DOI:
10.1039/C8RA04640A
(Paper)
RSC Adv., 2018,
8, 24376-24385
Design, synthesis and pharmacological evaluation of novel 2-chloro-3-(1H-benzo[d]imidazol-2-yl)quinoline derivatives as antitumor agents: in vitro and in vivo antitumor activity, cell cycle arrest and apoptotic response†
Received
31st May 2018
, Accepted 19th June 2018
First published on 6th July 2018
Abstract
A series of novel 2-chloro-3-(1H-benzo[d]imidazol-2-yl)quinoline derivatives (3a1−3d6) were designed and synthesized as antitumor agents. In vitro antitumor assay results showed that some compounds exhibited moderate to high inhibitory activities against HepG2, SK-OV-3, NCI-H460 and BEL-7404 tumor cell lines, and most compounds exhibited much lower cytotoxicities against HL-7702 normal cell line compared to 5-FU and cisplatin. In vivo antitumor assay results showed that the representative compound 3a1 exhibited effective inhibition on tumor growth in the HepG2 xenograft mouse model. Mechanistic studies suggested that 3a1 may exert antitumor activity by the up-regulation of Bax, intracellular Ca2+ release, ROS generation, p21, p27 and p53, downregulation of Bcl-2, activation of caspase-9 and caspase-3 and subsequent cleavage of PARP, and inhibition of CDK activity.
1. Introduction
As one of the second leading causes of mortality, cancer is a great concern to mankind. Due to negative lifestyle and food habits, which seriously increase the cancer risk, more and more people around the globe are diagnosed with this disease.1,2 Therefore, the discovery and development of promising new antitumor agents with high efficiency and low toxicity to healthy cells and tissues have become urgent and key focuses of pharmaceutical chemists worldwide. To realize this goal, a great deal of attention is being focused on molecules that act synchronously on multiple targets by combining two/more active pharmacophores covalently in a single-hybrid molecule with dual/multiple antitumor effects.3,4 This combination principle strategy encouraged us to design, synthesize and evaluate a new set of hybrid molecules by combining two pharmacophores present in a number of natural and synthetic agents: chloroquinoline and benzimidazole.
Quinoline and its derivatives display a variety of powerful biological activities including inhibition of cellular proliferation and developmental changes,5–7 which make the quinoline ring a valuable versatile synthetic scaffold for new antitumor agents. Among the various existing active scaffolds of quinolines, chloroquinoline is a type of alkaloid, which is as abundant as quinoline. Previous studies have indicated that chloroquinoline derivatives display preferable biological activities such as anticancer and antiproliferation activities.8,9 Chloroquinoline scaffold is thus chosen in the present study as an active antitumor pharmacy core, and some structural modifications are designed to explore the antitumor activity. In addition, a bicyclic compound, namely, benzimidazole, which is the fusion of benzene and imidazole, is a pharmacophore possessing a wide range of biological activities.10 Some benzimidazole derivatives have been found to display potent antitumor activities.11–13 Accordingly, the benzimidazole nucleus has been considered as an important pharmacophore in antitumor chemistry. It is thus expected that combining the chloroquinolone scaffold with the benzimidazole skeleton may lead to potent antitumor activity. Therefore, under the combination principle, a set of 2-chloroquinoline-benzimidazole derivatives 3(3a1−3d6) were synthesized, and their antitumor activities were evaluated. Moreover, previous studies have indicated that both quinoline and benzimidazole derivatives exert antitumor activities through modulation of cell cycle arrest and apoptotic responses.14–16 Therefore, mechanistic studies focused on apoptosis and cycle arrest induced by the representative compound 3a1 were also performed.
2. Results and discussion
2.1 Chemistry
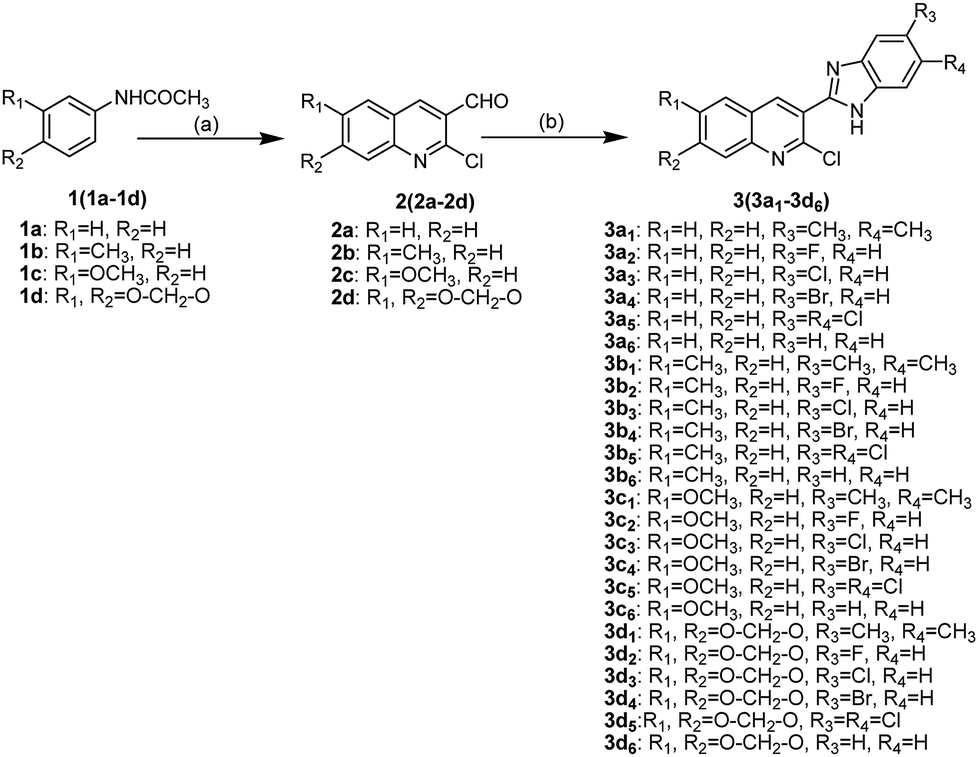
2-Chloro-3-(1H-benzo[d]imidazol-2-yl)quinoline derivatives 3(3a1−3d6) were synthesized as shown in Scheme 1. First, 2-chloro-quinoline-3-carbaldehyde derivatives 2(2a−2d) were synthesized through the Vilsmeier–Haack–Arnold reaction, which involves the combination of acetanilide derivatives 1(1a−1d) with N,N-dimethylformamide (DMF) in the presence of phosphorus oxychloride.15 2-Chloro-quinoline-3-benzimidazole derivatives 3(3a1−3d6) were then obtained in moderate yields by the condensation reactions of 2(2a−2d) with o-phenylenediamine derivatives in the presence of methanol. The structures of compounds 3(3a1−3d6) were confirmed using various spectroscopic methods including 1H NMR, 13C NMR and high-resolution mass spectrometry (HR-MS) (Part 2 of ESI†). In 1H NMR spectra, the hydrogen of NH fused in benzimidazole and the aromatic hydrogens (H-Ar) in quinolone and benzimidazole were identified by chemical shifts (δ) 12.66–13.26 and 7.14–8.79, respectively. In addition, the hydrogens of methyl groups were also identified at δ 3.93, 3.84, 2.54 and 2.34, whereas the hydrogens of O–CH2–O group were found around δ 6.30. In 13C NMR spectra, carbons in carbonyl and O–CH2–O groups were identified at δ 161.04–161.99 and around 95.40, respectively, whereas the carbons of methyl groups were found at δ 20.59–20.94 and 55.94–56.09. The signals (δ) at 99.47–148.19 were assigned to carbons in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN groups as well as the aromatic carbons in quinolone and benzimidazole. HR-MS provided exact formula weights and compositions of 3(3a1−3d6), which were well consistent with the results of 1H NMR and 13C NMR and the structures shown in Scheme 1, thus indicating that the structures of compounds 3(3a1−3d6) were confirmed.
C and CN groups as well as the aromatic carbons in quinolone and benzimidazole. HR-MS provided exact formula weights and compositions of 3(3a1−3d6), which were well consistent with the results of 1H NMR and 13C NMR and the structures shown in Scheme 1, thus indicating that the structures of compounds 3(3a1−3d6) were confirmed.
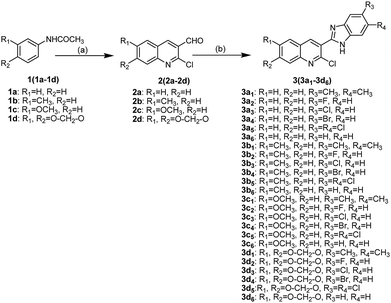 |
| | Scheme 1 Synthetic routes of compounds 3(3a1−3d6). Reagents and conditions: (a) POCl3, DMF; (b) o-phenylenediamine derivatives, methanol. | |
2.2. In vitro antiproliferative activity
The in vitro antiproliferative activities of the compounds 3(3a1−3d6) were evaluated against HepG2 (human liver cancer cells), SK-OV-3 (human ovarian cancer cells), NCI-H460 (human large cell lung cancer cells), BEL-7404 (human liver cancer cells) and HL-7702 (human liver normal cells) cell lines, using the methylthiazol tetrazolium (MTT) assay.15,17 Two commercial anticancer drugs 5-fluorouracil (5-FU) and cisplatin were used as positive controls. The results are shown in Table 1. Table 1 indicates that many compounds showed evident antiproliferative activities against HepG2, SKOV3, NCI-H460 and BEL-7404 cell lines, and some displayed better antiproliferative activities than 5-FU and cisplatin while exhibiting lower cytotoxicities against the HL-7702 normal cell line. In the HepG2 cell line assay, compounds 3a1, 3a3, 3a4, 3a5, 3c3, 3c4, 3c5 and 3c6 exhibited higher antiproliferative activities than 5-FU (IC50 = 31.98 μM), with IC50 values in the range of 7.54–28.24 μM. Compounds 3a1 and 3a5 exhibited better antiproliferative activities than cisplatin (IC50 = 10.12 μM). In the SKOV-3 cell line assay, compounds 3a1, 3a3, 3a5, 3c3, 3c5 and 3c6 showed higher antiproliferative activities than 5-FU (IC50 = 26.34 μM), with IC50 in the range of 8.83–23.00 μM. Compounds 3a1, 3a3 and 3c3 displayed better antiproliferative activities than cisplatin (IC50 = 15.60 μM). In the NCI-H460 cell line assay, compounds 3a1, 3a3, 3a4, 3a5, 3c1, 3c3, 3c4 and 3c5 showed higher antiproliferative activities than 5-FU (IC50 = 45.44 μM), with IC50 in the range of 14.03–38.25 μM. Compounds 3a1 and 3a5 displayed higher antiproliferative activities than cisplatin (IC50 = 20.36 μM). In the BEL-7404 cell line assay, compounds 3a1, 3a3, 3a4, 3a5, 3c1, 3c3, 3c4, 3c5 and 3c6 exhibited higher antiproliferative activities than 5-FU (IC50 = 40.21 μM), with IC50 values in the range of 9.06–39.94 μM. The compound 3c4 exhibited higher antiproliferative activity than cisplatin (IC50 = 14.72 μM). Based on the antiproliferation results shown in Table 1, it can be concluded that for compounds 3(3a1−3d6), the absence of substitution and the methoxy substitution at 6 position of quinolone moiety as well as dimethyl, chloro and bromo substitutions in the benzimidazole moiety had positive effects on the antiproliferative activity.
