University, 2017022).References
- Y. Li, X. Wang and Q. Yang, et al., Electrochim. Acta, 2017, 234, 63–70 CrossRef.
- P. Du, L. Lin and H. Wang, et al., Mater. Des., 2017, 127, 76–83 CrossRef.
- Y. Wang, W. Lai and N. Wang, et al., Energy Environ. Sci., 2017, 10, 941–949 RSC.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- B. Babakhani and D. G. Ivey, Electrochim. Acta, 2010, 55, 4014–4024 CrossRef.
- B. E. Conway, Electrochemical Supercapacitors, Kluwer Academic/Plenum Press, New York, 1999 Search PubMed.
- X. Pu, L. Li and M. Liu, et al., Adv. Mater., 2016, 28, 98–105 CrossRef PubMed.
- W. K. Chee, H. N. Lim and Z. Zainal, et al., J. Phys. Chem. C, 2016, 120, 4153–4172 CrossRef.
- Y. Xu, C.-Y. Chen and Z. Zhao, et al., Nano Lett., 2015, 15, 4605–4610 CrossRef PubMed.
- H. Zhang, K. Wang and X. Zhang, et al., J. Mater. Chem. A, 2015, 3, 11277–11286 RSC.
- E. Miniach, A. Śliwak and A. Moyseowicz, et al., Electrochim. Acta, 2017, 240, 53–62 CrossRef.
- C. Wu, J. Cai and Y. Zhu, et al., ACS Appl. Mater. Interfaces, 2017, 22, 19114–19123 CrossRef PubMed.
- W. Yang, H. Zhou and Z. Huang, et al., Electrochim. Acta, 2017, 245, 41–50 CrossRef.
- Z. Tian, H. Dou and B. Zhang, et al., Electrochim. Acta, 2017, 237, 109–118 CrossRef.
- Q. X. Xia, J. M. Yun and R. S. Mane, et al., Sustainable Energy Fuels, 2017, 1, 529–539 RSC.
- L. Deng, G. Zhang and L. Kang, et al., Electrochim. Acta, 2013, 112, 448–457 CrossRef.
- H. Chen, C. K. Hsieh and Y. Yang, et al., ChemElectroChem, 2017, 4, 2414–2422 CrossRef.
- Z. Wen, M. Li and F. Li, et al., Dalton Trans., 2016, 45, 936–942 RSC.
- X. Liu, R. Li and J. Zhang, et al., Ceram. Int., 2016, 42, 16364–16367 CrossRef.
- S. Zhu, L. Li and J. Liu, et al., ACS Nano, 2018, 12, 1033–1042 CrossRef PubMed.
- L. Chen, Y. Hernandez and X. Feng, et al., Angew. Chem., Int. Ed., 2012, 51, 2–17 CrossRef.
- S. Park, J. An and J. R. Potts, et al., Carbon, 2011, 49, 3019–3023 CrossRef.
- X. Fan, W. Peng and Y. Li, et al., Adv. Mater., 2008, 20, 4490–4493 CrossRef.
- S. Wakeland, R. Martinez and J. K. Grey, et al., Carbon, 2010, 48, 3463–3470 CrossRef.
- Z. Fan, K. Wang and T. Wei, et al., Carbon, 2010, 48, 1670–1692 CrossRef.
- Z.-J. Fan, K. Wang and J. Yan, et al., ACS Nano, 2013, 5, 191–198 CrossRef PubMed.
- M. J. Fernández-Merino, L. Guardia and J. I. Paredes, et al., J. Phys. Chem. C, 2010, 114, 6426–6432 CrossRef.
- Z. Lin, Y. Liu and Y. Yao, et al., J. Phys. Chem. C, 2011, 115, 7120–7125 CrossRef.
- N. Kovtyukhova, P. Ollivier and B. Martin, et al., Chem. Mater., 1999, 11, 771–779 CrossRef.
- Y. Bai, X. Yang and Y. He, et al., Electrochim. Acta, 2016, 187, 543–551 CrossRef.
- Z. Gao, J. Wang and Z. Li, et al., Chem. Mater., 2011, 23, 3509–3516 CrossRef.
- C. Zhu, S. Guo and Y. Fang, et al., ACS Nano, 2010, 4, 2429–2437 CrossRef PubMed.
- S. Stankovich, D. Dikin and R. Piner, et al., Carbon, 2007, 45, 1558–1565 CrossRef.
- H. A. Andreas and B. E. Conway, et al., Electrochim. Acta, 2006, 56, 6510–6520 CrossRef.
- M. P. Bichat, E. Raymundo-Piñero and B. Féguin, Carbon, 2010, 48, 4351–4561 CrossRef.
- B. Zhao, P. Liu and Y. Jiang, et al., J. Power Sources, 2012, 198, 423–427 CrossRef.
- Z. Yan, L. L. Ma and Y. Zhu, et al., ACS Nano, 2013, 7, 58–64 CrossRef PubMed.
- X. Yang, J. Zhu and L. Qiu, et al., Adv. Mater., 2011, 23, 2833–2838 CrossRef PubMed.
- K. Wang, N. Zhao and S. Lei, et al., Electrochim. Acta, 2015, 166, 1–11 CrossRef.
- B. Zhao, P. Liu and Y. Jiang, et al., J. Power Sources, 2012, 198, 423–427 CrossRef.
- Z.-D. Huang, B. Zhang and R. Liang, et al., Carbon, 2012, 50, 4239–4251 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04788j |
| This journal is © The Royal Society of Chemistry 2018 |