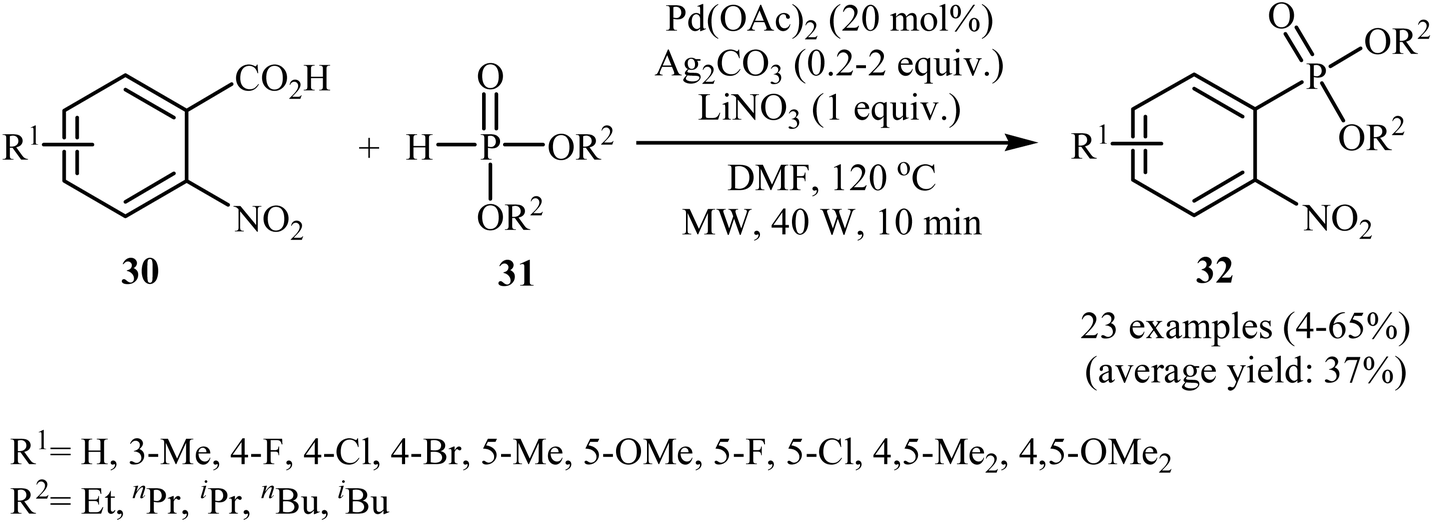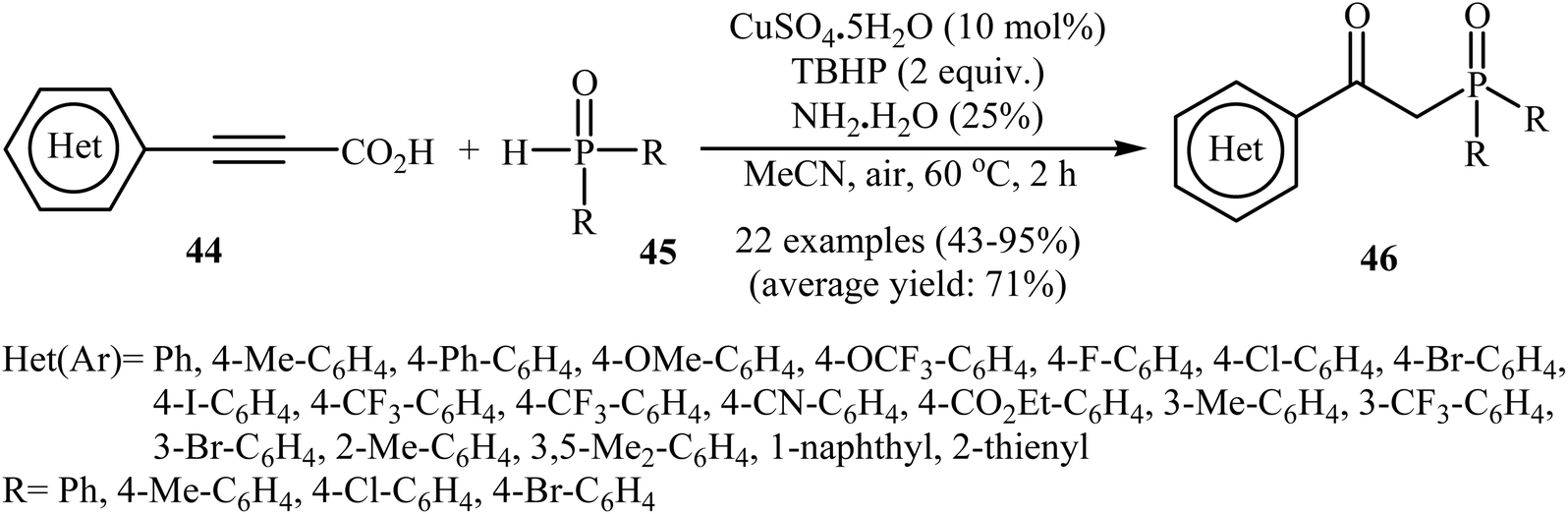DOI:
10.1039/C8RA04557G
(Review Article)
RSC Adv., 2018,
8, 26383-26398
Decarboxylative cross-coupling reactions for P(O)–C bond formation
Received
28th May 2018
, Accepted 3rd July 2018
First published on 24th July 2018
Abstract
Phosphorus-containing compounds are one of the most important classes of organic compounds, which have wide applications in organic chemistry, medicinal chemistry, agricultural chemistry, and materials chemistry. In particular, organophosphorus compounds bearing a P(O)–C bond have attracted significant attention in recent decades due to their widespread biological and pharmacological activities. In this review, we will highlight the most important developments in the construction of P(O)–C bonds through decarboxylative C–P cross-coupling reactions. The literature has been surveyed from 2011 to May 2018.
 Akram Hosseinian | Akram Hosseinian was born in Ahar, Iran, in 1973. She received her B.S. degree in Pure Chemistry from University of Tehran, Iran, and her M.S. degree in Inorganic Chemistry from Tarbiat Modares University, Tehran, Iran, in 2000 under the supervision of Prof. A. R. Mahjoub. She completed her PhD degree in 2007 under the supervision of Prof. A. R. Mahjoub. Now she is working at University of Tehran as an Associate Professor. Her research interests include inorganic and organic synthesis and new methodologies in nanomaterial synthesis. |
 Fatemeh Alsadat Hosseini Nasab | Fatemeh Alsadat Hosseini Nasab was born in Sirjan, Kerman, Iran, in 1980. She received her B.S. degree in Pure Chemistry from University of Shahid Bahonar, Kerman, Iran, and her M.S. degree in Organic Chemistry from Shahid Bahonar University, Kerman, Iran, in 2006 under the supervision of Dr M. Eslami. Now, she is working at Hormozgan University of Bandar Abbas as an Instructor. Her research interests include organic synthesis and new nanochemistry. |
 Sheida Ahmadi | Sheida Ahmadi was born in Miyaneh, Iran. She has received her B.S. degree in Applied Chemistry from Islamic Azad University, Tehran, Iran, in 1997, and her M.S. degree in inorganic chemistry from Shahid Bahonar University, Kerman, Iran, in 2007 under the supervision of Prof. S. J. Fatemi. She received her PhD degree in 2017 under the supervision of Prof. M. Hakimi in Payam Noor University, Tehran, Iran. Now, she is working at Payame Noor University of Tehran as an Assistant Professor. Her research interests include inorganic polymers, synthesis of inorganic complexes and bioinorganic chemistry. |
 Zahra Rahmani | Zahra Rahmani was born in Takab, Iran, in 1985. She received her B.S. degree in Applied Chemistry from University of Urmia, Iran, and her M.S. degree in Organic Chemistry from Payam Noor University, Zanjan, Iran, in 2012 under the supervision of Prof. E. Vessally. She is currently a PhD student in Organic Chemistry under the supervision of Prof. E. Vessalli in Islamic Azad University, Tabriz, Iran. Her doctoral thesis examines the interaction of boron nitride and aluminum nitride nanostructures with some sulfonamide drugs. Now, she is working at Zarshuran Gold Company of Takab as a Laboratory Supervisor. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in Pure Chemistry from University of Tabriz, Tabriz, Iran, and his M.S. degree in Organic Chemistry from Tehran University, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his PhD degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now, he is working at Payame Noor University as a full Professor of Organic Chemistry. His research interests include theoretical organic chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
1. Introduction
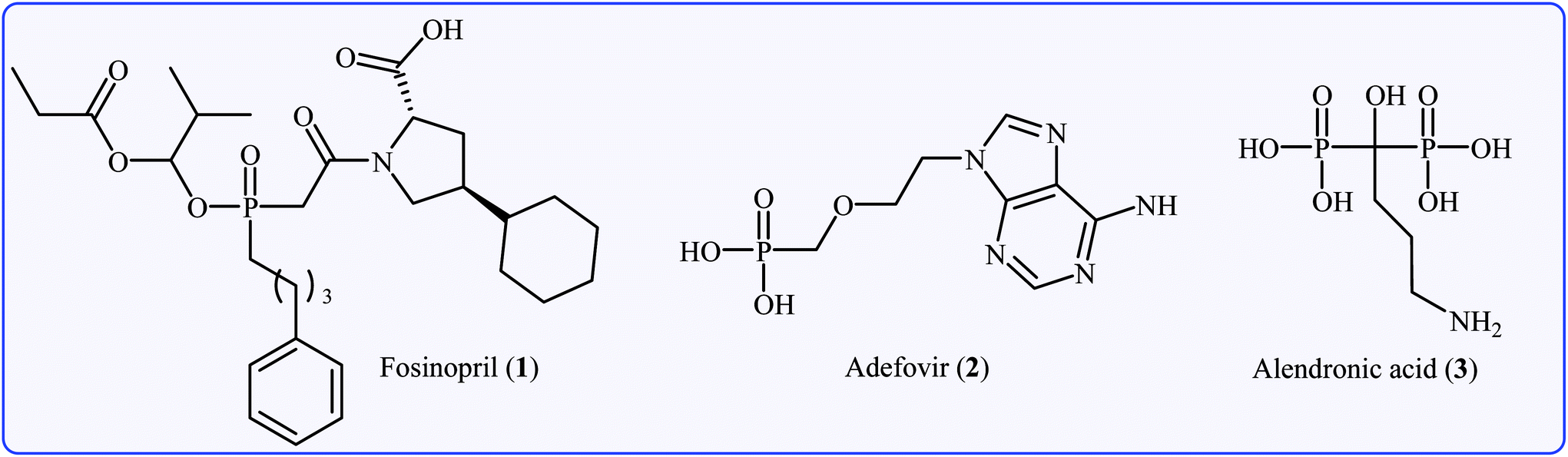
Organophosphorus compounds are among the most important classes of organic compounds, which are diversely found in natural products,1 pharmaceutical drugs,2 insecticides,3 and ligands.4 In particular, P(O)–C bond-containing compounds have attracted significant attention of medicinal chemists and pharmacologists in recent years owing to their broad spectrum of pharmacological profiles.5 Several currently marketed drugs contain this important structural unit, which is used for the treatment of various kinds of diseases. For example (Fig. 1), fosinopril 1 with the brand name Monopril is a phosphinate-containing angiotensin-converting enzyme inhibitor marketed worldwide for the treatment of hypertension (high blood pressure) and some types of chronic heart failures.6 Adefovir 2, with trade names Preveon and Hepsera, is a promising antiviral drug that is used to treat chronic hepatitis B; the drug works by blocking reverse transcriptase, an enzyme crucial for the hepatitis B virus to reproduce in the body.7 Alendronic acid 3 (Fosamax) is a bisphosphonate drug used to treat osteoporosis and several other bone diseases.8 As a consequence, the construction of P(O)–C bonds is an essential issue and has gained much attention from the research community interested in chemical synthesis.
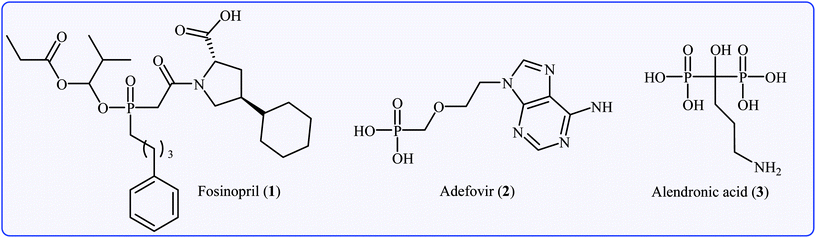 |
| | Fig. 1 Selected examples of drugs containing a P(O)–C bond. | |
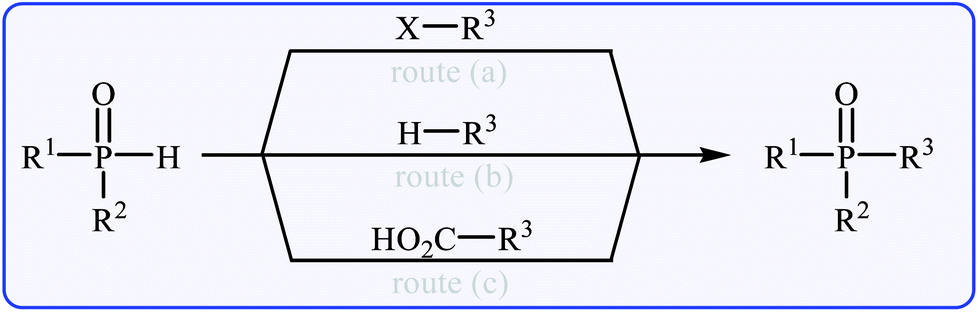
The most general and straightforward methods for the construction of P(O)–C bonds involve either C–P cross-coupling of organic halides or triflates with P(O)H compounds (Fig. 2, route a)9 or direct dehydrogenative C–P cross-coupling between R–H compounds and P(O)H compounds (Fig. 2, route b).10 However, each of these methods suffer from various inherent drawbacks, and the most important of these drawbacks are as follows: (i) most of the organic halides are environmentally toxic and liberate hazardous halogen by-products; (ii) triflates are unstable, expensive, and liberate potentially genotoxic triflic acid;11 and (iii) the selective activation of a specific C–H bond poses a particular challenge.12 An alternative and efficient protocol for the formation of P(O)–C bonds involves decarboxylative C–P cross-coupling between carboxylic acids and P(O)H compounds (Fig. 2, route c). To the best of our knowledge, a comprehensive review on decarboxylative C–P cross-coupling reactions has not been reported in the literature so far. In continuation of our recently published reviews on metal-catalyzed cross-coupling reactions13 and the green synthesis field,14 in this review, we will highlight the most important developments of decarboxylative C–P cross-coupling reactions from 2011 to May 2018. It is worth mentioning that a special emphasis is placed on the mechanistic aspects of the reactions.