Table 1 αIC50 values of compounds 3(3a1−3d6) toward four selected tumor cell lines and a normal cell linea
| Compounds |
IC50 (μM) |
| HepG2 |
SKOV-3 |
NCI-H460 |
BEL-7404 |
HL-7702 |
| αIC50 values are presented as mean ± SD (standard error of the mean) from three repeated experiments. |
| 3a1(3c) |
7.54 ± 0.77 |
9.40 ± 0.69 |
14.03 ± 0.94 |
15.40 ± 0.85 |
>100 |
| 3a2(3m) |
>100 |
>100 |
>100 |
>100 |
>100 |
| 3a3(3q) |
11.52 ± 0.89 |
10.40 ± 1.34 |
22.64 ± 1.02 |
22.20 ± 0.76 |
>100 |
| 3a4(3i) |
28.24 ± 1.24 |
34.14 ± 1.82 |
31.40 ± 1.47 |
29.99 ± 0.59 |
>100 |
| 3a5(3e) |
7.59 ± 1.64 |
21.35 ± 0.88 |
15.11 ± 0.57 |
19.39 ± 1.43 |
>100 |
| 3a6(3u) |
>100 |
>100 |
>100 |
>100 |
>100 |
| 3b1(3d) |
>100 |
>100 |
>100 |
>100 |
>100 |
| 3b2(3n) |
>100 |
>100 |
>100 |
>100 |
>100 |
| 3b3(3r) |
>100 |
>100 |
>100 |
>100 |
>100 |
| 3b4(3j) |
>100 |
>100 |
>100 |
>100 |
>100 |
| 3b5(3f) |
>100 |
>100 |
>100 |
>100 |
>100 |
| 3b6(3v) |
>100 |
>100 |
>100 |
>100 |
>100 |
| 3c1(3a) |
37.81 ± 1.05 |
36.92 ± 2.01 |
38.25 ± 1.58 |
39.94 ± 1.18 |
>100 |
| 3c2(3o) |
>100 |
>100 |
>100 |
>100 |
>100 |
| 3c3(3s) |
14.10 ± 1.55 |
8.83 ± 2.10 |
23.84 ± 0.84 |
26.69 ± 0.39 |
>100 |
| 3c4(3k) |
12.15 ± 1.05 |
32.66 ± 0.70 |
26.79 ± 1.06 |
11.29 ± 1.18 |
>100 |
| 3c5(3g) |
16.68 ± 1.19 |
17.88 ± 1.79 |
21.14 ± 1.41 |
21.06 ± 0.99 |
>100 |
| 3c6(3w) |
22.35 ± 1.25 |
23.00 ± 1.57 |
>100 |
>100 |
>100 |
| 3d1(3b) |
>100 |
>100 |
>100 |
>100 |
>100 |
| 3d2(3p) |
>100 |
>100 |
>100 |
>100 |
>100 |
| 3d3(3t) |
>100 |
>100 |
>100 |
>100 |
>100 |
| 3d4(3l) |
>100 |
>100 |
>100 |
>100 |
>100 |
| 3d5(3h) |
>100 |
>100 |
>100 |
>100 |
>100 |
| 3d6(3x) |
>100 |
41.57 |
>100 |
>100 |
>100 |
| 5-FU |
31.98 ± 0.56 |
26.34 ± 0.57 |
45.44 ± 0.94 |
40.21 ± 1.98 |
58.74 ± 2.31 |
| Cisplatin |
10.12 ± 0.71 |
15.60 ± 1.7 |
20.36 ± 0.50 |
14.72 ± 0.66 |
15.67 ± 0.32 |
Among the compounds 3(3a1−3d6), the compound 3a1 showed the best potential antiproliferative activity against these cancer cell lines. Thus, 3a1 was selected as a representative compound for in vivo antiproliferative activity and mechanistic studies.
2.3. Preliminary safety evaluation of 3a1
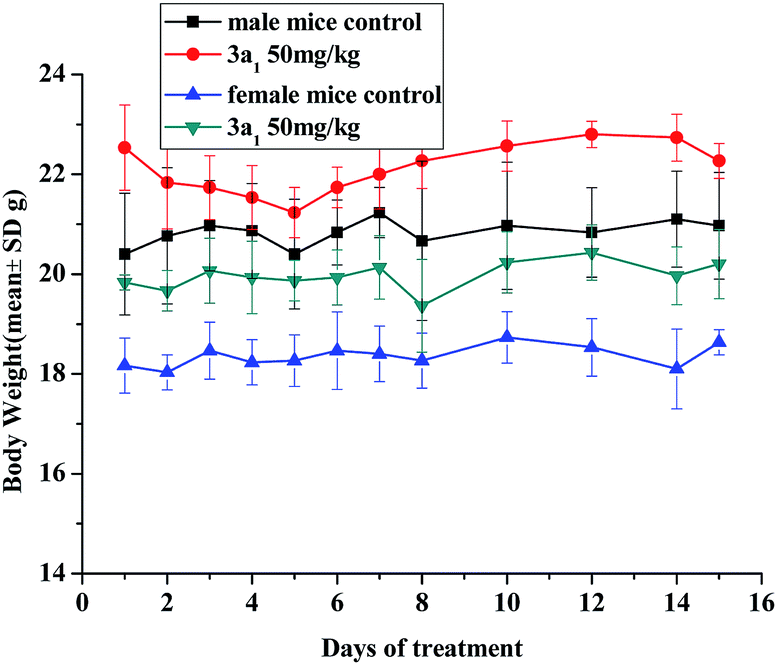
The in vivo toxicity of compound 3a1 was evaluated in Kun-ming (KM) mice. Through intraperitoneal injection, the mice received 3a1 (dissolved in 5% v/v DMSO/saline solvent) at a dose of 50 mg kg−1 per day for 8 consecutive days; then, for the next 6 days, the mice were treated with compound 3a1 at the same dose once every two days. The body weights of the animals were monitored daily and used as a parameter of systemic toxicity.
No adverse effects were observed in the KM mice injected with 3a1 during the 14 day monitoring period. As shown in Fig. 1, the average body weights of the mice treated with 3a1 showed similar growth rates to those of the control group. It is also worth noting that, as shown in Fig. 1, the injection of 3a1 even led to extra weight gain in the KM mice, indicating that no adverse effects were found after the treatment of these mice with 3a1.
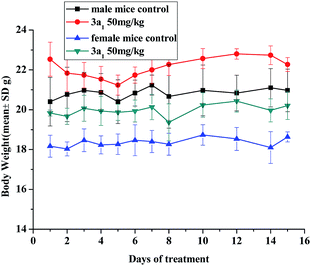 |
| | Fig. 1 Changes in the body weight of KM mice injected with compound 3a1. There are six mice in each experimental group. | |
2.4. In vivo antiproliferative activity of 3a1
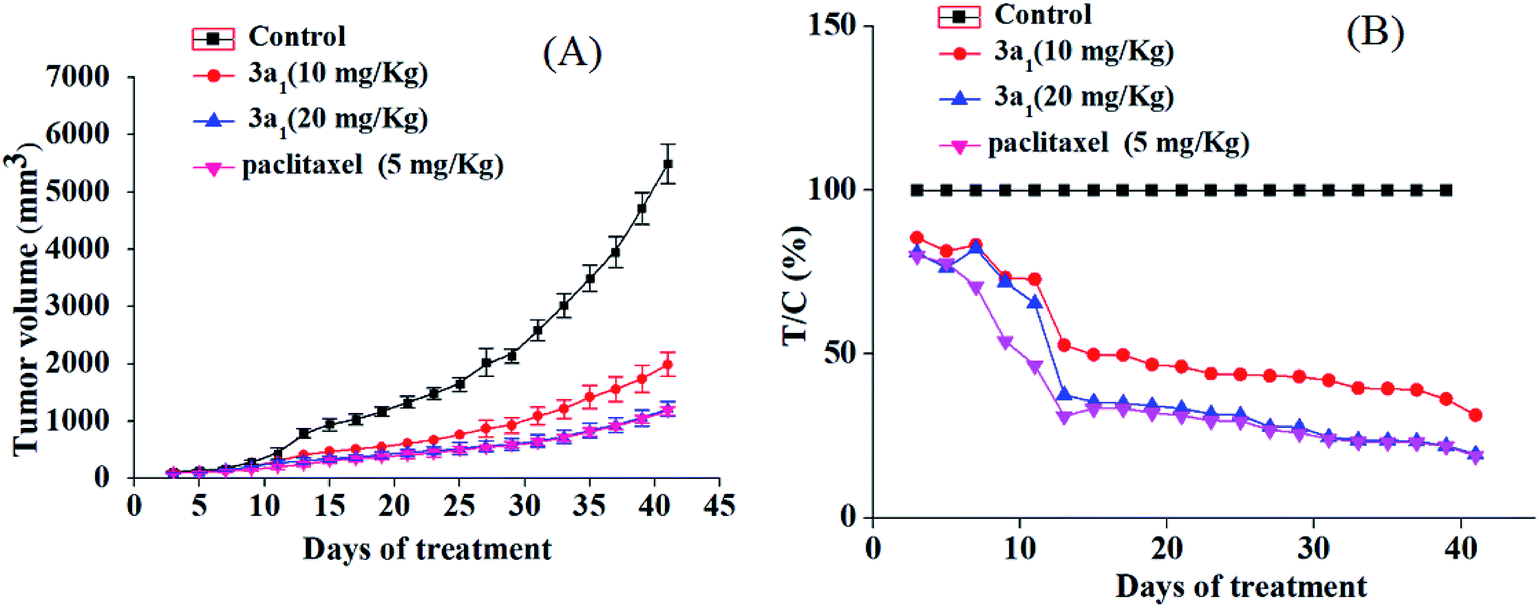
The potential in vitro antiproliferative activity and the absence of adverse effect of 3a1 encouraged us to perform in vivo antiproliferative assays to further evaluate its antitumor activity. The HepG2 xenograft model was selected in this study, and paclitaxel was used as the positive control. SPF BALB/c nude mice were randomly divided into vehicle control group, low dose of 3a1-treated group, high dose of 3a1-treated group, and positive control group. The mice in the vehicle control group were injected with 5% DMSO saline solution, and the mice in low and high dose of 3a1-treated group were injected with 3a1 (in 5% DMSO saline solution), whereas the mice in the positive control group were injected with paclitaxel (in 5% DMSO saline solution). Tumor size of mice was tested twice in a week in two dimensions, and the volume was measured to evaluate in vivo antitumor activity.
As shown in Fig. 2, compared with the observations for control, the treatment with 3a1 and paclitaxel significantly inhibited the increase in the tumour volume in mice, indicating that compound 3a1 exhibited potential antitumor effect. The treatment of high dose of 3a1 (20 mg kg−1) exhibited similar inhibition effect on the tumour volume as that of paclitaxel (5 mg kg−1). The T/C values of 3a1 injected in low (10 mg kg−1) and high doses (20 mg kg−1) were determined to be 31.3% and 19.5% (p < 0.01), respectively, at day 41. Paclitaxel at the 5 mg kg−1 dose showed a T/C value of 19.0%, which was almost equal to that of 3a1 at 20 mg kg−1 dose; the result indicated that 3a1 (at 20 mg kg−1 dose) showed almost equal in vivo antitumor activity with paclitaxel (at 5 mg kg−1 dose) in the HepG2 mouse model. In addition, no evident changes in the body weight and no other adverse effects were found in the mice treated with 3a1 (at 20 mg kg−1 dose) (Fig. S1-1†), suggesting that 3a1 may be a good candidate for an antitumor agent.
