
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA multifunctional microporous metal–organic framework: efficient adsorption of iodine and column-chromatographic dye separation†
Chan Yaoa,
Wei Wanga,
Shu-Ran Zhanga,
Hui-Ying Lia,
Yan-Hong Xu *ab,
Zhong-Min Suc and
Guang-Bo Che*a
*ab,
Zhong-Min Suc and
Guang-Bo Che*a
aKey Laboratory of Preparation and Applications of Environmental Friendly Materials, Jilin Normal University, Ministry of Education, Changchun 130103, Jilin, People's Republic of China. E-mail: xuyh198@163.com
bKey Laboratory of Functional Materials Physics and Chemistry of the Ministry of Education, Jilin Normal University, Siping 136000, Jilin, People's Republic of China
cInstitute of Functional Material Chemistry, Faculty of Chemistry, Northeast Normal University, Changchun, 130024, Jilin, People's Republic of China
First published on 26th October 2018
Abstract
In this work, a multifunctional microporous metal–organic framework (MOF), [Cd(ABTC)(H2O)2(DMA)]·4DMA (JLNU-4; JLNU = Jilin Normal University; H4ABTC = 3,3′,5,5′-azobenzenetetracarboxylic acid), has been synthesized based on the ligand H4ABTC under solvothermal conditions. JLNU-4 shows excellent uptake of iodine both in solution and in the vapor phase, owing to the existence of a microporous structure in JLNU-4. The adsorption kinetics during the process of iodine adsorption were analyzed via a series of qualitative and quantitative analyses, such as the Langmuir and Freündlich adsorption isotherms. In addition, according to UV/vis spectroscopy analysis and the colour variance of JLNU-4, the relatively small sized dye methylene blue (MB) could be efficiently adsorbed by JLNU-4, through size-exclusion effects. Particularly, JLNU-4 can serve as a column-chromatographic filler for the separation of dye molecules. Therefore, JLNU-4 is a multifunctional microporous MOF for iodine adsorption and column-chromatographic dye separation.
Introduction
The release of hazardous chemicals is continually growing due to the rapid development of modern society and technological progress, with examples including nuclear waste, toxic heavy metal ions, organic dye molecules, and so on, which have brought significant threats to human and animal health. Although the demand for efficient and non-carbon-emitting nuclear energy is growing rapidly, an urgent issue of safety concern relating to nuclear power production is the appropriate approach to the disposal of waste products generated from the nuclear fission of uranium.1 In particular, radioactive iodine (I2 containing 129I and/or 131I), as the main component of waste streams, has attracted considerable attention because it could cause severe tissue damage and cancer to humans through the respiratory system via both beta and gamma radiation.2 It can disperse rapidly in air and contaminate liquid as well as solid waste-processing systems.3 Additionally, the radioactive half-life is 8 days for 129I and as long as 1.57 × 107 years for 131I, which can be incorporated into the human metabolic system.4 Therefore, the design of stable and highly effective materials for the capture and storage of radiological iodine has become a critical issue to be addressed. In general, inorganic composite adsorbents, for instance silver-based zeolites or aerogels, have been utilized for radioactive iodine capture.5 However, their limited available surface areas result in low uptake capacities for iodine. Besides, the high cost and adverse environmental impact of silver are the main disadvantages of silver-based zeolites or aerogels, which hinder their utilization and development. It remains a labor-intensive endeavor to synthesize porous materials with more efficient, less costly materials that have steady high-iodine trapping abilities for iodine capture in the gas phase and liquid phase.Given the growing interest in exploring novel porous materials towards the effective capture and storage of iodine, various porous adsorbents, including inorganic materials,6 conjugated microporous polymers (CMPs),7 porous aromatic frameworks (PAFs),8 nanocomposites,9 covalent organic frameworks (COFs),10 and crystalline metal–organic frameworks (MOFs),11 have recently been constructed from well-functionalized organic building blocks for iodine capture. Among these porous materials, research into MOFs has been very fruitful, mainly because of their high specific surface areas, adjustable pore structures, regular pore shapes, and unique architectures and functionalities.12 Furthermore, the combination of appropriate organic ligands with varied geometries and functional metal ions or clusters could result in the rational design of MOFs with high porosities, tunable pore structures and desirable surface binding sites for the capture of iodine.13 Currently, Nenoff, Walton and their co-workers have demonstrated that HKUST-1,14 ZIF-8,2c,15 and MIL-53(Al)16 can adsorb iodine with varying capacities, reversibilities and stabilities. Nevertheless, there are limits to MOF materials with high iodine uptake capacities. The influences on the iodine adsorption capacity involve not only the pore size, but also effective sorption sites, binding interactions of iodine within the cavity of MOFs, and the effects of iodine–iodine interactions, which may form polyiodide anions.17 Owing to the large molecular mass of adsorbed iodine, the pores of MOFs would be highly disordered after iodine adsorption, and iodine tends to have a high activation barrier for diffusion in host structures.18 As a result, it is vital to analyze the adsorption kinetics of iodine during the adsorption process. Nowadays, it is still a great challenge to synthesize MOFs with high iodine uptake capacity, and where possible to make clear the nature of the host-iodine and iodine–iodine interaction(s) within MOF pores at a molecular level.