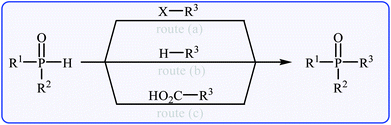 |
| | Fig. 2 C–P cross-coupling routes for the formation of P(O)–C bond. | |
2. Coupling of alkenyl acids with P(O)H compounds
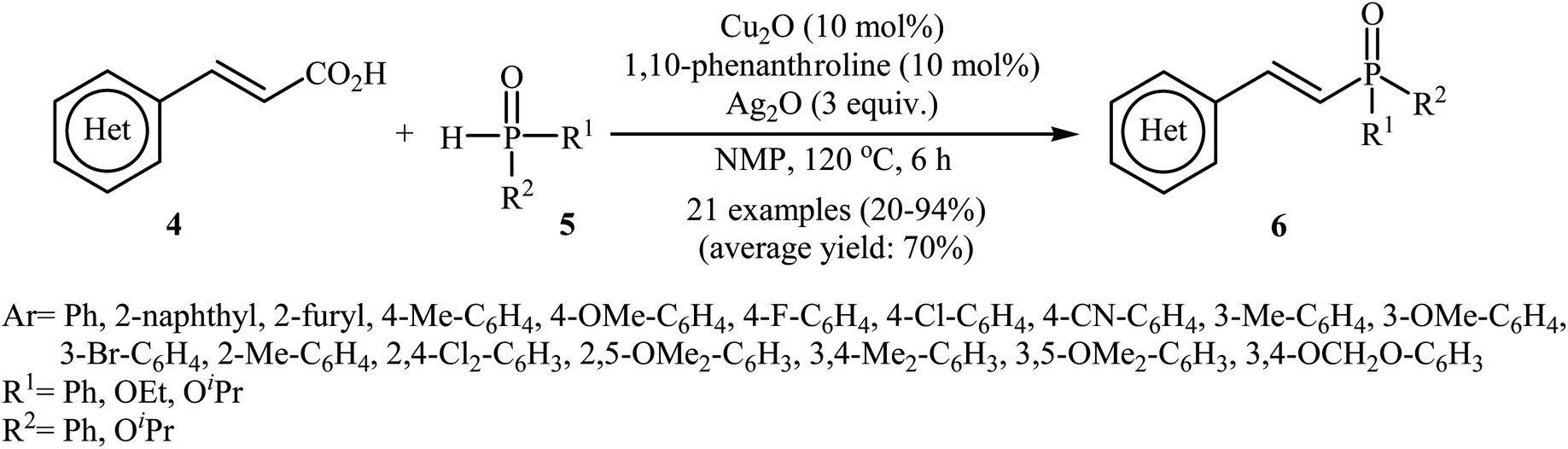
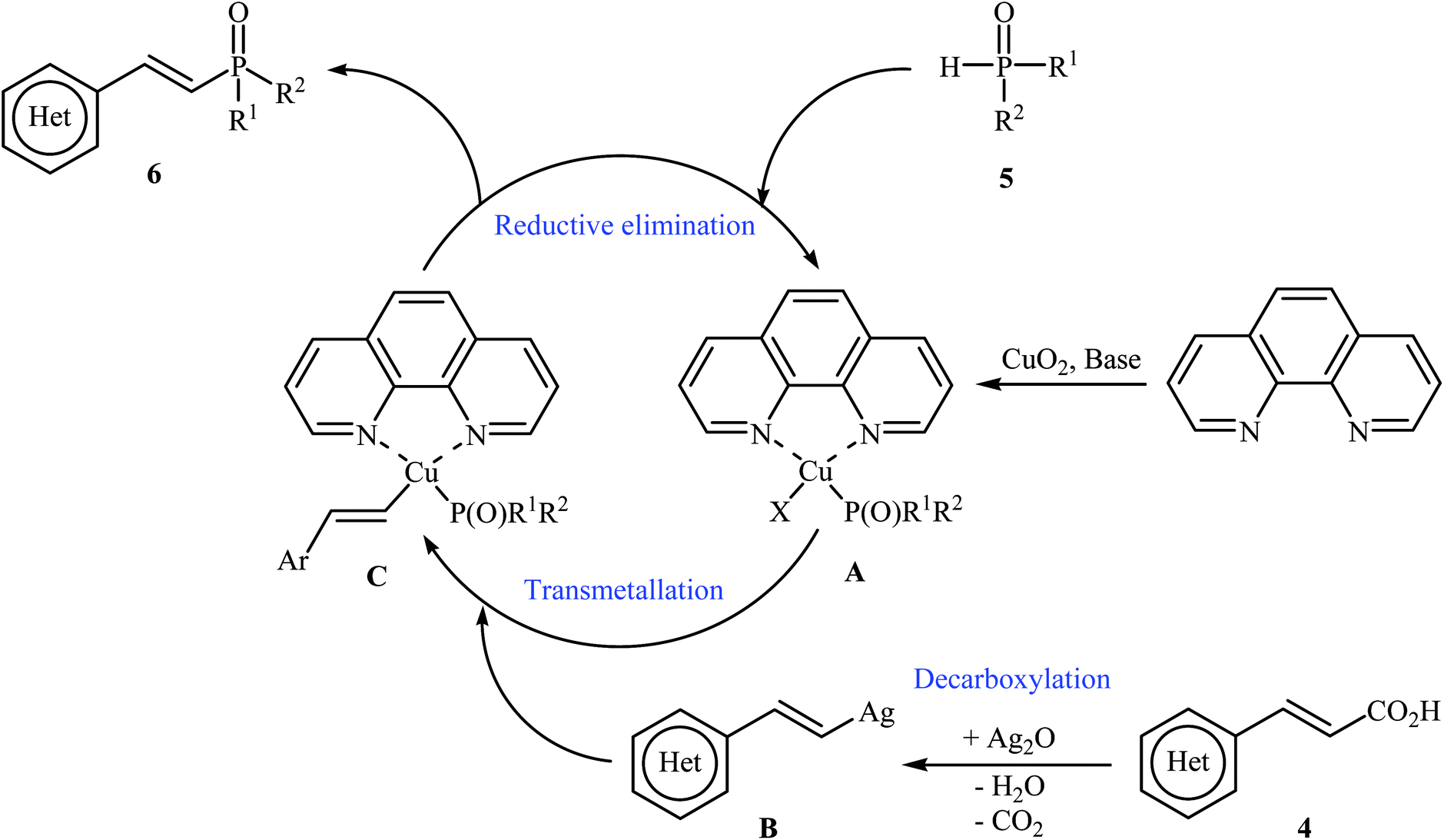
The first synthesis of alkenylphosphorus compounds through transition-metal-catalyzed decarboxylative coupling reactions of alkenyl acids with P(O)H compounds was reported by Hu and co-workers in 2011;15 they treated various cinnamic acids 4 with hydrogen phosphoryl compounds 5 in the presence of 10 mol% of low-cost, commercially-available Cu2O as the catalyst, 10 mol% of 1,10-phenanthroline as the ligand and 3.0 equiv. of Ag2O as the additive in NMP. The reactions were carried out at 120 °C for 6 h and provided the expected alkenylphosphorus compounds 6 in moderate to excellent yields (Scheme 1). It should be mentioned that all three kinds of P(O)–H compounds (H-phosphinates, H-phosphonates, and secondary phosphine oxides) were applicable to this coupling reaction. The decreasing order of the reactivities of hydrogen phosphoryl compounds with cinnamic acids under these reaction conditions was secondary phosphine oxides > H-phosphinates > H-phosphonates. Interestingly, the electronic character of the substituents in the phenyl ring periphery of cinnamic acids had negligible effect on the rate of the reaction. However, steric hindrance led to lower yields. A plausible mechanism that explains this transformation is depicted in Scheme 2. Initially, P(O)–H compound 5 reacted with the copper/phen catalyst to form the intermediate A. Meanwhile, the reaction of alkenyl acids 4 with Ag2O produced intermediate B through a decarboxylation process. This intermediate B upon transmetalation into intermediate A formed the organocopper intermediate C. Finally, reductive elimination of intermediate C afforded the observed alkenylphosphorus compounds 6 along with the initial copper species.
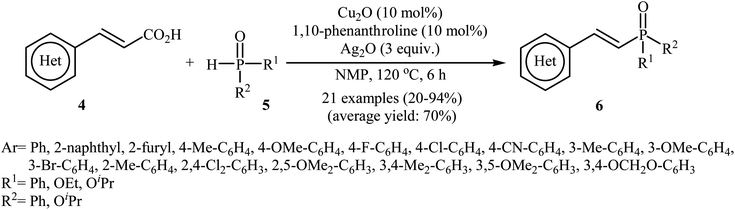 |
| | Scheme 1 Cu-catalyzed decarboxylative coupling reactions of alkenyl acids with P(O)H compounds developed by Hu and co-workers. | |
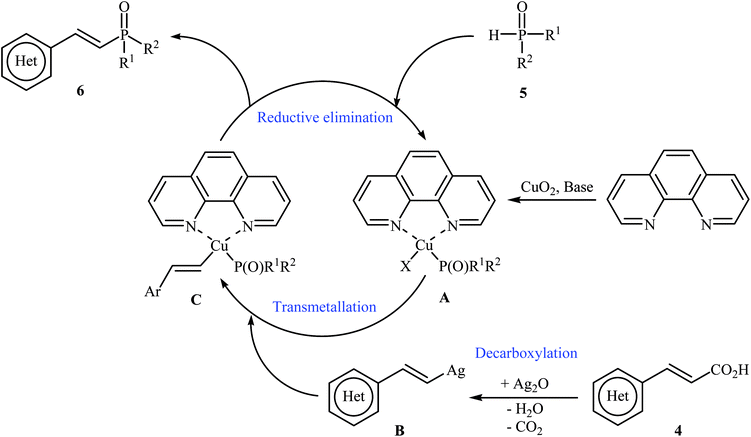 |
| | Scheme 2 Mechanistic proposal for the formation of alkenylphosphorus compounds 6. | |
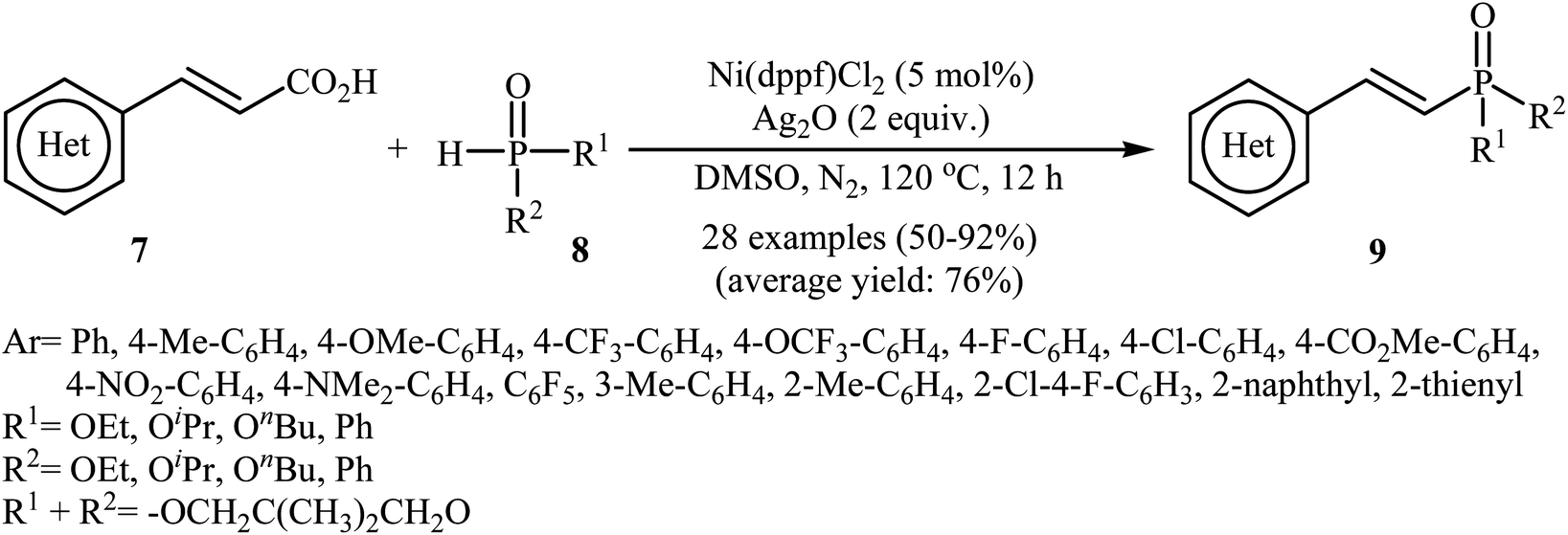
Next, Gao's group demonstrated for the first time the usefulness of nickel catalysts for decarboxylative coupling of alkenyl acids with P(O)–H compounds.16 Thus, in the presence of Ni(dppf)Cl2 as the catalyst and Ag2O as the additive in DMSO at 120 °C, coupling of cinnamic acids 7 with hydrogen phosphoryl compounds 8 afforded the corresponding (E)-1-alkenylphosphorus compounds 9 in moderate to high yields (Scheme 3). A variety of sensitive functional groups including NO2, NMe2, CO2Me, OMe, CF3, F, and Cl were tolerated by the reaction conditions employed. This made further derivatization of the products possible. It is noted that other nickel catalysts such as Ni(OAc)2, Ni(acac)2, NiBr2, and NiCl2 also promoted this coupling reaction; however, lower yields were observed. Based on density functional theory (DFT) calculations and experimental data, the authors found that in this coupling reaction, the phosphine ligand exhibited better catalytic performance than the nitrogen ligand in the reductive elimination step owing to stronger nucleophilicity and larger size.
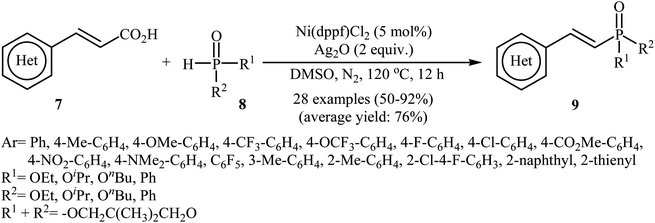 |
| | Scheme 3 Ni-catalyzed decarboxylative coupling of alkenyl acids 7 with P(O)–H compounds 8. | |
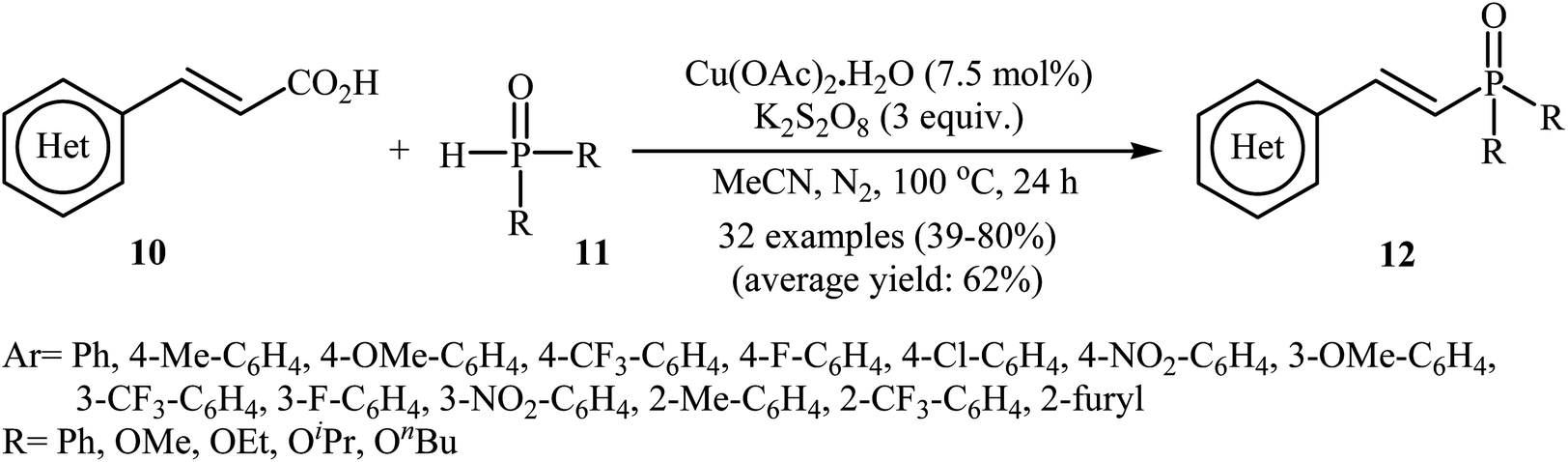
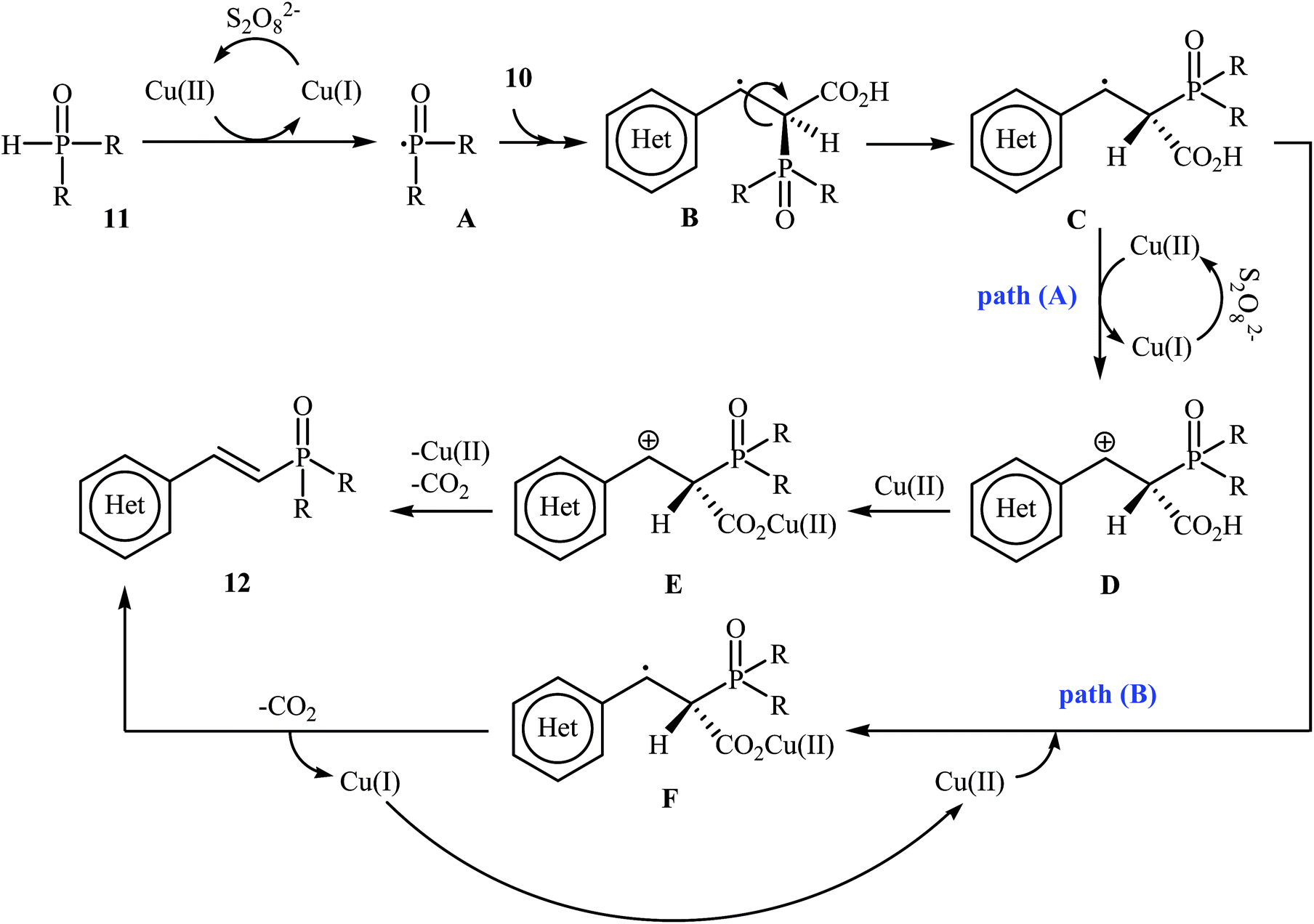
In a related investigation, Tang and Wang along with their co-workers found that the treatment of alkenyl acids 10 with P(O)–H compounds 11 (phosphine oxides and H-phosphonates) in the presence of Cu(OAc)2·H2O/K2S2O8/MeCN combination as a catalytic system afforded the corresponding (E)-1-alkenylphosphorus compounds 12 in moderate to high yields with excellent stereoselectivities (Scheme 4).17 Some important points regarding the reactions are listed below: (i) aliphatic alkenyl acids failed to enter the reaction; (ii) depending on the electronic effects of the substituents on the aryl ring of cinnamic acids, substrates with electron-donating groups gave higher yields than those with electron-withdrawing groups; and (iii) phosphine oxides gave lower yields of desired products compared with H-phosphonates. It is noteworthy that this catalytic system was also successfully applied in the cross-coupling of various P(O)–H compounds with styrenes and β-nitrostyrene. A plausible mechanism for this transformation starts with the formation of the phosphonyl radical A by oxidation of the starting phosphonate 11 with in situ generated CuII species [Cu(CH3CN)n]2+; its nucleophilic attack on the alkenyl acid 10 led to the formation of radical B, which could be converted into a more stable conformation C. Next, the oxidation of this intermediate C by the CuII species gave the carbocation intermediate D, which finally afforded the final product by a ligand-exchange-decarboxylation sequence (path A). In another possibility (path B), intermediate C gave complex D through ligand exchange, followed by decarboxylation to afford the corresponding (E)-1-alkenylphosphorus product (Scheme 5).