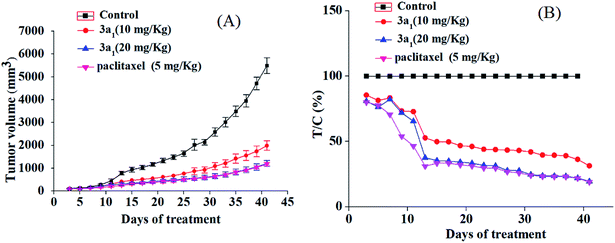 |
| | Fig. 2 In vivo anticancer activity of 3a1 in the HepG2 xenograft model. Effect of 3a1 (at doses of 10 and 20 mg kg−1), paclitaxel (at dose of 5 mg kg−1) and vehicle control (5% DMSO in saline, v/v) on tumor growth. Tumor growth was monitored by mean tumor volume (mm3) ± SD (n = 6) (A) and calculated as relative tumor increment rate (T/C, %) (B). | |
2.5. Study on cell cycle distribution
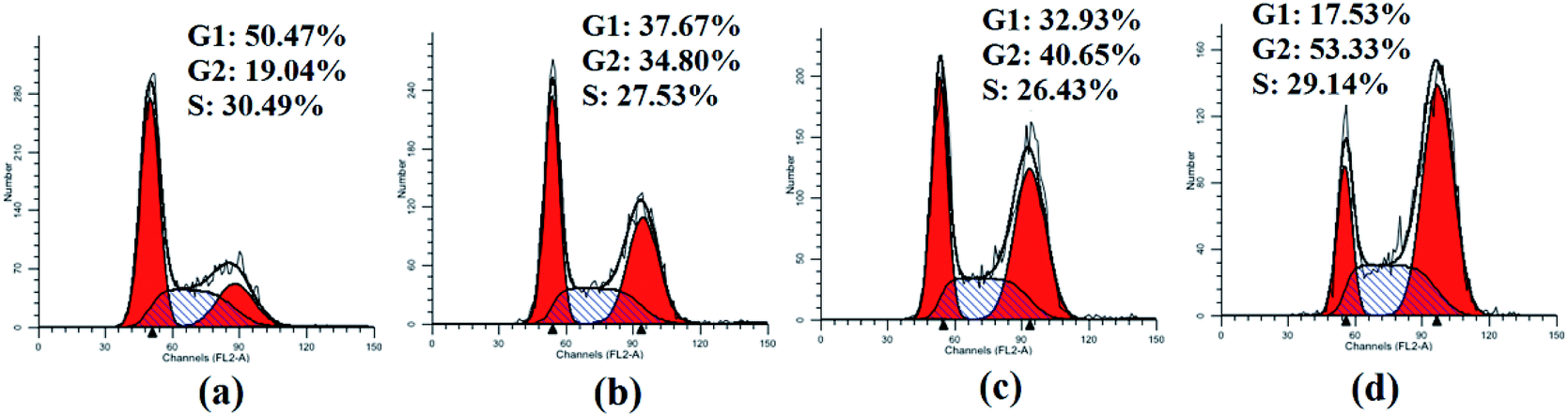
As significant control mechanisms, cell cycle checkpoints control and ensure the generation and performance of cell cycle events.18 To study the potential role of cell cycle arrest in tumor cell death induced by compounds 3, HepG2 cells were treated with the representative compound 3a1 at different concentrations (0, 6, 8 and 10 μM) for 48 h. As shown in Fig. 3, the treatment of HepG2 cells with 3a1 promoted cell cycle arrest at the G2 phase, which led to a clear increase in the G2-phase population (34.80% at 6 μM, 40.65% at 8 μM and 53.33% at 10 μM) compared with the observations in the control group (19.04%). Consequently, the G1-phase and S-phase populations of the HepG2 cells decreased. The results indicated that compound 3a1 clearly arrested the cell cycle of HepG2 cells at the G2/M stage in a concentration-dependent manner.
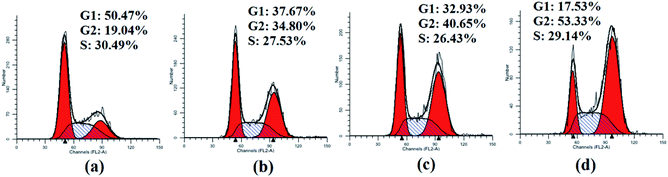 |
| | Fig. 3 Cell cycle analysis of HepG2 cells treated with 3a1. The HepG2 cells were treated with compound 3a1 at different concentrations, i.e., (a) 0 μM, (b) 6 μM, (c) 8 μM and (d) 10 μM for 48 h. | |
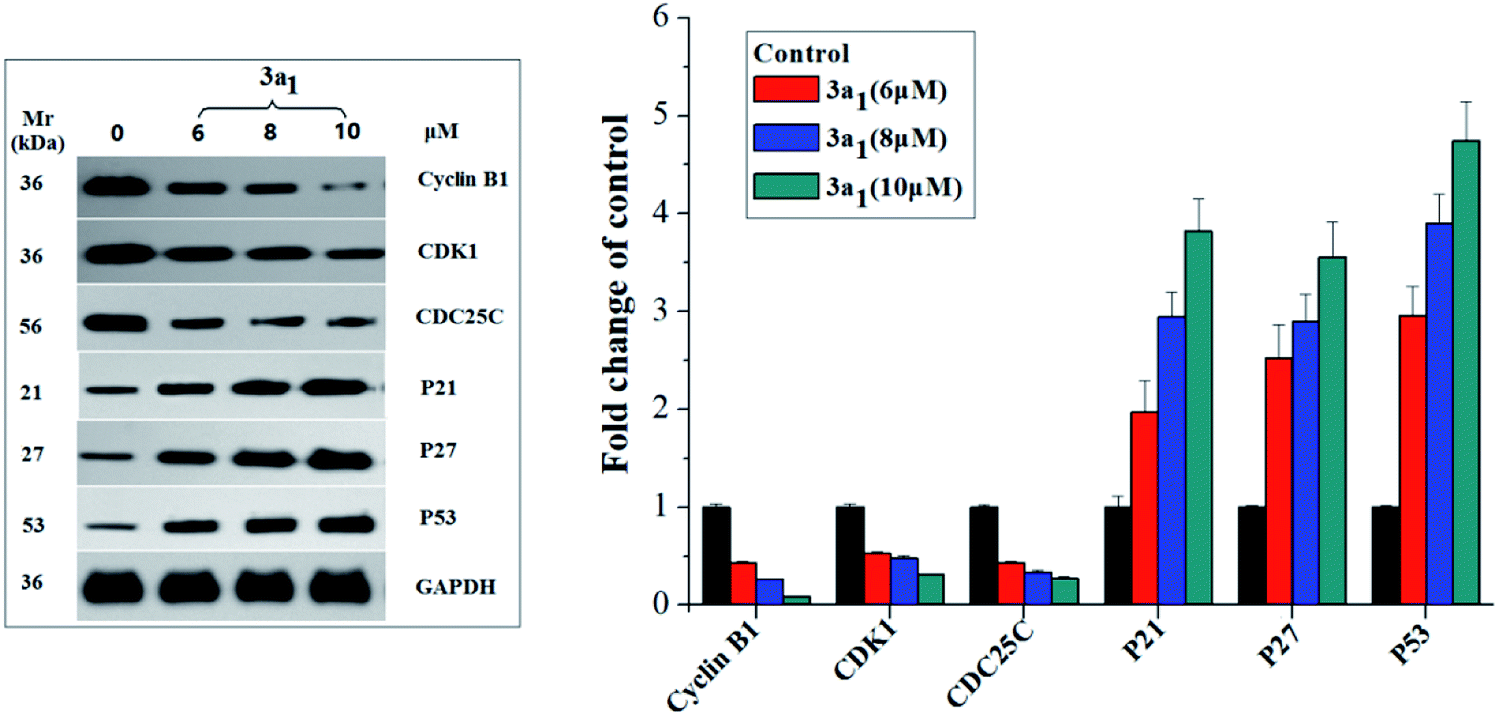
Cyclin B1/CDK1(CDC2) kinase complexes are main regulators of G2/M transition, and their generation is usually monitored by the positive mediator cell division cyclin 25 homolog C (CDC25C) phosphatase.19,20 CDC25C mediates the phosphorylation of CDK1 and consequently leads to the activation of cyclin B1/CDK1 complexes and execution of G2/M transition;19,20 phosphorylation of CDC25C accompanied by DNA damage and cellular activities resulting in G2/M arrest causes its degradation and sequestration to the cytoplasm, which consequently leads to the destabilization of the cyclin–CDK complexes.21 The p53, p27 and p21 genes encoded by some antioncogenes are also involved in the G2/M arrest through increasing the accumulation of the dormant cyclin B–CDK complex.22 Therefore, in the present study, the expression levels of cyclin B1, CDK1, CDC25C, p53, p27 and p21 in the HepG2 cells were assayed using western blot. As shown in Fig. 4, compared to the observations for the control, the lysates of the HepG2 cells treated with 3a1 down-regulated the levels of cyclin B1, CDK1 and CDC25C while up-regulating the levels of p53, p21 and p27. Thus, compound 3a1 induced G2/M phase cell cycle arrest through inhibition of CDK activity and increase in the expression levels of p53, p27 and p21.