The porous structures of MOFs also inspire us to continuously explore their application in other fields as an excellent platform for host–guest chemistry, for example, the adsorption and separation of dye molecules. It has been extensively recognized that organic dyes have been widely used in many industrial fields, such as paper-making, printing, plastics, textiles, pharmaceuticals, and so on.19 But their toxicity and even carcinogenicity have posed a huge threat to the aqueous environment and human health, which cannot be ignored.20 In the meantime, most dyes are difficult to degrade because of their stability to light and oxidants. In this regard, low-cost and eco-friendly technology for the adsorption and separation of dyes is urgently required, which is more competitive than some other methods. Recently, MOFs have been applied in adsorbing or separating dye molecules, which usually consist of two types of process. (i) Dye removal depending on the size-exclusion effect, in other words, the kind of dyes to be absorbed relies on the pore size of the MOF.21 Sun et al. have synthesized two Cd-tib-carboxylate (tib = 1,3,5-tris(1-imidazolyl)benzene) MOFs under solvothermal conditions, which could selectively adsorb methyl orange molecules.22 Lan and co-workers have reported a series of mesoporous MOFs with tailored pore sizes, which can incorporate a dye with a smaller size than that of a MOF pore, followed by column-chromatographic separation for large dye molecules.23 (ii) Charged MOFs adsorb dyes with opposite charges based on ionic exchange and electrostatic interactions.24 Shi et al. have prepared negatively charged uranyl-MOFs for selectively removing positively charged organic dyes over negatively charged and neutral ones.25 In a previous work, we also reported a microporous anionic MOF, [Zn(ABTC)0.5(NO3)][(CH3)2NH2]·DMA·3H2O (H4ABTC = 3,3′,5,5′-azobenzenetetracarboxylic acid; DMA = N,N-dimethylacetamide), which could efficiently adsorb cationic dyes and serve as a column-chromatographic filler for the separation of dye molecules.26
As a continuation of our work, we obtain a 3D microporous MOF, [Cd(ABTC)(H2O)2(DMA)]·4DMA (JLNU-4), synthesized via the solvothermal reaction of Cd(NO3)2·4H2O and H4ABTC. It is shown that neutral iodine can be trapped in the vapor and solution phase by JLNU-4. In addition, we have carried out a series of qualitative and quantitative analyses during the process of I2 adsorption. Meanwhile, JLNU-4 can selectively adsorb the dye molecule methylene blue (MB) via size-exclusion effects, due to the microporous structure, which makes it an excellent candidate for column-chromatographic dye adsorption and separation.
Experimental
Materials and measurements
Chemicals were obtained from commercial sources and were used without further purification. Fourier transform infrared (FT-IR) spectra were recorded over the range 4000–400 cm−1 on a Perkin-Elmer FT-IR-Frontier infrared spectrometer using KBr pellets. Elemental microanalyses (C, H, and N) were performed on a EuroEA-3000 elemental analyzer. Thermogravimetric analysis (TGA) was performed on a Q5000IR analyser (TA Instruments) with an automated vertical overhead thermobalance heated from 40 to 1000 °C at a ramp rate of 5 °C min−1 under a nitrogen atmosphere. Powder X-ray diffraction (PXRD) was performed on a Rigaku model RINT Ultima III diffractometer over the range of 5–60° at 293 K by depositing powder on a glass substrate. The solution UV/vis adsorption spectra were recorded using Shimadzu UV-3600 apparatus. X-ray photoelectron spectra (XPS) were recorded on an ESCALAB250Xi electron spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). Nitrogen sorption isotherms were measured at 77 K with an ASIQ (iQ-2) volumetric adsorption analyzer. Before gas adsorption measurements, the sample was immersed in methanol for 24 h, and the extract was decanted. Fresh methanol was subsequently added, and the crystals were left for an additional 24 h to remove nonvolatile solvates. The sample was collected via decanting and treated with dichloromethane similarly to remove methanol solvates. After the removal of dichloromethane via decanting, the sample was activated through drying under a dynamic vacuum at room temperature overnight. Before measurements, the sample was dried again by using the ‘outgas’ function of the surface area analyzer for 12 h at room temperature. The Brunauer–Emmett–Teller (BET) method was utilized to calculate the specific surface areas and pore volumes. Non-local density functional theory (NLDFT) was applied for estimations of pore size and pore size distribution. Energy dispersive spectroscopy (EDS) images were obtained on a JEOL model JSM-6700 operating at an accelerating voltage of 5.0 kV. The samples were prepared for EDS via drop-casting a tetrahydrofuran suspension onto a mica substrate and then coating with gold.Synthesis of [Cd(ABTC)(H2O)2(DMA)]·4DMA (JLNU-4)