 |
| | Scheme 4 Tang–Wang's synthesis of (E)-1-alkenylphosphorus compounds 12. | |
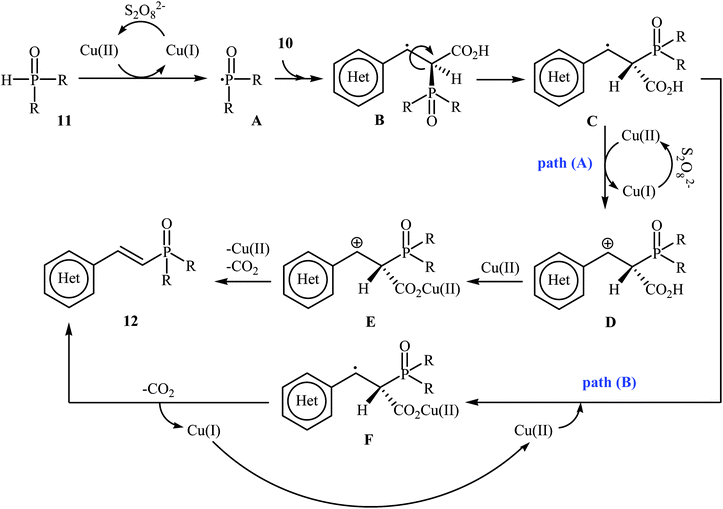 |
| | Scheme 5 Mechanistic proposal for the reactions in Scheme 4. | |
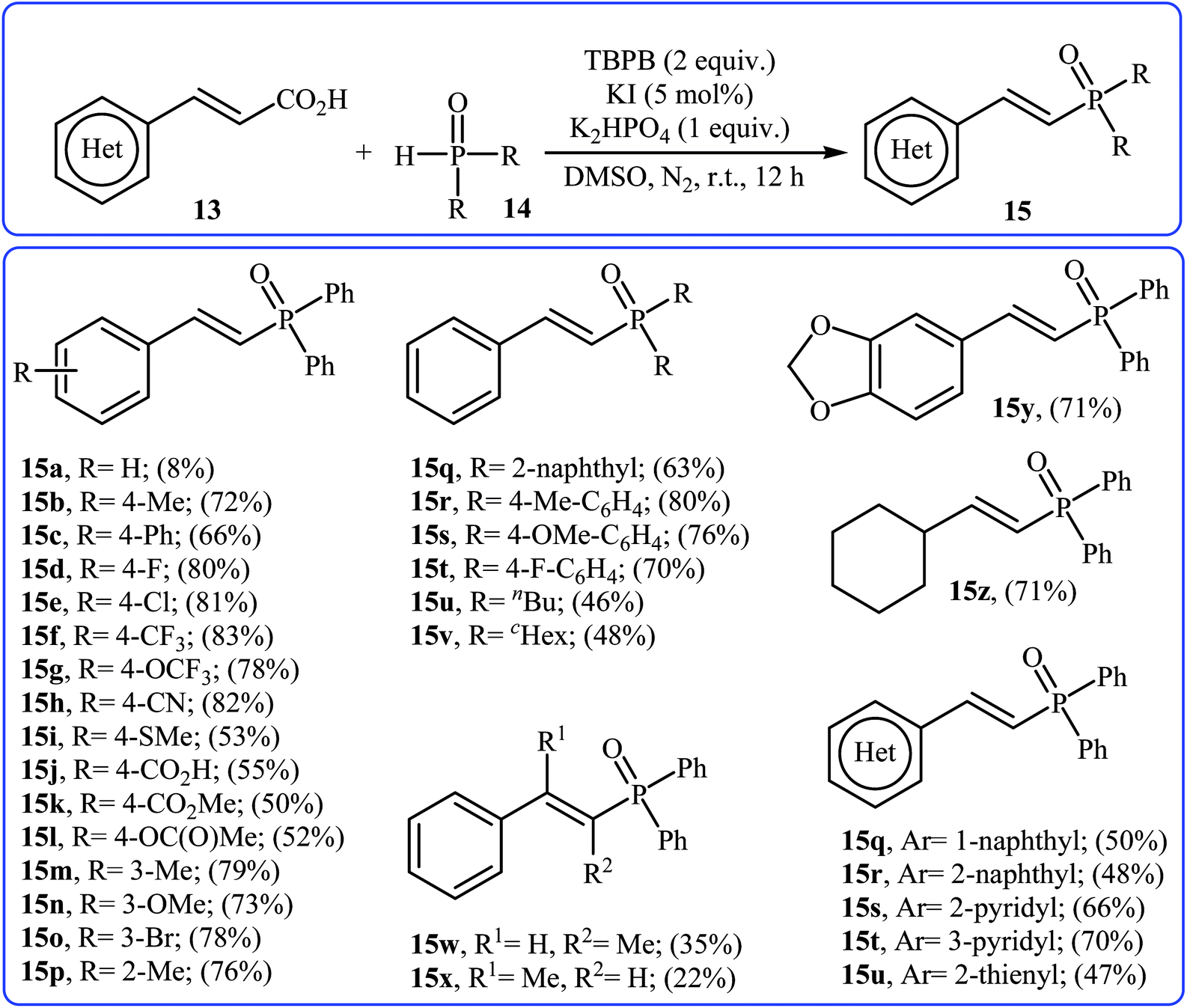
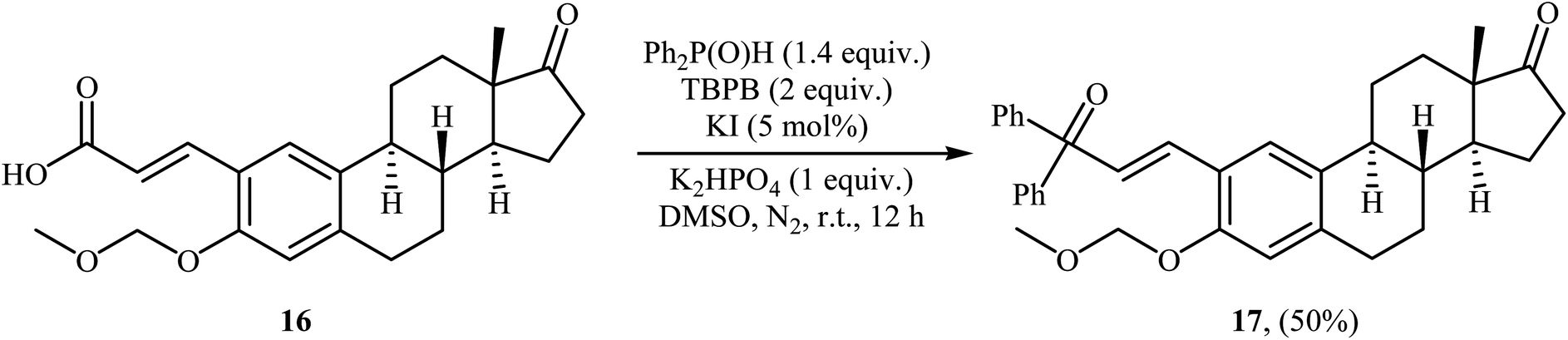
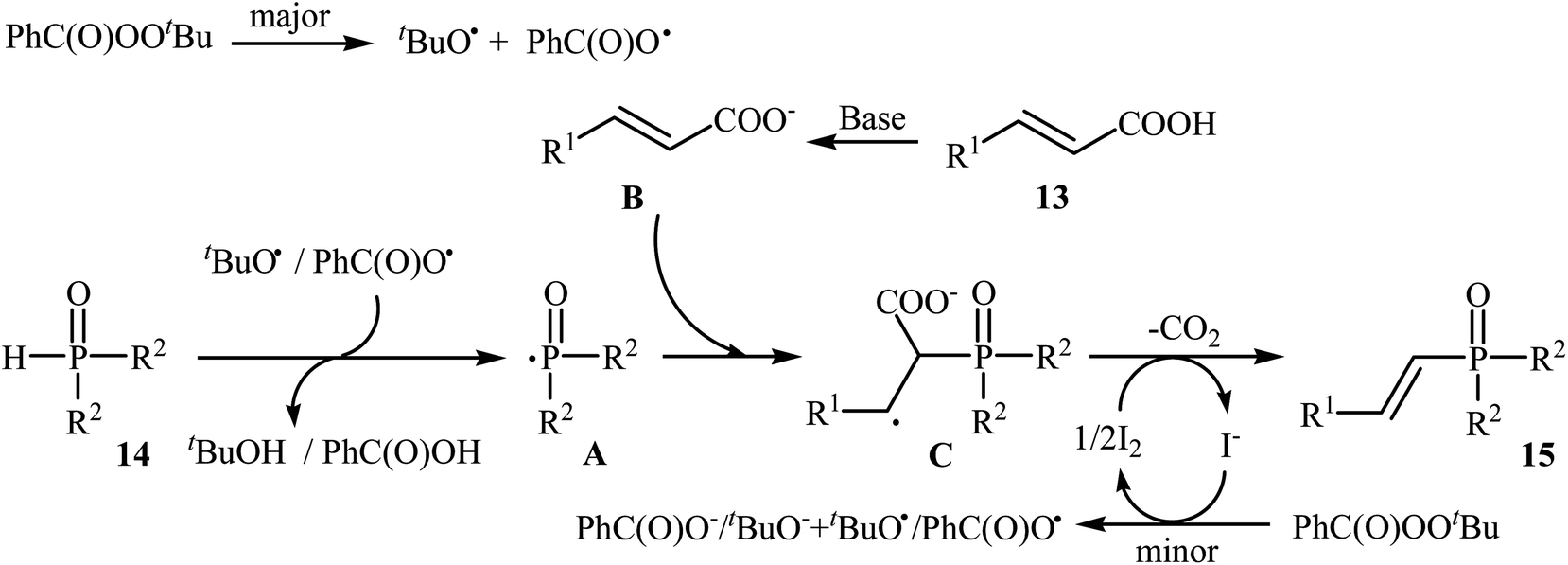
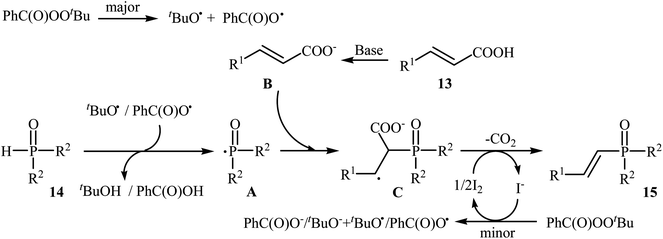
Very recently, Liu and co-workers described efficient transition-metal-free phosphorylation of alkenyl acid derivatives with P(O)H compounds via radical-promoted decarboxylation under mild conditions.18 They carefully tested several oxidants, bases, additives and solvents, and the system of TBPB/K2HPO4/KI/DMSO was found to be superior. Under optimized conditions, various aromatic and heteroaromatic alkenyl acids 13, including those exhibiting substitution on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, and symmetrical secondary phosphine oxides 14 reacted to give good yields of the corresponding products 15 (Scheme 6). In addition to mild reaction conditions and excellent functional group tolerance (F, Cl, Br, CN, CO2H, CO2Me, OCOMe, OMe, and SMe), the major advantage of this method is the formation of (E)-1-alkenylphosphorus compounds 15 on a gram-scale. The authors also showed the application of this procedure for the high yielding syntheses of the estrone derivative 17, a steroidal progestin used in clinic, with estrogenic activity (Scheme 7). They assumed that the mechanism of this coupling reaction involved the initial formation of tert-butoxyl and benzoyloxyl radicals via decomposition of TBPB, which was promoted with the assistance of iodide anions. Next, the abstraction of a hydrogen atom from H(O)PR1R2 14 by a tert-butoxyl or benzoyloxyl radical afforded the phosphorus radical A, which underwent radical addition to the cinnamic acid anion B (generated from the reaction of the starting acid 13 with a base) to provide the radical intermediate C. Finally, decarboxylation of intermediate C followed by single-electron oxidation of the radical anion afforded the expected products 15 (Scheme 8).
C bond, and symmetrical secondary phosphine oxides 14 reacted to give good yields of the corresponding products 15 (Scheme 6). In addition to mild reaction conditions and excellent functional group tolerance (F, Cl, Br, CN, CO2H, CO2Me, OCOMe, OMe, and SMe), the major advantage of this method is the formation of (E)-1-alkenylphosphorus compounds 15 on a gram-scale. The authors also showed the application of this procedure for the high yielding syntheses of the estrone derivative 17, a steroidal progestin used in clinic, with estrogenic activity (Scheme 7). They assumed that the mechanism of this coupling reaction involved the initial formation of tert-butoxyl and benzoyloxyl radicals via decomposition of TBPB, which was promoted with the assistance of iodide anions. Next, the abstraction of a hydrogen atom from H(O)PR1R2 14 by a tert-butoxyl or benzoyloxyl radical afforded the phosphorus radical A, which underwent radical addition to the cinnamic acid anion B (generated from the reaction of the starting acid 13 with a base) to provide the radical intermediate C. Finally, decarboxylation of intermediate C followed by single-electron oxidation of the radical anion afforded the expected products 15 (Scheme 8).