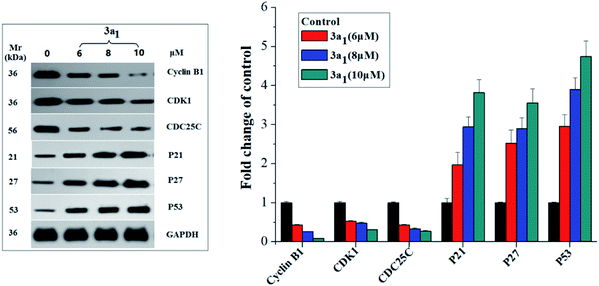 |
| | Fig. 4 Effects of compounds 3a1 on the expression levels of relevant proteins in HepG2 cells. western blot analysis was performed with antibodies against cyclin B1, CDK1, CDC25C, p21, p23, and p53. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the loading standard. | |
2.6. Apoptosis assays
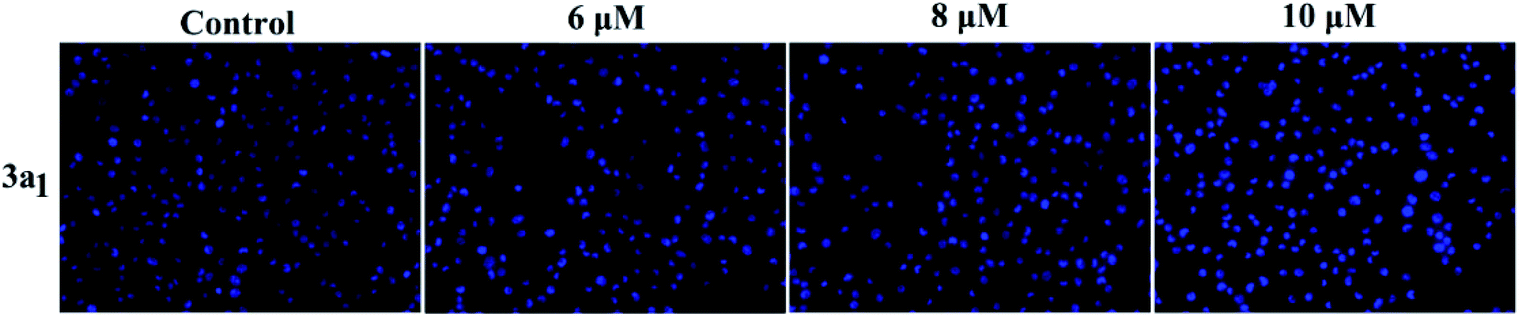
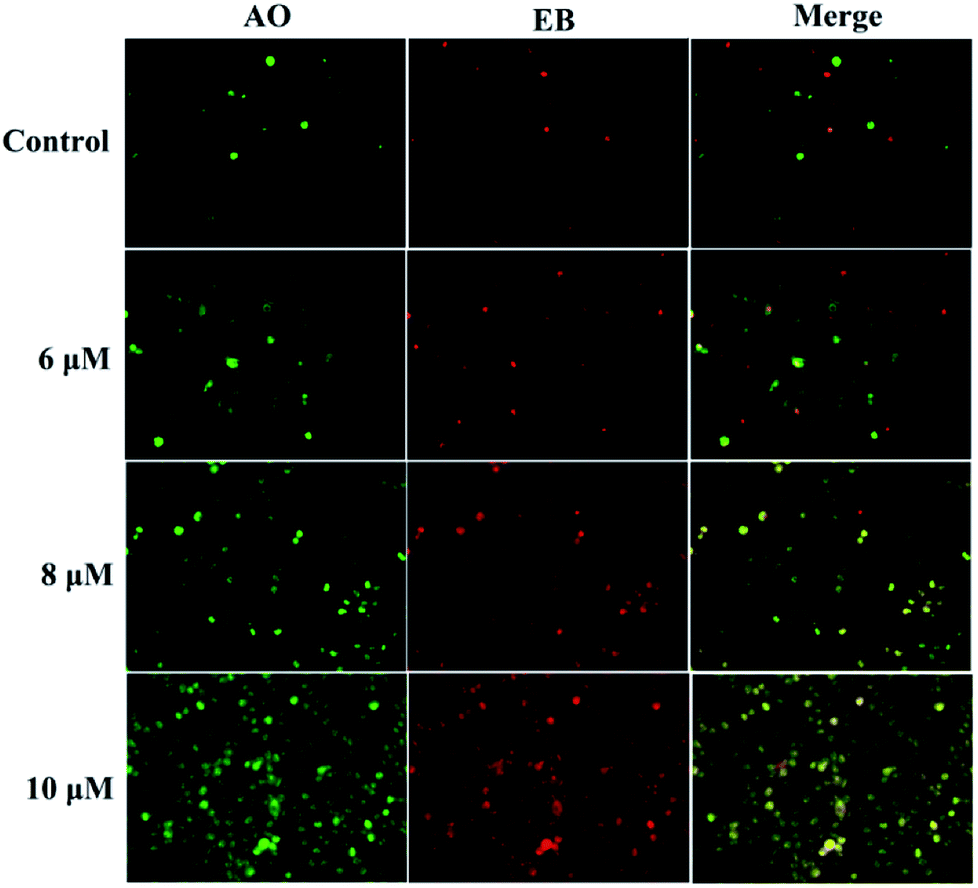
Apoptosis is another important mechanism in cancer treatment, and it is a key pathway leading to cell death.23,24 Thus, the apoptosis-inducing effect of compound 3a1 was investigated by staining with the fluorescent probes Hoechst 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 258 and acridine orange/ethidium bromide (AO/EB). In the Hoechst 33258 staining assay (Fig. 5), normal HeGp2 cells emitted blue fluorescence, whereas the cells treated with 3a1 for 12 h exhibited stronger blue fluorescence and displayed characteristic apoptotic morphologies. This phenomenon indicated that compound 3a1 induced the apoptosis of HeGp2 cells.21 In the AO/EB staining assay, the morphology of 3a1-treated HeGp2 cells exhibited evident changes (Fig. 6). The cell nuclei were stained yellow/orange, and the morphology changes suggested pycnosis, membrane blebbing and cell budding, all of which are clear characteristics of apoptosis.21 Therefore, on the basis of the above-mentioned observations, it can be concluded that the compound 3a1 induced apoptosis of HeGp2 cells.
258 and acridine orange/ethidium bromide (AO/EB). In the Hoechst 33258 staining assay (Fig. 5), normal HeGp2 cells emitted blue fluorescence, whereas the cells treated with 3a1 for 12 h exhibited stronger blue fluorescence and displayed characteristic apoptotic morphologies. This phenomenon indicated that compound 3a1 induced the apoptosis of HeGp2 cells.21 In the AO/EB staining assay, the morphology of 3a1-treated HeGp2 cells exhibited evident changes (Fig. 6). The cell nuclei were stained yellow/orange, and the morphology changes suggested pycnosis, membrane blebbing and cell budding, all of which are clear characteristics of apoptosis.21 Therefore, on the basis of the above-mentioned observations, it can be concluded that the compound 3a1 induced apoptosis of HeGp2 cells.
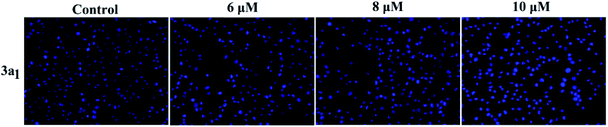 |
| | Fig. 5 Effects of the compound 3a1 on the morphology of HepG2 cells. The cells were stained with the Hoechst 33258 dye. | |
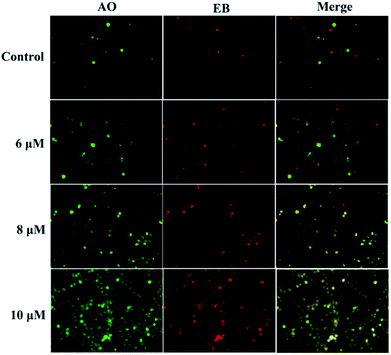 |
| | Fig. 6 Compound 3a1 induced apoptosis in HepG2 cells. The cells were visualized via AO/EB staining coupled with fluorescence microscopy. | |
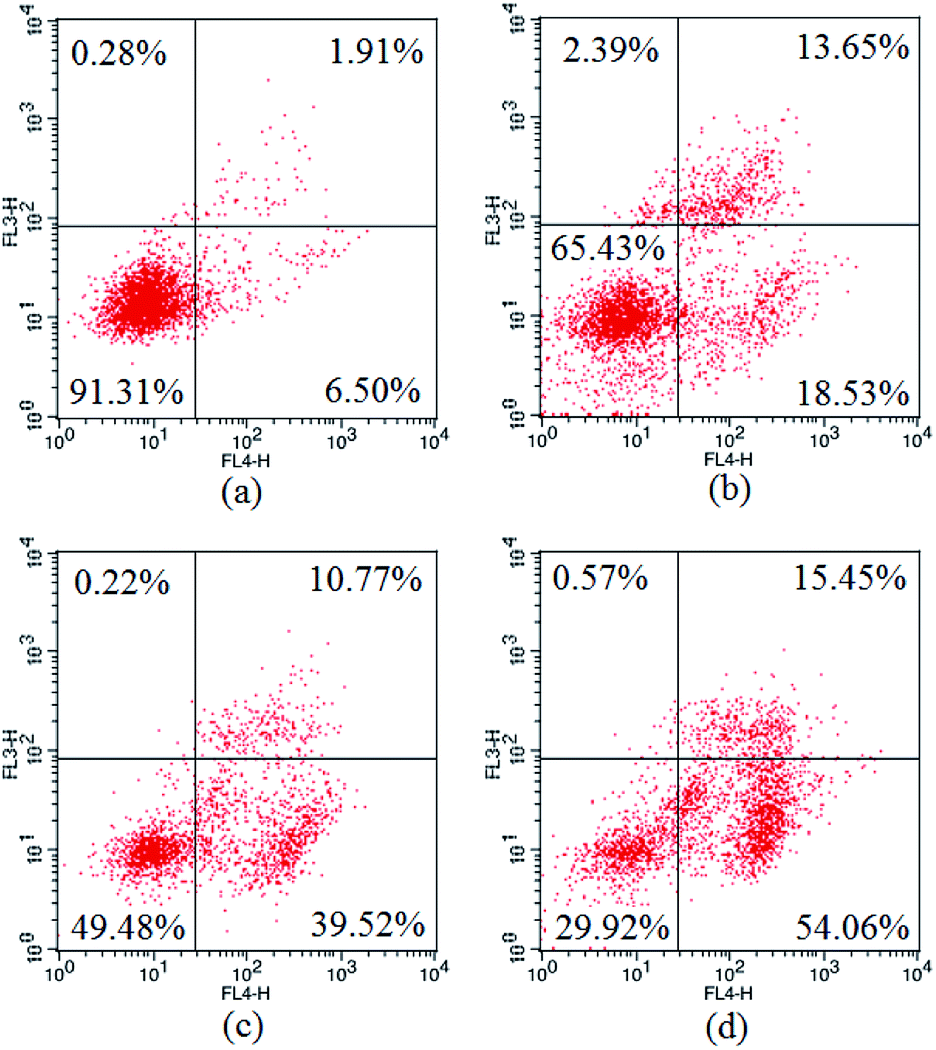
The apoptosis ratios of HepG2 cells treated with 3a1 were then quantitatively assayed by flow cytometry. As shown in Fig. 7, the treatment of HepG2 cells with compound 3a1 at different concentrations (6, 8 and 10 μM) led to significant increase in the apoptotic cell population in a concentration-dependent manner. The results indicated that compound 3a1 induced apoptosis of HepG2 cells in a dose-dependent manner.