A mixture of H4ABTC (0.01 g, 0.03 mmol), Cd(NO3)2·4H2O (0.20 g, 0.65 mmol), DMA (4 mL) and CH3OH (4 mL) was sealed in a Teflon-lined stainless steel container and heated in an autoclave at 100 °C for 96 h. After the autoclave was cooled to room temperature, yellow crystals were obtained and isolated via washing with DMA and drying at room temperature; yield: 65% based on H4ABTC. Elemental microanalysis for C36H51N7O15Cd2: calculated (%): C, 41.31; H, 4.91; N, 9.37; found (%): C, 41.25; H, 4.98; N, 9.46. IR (cm−1, ESI Fig. S1a†): 3082 (m), 2933 (m), 1642 (s), 1507 (s), 1443 (m), 1401 (s), 1366 (s), 1260 (s), 1187 (m), 1019 (m), 971 (w), 787 (s), 724 (w), 590 (w), 477 (w).X-ray crystallography
Single crystal X-ray diffraction data in this work were recorded on a Bruker APEXII CCD diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71069 Å) at 293 K. Absorption corrections were applied using a multi-scan technique. All structures were solved via the direct method of SHELXS-97 (ref. 27a) and refined via full-matrix least-squares techniques using the SHELXL-97 program27b within WINGX.27c Non-hydrogen atoms were refined with anisotropic temperature parameters. The SQUEEZE program implemented in PLATON was used to remove electron densities from the compounds. Thus, all electron densities from free solvent molecules have been “squeezed” out. The CCDC number is 1588369. Detailed crystallographic data and structure refinement parameters are summarized in Table S1 (ESI†).Results and discussion
The yellow block crystals of JLNU-4 have been obtained via the solvothermal reaction of Cd(NO3)2·4H2O and H4ABTC in DMA. The formula of JLNU-4 is [Cd(ABTC)(H2O)2(DMA)]·4DMA, taking single-crystal X-ray diffraction studies, TGA (Fig. S2, ESI†) and elemental analysis into consideration. Single-crystal X-ray diffraction studies reveal that JLNU-4 crystallizes in the monoclinic space group P21/c (Table S1, ESI†). The crystallographically independent unit contains two Cd2+ ions, a type of ligand fragment, two coordinated H2O molecules and one coordinated DMA molecule (Fig. 1a). The Cd1 atom is seven-coordinate through seven oxygen atoms from four H4ABTC ligands. The Cd2 atom is six-coordinate through three oxygen atoms from three H4ABTC ligands, one oxygen atom from a coordinated DMA molecule and two oxygen atoms from two coordinated H2O molecules. The two kinds of Cd atom are linked by three carboxylate groups to form a binuclear [Cd2(COO)4(H2O)2(DMA)] cluster (Fig. S3, ESI†). The Cd–O bond lengths are all within the normal ranges (Table S2, ESI†). The adjacent [Cd2(COO)4(H2O)2(DMA)] clusters are connected to each other by the ligand, giving rise to an infinite 3D framework with microporous characteristics (Fig. 1b and S4, ESI†). The approximate sizes of the channels are 10.4 × 8.7 Å2 and 7.8 × 7.5 Å2 along the b axis. From topological analysis, each [Cd2(COO)4(H2O)2(DMA)] cluster is considered as a 4-connected node and, in turn, each H4ABTC ligand unit also acts as a 4-connected node (Fig. S5, ESI†), leading to a (4,4)-connected 3D network with pts topology (Schläfli symbol: (42·84); Fig. 1c–e).28 Although the ligand and topological structure are the same as in Zhu's work,29 the structural details, such as the space group, the coordination environment of the Cd atoms, the pore size, and the synthetic method are all distinguishing.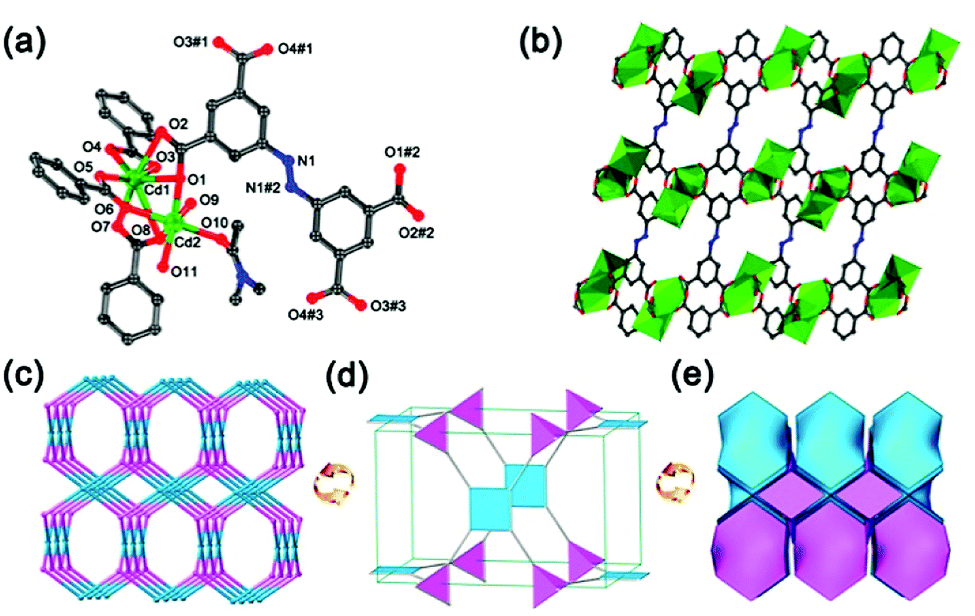 | ||
| Fig. 1 (a) The coordination environments of Cd atoms in JLNU-4. Symmetry code: #1: −x + 1, y + 0.5, −z + 0.5; #2: −x + 1, −y + 1, −z; #3: x, −y + 0.5, z − 0.5. (b) Infinite 3D framework with channels viewed along the b axis. (c) Ball-and-stick and (d) polyhedral views of the (4,4)-connected topology network in JLNU-4. (e) The (4,4)-connected augmented net in the form of natural tiling. All hydrogen atoms are omitted for clarity. | ||
The PXRD patterns of both simulated and as-synthesized JLNU-4 match well in key positions, thus indicating that it possesses good phase purity (Fig. S6, ESI†). The PXRD pattern of activated JLNU-4 is almost identical with that of the as-synthesized one, as illustrated in Fig. S6 in the ESI,† implying that the basic 3D framework is retained. The calculated free volume in fully desolvated JLNU-4 is 56.3% from PLATON.30 N2 sorption studies demonstrate the existence of permanent porosity in JLNU-4. The Brunauer–Emmett–Teller (BET) surface area of JLNU-4 is estimated to be 64 m2 g−1 (Fig. S7a, ESI†). The pore size distribution plot has been drawn by employing the NLDFT method, as shown in Fig. S7b (ESI†), representing that micropores exist in JLNU-4.
Considering the microporous properties of JLNU-4, its capacity towards molecular iodine capture both in solvent and vapor phases was explored. When fresh samples of JLNU-4 (20 mg) were immersed in a hexane (3 mL) solution of iodine (4 mg mL−1),31–43 the colour of the solution gradually faded from dark purple to light purple with the extension of time (Fig. 2a, top). At the same time, the colour of the crystals changed from yellow to brown to black over 24 h (Fig. 2b). To further qualitatively investigate the process of iodine capture in JLNU-4, UV/vis spectra were monitored at room temperature. The intensities of the UV/vis adsorption spectra decreased with prolonged time (Fig. 2c).