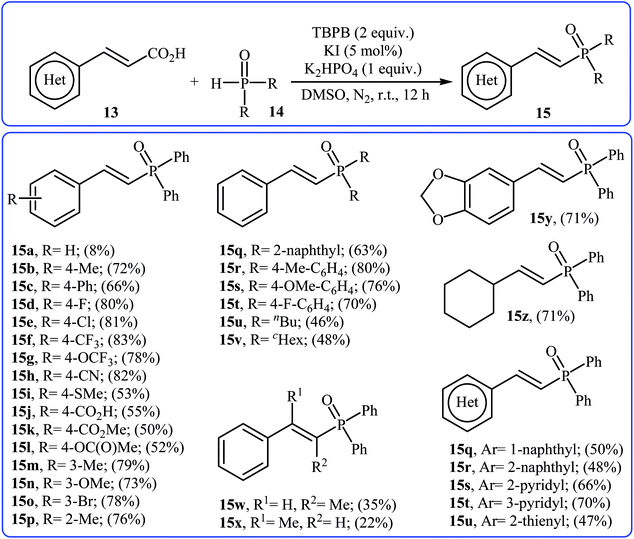 |
| | Scheme 6 Transition-metal-free phosphorylation of alkenyl acid derivatives with P(O)H compounds reported by Liu. | |
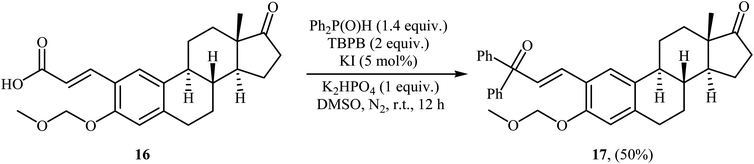 |
| | Scheme 7 Synthesis of estrone derivative 17 through a decarboxylative C–P cross-coupling reaction. | |
 |
| | Scheme 8 Mechanism that accounts for the formation of (E)-1-alkenylphosphorus compounds 15. | |
3. Coupling of alkynyl acids with P(O)H compounds
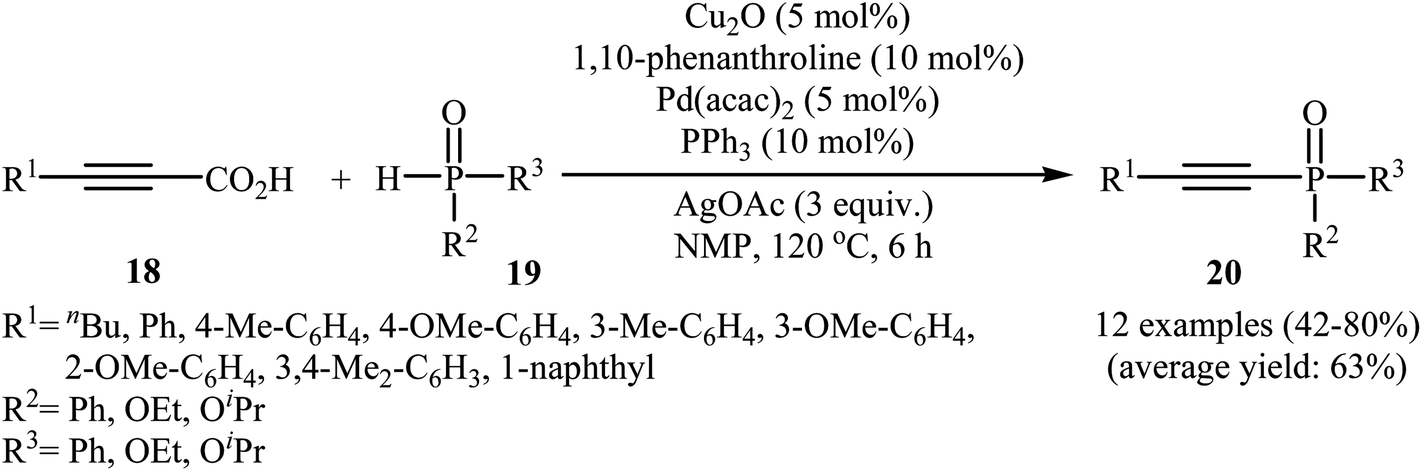
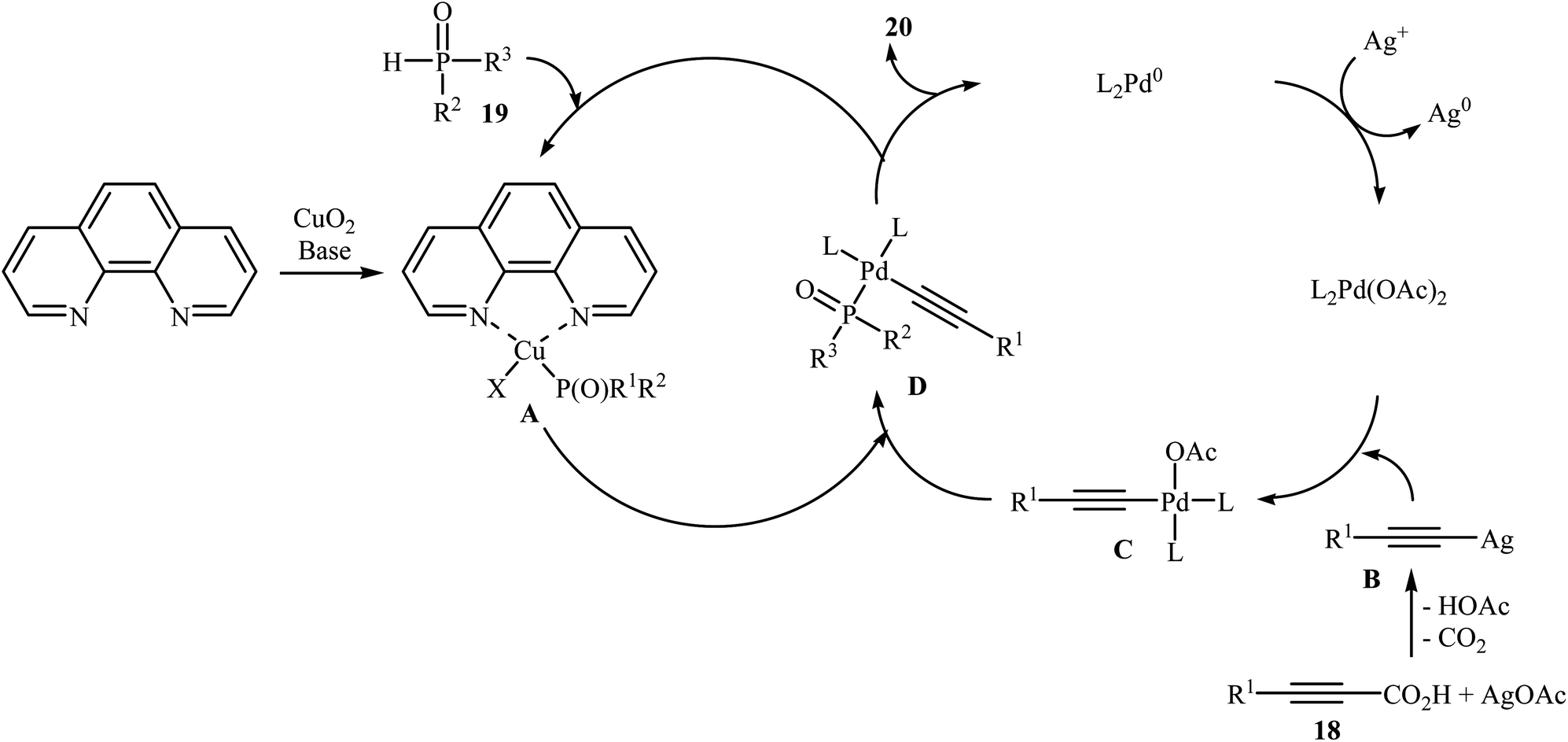
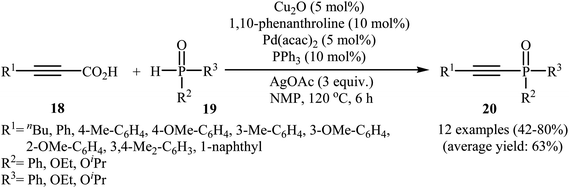
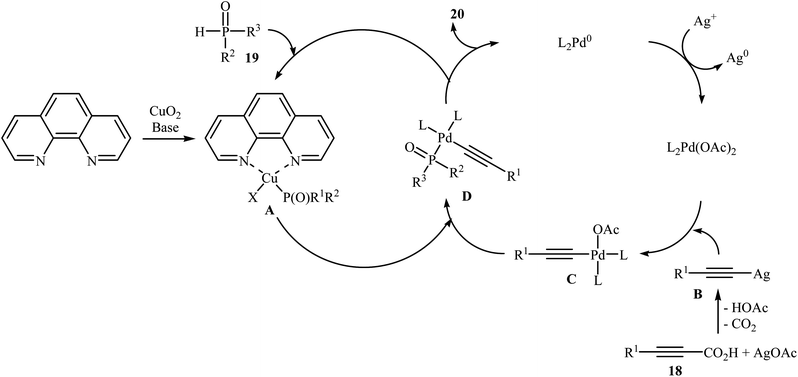
In 2011, Liang and Yang along with their co-workers reported the first example of decarboxylative C–P cross-coupling of alkynyl acids with P(O)H compounds.15 They showed that electron-rich alkynyl acids 18 underwent rapid C–P cross-coupling with various hydrogen phosphoryl compounds 19 in the presence of Cu2O/Pd(acac)2/1,10-phenanthroline/PPh3/AgOAc combination as a catalytic system in NMP at 120 °C. The corresponding alkynylphosphorus compounds 20 were obtained in moderate to high yields (Scheme 9). The reaction is noteworthy since both alkyl and aryl alkynyl carboxylic acids and all three kinds of P(O)–H compounds were tolerated. It was noted that the presence of both copper and palladium catalysts is essential in this reaction; unsatisfactory yields of the expected products were obtained in the absence of any of these catalysts. The proposed mechanism by the authors is illustrated in Scheme 10. First, a CuII–phosphine-complex A was formed via the reaction of P(O)H compound 19 with the copper/phen catalyst. In parallel, decarboxylation of alkynyl acid 18 by silver gave alkynyl–silver species B. The in situ generated PdII complex (by oxidation of Pd0 with silver) transmetallated with the alkynyl–silver species B to generate the alkynyl–palladium intermediate C. The transmetallation of this highly active intermediate C with the CuII-complex A then gave the intermediate D. Finally, reductive elimination of intermediate D resulted in the formation of the expected alkynylphosphorus compounds 20. Later, Gao's group showed that alkynyl phosphonate derivatives were also formed from the corresponding alkynyl acids and diisopropyl phosphonate by a simple process employing Ni(NO3)2·6H2O as the catalyst and Ag2CO3 as the oxidant in DMSO at 100 °C. However, the yields of the desired products were low (36–45% for three examples).16
 |
| | Scheme 9 Cu/Pd-catalyzed decarboxylative C–P cross-coupling of alkynyl acids 18 with P(O)H compounds 19. | |
 |
| | Scheme 10 Mechanism proposed to explain the synthesis of alkynylphosphorus compounds 20. | |
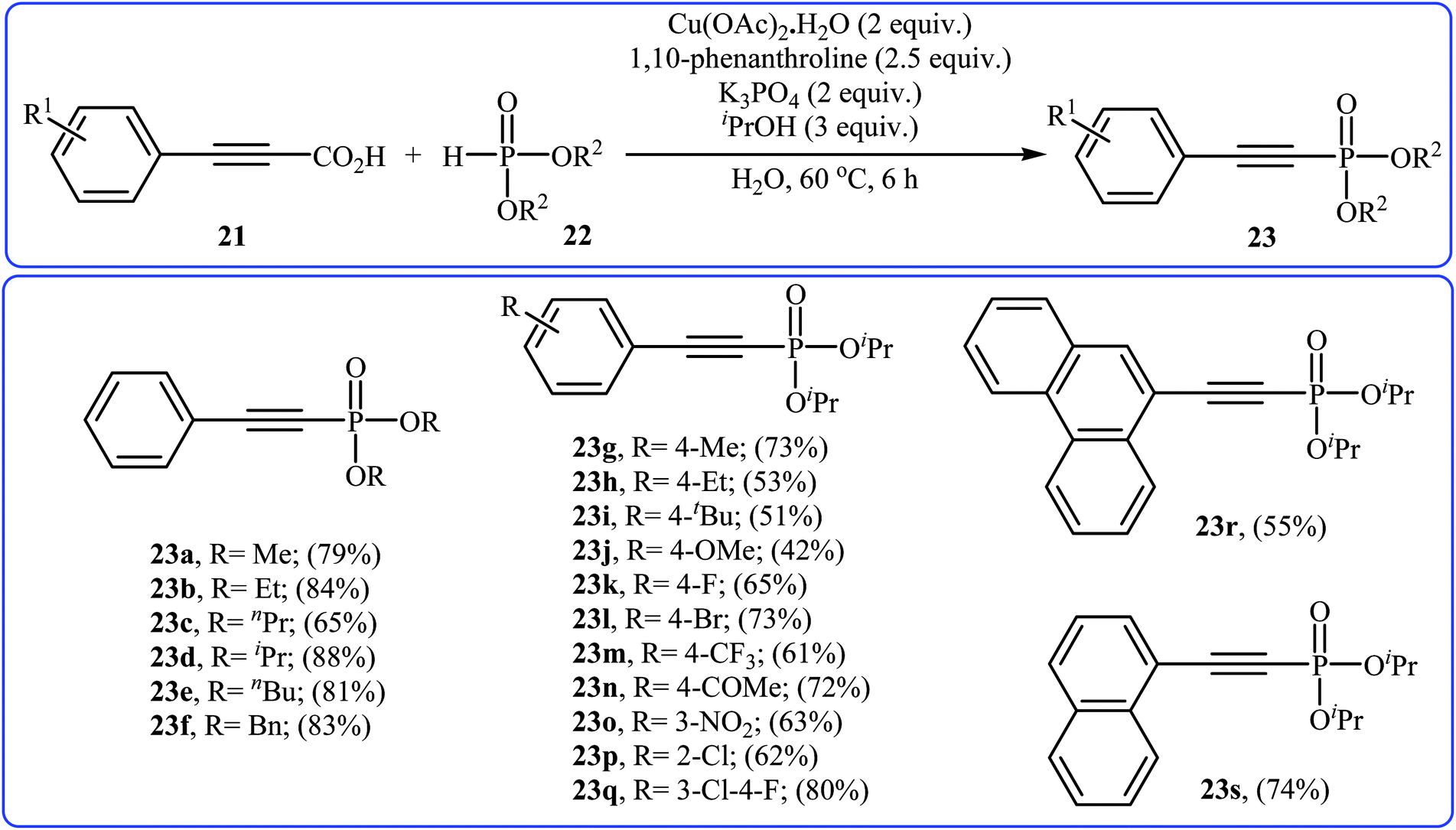
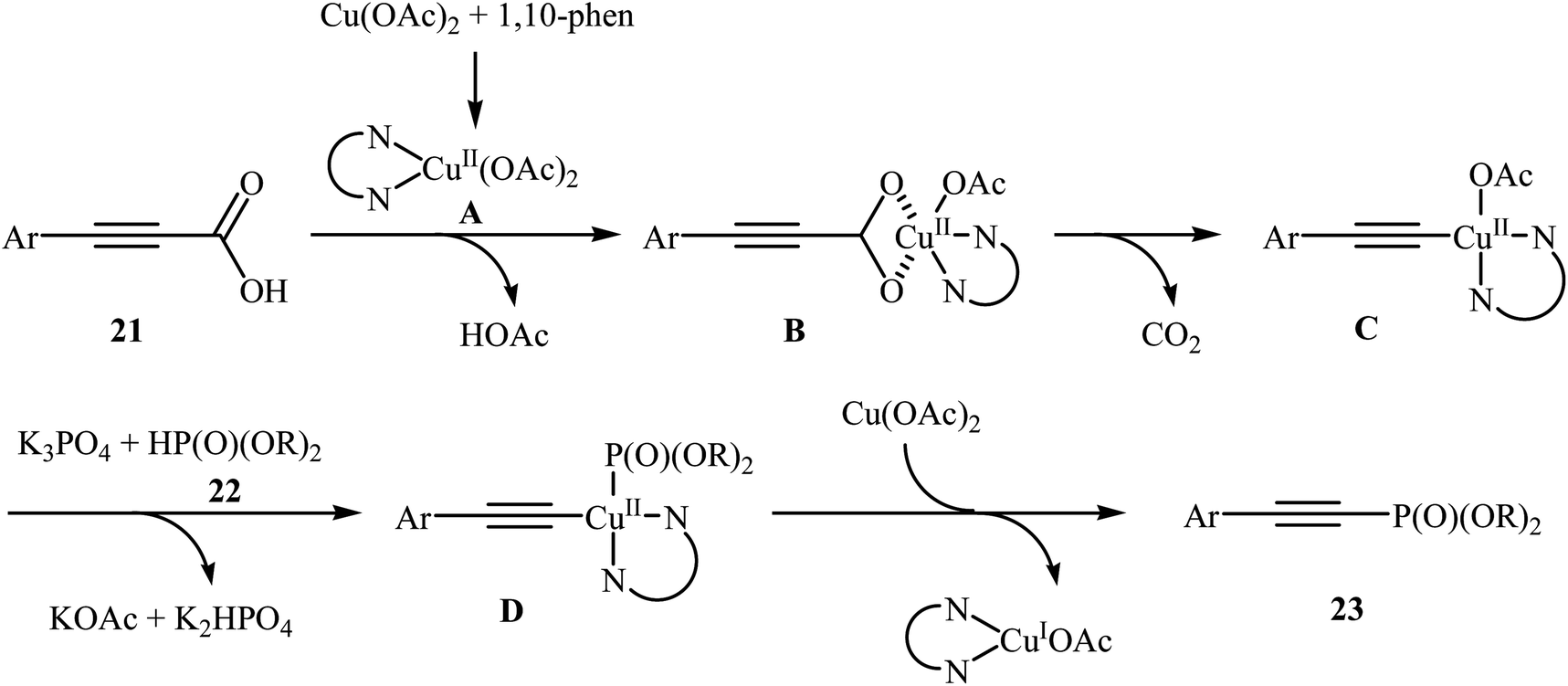
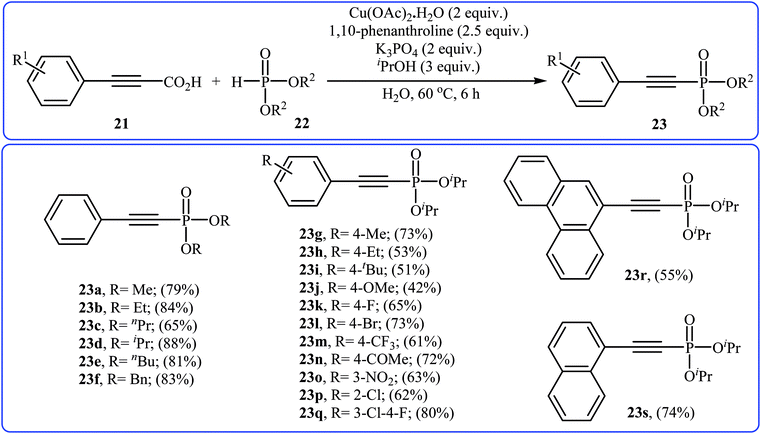
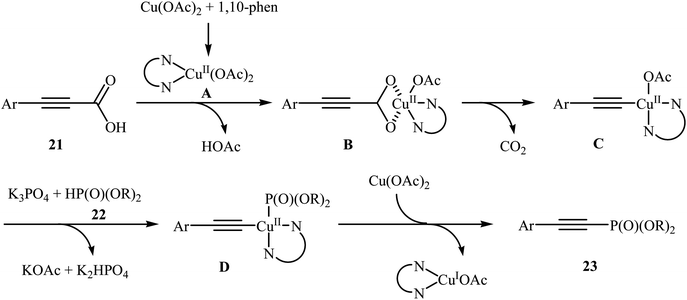
In 2014, Li and co-workers reported that the combination of Cu(OAc)·H2O/1,10-phenanthroline could efficiently catalyze the decarboxylative cross-coupling of alkynyl acids 21 with dialkyl H-phosphonates 22 under relatively mild conditions in the most significant green solvent water (Scheme 11).19 Various aryl alkynyl acids and several dialkyl H-phosphonates were well tolerated in this catalytic system with moderate to high yields (40–88%). However, alkyl alkynyl acids failed to participate in this reaction. Interestingly, other copper salts [e.g., CuCl2, Cu(OTf)2, and CuSO4·5H2O] were also found to promote the reaction albeit with reduced efficiencies. According to the mechanism proposed by the authors (Scheme 12), this reaction consists of the following key steps: (i) initial formation of active copper(II) intermediate A via coordination of 1,10-phenanthroline to Cu(OAc)2; (ii) ligand exchange between intermediate A and alkynyl acid 21 to afford the copper(II) intermediate B; (iii) decarboxylative reaction of intermediate B to give the copper(II) intermediate C; (iv) reaction of the intermediate C with a phosphonate anion (generated from H-phosphonate 22 and K3PO4) to form the copper(II) intermediate D and (v) reductive elimination of intermediate D to give the desired products 23.