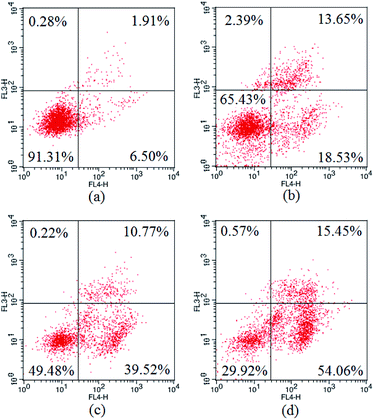 |
| | Fig. 7 Apoptosis ratios of HepG2 cells. (a) Negative control, (b) treated with 6 μM 3a1, (c) treated with 8 μM 3a1, (d) treated with 10 μM 3a1. The cells were analyzed using the Annexin V/PI assay in a flow cytometer. | |
2.7. ROS generation
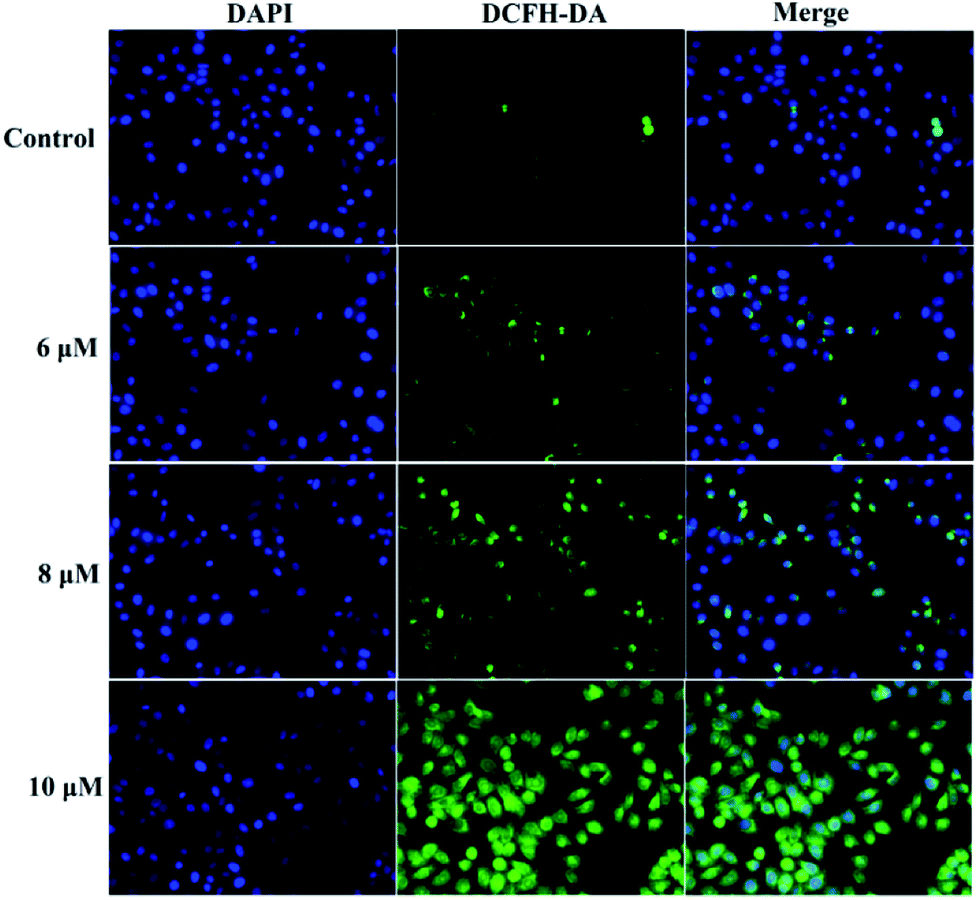
A previous study proved that the generation of intracellular reactive oxygen species (ROS) can induce apoptosis.25 To investigate the role of ROS in 3a1-induced apoptosis in the HepG2 cells, intracellular ROS levels were determined through fluorescence microscopy using 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) and 2,7-dichlorofluorescein diacetate (DCFH-DA) as fluorescent probes. Fig. 8 demonstrates that both control cells and HepG2 cells treated with compound 3a1 displayed blue fluorescence in the cytoplasm in the DAPI staining assay, whereas the HepG2 cells treated with compound 3a1 exhibited stronger green fluorescence signals compared to the control in the DCFH-DA staining assay, indicating that 3a1 can up-regulate the levels of intracellular ROS. These results indicated that the generation of ROS induced by 3a1 may be an underlying cause for apoptosis.24
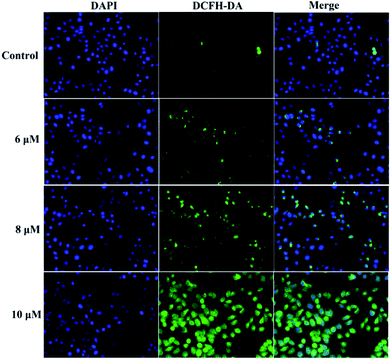 |
| | Fig. 8 Compound 3a1 affected the levels of intracellular ROS in HepG2 cells. | |
2.8. Intracellular Ca2+ release
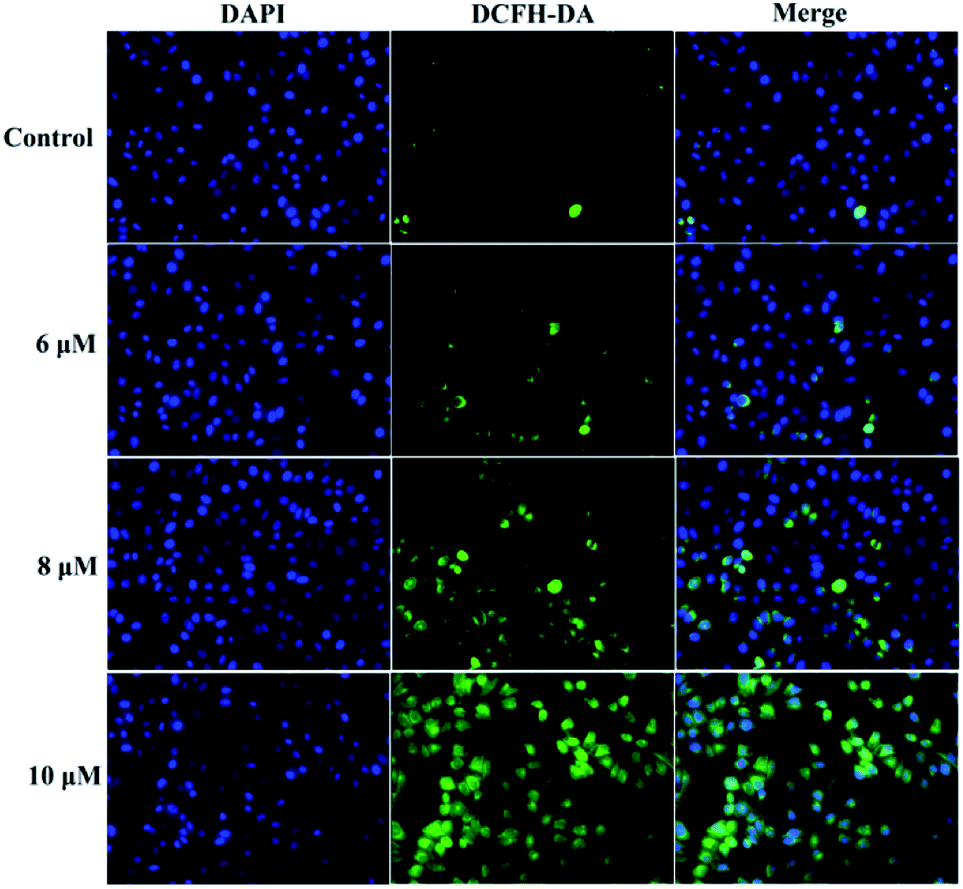
It is known that intracellular calcium takes part in cell apoptosis, and an overload of calcium can induce apoptosis.26 Thus, the role of calcium signaling in cellular apoptosis induced by 3a1 was studied. The HepG2 cells were treated with 3a1 for 24 h, and the intracellular Ca2+ level was assayed by fluorescence microscopy using DCFH-DA as a calcium probe. Fig. 9 shows that the treatment of HepG2 cells with 3a1 caused an increase in the intracellular Ca2+ concentration, indicating that 3a1-induced apoptosis was related to the up-regulation of intracellular Ca2+ level.
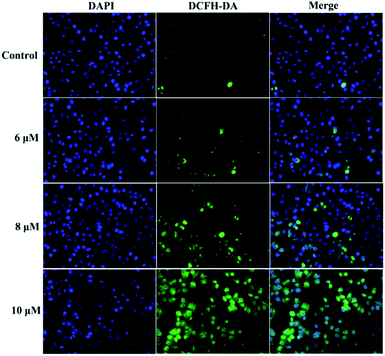 |
| | Fig. 9 Compound 3a1 caused an increase in the level of intracellular Ca2+ in HepG2 cells. | |
2.9. Caspase-dependent apoptosis in HepG2 cells
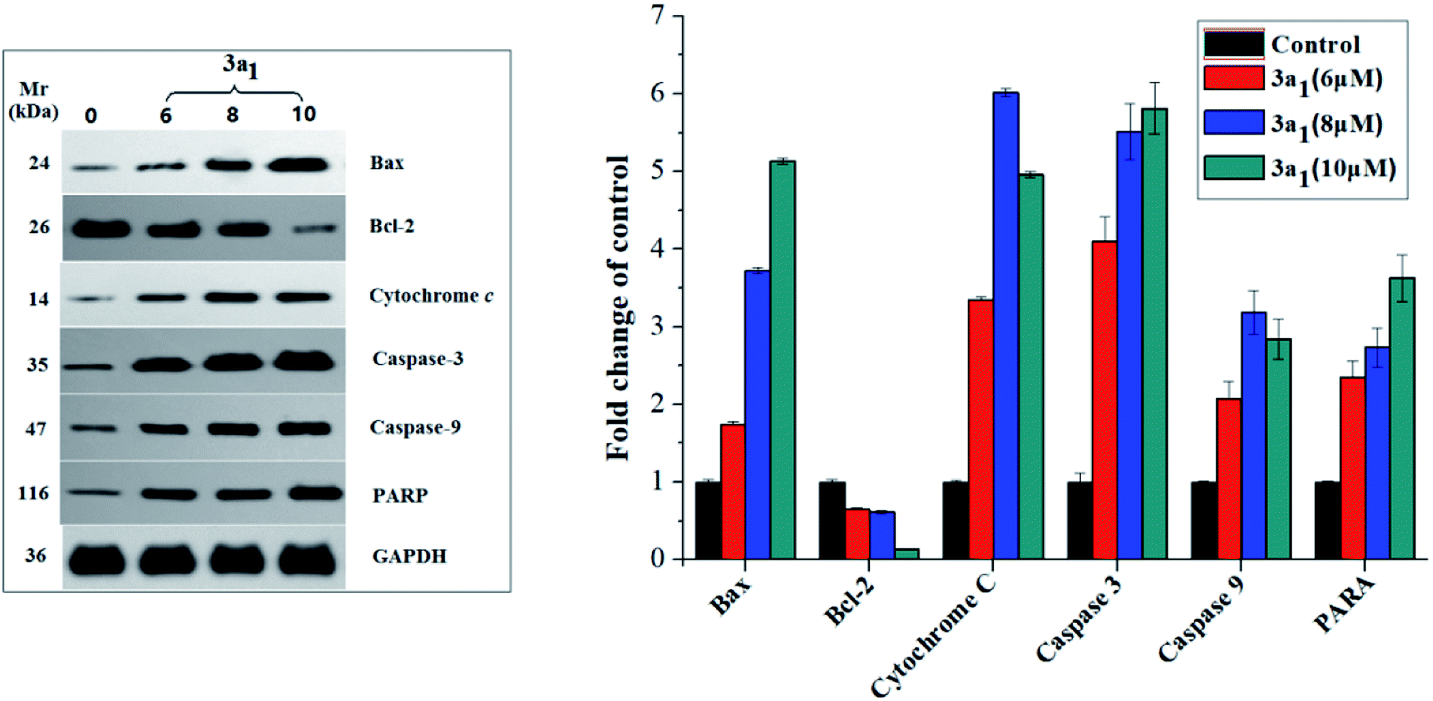
As well-known anti- and pro-apoptotic proteins, Bcl-2/Bax family proteins play important roles in the inhibition or promotion of cell death.27,28 A previous study proved that the ratio of Bcl-2/Bax expression is vital in the regulation of apoptosis.29 Bcl-2/Bax family proteins mainly participate in apoptosis by interfering with caspases, which are the pivotal effectors of programmed cell death.30,31 Caspases are a family of cysteinyl aspartate specific proteases, which participate in apoptosis, and they can be divided into initiators (caspases 8, 9 and 10) and executioners (caspases 3, 6 and 7).30,31 The caspase cascade is mediated by the proteolysis of inactive procaspases and propagated by the cleavage of downstream caspases and substrates including poly(ADP-ribose) polymerase (PARP).21 To investigate the mechanism of 3a1-induced apoptosis in cancer cells and to find if caspases were involved in 3a1-induced apoptosis, the expression levels of Bax, Bcl-2, cytochrome c, caspase-9, caspase-3 and PARP in HepG2 cells treated with 3a1 were assayed by western blot. As shown in Fig. 10, the treatment of HepG2 cells with 3a1 led to evident increase in the expression levels of Bax, caspase-9, caspase-3 and PARP and reduction in the expression level of Bcl-2 accompanied by the release of mitochondrial cytochrome c into the cytosol compared to the observations for control. These results indicated that compound 3a1 could down-regulate the expression of Bcl-2, leading to the release of cytochrome c, activation of caspase-9 and induction of apoptosis, thus demonstrating that compound 3a1 exerted pro-apoptotic effects through a mitochondrial-mediated pathway and caspase cascade.