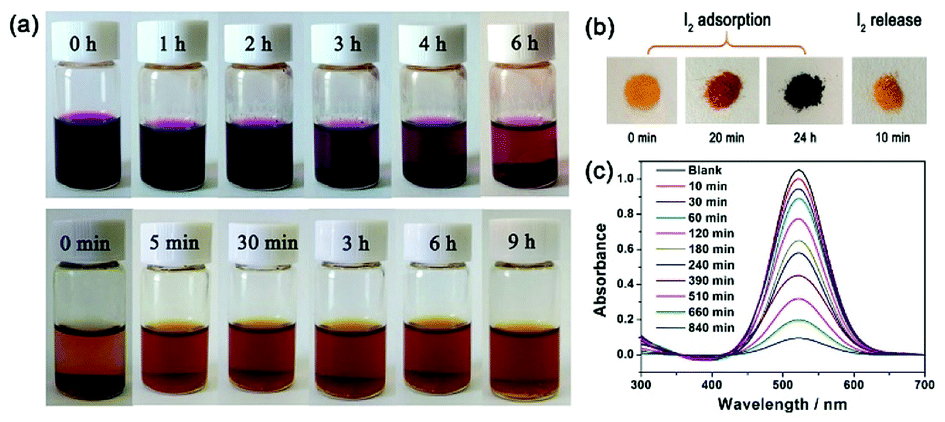 | ||
| Fig. 2 (a) Photographs of JLNU-4 during the iodine adsorption process in hexane solution (top) and the iodine release process in ethanol solution (bottom). (b) Photographs showing the visual color change of JLNU-4 during iodine adsorption and release. (c) UV/vis spectra upon immersion of JLNU-4 (20 mg) in a hexane solution of iodine (0.4 mg mL−1). | ||
Subsequently, quantitative analysis was carried out to gain a deeper insight into the adsorption kinetics of iodine. At an early stage, the adsorption capacity increased quickly with the prolongation of contact time and tended to increase gradually after 10 h, illustrating that the adsorption kinetics were related to contact time (Fig. S8 and S9, ESI†). The removal efficiency was raised to as much as 92.9%. The adsorption kinetics were analysed through the frequently used pseudo-first-order and pseudo-second-order kinetic models.44 The equation of the linear form for the pseudo-first-order kinetic model was eqn (1) as follows, where Qt and Qe represent the mass percent of iodine adsorbed at time t and equilibrium (%), and k1 is the rate constant of the adsorption process (h−1). Eqn (2) was the linear form of the pseudo-second-order kinetic model, in which Qt and Qe were the mass percentages of iodine adsorbed at time t and equilibrium (%), and k2 was the pseudo-second-order rate constant of the adsorption process (h−1). According to the simulation analysis, we found that the adsorption data fitted well to the pseudo-second-order kinetic model with a good linear correlation coefficient (R2) value of 0.9837 (Table S3, ESI†). This also proved that the pseudo-second-order kinetic model governed the process of iodine adsorption.
ln(Qe − Qt) = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Qe − k1t Qe − k1t
| (1) |
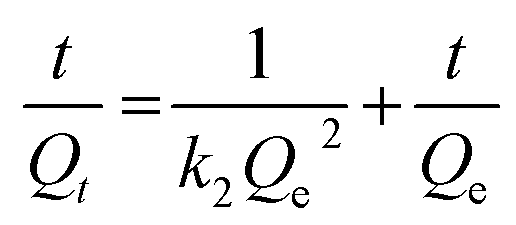 | (2) |
Besides, the adsorption isotherms, Langmuir or Freündlich, could determine the saturated iodine adsorption capacity of JLNU-4. The Langmuir and Freündlich adsorption isotherm models are given in eqn (3) and (4), respectively. In the equations, kL (mg−1) and Qm (mg g−1) are the Langmuir isotherm constants, kF (mg−1) and n are the Freündlich isotherm constants, Ce is the concentration at equilibrium (mg mL−1), and Qe is the amount of iodine adsorbed at equilibrium (mg g−1). There were two different adsorption stages in the plot of equilibrium concentration versus quantity of adsorbed iodine at equilibrium (Fig. S10 and S11, ESI†). In the first instance, the plots were nearly linear at low concentrations, that is to say the equilibrium uptake increased linearly with the increase in iodine solution concentration. After that, the adsorption reached the maximum value and the adsorption process became independent of the concentration. The Langmuir adsorption isotherm could better judge the saturated iodine adsorption capacity with a good linear correlation coefficient (R2) value of 0.98043 from the simulation analysis (Table S4, ESI†). The saturated iodine adsorption capacity of JLNU-4 in the solvent phase could reach 680 mg g−1, as depicted in Fig. S10† in the ESI.† Moreover, the release of the trapped iodine molecules into JLNU-4 occured very soon via the immersion of the iodine-loaded samples in ethanol. The color of the solvent changed from colorless to dark brown when the iodine-loaded samples were immersed in ethanol (Fig. 2a, bottom), suggesting that iodine molecules were released from JLNU-4. The color of the crystals almost recovered its original yellow after the initial ten minutes of the release process (Fig. 2b). There were no major changes to either the external appearance or the XRPD patterns after immersing JLNU-4 in hexane solution, even after the process of iodine adsorption and release (Fig. S12, ESI†). This confirmed the reversibility of iodine adsorption and release, and the high stability of the host, JLNU-4.
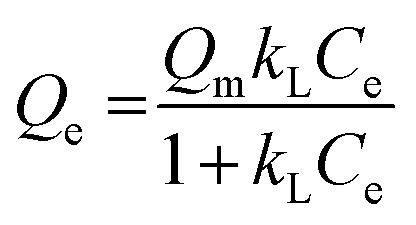 | (3) |
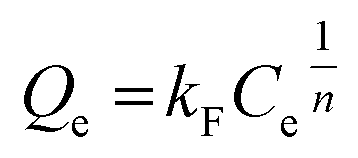 | (4) |
It is widely acknowledged that iodine is volatile, thus iodine adsorption in the vapor phase is critically important. JLNU-4 (30 mg) was exposed to nonradioactive iodine vapor in a sealed vessel at 350 K and under ambient pressure; these are typical fuel reprocessing conditions.45–50 During the adsorption process, gravimetric measurements were performed at time intervals (Table S5, ESI†). The maximum iodine uptake of JLNU-4 reached saturation after 10 h (Fig. S13, ESI†). The saturated iodine adsorption value was 64 wt% (636.7 mg g−1). There was significant weight loss from 90 to 300 °C in the TGA of iodine-loaded JLNU-4, which might be caused by the decomposition of iodine (Fig. S2, ESI†). The XPS spectra of JLNU-4 after iodine adsorption in hexane solution and iodine vapor adsorption verified the existence of iodine, which meant that the adsorption took the form of physical adsorption (Fig. S14, ESI†).