 |
| | Scheme 11 Decarboxylative cross-coupling of alkynyl acids 21 with dialkyl H-phosphonates 22 mediated by Cu(OAc)·H2O. | |
 |
| | Scheme 12 Proposed mechanistic pathways for the formation of alkynylphosphorus compounds 23. | |
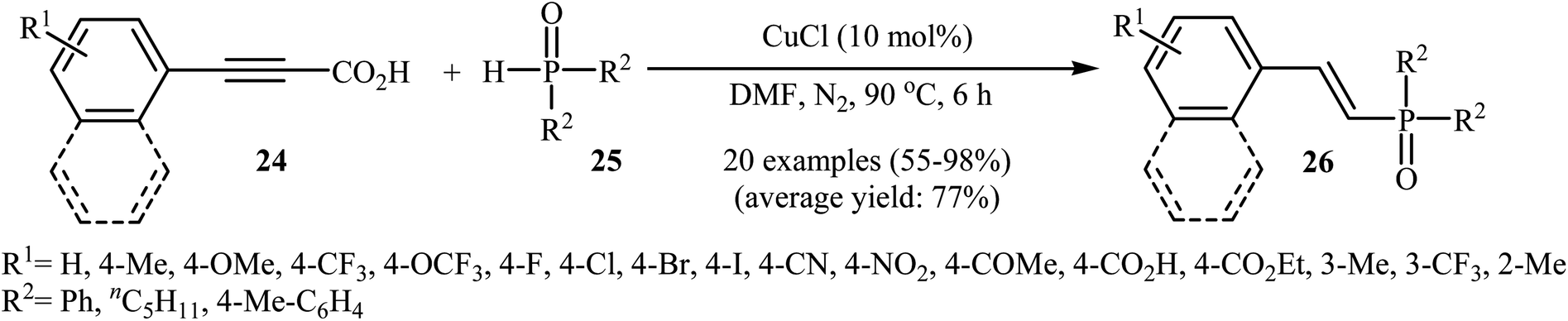
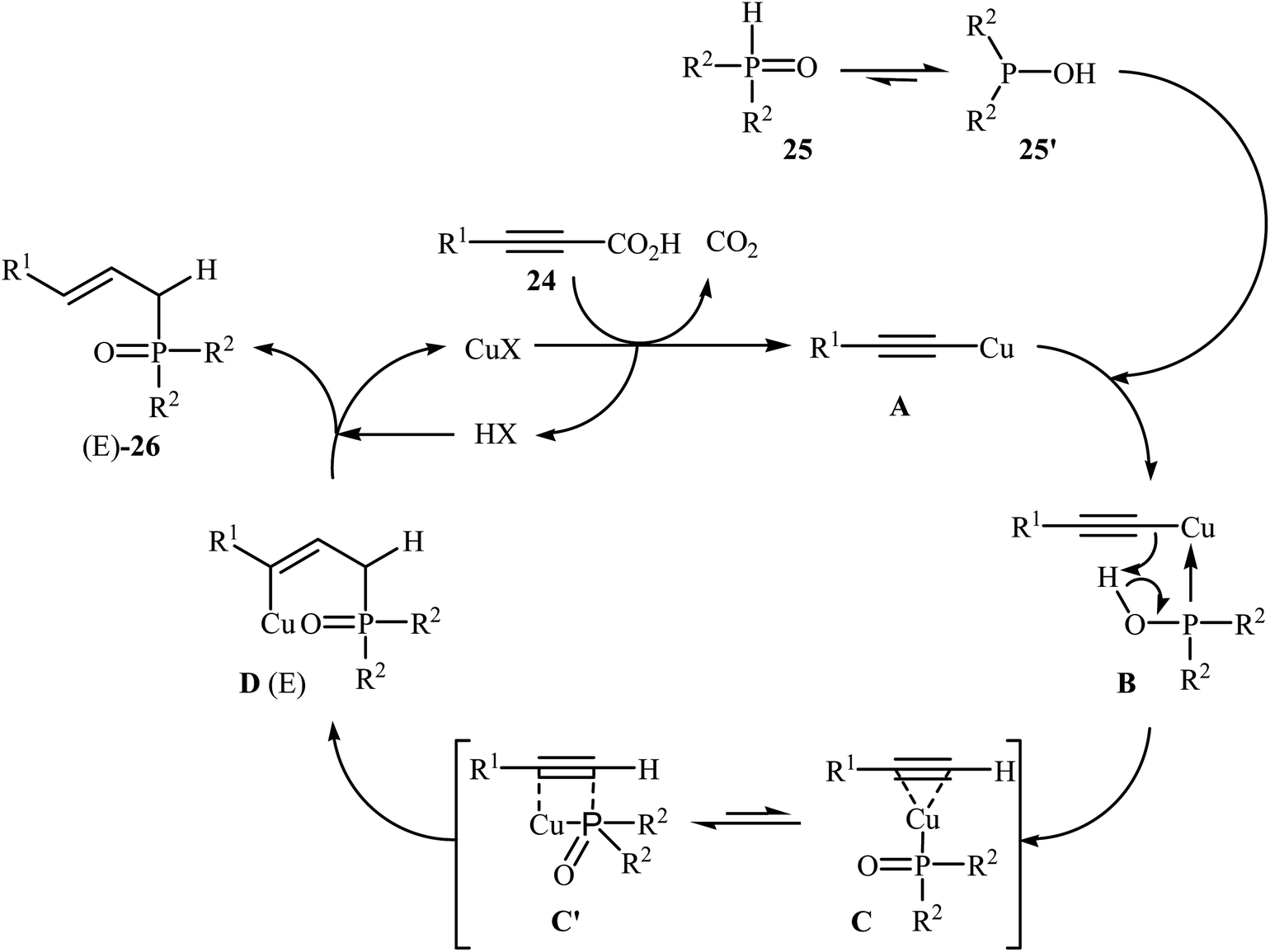
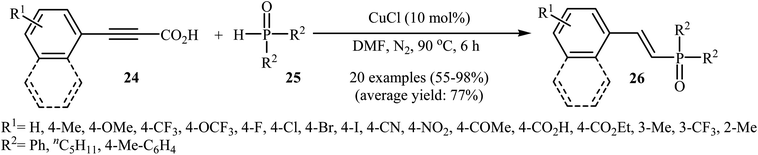
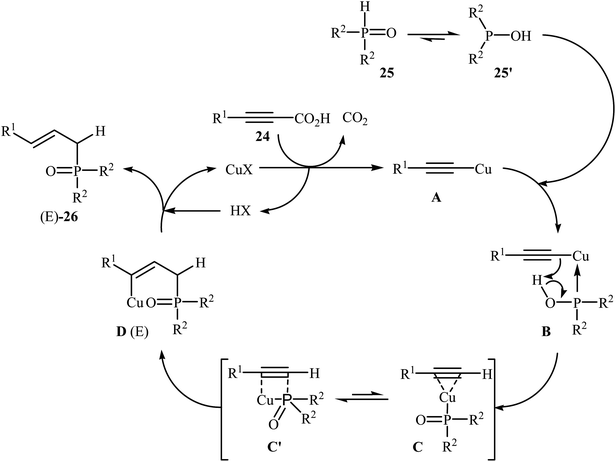
At almost the same time, Hu, Gao, and Zhao demonstrated elegant and innovative copper-catalyzed decarboxylative cross-coupling of alkynyl acids with hydrogen phosphoryl compounds for the formation of alkenylphosphorus compounds.20 Thus, in the presence of a catalytic amount of CuCl (10 mol%) in DMF under nitrogen atmosphere, cross-coupling of aryl alkynyl acids 24 with H-phosphine oxides 25 afforded the corresponding (E)-alkenylphosphine oxide derivatives 26 in moderate to almost quantitative yields (Scheme 13). The reaction could also be conducted successfully on a gram scale. However, the reaction did not give good yields with alkyl and heteroaryl alkynyl acids. The proposed mechanism of the reaction by the authors is depicted in Scheme 14. It consists of the following key steps: (i) initial formation of the alkynyl copper intermediate A via decarboxylation of alkynyl acid 24 with CuCl and the generation of one molecule of CO2 and one molecule of HCl at the same time; (ii) coordination of P(O)H compound 25 (in the form of trivalent phosphine oxide 25′) to the intermediate A to form the intermediate B; (iii) migration of a proton from the oxygen atom to the C sp atom in the intermediate B to give a three-membered-ring transition state C; (iv) conversion of intermediate C (in the form of four-centered transition state C′) to a thermodynamically more stable alkenyl copper intermediate D with the E configuration; and (v) protonolysis of intermediate D with alkynyl acid 24 or HCl to generate the desired product 26.