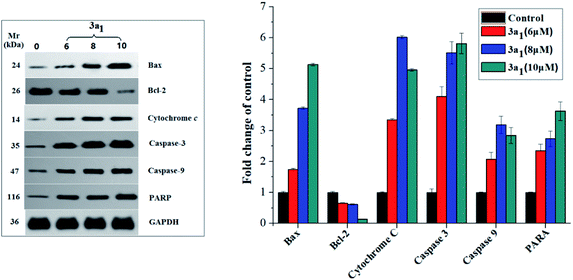 |
| | Fig. 10 Effect of compound 3a1 on the expression levels of Bax, Bcl-2, cytochrome c, caspase-3, caspase-9 and PARP in HepG2 cells. GAPDH was used as a loading standard. | |
3. Conclusions
In summary, we designed and synthesized a set of 2-chloro-3-(1H-benzo[d]imidazol-2-yl)quinoline derivatives and evaluated the antiproliferative activities of these compounds against four cancer cell lines (HepG2, SKOV-3, NCI-H460 and BEL-7404). We identified some compounds exhibiting high in vitro anticancer activities and low toxicities against normal HL-7702 cells. In vivo antitumor activity study and animal toxicology test showed that 3a1 exhibited potent antitumor activity in the HepG2 xenograft mouse model. Mechanistic studies showed that 3a1 caused apoptosis of tumor cells possibly through regulation of the expression levels of apoptotic proteins (such as Bax, Bcl-2, cytochrome c, caspase-9, caspase-3, and PARP), intracellular Ca2+ release and ROS regulation. Cell cycle studies indicated that compound 3a1 induced cell cycle arrest at the G2/M phase by inhibition of CDK activity and activation of the p53 protein. This study suggests that the rational design of 2-chloro-3-(1H-benzo[d]imidazol-2-yl)quinoline derivatives under the combination principle is a feasible method to discover new antitumor therapeutics.
4. Experimental
4.1. Materials
All chemicals of reagent grade were commercially available and used without further purification. NMR spectra were assayed on a BRUKER AVANCE AV 400/500 spectrometer using tetramethyl silane (TMS) as the internal standard. Mass spectra were obtained on a BRUKER ESQUIRE HCT spectrometer. The GelRed nucleic acid stain was purchased from Biotium.
4.2. General synthesis procedure for compounds 3(3a1−3d6)
2-Chloro-quinoline-3-carbaldehyde derivatives 2(2a−2d) were synthesized by condensation of acetanilide derivatives 1(1a−1d) with DMF in the presence of phosphorus oxychloride, according to our previous study.15 2-Chloro-quinoline 3-carbaldehyde derivatives 2(2a−2d) (1 mmol), o-phenylenediamine derivatives (1 mmol) and 3 mL methanol were mixed in a pressure tube and heated at 90 °C for 4 h. After the reaction, the mixture was cooled to room temperature and filtered to obtain the product as yellow powder. If necessary, the target compounds 3(3a1−3d6) were purified by column chromatography (silica 300–400 mesh) with acetic ether–petroleum benzine (v/v: 1/5) as an eluent.
3a1. Yield, 64.8%; 1H NMR (500 MHz, DMSO-d6) δ 12.68 (s, 1H, NH), 8.97 (s, 1H, H-Ar), 8.20 (s, 1H, H-Ar), 8.06 (s, 1H, H-Ar), 7.92 (d, J = 8.4 Hz, 1H, H-Ar), 7.75 (d, J = 8.1 Hz, 1H, H-Ar), 7.45 (s, 2H, H-Ar), 2.36 (s, 6H, –CH3); 13C NMR (126 MHz, DMSO-d6) δ 147.68, 147.21, 141.58, 132.36, 129.01, 128.44, 128.16, 126.87, 125.14, 20.50; ESI-HRMS, calculated m/z for C18H14ClN3 [M + H]+: 308.0955, found: 308.0972.
3a2. Yield, 57.3%; 1H NMR (500 MHz, DMSO-d6) δ 13.08 (s, 1H, NH), 9.01 (s, 1H, H-Ar), 8.20 (d, J = 7.6 Hz, 1H, H-Ar), 8.07 (d, J = 8.5 Hz, 1H, H-Ar), 7.95 (d, J = 8.4 Hz, 1H, H-Ar), 7.77 (s, 1H, H-Ar), 7.70 (s, 1H, H-Ar), 7.50 (s, 1H, H-Ar), 7.16 (d, J = 8.5 Hz, 1H, H-Ar); 13C NMR (126 MHz, DMSO-d6) δ 147.48, 147.36, 141.99, 132.66, 129.11, 128.58, 128.19, 126.79, 124.57; ESI-HRMS, calculated m/z for C16H9ClFN3 [M + H]+: 298.0547, found: 298.0543.
3a3. Yield, 56.9%; 1H NMR (400 MHz, DMSO-d6) δ 13.17 (s, 1H, NH), 9.03 (s, 1H, H-Ar), 8.21 (d, J = 8.0 Hz, 1H, H-Ar), 8.08 (d, J = 8.4 Hz, 1H, H-Ar), 7.95 (s, 1H, H-Ar), 7.77 (s, 2H, H-Ar), 7.70 (s, 1H, H-Ar), 7.32 (s, 1H, H-Ar); 13C NMR (101 MHz, DMSO-d6) δ 147.43, 147.40, 142.16, 132.75, 129.15, 128.61, 128.20, 126.77, 124.41; ESI-HRMS, calculated m/z for C16H9Cl2N3 [M − H]−: 312.0096, found: 312.0098.
3a4. Yield, 55.4%; 1H NMR (500 MHz, DMSO-d6) δ 13.18 (s, 1H, NH), 9.02 (s, 1H, H-Ar), 8.21 (d, J = 7.7 Hz, 1H, H-Ar), 8.07 (d, J = 7.9 Hz, 1H, H-Ar), 7.95 (s, 1H, H-Ar), 7.91 (s, 1H, H-Ar), 7.77 (s, 1H, H-Ar), 7.67 (s, 1H, H-Ar), 7.44 (s, 1H, H-Ar); 13C NMR (126 MHz, DMSO-d6) δ 147.44, 147.42, 142.14, 132.75, 129.14, 128.61, 128.20, 126.76, 124.38; ESI-HRMS, calculated m/z for C16H9BrClN3 [M − H]−: 355.9590, found: 355.9625.
3a5. Yield, 55.1%; 1H NMR (400 MHz, DMSO-d6) δ 13.30 (s, 1H, NH), 9.03 (s, 1H, H-Ar), 8.21 (d, J = 7.6 Hz, 1H, H-Ar), 8.08 (d, J = 8.4 Hz, 2H, H-Ar), 7.96 (s, 2H, H-Ar), 7.77 (s, 1H, H-Ar); 13C NMR (101 MHz, DMSO-d6) δ 150.82, 147.47, 147.32, 142.32, 132.89, 129.19, 128.67, 128.21, 126.71, 124.03; ESI-HRMS, calculated m/z for C16H8Cl3N3 [M − H]−: 345.9706, found: 345.9747.
3a6. Yield, 63.2%; 1H NMR (400 MHz, DMSO-d6) δ 12.97 (s, 1H, NH), 9.01 (s, 1H, H-Ar), 8.21 (d, J = 7.4 Hz, 1H, H-Ar), 8.08 (d, J = 8.0 Hz, 1H, H-Ar), 7.94 (s, 1H, H-Ar), 7.77 (s, 2H, H-Ar), 7.67 (s, 1H, H-Ar), 7.30 (s, 2H, H-Ar); 13C NMR (101 MHz, DMSO-d6) δ 148.21, 147.63, 147.31, 141.97, 132.58, 129.09, 128.55, 128.19, 126.83, 124.92; ESI-HRMS, calculated m/z for C16H10ClN3 [M − H]−: 278.0485, found: 278.0489.
3b1. Yield, 58.1%; 1H NMR (500 MHz, DMSO-d6) δ 12.66 (s, 1H, NH), 8.84 (s, 1H, H-Ar), 7.94 (s, 2H, H-Ar), 7.84 (s, 1H, H-Ar), 7.46 (s, 2H, H-Ar), 2.54 (s, 3H, –CH3), 2.36 (s, 6H, –CH3); 13C NMR (126 MHz, DMSO-d6) δ 147.32, 146.78, 145.84, 140.84, 138.21, 134.49, 127.90, 127.54, 126.88, 125.09, 21.62, 20.50; ESI-HRMS, calculated m/z for C19H16ClN3 [M + H]+: 322.1111, found: 322.1129.
3b2. Yield, 64.6%; 1H NMR (400 MHz, DMSO-d6) δ 12.14 (s, 1H, NH), 8.87 (s, 1H, H-Ar), 7.96 (d, J = 8.6 Hz, 1H, H-Ar), 7.94 (s, 1H, H-Ar), 7.77 (d, J = 8.6 Hz, 1H, H-Ar), 7.70 (s, 1H, H-Ar), 7.51 (s, 1H, H-Ar), 7.16 (s, 1H, H-Ar), 2.54 (s, 3H, –CH3); 13C NMR (101 MHz, DMSO-d6) δ 146.57, 145.98, 141.23, 138.35, 134.76, 127.92, 127.61, 126.79, 124.52, 21.62; ESI-HRMS, calculated m/z for C17H11ClFN3 [M + H]+: 312.0704, found: 312.0702.
3b3. Yield, 55.9%; 1H NMR (400 MHz, DMSO-d6) δ 13.13 (s, 1H, NH), 8.88 (s, 1H, H-Ar), 7.97 (d, J = 8.7 Hz, 1H, H-Ar), 7.95 (s, 1H, H-Ar), 7.78 (d, J = 8.6 Hz, 2H, H-Ar), 7.70 (s, 1H, H-Ar), 7.32 (d, J = 8.6 Hz, 1H, H-Ar), 2.54 (s, 3H, –CH3); 13C NMR (101 MHz, DMSO-d6) δ 146.53, 146.04, 141.38, 138.40, 134.86, 127.94, 127.64, 126.77, 124.36, 21.64; ESI-HRMS, calculated m/z for C17H11Cl2N3 [M − H]−: 326.0252, found: 326.0255.