From an environmental point of view, removing dyes from effluents before discharging them into natural water bodies is extremely vital. Organic dyes, with a variety of colors and sizes, are a big group of organic molecules, which are frequently used in numerous manufacturing industries and subsequently cause various environmental problems.51 Motivated by the existing pores in JLNU-4, we selected several organic dyes with different molecular sizes for dye sorption on the basis of the size-exclusion effect. There was no need for any further activation process compared with other reported MOFs for dye sorption.52 To measure whether JLNU-4 had the ability for dye molecule adsorption, we prepared fresh JLNU-4 to capture dyes in DMA. Crystals of JLNU-4 (100 mg) were soaked in DMA solutions of a series of dyes (10 mL, 1.0 mmol). A color change in JLNU-4 was clearly observed in methylene blue (MB) by the naked eye after soaking for about one hour (Fig. 3). As illustrated in the inset of Fig. 3, the uniform distribution of MB throughout JLNU-4 indicated that the organic dye molecules deeply penetrated into the pores instead of only being adsorbed on the external surface of JLNU-4.53 This may be caused by π–π* interactions between aromatic moieties of the frameworks and dyes.22 We have also measured the N2 gas-sorption for MB loaded JLNU-4 (Fig. S15, ESI†). The adsorption capacity of MB loaded JLNU-4 significantly decreased compared to JLNU-4 (Fig. S7a, ESI†). The obvious sorption hysteresis can be interpreted as arising from the network swelling or restricted access of adsorbate molecules through narrow pore openings. The BET surface area of MB loaded JLNU-4 is estimated to be 8.1 m2 g−1, which is also lower than that of JLNU-4 (64 m2 g−1). Therefore, MB has been adsorbed into the pores of JLNU-4. Owing to the microporous structure of JLNU-4, only the relatively small sized MB, in comparison to other dye molecules, could be adsorbed into the pores. The adsorption capabilities of JLNU-4 for MB were quantitatively determined through UV/vis spectroscopy analysis (Fig. 3). The concentration of MB in DMA observably decreased with the passage of time, which showed that JLNU-4 could completely and rapidly adsorb MB. At the same time, the PXRD patterns affirmed that JLNU-4 could remain stable after adsorbing MB in DMA (Fig. S16, ESI†). Moreover, EDS data (Fig. S17, ESI†) and XPS (Fig. S18, ESI†) data testify the existence of chlorine atoms in MB loaded JLNU-4. Therefore, MB molecules were absorbed as a chloride salt.
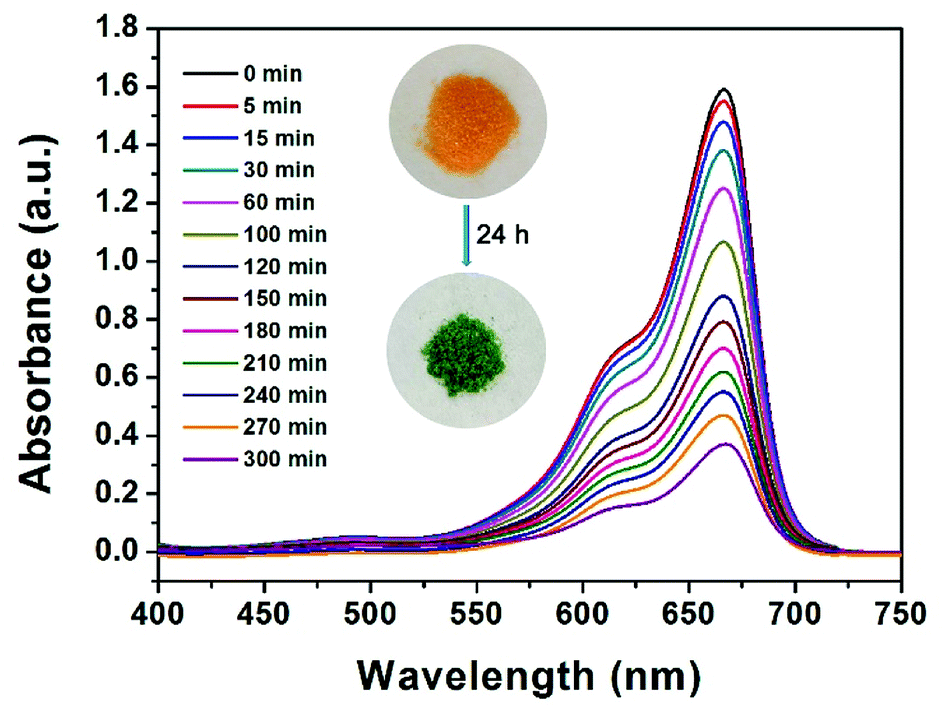 | ||
| Fig. 3 UV/vis spectra of DMA solutions of MB with freshly prepared JLNU-4. Inset: photographs of the color change of crystals after immersion in a solution of MB for a period of time. | ||
Inspired by the dye adsorption properties of JLNU-4, we prepared a chromatography column filled with JLNU-4 and a mixture of MB and CR (1:1, 1.0 mmol, CR = congo red) in DMA solution. The two dyes could be successfully separated via passing them through the MOF-filled column. As shown in Fig. 4(I), JLNU-4 filled the chromatography column made of a glass dropper. When the dye mixture of MB and CR naturally passed through the JLNU-4-filled column under gravity, it was clearly observed that the color of the column became dark green (Fig. 4(II)). Next, the pure DMA stream passed through the column several times. As we expected, the larger and unincorporated CR was rapidly transported through the column along with the DMA stream while the small-sized MB was captured inside the JLNU-4-filled column (Fig. 4(III–VII)). UV-vis spectra of the effluent as the dye mixture of MB and CR passed through the JLNU-4-filled chromatographic column reveal the successful separation of MB and CR (Fig. S19, ESI†). The continuing decrease of the characteristic absorbance of MB reveals its successful incorporation, whereas the unchanged peak of BR suggests its exclusion by the MOF pores. Therefore, JLNU-4 could be used for dye separation via a combination of chromatography column and the size-exclusion effect. This also demonstrated that MOFs with suitable pore sizes could be an outstanding selection for column-packing materials, which might be a substitute for traditional silica gels for size-dependent large molecule separation.