 |
| | Scheme 13 Cu-catalyzed decarboxylative cross-coupling of alkynyl acids 24 with H-phosphine oxides 25 for the stereoselective synthesis of (E)-alkenylphosphine oxides 26. | |
 |
| | Scheme 14 Mechanistic explanation of the synthesis of (E)-alkenylphosphine oxide derivatives 26. | |
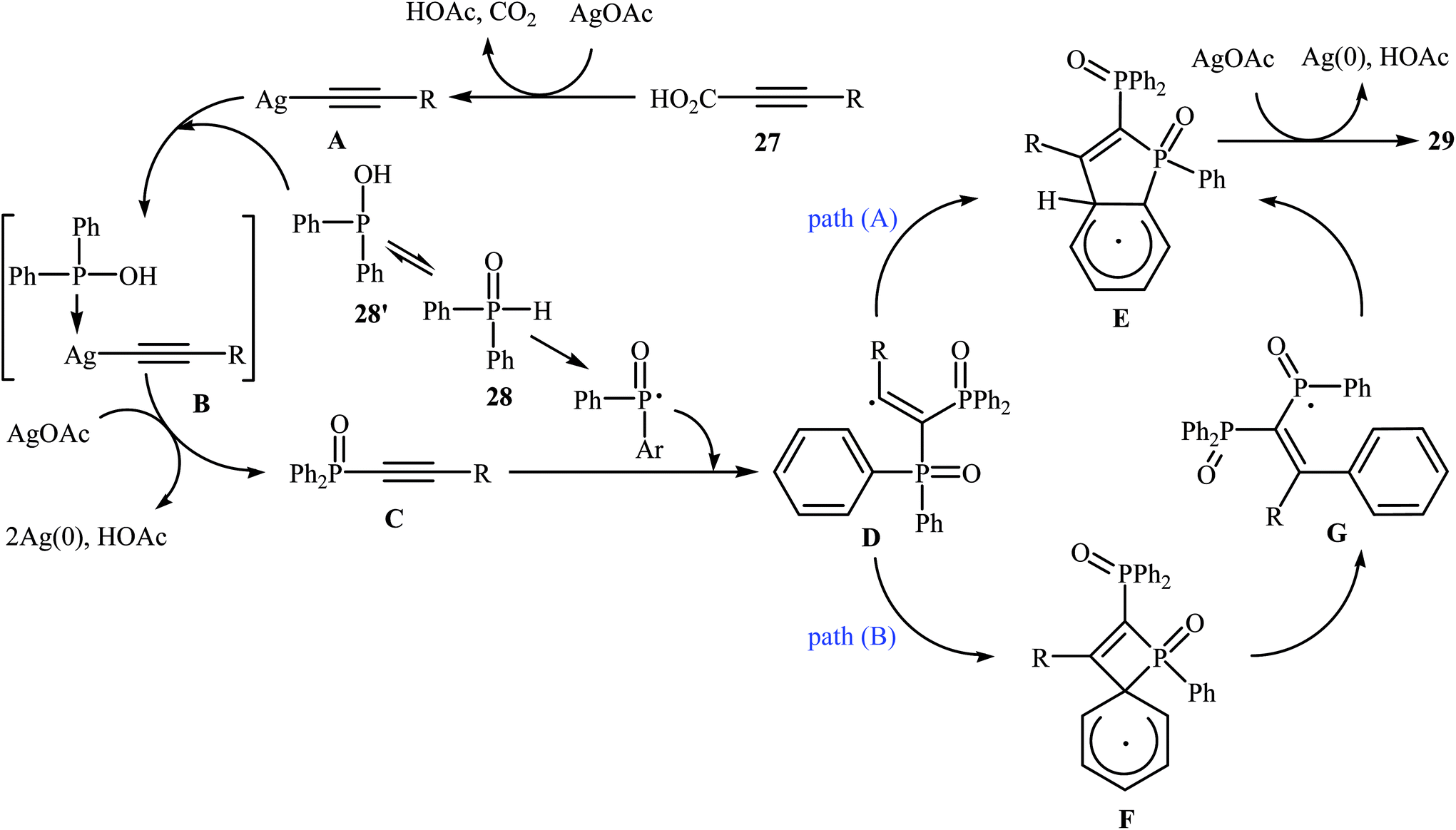
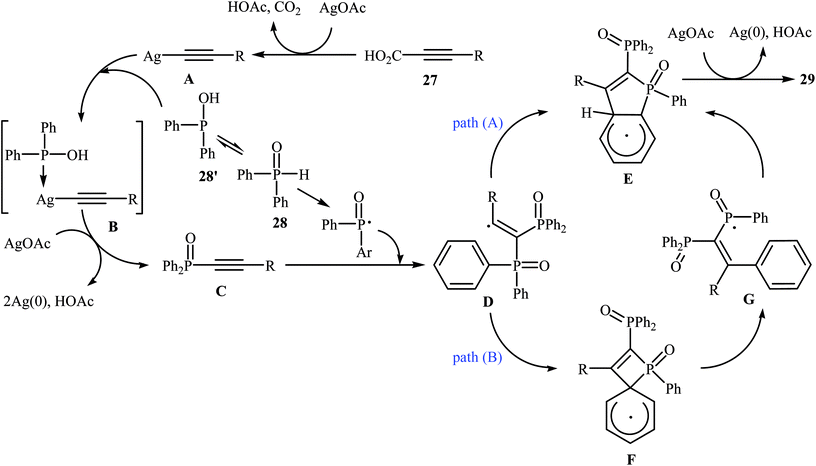
Recently, the same research team reported an interesting silver-mediated cascade decarboxylative C–P cross-coupling and annulation reaction of alkynyl acids 27 with diarylphosphine oxides 28 to form structurally sophisticated 2-phosphinobenzo[b]phosphole oxide frameworks 29 (Scheme 15).21 The reaction was performed under an argon atmosphere using AgOAc as the oxidant in DMF; it tolerated a number of functional groups (e.g., OMe, OCF3, F, Cl, Br, CN, CHO, COMe, CO2H, and CO2Et) and generally provided highly substituted 2-phosphinobenzo[b]phosphole oxides 29 in moderate to high yields. According to mechanistic studies, this reaction proceeded through the formation of an arylethynyl silver intermediate A from the decarboxylative reaction of alkynyl acid 27 and AgOAc under heat, following the coordination of this intermediate A with 28 (in the form of the trivalent phosphine oxide 28′) to give the intermediate B, which then underwent silver-mediated oxidative cross-coupling to produce the critical intermediate C via two single-electron oxidations. Subsequently, the addition of the diphenylphosphonyl radical (generated from 28) to the intermediate C afforded the alkenyl radical intermediate D, which underwent intramolecular addition to the phenyl ring at the ortho position of the phosphorus atom to afford the intermediate E. Then, the oxidation of this intermediate by AgOAc along with the removal of a proton led to the final product 29. In another possibility, the alkenyl radical D attacked the phenyl ring at the carbon position directly attached to the phosphorus atom to form the intermediate F, followed by C–P bond cleavage to give the phosphorus radical G. Next, the addition of G to the phenyl ring generated the intermediate E, which after oxidation, afforded 2-phosphinobenzo[b]phosphole oxide frameworks 29 (Scheme 16).
 |
| | Scheme 15 Ag-mediated cascade reaction of arylpropiolic acids 27 with diarylphosphine oxides 28 developed by Gao. | |
 |
| | Scheme 16 Mechanistic proposal for the reaction in Scheme 14. | |
4. Coupling of benzoic acids with P(O)H compounds
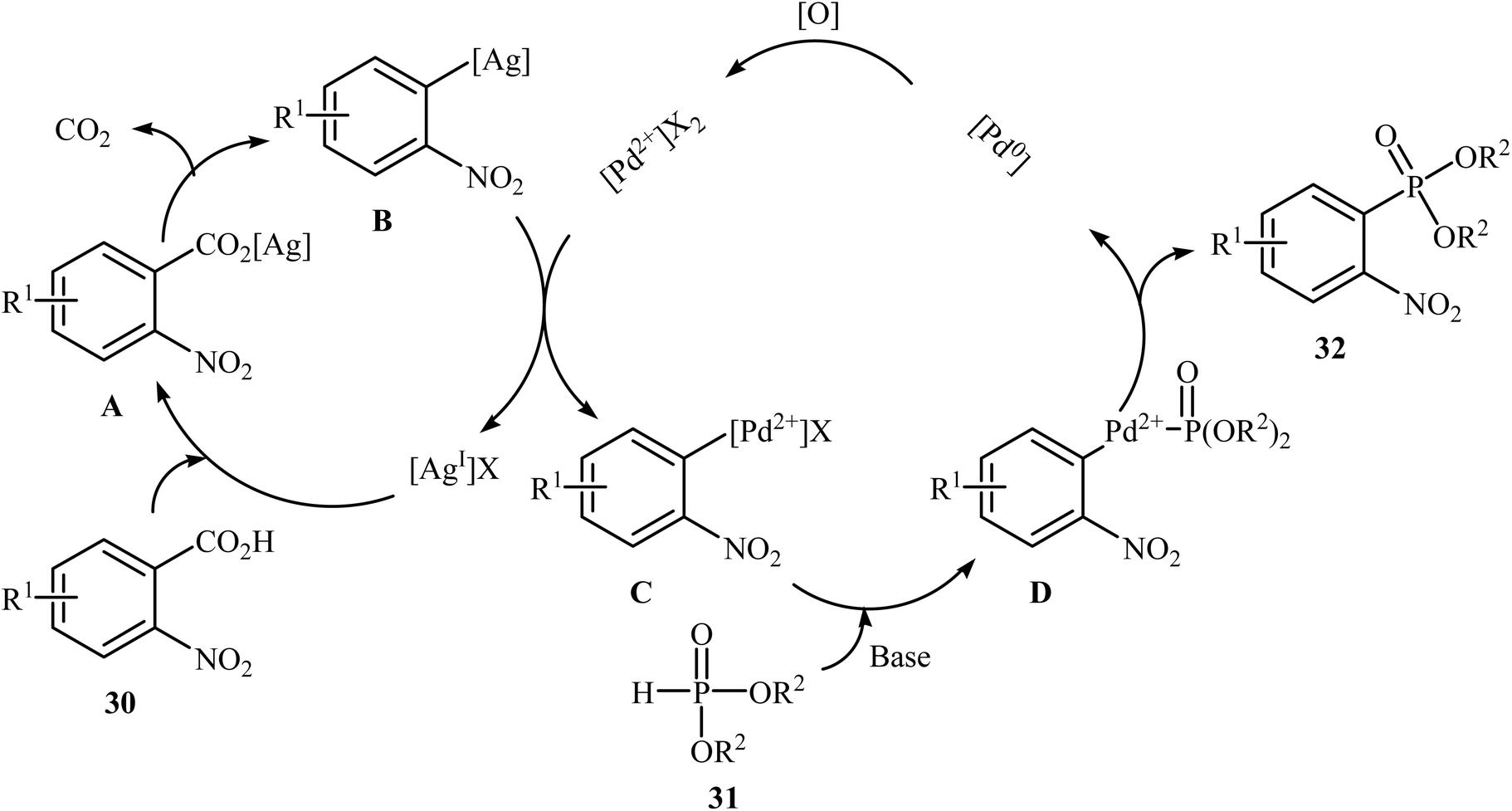
Decarboxylative C–P cross-coupling of benzoic acids with P(O)H compounds has been scarcely studied; in fact, only one example of such a reaction has been reported so far. In this report, Xiao and co-workers showed that the treatment of ortho-nitrobenzoic acids 30 with H-phosphonates 31 in the presence of a bimetallic palladium/silver catalytic system [Pd(OAc)2/Ag2CO3] in DMF resulted in the formation of functionalized aryl phosphonates 32 in low to good yields (Scheme 17).22 However, meta- and para-substituted benzoic acids were inert to this coupling reaction. The results showed that the reaction was not very efficient and general since it had a number of limitations and exceptions. Thus, the development of a novel and efficient catalytic system that improves the efficiency of this interesting coupling reaction is highly desirable. According to the mechanism proposed by the authors (Scheme 18), this reaction starts with the reaction of silver salt with benzoic acid 30, leading to the formation of the silver benzoate species A. Subsequently, decarboxylation of this intermediate yields an aryl-Ag species B; its transmetalation with Pd2+X2 forms an aryl-Pd intermediate C along with regeneration of the Ag+ salt. Next, ligand exchange of intermediate C with H-phosphonate 31 generates the Pd2+ complex D. Finally, reductive elimination of D affords the observed phosphonation product 32 and Pd0. Pd0 is then oxidized to Pd2+ to close the catalytic cycle.
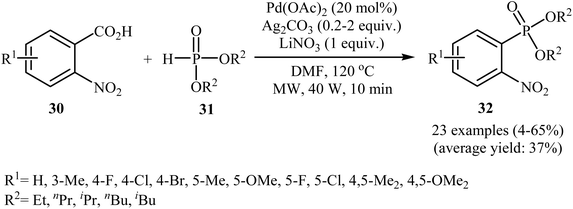 |
| | Scheme 17 Pd/Ag-catalyzed decarboxylative C–P coupling of o-nitrobenzoic acids 30 with H-phosphonates 31. | |
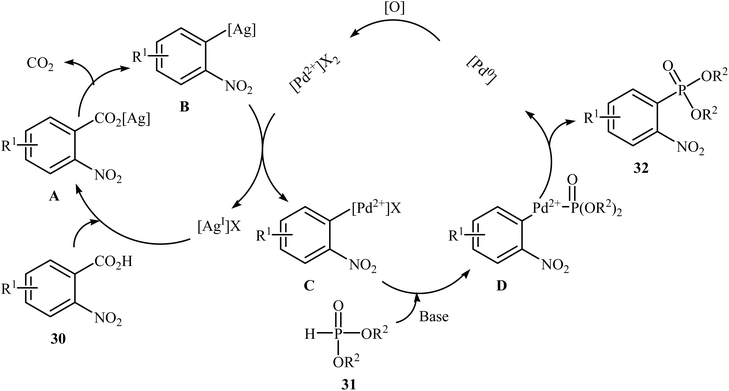 |
| | Scheme 18 Plausible mechanism for the formation of aryl phosphonates 32. | |
5. Coupling of amino acids with P(O)H compounds
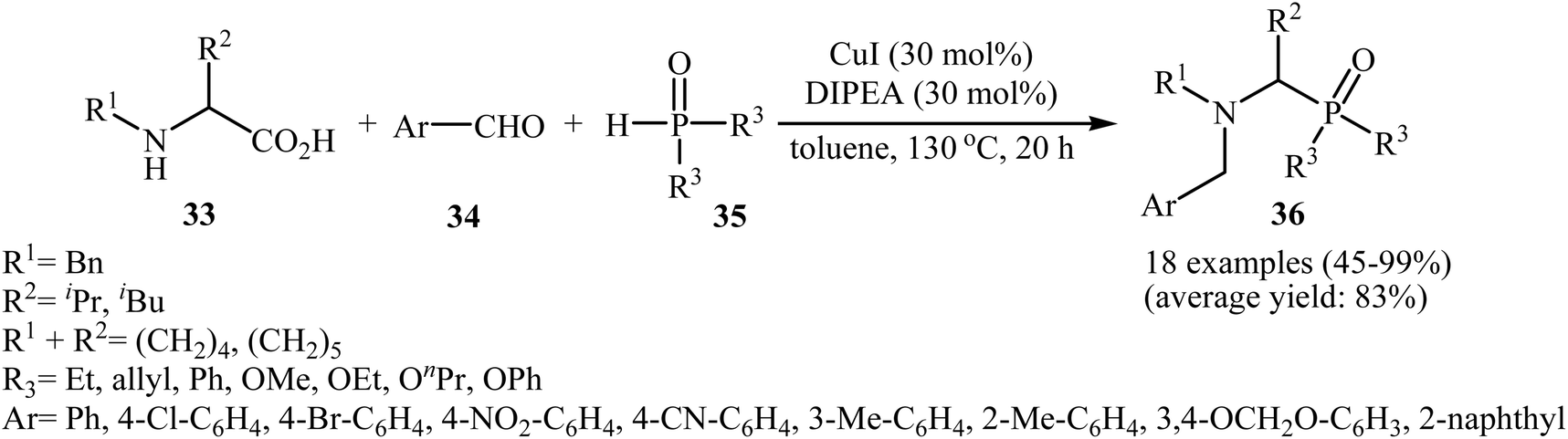
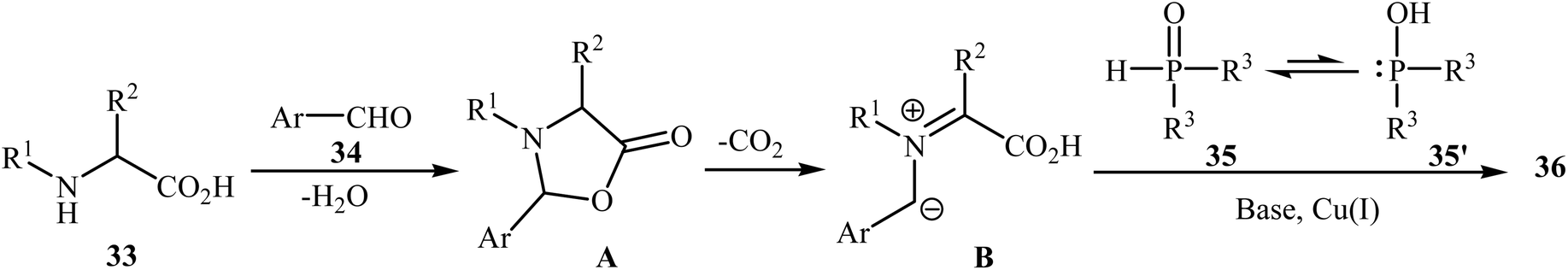
After the pioneering study by Hu and co-workers on decarboxylative coupling of N-benzyl-protected proline with diphenylphosphine oxide employing CuCl as the catalyst,15 the first general report on the synthesis of biologically important tertiary amino phosphorus compounds 36 through three-component decarboxylative coupling of natural α-amino acids 33 with aromatic aldehydes 34 and P(O)H compounds 35 was published in 2011 by Yang et al.23 They carefully studied the effects of reaction variables such as catalyst, additive, solvent, and temperature and found that performing the reaction in toluene at 130 °C using CuI/DIPEA (N,N-diisopropylethylamine) combination as the catalytic system was the optimum reaction condition. Various α-amino acids, H-phosphonates, secondary phosphine oxides, and both electron-poor and electron-rich aldehydes reacted well under the optimized reaction conditions to produce the corresponding tertiary amino phosphorus compounds 36 in moderate to quantitative yields (Scheme 19). The proposed mechanism by the authors is depicted in Scheme 20. First, an oxazolidin-5-one intermediate A was formed via condensation of an α-amino acid 33 with an aldehyde 34; this intermediate underwent decarboxylation to generate the azomethine ylide B. Finally, the addition of P(O)H compound 35 (in the form of the trivalent phosphine oxide 3′) to this dipole B gave the expected product 36.