3b4. Yield, 61.0%; 1H NMR (400 MHz, DMSO-d6) δ 13.16 (s, 1H, NH), 8.88 (s, 1H, H-Ar), 7.97 (d, J = 8.8 Hz, 1H, H-Ar), 7.95 (s, 1H, H-Ar), 7.78 (d, J = 8.6 Hz, 1H, H-Ar), 7.73 (s, 1H, H-Ar), 7.68 (s, 1H, H-Ar), 7.43 (d, J = 8.3 Hz, 1H, H-Ar), 2.54 (s, 3H, –CH3); 13C NMR (101 MHz, DMSO-d6) δ 146.53, 146.04, 141.40, 138.41, 134.87, 127.94, 127.65, 126.77, 124.34, 21.64; ESI-HRMS, calculated m/z for C17H11BrClN3 [M − H]−: 369.9747, found: 369.9774.
3b5. Yield, 59.9%; 1H NMR (500 MHz, DMSO-d6) δ 13.26 (s, 1H, NH), 8.88 (s, 1H, H-Ar), 7.96 (d, J = 8.6 Hz, 2H, H-Ar), 7.94 (s, 2H, H-Ar), 7.78 (s, 1H, H-Ar), 2.54 (s, 3H, –CH3); 13C NMR (126 MHz, DMSO-d6) δ 150.92, 146.41, 146.14, 141.46, 138.44, 134.94, 127.94, 127.66, 126.71, 123.99, 21.61; ESI-HRMS, calculated m/z for C17H11BrClN3 [M − H]−: 359.9862, found: 359.9903.
3b6. Yield, 61.7%; 1H NMR (400 MHz, DMSO-d6) δ 12.96 (s, 1H, NH), 8.87 (s, 1H, H-Ar), 7.97 (d, J = 8.7 Hz, 1H, H-Ar), 7.95 (s, 1H, H-Ar), 7.77 (d, J = 8.6 Hz, 1H, H-Ar), 7.69 (s, 2H, H-Ar), 7.29 (d, J = 6.0 Hz, 2H, H-Ar), 2.54 (s, 3H, –CH3); 13C NMR (101 MHz, DMSO-d6) δ 148.32, 146.73, 145.95, 141.19, 138.32, 134.69, 127.93, 127.60, 126.83, 124.87, 21.64; ESI-HRMS, calculated m/z for C17H12ClN3 [M + H]+: 294.0798, found: 294.0790.
3c1. Yield, 59.0%; 1H NMR (500 MHz, DMSO-d6) δ 12.78 (s, 1H, NH), 8.84 (s, 1H, H-Ar), 7.96 (d, J = 9.2 Hz, 1H, H-Ar), 7.60 (s, 1H, H-Ar), 7.55 (d, J = 9.1 Hz, 1H, H-Ar), 7.46 (s, 2H, H-Ar), 3.93 (s, 3H, –OCH3), 2.36 (s, 6H, –CH3); 13C NMR (126 MHz, DMSO-d6) δ 158.70, 147.27, 144.89, 143.27, 140.29, 131.65, 129.64, 128.16, 125.06, 124.67, 106.81, 56.24, 20.49; ESI-HRMS, calculated m/z for C19H16ClN3O [M + H]+: 338.1060, found: 338.1076.
3c2. Yield, 56.4%; 1H NMR (500 MHz, DMSO-d6) δ 13.05 (s, 1H, NH), 8.87 (s, 1H, H-Ar), 7.97 (d, J = 9.1 Hz, 1H, H-Ar), 7.76 (s, 1H, H-Ar), 7.59 (s, 1H, H-Ar), 7.57 (d, J = 9.1 Hz, 1H, H-Ar), 7.44 (s, 1H, H-Ar), 7.15 (s, 1H, H-Ar), 3.93 (s, 3H, –OCH3); 13C NMR (126 MHz, DMSO-d6) δ 158.70, 143.38, 129.65, 128.11, 124.92, 124.66, 106.76, 56.23; ESI-HRMS, calculated m/z for C17H11ClFN3O [M + H]+: 328.0653, found: 328.0640.
3c3. Yield 55.1%, 1H NMR (400 MHz, DMSO-d6) δ 13.12 (s, 1H, NH), 8.88 (s, 1H, H-Ar), 7.98 (d, J = 9.0 Hz, 1H, H-Ar), 7.76 (s, 1H, H-Ar), 7.70 (s, 1H, H-Ar), 7.60 (s, 1H, H-Ar), 7.58 (d, J = 9.0 Hz, 1H, H-Ar), 7.32 (d, J = 8.6 Hz, 1H, H-Ar), 3.93 (s, 3H, –OCH3);13C NMR (101 MHz, DMSO-d6) δ 158.74, 149.73, 144.65, 143.45, 140.73, 129.68, 128.10, 125.02, 124.51, 106.81, 56.26; ESI-HRMS m/z calc. for C17H11Cl2N3O [M − H]−: 342.0201; found: 342.0205.
3c4. Yield, 57.1%; 1H NMR (400 MHz, DMSO-d6) δ 13.14 (s, 1H, NH), 8.88 (s, 1H, H-Ar), 7.98 (d, J = 9.1 Hz, 1H, H-Ar), 7.90 (s, 1H, H-Ar), 7.68 (s, 1H, H-Ar), 7.60 (s, 1H, H-Ar), 7.58 (d, J = 9.1 Hz, 1H, H-Ar), 7.43 (d, J = 8.4 Hz, 1H, H-Ar), 3.93 (s, 3H, –OCH3); 13C NMR (101 MHz, DMSO-d6) δ 158.73, 144.64, 143.45, 140.75, 129.68, 128.10, 125.04, 124.47, 106.80, 56.26; ESI-HRMS, calculated m/z for C17H11Cl2N3O [M − H]−: 385.9696, found: 385.9715.
3c5. Yield, 57.5%; 1H NMR (400 MHz, DMSO-d6) δ 13.26 (s, 1H, NH), 8.88 (s, 1H, H-Ar), 7.97 (s, 3H, H-Ar), 7.60 (s, 2H, H-Ar), 3.93 (s, 3H, –OCH3); 13C NMR (101 MHz, DMSO-d6) δ 158.77, 150.93, 144.53, 143.52, 140.87, 129.69, 128.05, 125.15, 124.12, 106.85, 56.27; ESI-HRMS, calculated m/z for C17H11Cl2N3O [M − H]−: 375.9811, found: 378.9859.
3c6. Yield, 65.1%; 1H NMR (400 MHz, DMSO-d6) δ 12.68 (s, 1H, NH), 9.10 (s, 1H, H-Ar), 7.75 (s, 1H, H-Ar), 7.66 (s, 1H, H-Ar), 7.52 (s, 1H, H-Ar), 7.39 (d, J = 9.0 Hz, 1H, H-Ar), 7.29 (s, 1H, H-Ar), 7.22 (s, 1H, H-Ar), 7.20 (s, 1H, H-Ar), 3.84 (s, 3H, –OCH3); 13C NMR (101 MHz, DMSO-d6) δ 160.79, 155.14, 148.34, 143.27, 139.05, 134.88, 133.84, 122.73, 122.36, 121.76, 120.64, 120.25, 118.77, 117.06, 113.27, 110.27, 56.00; ESI-HRMS, calculated m/z for C17H12ClN3O [M + H]+: 310.0747, found: 310.0741.
3d1. Yield, 64.5%; 1H NMR (400 MHz, DMSO-d6) δ 12.59 (s, 1H, NH), 8.75 (s, 1H, H-Ar), 7.54 (s, 1H, H-Ar), 7.42 (s, 3H, H-Ar), 6.29 (s, 2H, O–CH2–O), 2.34 (s, 6H, –CH3); 13C NMR (101 MHz, DMSO-d6) δ 152.93, 149.06, 147.43, 145.92, 145.06, 140.02, 124.16, 122.81, 104.52, 103.46, 103.22, 20.54; ESI-HRMS, calculated m/z for C19H14ClN3O2 [M + H]+: 352.0853, found: 352.0880.
3d2. Yield, 58.9%; 1H NMR (500 MHz, DMSO-d6) δ 12.96 (s, 1H, NH), 8.77 (s, 1H, H-Ar), 7.68 (s, 1H, H-Ar), 7.54 (s, 1H, H-Ar), 7.47 (s, 1H, H-Ar), 7.43 (s, 1H, H-Ar), 7.14 (s, 1H, H-Ar), 6.30 (s, 2H, O–CH2–O); 13C NMR (126 MHz, DMSO-d6) δ 153.14, 149.15, 146.17, 144.91, 140.25, 124.12, 122.24, 104.55, 103.52, 103.28, 100.00; ESI-HRMS, calculated m/z for C17H9ClFN3O2 [M + H]+: 342.0446, found: 342.0429.
3d3. Yield, 59.1%; 1H NMR (400 MHz, DMSO-d6) δ 13.05 (s, 1H, NH), 8.79 (s, 1H, H-Ar), 7.78 (s, 1H, H-Ar), 7.65 (s, 1H, H-Ar), 7.56 (s, 1H, H-Ar), 7.44 (s, 1H, H-Ar), 7.30 (d, J = 8.1 Hz, 1H, H-Ar), 6.30 (s, 2H, O–CH2–O); 13C NMR (101 MHz, DMSO-d6) δ 153.22, 149.19, 146.25, 144.87, 140.40, 124.12, 122.08, 104.57, 103.54, 103.32; ESI-HRMS, calculated m/z for C17H9Cl2N3O2 [M − H]−: 355.9994, found: 355.9999.
3d4. Yield, 68.6%; 1H NMR (400 MHz, DMSO-d6) δ 13.04 (s, 1H, NH), 8.79 (s, 1H, H-Ar), 7.87 (s, 1H, H-Ar), 7.64 (s, 1H, H-Ar), 7.55 (s, 1H, H-Ar), 7.44 (s, 1H, H-Ar), 7.41 (d, J = 8.6 Hz, 1H, H-Ar), 6.30 (s, 2H, O–CH2–O); 13C NMR (101 MHz, DMSO-d6) δ 153.22, 149.18, 146.25, 144.86, 140.40, 124.11, 122.04, 104.57, 103.54, 103.31; ESI-HRMS, calculated m/z for C17H9BrClN3O2 [M − H]−: 399.9489, found: 399.9508.
3d5. Yield, 60.5%; 1H NMR (500 MHz, DMSO-d6) δ 13.15 (s, 1H, NH), 8.79 (s, 1H, H-Ar), 7.96 (s, 2H, H-Ar), 7.56 (s, 1H, H-Ar), 7.44 (s, 1H, H-Ar), 6.30 (s, 2H, O–CH2–O); 13C NMR (126 MHz, DMSO-d6) δ 153.32, 151.05, 149.23, 146.38, 144.79, 140.46, 131.96, 129.12, 124.07, 121.69, 104.58, 103.57, 103.33; ESI-HRMS, calculated m/z for C17H8Cl3N3O2 [M − H]−: 389.9604, found: 389.9649.