 | ||
| Fig. 4 Photographic records of the JLNU-4-filled column-chromatographic separation process for MB and CR dyes showing: (I) a JLNU-4-filled column, (II–VI) the separation process with the gradual color change, and (VII) complete separation with only MB adsorbed. | ||
Conclusions
In summary, a 3D microporous MOF, JLNU-4, has been rationally designed and constructed via a solvothermal reaction. Bearing the microporous structure of JLNU-4 in mind, we carried out iodine adsorption experiments on JLNU-4. The results have proved that JLNU-4 could capture neutral iodine molecules in the solution phase as well as in the vapor phase. The saturated iodine adsorption capacity of JLNU-4 was 680 mg g−1 in the solvent phase. A series of qualitative and quantitative analyses were performed to analyze the adsorption kinetics in the process of iodine adsorption. Meanwhile, JLNU-4 can not only be used for the selective adsorption of MB but also can be applied as a column-chromatography filler for separating dye molecules. Therefore, JLNU-4 is a prominent and effective adsorbent for iodine adsorption and dye separation. This research has presented progressive evolution in the construction of MOF-based multifunctional materials. The exploration of more novel applications for multifunctional MOF materials is currently underway and will be useful under more realistic circumstances in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support of the National Natural Science Foundation of China (Grants no. 21501065), Changbai Mountain Scholars Program (Grants no. 2013073), the Science and Technology Program of Jilin Province (Grants no. 20160101319JC, 20180520159JH and 20180520152JH), the Science and Technology Plan Funds of Siping City, China (No. 2017092), and the Thirteenth Five-Year Program for Science and Technology of Education Department of Jilin Province (JJKH20180760KJ) is acknowledged.Notes and references
- G. Steinhauser, Environ. Sci. Technol., 2014, 48, 4649–4663 CrossRef CAS PubMed.
- (a) D. Shetty, J. Raya, D. S. Han, Z. Asfari, J.-C. Olsen and A. Trabolsi, Chem. Mater., 2017, 29, 8968–8972 CrossRef CAS; (b) N. Yoshida and J. Kanda, Science, 2012, 336, 1115–1116 CrossRef CAS PubMed; (c) D. F. Sava, M. A. Rodriguez, K. W. Chapman, P. J. Chupas, J. A. Greathouse, P. S. Crozier and T. M. Nenoff, J. Am. Chem. Soc., 2011, 133, 12398–12401 CrossRef CAS PubMed; (d) W. J. Weber and F. P. Roberts, Nucl. Technol., 1983, 60, 178–198 CrossRef CAS.
- C. Pei, T. Ben, S. Xua and S. Qiu, J. Mater. Chem. A, 2014, 2, 7179–7187 RSC.
- (a) G. Audi, O. Bersillon, J. Blachot and A. H. Wapstra, Nucl. Phys. A, 2003, 729, 3–128 CrossRef; (b) J. Wang, J. Luo, X. Luo, J. Zhao, D.-S. Li, G. Li, Q. S. Huo and Y. Liu, Cryst. Growth Des., 2015, 15, 915–920 CrossRef CAS.
- (a) K. W. Chapman, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2010, 132, 8897–8899 CrossRef CAS PubMed; (b) K. S. Subrahmanyam, D. Sarma, C. D. Malliakas, K. Polychronopoulou, B. J. Riley, D. A. Pierce, J. Chun and M. G. Kanatzidis, Chem. Mater., 2015, 27, 2619–2626 CrossRef CAS.
- (a) T. Hertzsch, F. Budde, E. Weber and J. Hulliger, Angew. Chem., Int. Ed., 2002, 41, 2281–2284 CrossRef CAS; (b) L. Mohanambe and S. Vasudevan, Inorg. Chem., 2004, 43, 6421–6425 CrossRef CAS PubMed; (c) J. L. Krumhansl and T. M. Nenoff, Appl. Geochem., 2011, 26, 57–64 CrossRef CAS.
- (a) Y. Zhu, Y.-J. Ji, D.-G. Wang, Y. Zhang, H. Tang, X.-R. Jia, M. Song, G. Yu and G.-C. Kuang, J. Mater. Chem. A, 2017, 5, 6622–6629 RSC; (b) Y. Liao, J. Weber, B. M. Mills, Z. Ren and C. F. J. Faul, Macromolecules, 2016, 49, 6322–6333 CrossRef CAS; (c) A. Sigen, Y. Zhang, Z. Li, H. Xia, M. Xue, X. Liu and Y. Mu, Chem. Commun., 2014, 50, 8495–8498 RSC; (d) T. Geng, Z. Zhua, W. Zhang and Y. Wang, J. Mater. Chem. A, 2017, 5, 7612–7617 RSC; (e) X. Qian, Z.-Q. Zhu, H.-X. Sun, F. Ren, P. Mu, W. Liang, L. Chen and A. Li, ACS Appl. Mater. Interfaces, 2016, 8, 21063–21069 CrossRef CAS PubMed.
- Z. Yan, Y. Yuan, Y. Tian, D. Zhang and G. Zhu, Angew. Chem., Int. Ed., 2015, 54, 12733–12737 CrossRef CAS PubMed.
- G. Massasso, M. Rodriguez-Castillo, J. Long, A. Grandjean, B. Onida, Y. Guari, Ch. Guerina and J. Larionova, J. Mater. Chem. A, 2015, 3, 179–188 RSC.
- (a) Y. Lin, X. Jiang, S. T. Kim, S. B. Alahakoon, X. Hou, Z. Zhang, C. M. Thompson, R. A. Smaldone and C. Ke, J. Am. Chem. Soc., 2017, 139, 7172–7175 CrossRef CAS PubMed; (b) A. P. Katsoulidis, J. Q. He and M. G. Kanatzidis, Chem. Mater., 2012, 24, 1937–1943 CrossRef CAS.