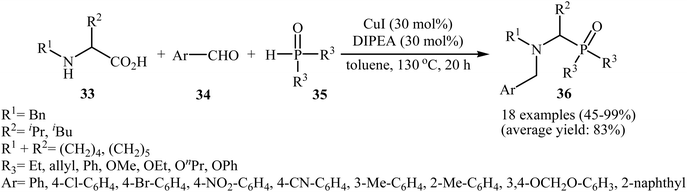 |
| | Scheme 19 Cu-catalyzed three-component decarboxylative coupling of natural α-amino acids 33 with aromatic aldehydes 34 and P(O)H compounds 35. | |
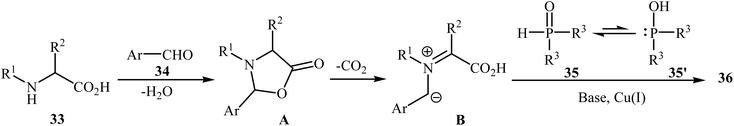 |
| | Scheme 20 Mechanism that accounts for the formation of tertiary amino phosphorus compounds 36. | |
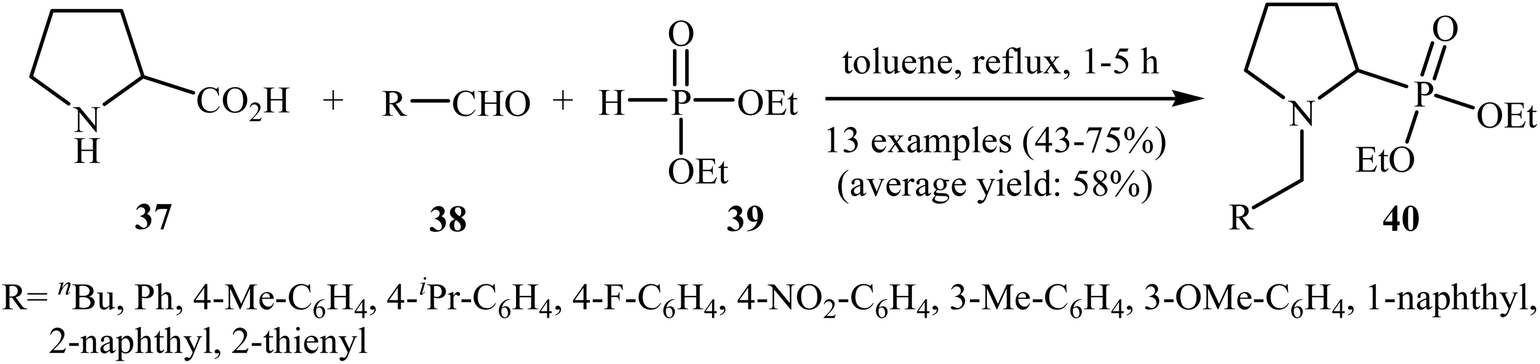
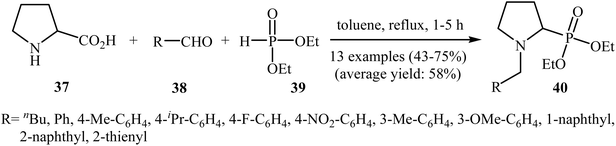
In a closely related investigation, Kaboudin's group showed that many N-benzyl-2-(di(ethyl)phosphoryl)pyrrolidine derivatives 40 were successfully formed from the reaction of proline 37 with aldehydes 38 and diethyl phosphite 39 in the absence of any transition metal catalyst by simple heating in toluene (Scheme 21).24 Various aromatic, heteroaromatic, and aliphatic aldehydes were used to establish the general applicability of the method. The results demonstrated that the aromatic aldehydes afforded better yields compared to the aliphatic aldehydes. However, ortho-substituted benzaldehydes failed to enter this reaction.
 |
| | Scheme 21 Kaboudin's synthesis of N-benzyl-2-(di(ethyl)phosphoryl)pyrrolidine derivatives 40. | |
6. Oxidative decarboxylative coupling of carboxylic acids with P(O)H compounds
6.1. Alkynyl acids
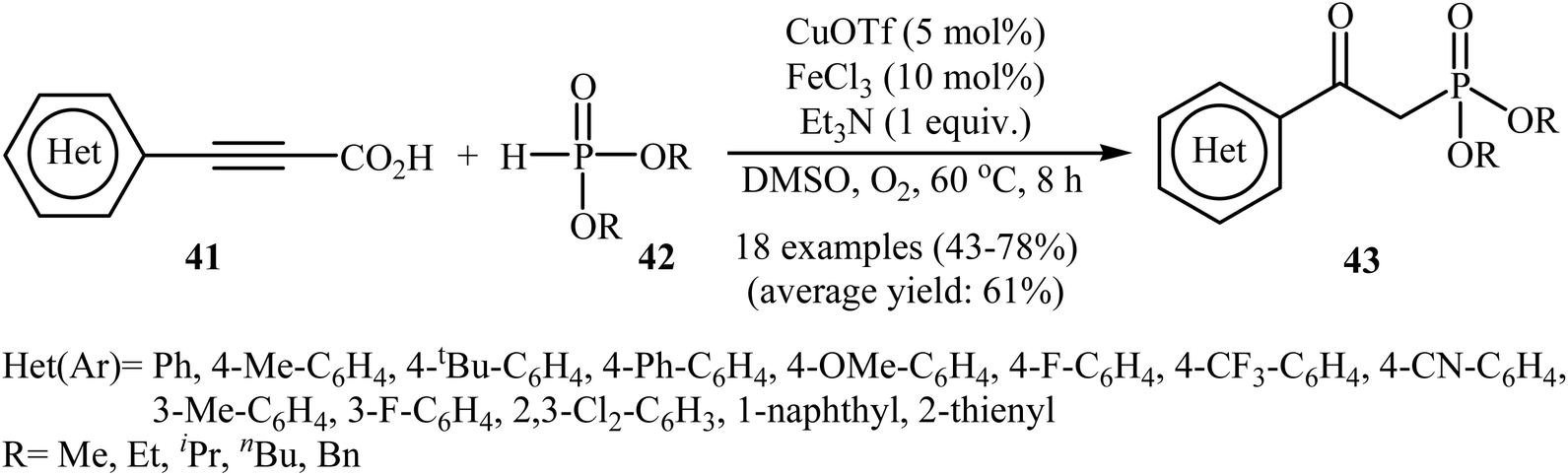
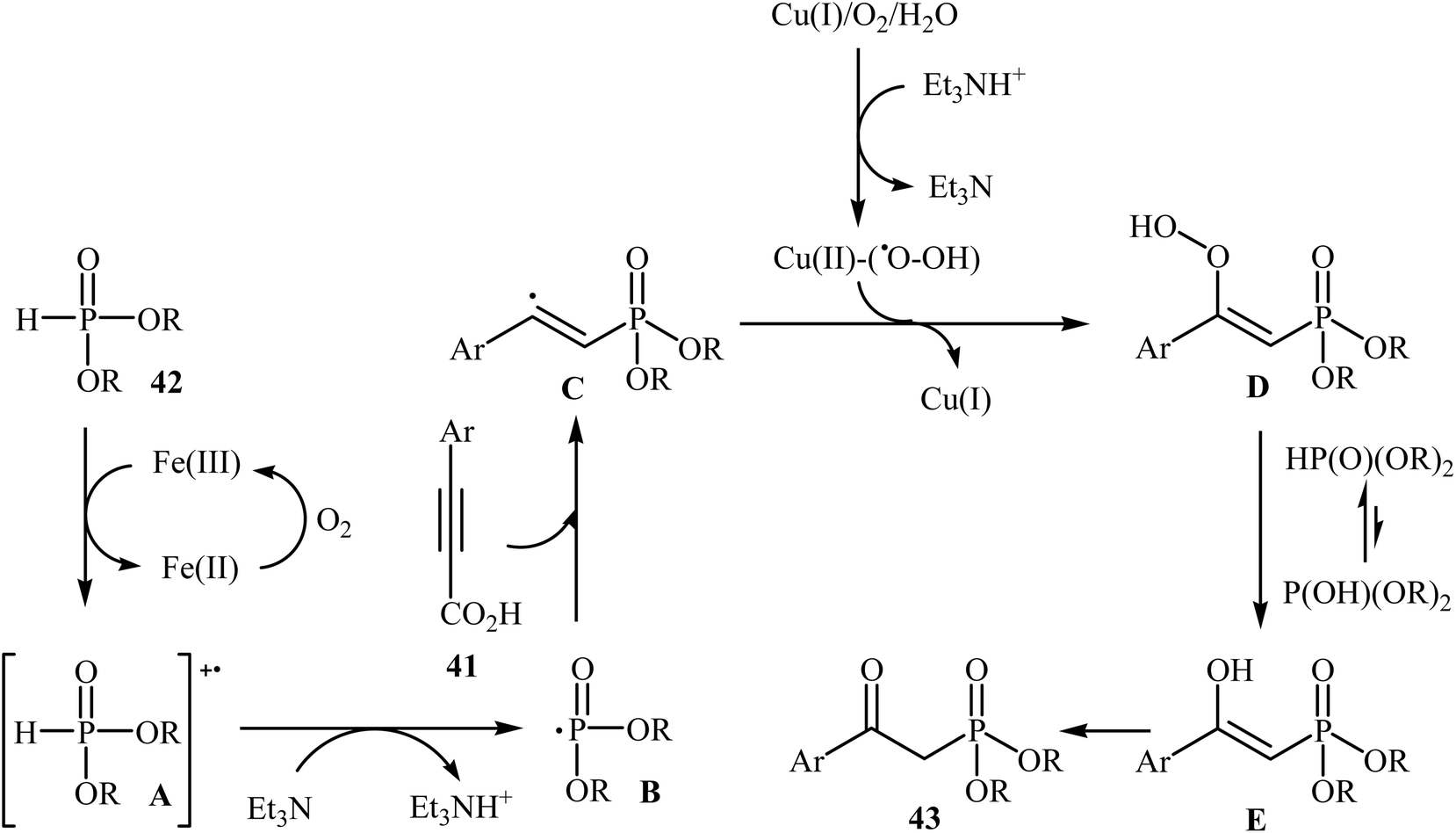
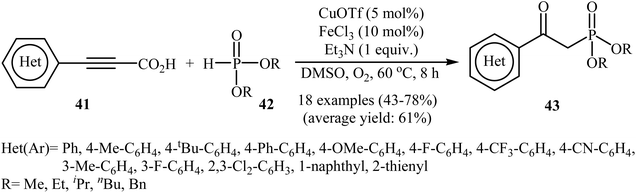
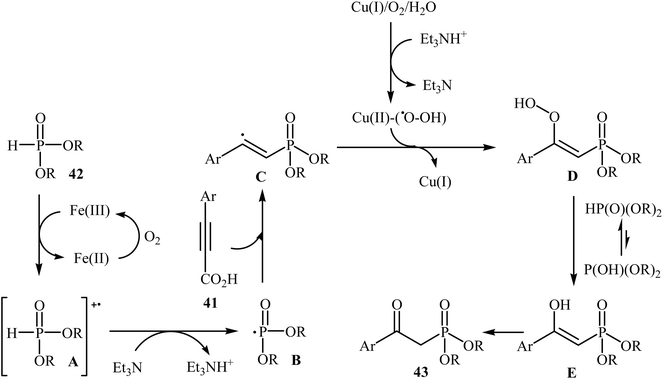
The possibility of metal-catalyzed oxidative decarboxylative coupling of alkynyl acids with P(O)H compounds was first demonstrated by Song and his research team in 2015,25 and they synthesized a series of β-ketophosphonates 43 from the reaction of propiolate acids 41 with H-phosphonates 42 in the presence of only 5 mol% of CuOTf as the catalyst, 10 mol% of FeCl3 as the co-catalyst, and 1.0 equiv. of Et3N as the base in DMSO under oxygen atmosphere (balloon). This Cu/Fe-cocatalyzed reaction tolerated both aryl as well as heteroaryl alkynyl acids and various symmetrical H-phosphonates and provided β-ketophosphonates 43 in fair to good yields (Scheme 22). However, aliphatic alkynyl acids did not function well under these reaction conditions. It should be mentioned that this catalytic system was also successfully applied in the oxyphosphorylation of terminal alkynes with P(O)H compounds. The mechanistic pathway of this oxidative decarboxylative coupling reaction is shown in Scheme 23, and it involved the initial formation of the dialkyl phosphonate cation radical A through the transfer of a single electron from iron(II) species to H-phosphonate 42 in the presence of molecular oxygen. Next, deprotonation of this cation radical A in the presence of a base gave the dialkyl phosphonyl radical B, which then attacked the triple bond of the alkynyl acid 41 to produce the radical intermediate C. Subsequently, the reaction of this radical with Cu(II)–(˙OOH) species (generated from Cu(I) under dioxygen atmosphere) gave the hydroperoxide species D. Finally, the removal of oxygen from D afforded the observed β-ketophosphonates 43 through the intermediate E.
 |
| | Scheme 22 Oxidative decarboxylative coupling of alkynyl acids 41 with P(O)H compounds 42 developed by Song. | |
 |
| | Scheme 23 Mechanistic proposal for the reaction in Scheme 22. | |
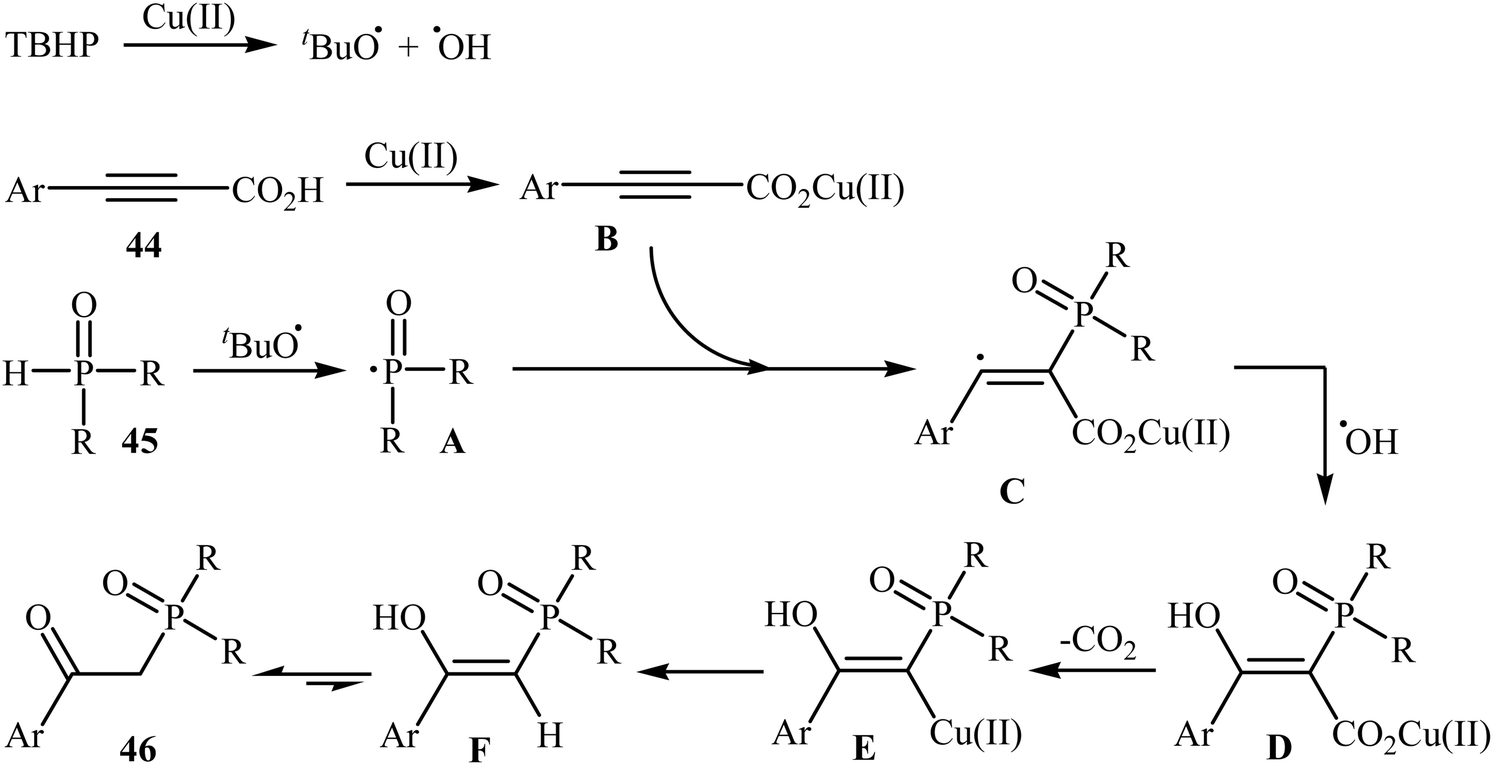
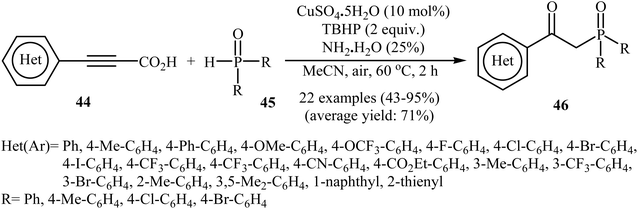
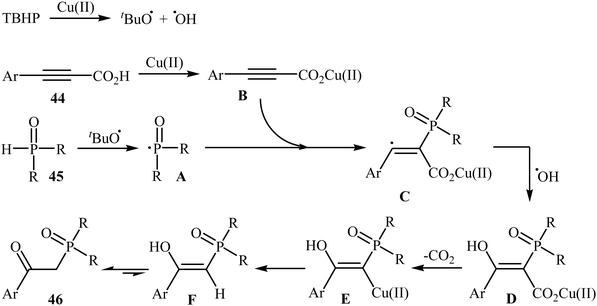
Concurrently, Zhang and co-workers presented efficient copper-catalyzed oxidative decarboxylative coupling of alkynyl acids 44 with H-phosphine oxides 45 to form β-ketophosphine oxide derivatives 46.26 The results of this investigation revealed that the optimum condition for this reaction was the combination of CuSO4·5H2O (10 mol%), TBHP (2 equiv.), and NH3·H2O (25%) as the catalytic system using acetonitrile as the solvent at 60 °C. Under optimized conditions, the reaction tolerated various symmetrical and unsymmetrical H-phosphine oxides and a range of aromatic and heteroaromatic alkynyl acids and gave the expected products in moderate to excellent yields (Scheme 24). However, aliphatic alkynyl acids failed to enter the reaction. Mechanistically, this reaction was proposed to be initiated by the formation of tert-butoxy and hydroxyl radicals via decomposition of TBPB in the presence of Cu(II). Then, the abstraction of a hydrogen atom from H-phosphine oxide 45 by a tert-butoxyl radical afforded the phosphorus radical A. Meanwhile, the reaction of alkynyl acid 44 with Cu(II) generated a salt of cupric carboxylate B. Subsequently, the addition of the phosphorus radical A to the α-position of the triple bond of the cupric carboxylate B gave the radical intermediate C, which was ultimately trapped by a hydroxyl radical to provide the intermediate D. The formation of the alkenyl copper intermediate E occurred next, followed by its protonolysis to give the intermediate F. Finally, isomerization of F led to the formation of the desired product 46 (Scheme 25).