3d6. Yield, 62.2%; 1H NMR (400 MHz, DMSO-d6) δ 12.96 (s, 1H, NH), 8.78 (s, 1H, H-Ar), 7.67 (s, 2H, H-Ar), 7.55 (s, 1H, H-Ar), 7.44 (s, 1H, H-Ar), 7.27 (s, 2H, H-Ar), 6.30 (s, 2H, O–CH2–O); 13C NMR (101 MHz, DMSO-d6) δ 153.07, 149.12, 148.42, 146.10, 145.04, 140.27, 124.14, 122.89, 122.55, 104.55, 103.50, 103.27; ESI-HRMS, calculated m/z for C17H10ClN3O2 [M + H]+: 324.0540, found: 324.0532.
4.3. Biological assays
The procedures for biological assays are described in the ESI† (Part 1). The materials, instrumentation, and methods for the cytotoxicity assay, cell cycle analysis, cell apoptosis analysis and western blot assays were reported previously.17,32–34
Ethical statement
The in vivo antitumor studies were carried out at the Institute of Radiation Medicine, Chinese Academy of Medical Sciences (Tian Jin, China). The handling of animals and experimental design were approved by the Ethics Committee and Animal Care Committee of the Institute.
Conflicts of interest
The authors declare no competing financial interest.
Abbreviations used
| Bax | Bcl2-associated X protein |
| PI | Propidium iodide |
| EB | Ethidium bromide |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| CDC | Cell division cyclin |
| CDK | Cyclin-dependent kinase |
| CDC25C | Cell division cyclin 25 homolog C |
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 21501032), the Innovative Team & Outstanding Talent Program of Colleges and Universities in Guangxi (2017-38), Guangxi Natural Science Foundation (Nos. 2016GXNSFAA380300 and 2014GXNSFBA118050), Guangxi New Century Ten, Hundred and Thousand Talents Project ([2017]42) and the State Key Laboratory Cultivation Base for Chemistry and Molecular Engineering of Medicinal Resources (Ministry of Science and Technology of China, No. CMEMR2014-B14).
Notes and references
- World Cancer Report (World Health Organization, International Agency for Cancer Research), ed. B. Stewart and P. Kleihues, IARC Press, Lyon, 2003 Search PubMed.
- D. M. Parkin, Global cancer statistics in the year 2000, Lancet Oncol., 2001, 2, 533–543 CrossRef PubMed.
- A. Anighoro, J. Bajorath and G. Rastelli, Polypharmacology: challenges and opportunities in drug discovery, J. Med. Chem., 2014, 57, 7874–7887 CrossRef PubMed.
- S. Fortin and G. Berube, Advances in the development of hybrid anticancer drugs, Expert Opin. Drug Discovery, 2013, 8, 1029–1047 CrossRef PubMed.
- S. Kumar, S. Bawa and H. Gupta, Biological activities of quinoline derivatives, Mini-Rev. Med. Chem., 2009, 9, 1648–1654 CrossRef PubMed.
- C. H. Tseng, C. C. Tzeng, C. L. Yang, P. J. Lu, H. L. Chen and H. Y. Li, et al., Synthesis and antiproliferative evaluation of certain indeno[1,2-c]quinoline derivatives. Part 2, J. Med. Chem., 2010, 53, 6164–6179 CrossRef PubMed.
- C. H. Tseng, Y. L. Chen, P. J. Lu, C. N. Yang and C. C. Tzeng, Synthesis and antiproliferative evaluation of certain indeno[1,2-c]quinoline derivatives, Bioorg. Med. Chem., 2008, 16, 3153–3162 CrossRef PubMed.
- P. M. Njaria, J. Okombo, N. M. Njuguna and K. Chibale, Chloroquine-containing compounds: a patent review (2010–2014), Expert Opin. Ther. Pat., 2015, 25, 1003–1024 CrossRef PubMed.
- T. Kimura, Y. Takabatake, A. Takahashi and Y. Isaka, Chloroquine in cancer therapy: a double-edged sword of autophagy, Cancer Res., 2013, 73, 3–7 CrossRef PubMed.
- L. L. Brunton, J. S. Lazo and K. L. Parker, The pharmacological basis of therapeutics, Mc Graw-Hill, New York, 11th edn, 2006 Search PubMed.
- V. Králová, V. Hanušová, E. Rudolf, K. Čáňová and L. Skálová, Flubendazole induces mitotic catastrophe and senescence in colon cancer cells in vitro, J. Pharm. Pharmacol., 2016, 68, 208–218 CrossRef PubMed.
- N. S. El-Gohary and M. I. Shaaban, Synthesis, antimicrobial, antiquorum-sensing and antitumor activities of new benzimidazole analogs, Eur. J. Med. Chem., 2017, 137, 439–449 CrossRef PubMed.
- F. Gümüş, G. Eren, L. Açık, A. Çelebi, F. Öztürk, Ş. Yılmaz, R. I. Saǧkan, S. Gür, A. Özkul, A. Elmal and Y. Elerman, Synthesis, Cytotoxicity, and DNA Interactions of New Cisplatin Analogues Containing Substituted Benzimidazole Ligands, J. Med. Chem., 2009, 52, 1345–1357 CrossRef PubMed.
- C.-C. Chiu, H.-L. Chou, B.-H. Chen, K.-F. Chang, C.-H. Tseng, Y. Fong, T.-F. Fu, H.-W. Chang, C.-Y. Wu, E.-M. Tsai, S.-R. Lin and Y.-L. Chen, BPIQ, a novel synthetic quinoline derivative, inhibits growth and induces mitochondrial apoptosis of lung cancer cells in vitro and in zebrafish xenograft model, BMC Cancer, 2015, 15, 962 CrossRef PubMed.
- Y. C. Yu, W. B. Kuang, R. Z. Huang, Y. L. Fang, Y. Zhang, Z. F. Chen and X. L. Ma, Design, synthesis and pharmacological evaluation of new 2-oxoquinoline derivatives containing α-aminophosphonates as potential antitumor agents, MedChemComm, 2017, 8, 1158–1172 RSC.
- V. Lakshma Nayak, N. Nagesh, A. Ravikumar, C. Bagul, M. V. P. S. Vishnuvardhan, V. Srinivasulu and A. Kamal, 2-aryl benzimidazole conjugate induced apoptosis in human breast cancer MCF-7 cells through caspase independent pathway, Apoptosis, 2017, 22, 118–134 CrossRef PubMed.
- B. Q. Zou, X. Lu, Q. P. Qin, Y. X. Bai, Y. Zhang, M. Wang, Y. C. Liu, Z. F. Chen and H. Liang, Three novel transition
metal complexes of 6-methyl-2-oxo-quinoline-3-carbaldehyde thiosemicarbazone: synthesis, crystal structure, cytotoxicity, and mechanism of action, RSC Adv., 2017, 7, 17923–17933 RSC.
- J. A. Choi, J. Y. Kim, J. Y. Lee, C. M. Kang, H. J. Kwon, Y. D. Yoo, T. W. Kim, Y. S. Lee and S. J. Lee, Induction of cell cycle arrest and apoptosis in human breast cancer cells by quercetin, Int. J. Oncol., 2001, 19, 837–844 Search PubMed.
- M. B. Kastan and J. Bartek, Cell-cycle checkpoints and cancer, Nature, 2004, 432, 316–322 CrossRef PubMed.
- S. U. Dighe, S. Khan, I. Soni, P. Jain, S. Shukla, R. Yadav, P. Sen, S. M. Meeran and S. Batra, Synthesis of β-Carboline-Based N-Heterocyclic Carbenes and Their Antiproliferative and Antimetastatic Activities against Human Breast Cancer Cells, J. Med. Chem., 2015, 58, 3485–3499 CrossRef PubMed.
- R. Boutros, V. Lobjois and B. Ducommun, CDC25 phosphatases in cancer cells: key players? Good targets?, Nat. Rev. Cancer, 2007, 7, 495–507 CrossRef PubMed.
- F. Belluti, G. Fontana, L. D. Bo, N. Carenini, C. Giommarelli and F. Zunino, Design, synthesis and anticancer activities of stilbene-coumarin hybrid compounds: Identification of novel proapoptotic agents, Bioorg. Med. Chem., 2010, 18, 3543–3550 CrossRef PubMed.
- M. O. Hengartner, The biochemistry of apoptosis, Nature, 2000, 407, 770–776 CrossRef PubMed.
- M. Y. Ye, G. Y. Yao, Y. M. Pan, Z. X. Liao, Y. Zhang and H. S. Wang, Synthesis and antitumor activities of novel α-aminophosphonate derivatives containing an alizarin moiety, Eur. J. Med. Chem., 2014, 83, 116–128 CrossRef PubMed.
- R. Martin, J. Carvalho, E. Ibeas, M. Hernández, V. Ruiz-Gutierrez and M. Luisa-Nieto, Acidic triterpenes compromise growth and survival of astrocytoma cell lines by regulating reactive oxygen species accumulation, Cancer Res., 2007, 67, 3741–3751 CrossRef PubMed.
- L. Gomez, P. A. Thiebaut, M. Paillard, S. Ducreux, M. Abrial, C. C. D. Silva, A. Durand, M. R. Alam, F. V. Coppenolle, S. S. Sheu and M. Ovize, The SR/ER-mitochondria calcium crosstalk is regulated by GSK3β during reperfusion injury, Cell Death Differ., 2016, 23, 313–322 CrossRef PubMed.
- Y. Tsujimoto, J. Cossman, E. Jaffe and C. M. Croce, Involvement of the bcl-2 gene in human follicular lymphoma, Science, 1985, 228, 1440–1443 CrossRef PubMed.
- D. M. Hockenbery, M. Zutter, W. Hickey, M. Nahm and S. J. Korsmeyer, BCL2 protein is topographically restricted in tissues characterized by apoptotic cell death, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 6961–6965 CrossRef.
- S. Qiao, T. Nagasaka, T. Harada and N. Nakashima, p53, Bax and Bcl-2 expression, and apoptosis in gestational trophoblast of complete hydatidiform mole, Placenta, 1998, 19, 361–369 CrossRef PubMed.
- K. W. Yip and J. C. Reed, Bcl-2 family proteins and cancer, Oncogene, 2008, 27, 6398–6406 CrossRef PubMed.
- W. A. Siddiqui, A. Ahad and H. Ahsan, The mystery of BCL2 family: Bcl-2 proteins and apoptosis: an update, Arch. Toxicol., 2015, 89, 289–317 CrossRef PubMed.
- S. X. Hua, R. Z. Huang, M. Y. Ye, Y. M. Pan, G. Y. Yao, Y. Zhang and H. S. Wang, Design, synthesis and in vitro evaluation of novel ursolic acid derivatives as potential anticancer agents, Eur. J. Med. Chem., 2015, 95, 435–452 CrossRef PubMed.
- Z. F. Chen, Q. P. Qin, J. L. Qin, Y. C. Liu, K. B. Huang, Y. L. Li, T. Meng, G. H. Zhang, Y. Peng, X.
J. Luo and H. Liang, Stabilization of G-quadruplex DNA, inhibition of telomerase activity and tumor cell apoptosis of organoplatinum(II) complexes with oxoisoaporphine, J. Med. Chem., 2015, 58, 2159–2179 CrossRef PubMed.
- Y. L. Chu, C. T. Ho, J. G. Chung, R. Raghu, Y. C. Lo and L. Y. Sheen, Allicin induces anti-human liver cancer cells through p53 gene modulating apoptosis and autophagy, J. Agric. Food Chem., 2013, 61, 9839–9848 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The procedures for biological assays, 1H NMR, 13C NMR and HR-MS. See DOI: 10.1039/c8ra04640a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abd
*abd