- (a) D. Y. Lee, E.-K. Kim, N. K. Shrestha, D. W. Boukhvalov, J. K. Lee and S.-H. Han, ACS Appl. Mater. Interfaces, 2015, 7, 18501–18507 CrossRef CAS PubMed; (b) F. Dai, W. Fan, X. Yuan, Z. Huang, Y. Wang, X. Xin, H. Lin, L. Zhang, R. Wang and D. Sun, Chem. Commun., 2017, 53, 5694–5697 RSC.
- (a) H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed; (b) S. Kitagawa, R. Kitaura and S. I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed; (c) G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- (a) D. Zhao, D. J. Timmons, D. Yuan and H.-C. Zhou, Acc. Chem. Res., 2011, 44, 123–133 CrossRef CAS PubMed; (b) W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang and T. Gentle III, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC; (c) F. A. A. Paz, J. Klinowski, S. M. Vilela, J. P. Tome, J. A. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088–1110 RSC; (d) A. Schoedel, M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2016, 116, 12466–12535 CrossRef CAS PubMed.
- D. F. Sava, K. W. Chapman, M. A. Rodriguez, J. A. Greathouse, P. S. Crozier, H. Zhao, P. J. Chupas and T. M. Nenoff, Chem. Mater., 2013, 25, 2591–2596 CrossRef CAS.
- J. T. Hughes, D. F. Sava, T. M. Nenoff and A. Navrotsky, J. Am. Chem. Soc., 2013, 135, 16256–16259 CrossRef CAS PubMed.
- A. S. Munn, F. Millange, M. Frigoli, N. Guillou, C. Falaise, V. Stevenson, C. Volkringer, T. Loiseau, G. Cibin and R. I. Walton, CrystEngComm, 2016, 18, 8108–8114 RSC.
- R.-X. Yao, X. Cui, X.-X. Jia, F.-Q. Zhang and X.-M. Zhang, Inorg. Chem., 2016, 55, 9270–9275 CrossRef CAS PubMed.
- X. Zhang, I. da Silva, H. G. W. Godfrey, S. K. Callear, S. A. Sapchenko, Y. Cheng, I. Vitórica-Yrezábal, M. D. Frogley, G. Cinque, C. C. Tang, C. Giacobbe, C. Dejoie, S. Rudić, A. J. Ramirez-Cuesta, M. A. Denecke, S. Yang and M. Schröder, J. Am. Chem. Soc., 2017, 139, 16289–16296 CrossRef CAS PubMed.
- (a) F.-Y. Yi, W. Zhu, S. Dang, J.-P. Li, D. Wu, Y.-H. Li and Z.-M. Sun, Chem. Commun., 2015, 51, 3336–3339 RSC; (b) C.-F. Zhang, L.-G. Qiu, F. Ke, Y.-J. Zhu, Y.-P. Yuan, G.-S. Xu and X. Jiang, J. Mater. Chem. A, 2013, 1, 14329–14334 RSC.
- (a) W. Fan, W. Gao, C. Zhang, W. W. Tjiu, J. Pan and T. Liu, J. Mater. Chem., 2012, 22, 25108–25115 RSC; (b) A. A. Adeyemo, I. O. Adeoye and O. S. Bello, Toxicol. Environ. Chem., 2012, 94, 1846–1863 CrossRef CAS.
- (a) B.-X. Dong, M. Tang, W.-L. Liu, Y.-C. Wu, Y.-M. Pan, F.-Y. Bu and Y.-L. Teng, Cryst. Growth Des., 2016, 16, 6363–6370 CrossRef CAS; (b) L.-H. Cao, Y.-L. Wei, C. Ji, M.-L. Ma, S.-Q. Zang and T. C. W. Mak, Chem.–Asian J., 2014, 9, 3094–3098 CrossRef CAS PubMed; (c) Y. Zhu, Y.-M. Wang, S.-Y. Zhao, P. Liu, C. Wei, Y.-L. Wu, C.-K. Xia and J.-M. Xie, Inorg. Chem., 2014, 53, 7692–7699 CrossRef CAS PubMed.
- Y.-L. Li, Y. Zhao, P. Wang, Y.-S. Kang, Q. Liu, X.-D. Zhang and W.-Y. Sun, Inorg. Chem., 2016, 55, 11821–11830 CrossRef CAS PubMed.
- Y.-Q. Lan, H.-L. Jiang, S.-L. Li and Q. Xu, Adv. Mater., 2011, 23, 5015–5020 CrossRef CAS PubMed.
- (a) Y. Shen, C.-C. Fan, Y.-Z. Wei, J. Du, H.-B. Zhu and Y. Zhao, Dalton Trans., 2016, 45, 10909–10915 RSC; (b) Z. Zhu, Y.-L. Bai, L. Zhang, D. Sun, J. Fang and S. Zhu, Chem. Commun., 2014, 50, 14674–14677 RSC; (c) Y.-X. Tan, Y. Zhang, Y.-P. He, Y.-J. Zheng and J. Zhang, Inorg. Chem., 2014, 53, 12973–12976 CrossRef CAS PubMed; (d) D.-M. Chen, W. Shi and P. Cheng, Chem. Commun., 2015, 51, 370–372 RSC; (e) J. Li, H.-R. Fu, J. Zhang, L.-S. Zheng and J. Tao, Inorg. Chem., 2015, 54, 3093–3095 CrossRef CAS PubMed; (f) C.-Y. Sun, X.-L. Wang, C. Qin, J.-L. Jin, Z.-M. Su, P. Huang and K.-Z. Shao, Chem.–Eur. J., 2013, 19, 363–3645 Search PubMed.
- K.-Q. Hu, X. Jiang, C.-Z. Wang, L. Mei, Z.-N. Xie, W.-Q. Tao, X.-L. Zhang, Z.-F. Chai and W.-Q. Shi, Chem.–Eur. J., 2017, 23, 529–532 CrossRef CAS PubMed.
- S.-R. Zhang, J. Li, D.-Y. Du, J.-S. Qin, S.-L. Li, W.-W. He, Z.-M. Su and Y.-Q. Lan, J. Mater. Chem. A, 2015, 3, 23426–23434 RSC.
- (a) G. M. Sheldrick, SHELXS-97, Programs for X-ray Crystal Structure Solution, University of Göttingen: Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, SHELXL-97, Programs for X-ray Crystal Structure Refinement, University of Göttingen: Göttingen, Germany, 1997 Search PubMed; (c) L. J. Farrugia, WINGX, A Windows Program for Crystal Structure Analysis, University of Glasgow, Glasgow, UK, 1988 Search PubMed.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS.
- M. Xue, G. Zhu, Y. Li, X. Zhao, Z. Jin, E. Kang and S. Qiu, Cryst. Growth Des., 2008, 8, 2478–2483 CrossRef CAS.
- (a) P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef; (b) A. L. Spek, PLATON, Utrecht University, The Netherlands, 2001 Search PubMed.
- G. Massasso, J. Long, J. Haines, S. Devautour-Vinot, G. Maurin, A. Grandjean, B. Onida, B. Donnadieu, J. Larionova, C. Guérin and Y. Guari, Inorg. Chem., 2014, 53, 4269–4271 CrossRef CAS PubMed.
- W.-W. He, S.-L. Li, G.-S. Yang, Y.-Q. Lan, Z.-M. Su and Q. Fu, Chem. Commun., 2012, 48, 10001–10003 RSC.
- H. J. Choi and M. P. Suh, J. Am. Chem. Soc., 2004, 126, 15844–15851 CrossRef CAS PubMed.
- M.-H. Zeng, Q.-X. Wang, Y.-X. Tan, S. Hu, H.-X. Zhao, L.-S. Long and M. Kurmoo, J. Am. Chem. Soc., 2010, 132, 2561–2563 CrossRef CAS PubMed.
- M. S. Deshmukh, A. Chaudhary, P. N. Zolotarev and R. Boomishankar, Inorg. Chem., 2017, 56, 11762–11767 CrossRef CAS PubMed.
- Z. Yin, Q.-X. Wang and M.-H. Zeng, J. Am. Chem. Soc., 2012, 134, 4857–4863 CrossRef CAS PubMed.
- Z.-Q. Jiang, F. Wang and J. Zhang, Inorg. Chem., 2016, 55, 13035–13038 CrossRef CAS PubMed.
- Q.-K. Liu, J.-P. Ma and Y.-B. Dong, Chem. Commun., 2011, 47, 7185–7187 RSC.
- P. Shen, W.-W. He, D.-Y. Du, H.-L. Jiang, S.-L. Li, Z.-L. Lang, Z.-M. Su, Q. Fu and Y.-Q. Lan, Chem. Sci., 2014, 5, 1368–1374 RSC.
- M. Arici, O. Z. Yeşilel, M. Taş and H. Demiral, Cryst. Growth Des., 2017, 17, 2654–2659 CrossRef CAS.
- Z.-J. Zhang, W. Shi, Z. Niu, H.-H. Li, B. Zhao, P. Cheng, D.-Z. Liao and S.-P. Yan, Chem. Commun., 2011, 47, 6425–6427 RSC.
- J.-P. Ma, Y. Yu and Y.-B. Dong, Chem. Commun., 2012, 48, 2946–2948 RSC.
- L. Chen, K. Tan, Y.-Q. Lan, S.-L. Li, K.-Z. Shao and Z.-M. Su, Chem. Commun., 2012, 48, 5919–5921 RSC.
- (a) G. Li, C. Yao, J. Wang and Y. Xu, Sci. Rep., 2017, 7, 13972 CrossRef PubMed; (b) P. Arab, M. G. Rabbani, A. K. Sekizkardes, T. İsllamoğlu and H. M. El-Kaderi, Chem. Mater., 2014, 26, 1385–1392 CrossRef CAS.
- R. J. Marshall, S. L. Griffin, C. Wilson and R. S. Forgan, Chem.–Eur. J., 2016, 22, 4870–4877 CrossRef CAS PubMed.
- Z. Wang, Y. Zhang, T. Liu, M. Kurmoo and S. Gao, Adv. Funct. Mater., 2007, 17, 1523–1536 CrossRef CAS.
- Y. Rachuri, K. K. Bisht and E. Suresh, Cryst. Growth Des., 2014, 14, 3300–3308 CrossRef CAS.
- L. Dobrzańska, G. O. Lloyd, H. G. Raubenheimer and L. J. Barbour, J. Am. Chem. Soc., 2006, 128, 698–699 CrossRef PubMed.
- B. F. Abrahams, M. Moylan, S. D. Orchard and R. Robson, Angew. Chem., Int. Ed., 2003, 42, 1848–1851 CrossRef CAS PubMed.
- N.-X. Zhu, C.-W. Zhao, J.-C. Wang, Y.-A. Li and Y.-B. Dong, Chem. Commun., 2016, 52, 12702–12705 RSC.
- Q. Zhang, F. Liu, K. T. Nguyen, X. Ma, X. Wang, B. Xing and Y. Zhao, Adv. Funct. Mater., 2012, 22, 5144–5156 CrossRef CAS.
- (a) M. Du, X. Wang, M. Chen, C.-P. Li, J.-Y. Tian, Z.-W. Wang and C.-S. Liu, Chem.–Eur. J., 2015, 21, 9713–9719 CrossRef CAS PubMed; (b) E. Haque, V. Lo, A. I. Minett, A. T. Harris and T. L. Church, J. Mater. Chem. A, 2014, 2, 193–203 RSC.
- P.-Z. Li, J. Su, J. Liang, J. Liu, Y. Zhang, H. Chen and Y. Zhao, Chem. Commun., 2017, 53, 3434–3437 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: IR, TGA, PXRD, N2 gas-sorption isotherm, pore size distribution, iodine adsorption kinetic, Langmuir and Freündlich adsorption isotherm, XPS and UV/vis spectra data, and additional figures and tables for JLNU-4. CCDC 1588369. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra04648d |
| This journal is © The Royal Society of Chemistry 2018 |