 |
| | Scheme 24 CuSO4·5H2O-catalyzed oxidative decarboxylative coupling of alkynyl acids 44 with H-phosphine oxides 45 into β-ketophosphine oxides 46. | |
 |
| | Scheme 25 Plausible mechanism for the formation of β-ketophosphine oxides 46. | |
The synthesis of β-oxophosphine oxides in moderate to good yields (40–73%) was also reported by Zeng and co-workers through the reaction of corresponding alkynyl acids with H-phosphine oxides using Ag2CO3 as an efficient catalyst in ethanol at room temperature and air conditions.27 Other silver salts such as Ag2O, AgOAc, and AgOTf were also found to promote this reaction, albeit with lower yields. Under optimized conditions, both dialkyl and diaryl phosphine oxides were well tolerated. The reaction could also be conducted successfully on a gram scale.
6.2. Alkenyl acids
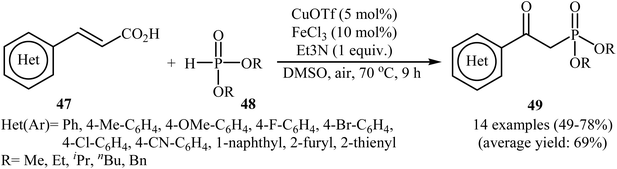
In 2015, an innovative and efficient synthetic protocol was reported by Zhou and co-workers for the oxidative decarboxylative coupling of various alkenyl acids 47 with H-phosphonates 48 to afford a diverse range of β-ketophosphonates 49 using CuOTf/FeCl3/Et3N combination as the catalytic system in DMSO (Scheme 26).28 The reactions were performed under air and tolerated a series of sensitive functional groups including fluoro, chloro, bromo, methoxy, and cyano functionalities; the target products were obtained in moderate to good yields (49–79%). The authors showed that both H-phosphinate oxides and phosphine oxides were also compatible in this transformation and gave desired products in moderate yields. Moreover, they successfully extended this chemistry to the synthesis of a variety of functionalized β-ketophosphonates via a domino Knoevenagel-decarboxylation–oxyphosphorylation sequence from aromatic aldehydes and H-phosphonates.
 |
| | Scheme 26 Cu/Fe-catalyzed oxidative decarboxylative coupling of various alkenyl acids 47 with H-phosphonates 48. | |
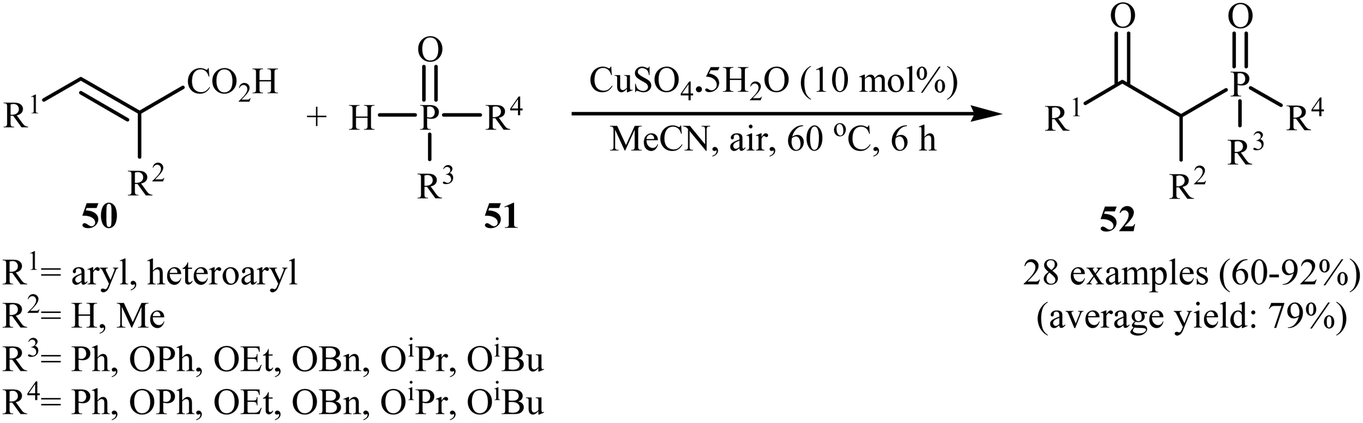
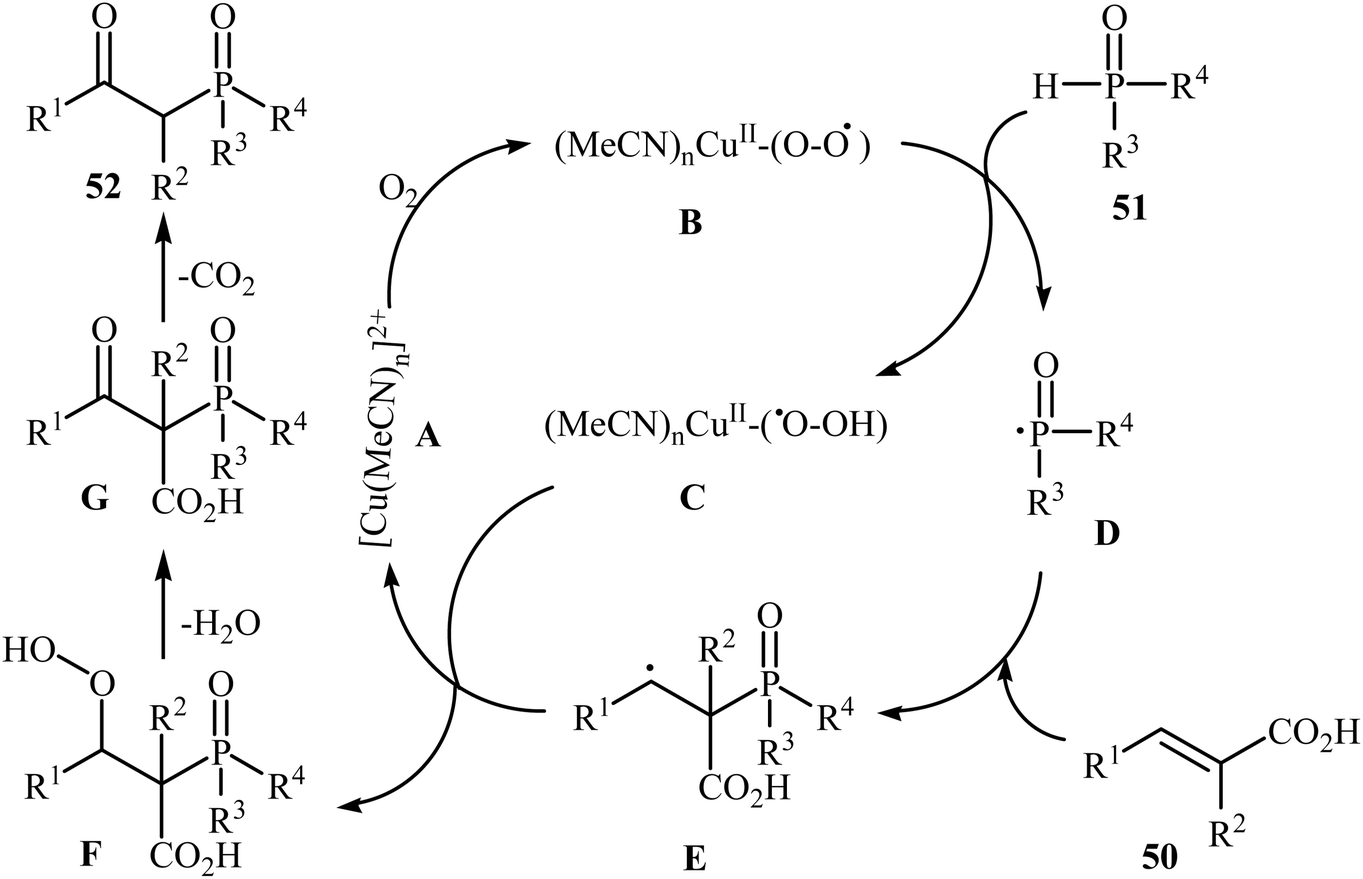
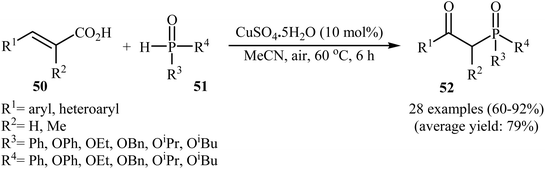
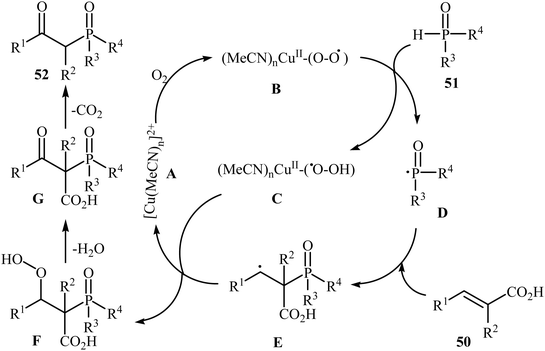
The synthesis of β-keto phosphorus compounds 52 in good to excellent yields (up to 92%) was also reported by Chen's research team through the oxidative decarboxylative coupling reaction of cinnamic acids 50 with P(O)H compounds 51 by employing CuSO4·5H2O as an efficient catalyst and air oxygen as an environmentally benign oxidant (Scheme 27).29 Solvent has a dramatic role in this reaction; among the common solvents (e.g., THF, DCE, DMF, DMSO, MeCN, EtOH, and 1,4-dioxane), acetonitrile was the most effective solvent. Interestingly, the other solvents were completely ineffective. The authors proposed a mechanism for this decarboxylation–oxyphosphorylation reaction in which an acetonitrile-soluble complex [Cu(MeCN)n]2+ A, generated from the copper(II) ion and MeCN, underwent a reaction with air oxygen to form a copper-active-oxygen complex B, which then quickly attacked the P(O)H compound 51 to produce the hydroperoxide complex C as well as the phosphonyl radical D. Subsequently, the addition of radical D to cinnamic acid 50 afforded the radical E, which in the presence of the hydroperoxide complex C yielded a hydroperoxide intermediate F. Next, dehydration of this intermediate F gave the intermediate G. Finally, energetically favorable decarboxylation of intermediate G afforded the final products 52 (Scheme 28).
 |
| | Scheme 27 Chen's synthesis of β-keto phosphorus compounds 52. | |
 |
| | Scheme 28 Plausible mechanism for the formation of β-keto phosphorus compounds 52. | |
7. Summary and conclusion
Carbon–phosphorus bond construction represents a key step in the synthesis of various medicines, insecticides, ligands, and modified nucleosides and nucleotides. In particular, P(O)–C bond formation reactions have attracted much attention owing to their presence in a number of commercialized drugs such as fosinopril, adefovir, alendronic acid, cidofovir, and brincidofovir. General methods to construct the P(O)–C bond involve transition-metal-catalyzed cross-coupling of organic halides, triflates, or tosylates with P(O)H compounds or the nucleophilic substitution reaction of organometallic reagents with electrophilic P-reagents. Despite significant progress, these methods still suffer from some limitations such as the use of metal reagents, expensive and unstable triflates, environmentally toxic organic halides, and poor tolerance of functional groups. In the last seven years, the decarboxylative C–P cross-coupling reaction between carboxylic acids and P(O)H compounds has emerged as one of the most reliable and robust tools for the construction of P(O)–C bonds.
As illustrated, all three kinds of P(O)–H compounds (H-phosphinates, H-phosphonates, and secondary phosphine oxides) were applicable to this novel coupling reaction. Interestingly, most of the reactions covered in this review could be scaled up to provide multigram quantities of the desired coupling products. These results showed the potential applications of these reactions in industry. Despite all these successes, many challenges still remain to be overcome: (i) the number of reported examples in this interesting research arena is limited. There is still a further need to study the scope and limitations of this approach for the formation of P(O)–C bonds; (ii) most of the reported decarboxylative C–P cross-coupling reactions are performed at high temperatures; thus, the exploration of milder processes is highly desirable, and (iii) other reactions such as coupling of allylic and benzylic carboxylic acids with various P(O)–H compounds should be explored. We conclude this review by hoping that it will stimulate researchers to carry out further research in this domain.
Conflicts of interest
There are no conflicts to declare.
References
- S. C. Fields, Tetrahedron, 1999, 55, 12237–12273 CrossRef.
-
(a) B. Becker and T. Gage, AMA Arch. Ophthalmol., 1960, 63, 102–107 CrossRef;
(b) J. R. Kouvaris, V. E. Kouloulias and L. J. Vlahos, Oncologist, 2007, 12, 738–747 CrossRef PubMed;
(c) M. Aapro, A. Carides, B. L. Rapoport, H.-J. Schmoll, L. Zhang and D. Warr, Oncologist, 2015, 20, 450–458 CrossRef PubMed;
(d) J. Dunning, S. B. Kennedy, A. Antierens, J. Whitehead, I. Ciglenecki, G. Carson, R. Kanapathipillai, L. Castle, R. Howell-Jones and R. Pardinaz-Solis, PLoS One, 2016, 11, e0162199 CrossRef PubMed.
- M. Kazemi, A. M. Tahmasbi, R. Valizadeh, A. A. Naserian and A. Soni, Agricultural Science Research Journal, 2012, 2, 512–522 Search PubMed.
-
(a) J. Ansell and M. Wills, Chem. Soc. Rev., 2002, 31, 259–268 RSC;
(b) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3070 CrossRef PubMed;
(c) S. Castillón, C. Claver and Y. Díaz, Chem. Soc. Rev., 2005, 34, 702–713 RSC;
(d) L. Kollár and G. Keglevich, Chem. Rev., 2010, 110, 4257–4302 CrossRef PubMed.
- S. Demkowicz, J. Rachon, M. Daśko and W. Kozak, RSC Adv., 2016, 6, 7101–7112 RSC.
- P. Todd and K. Goa, Drugs, 1992, 43, 346–381 CrossRef PubMed.
- S. J. Hadziyannis, N. C. Tassopoulos, E. J. Heathcote, T. T. Chang, G. Kitis, M. Rizzetto, P. Marcellin, S. G. Lim, Z. Goodman and J. Ma, Gastroenterology, 2006, 131, 1743–1751 CrossRef PubMed.
- O. Johnell, B. Jönsson, L. Jönsson and D. Black, PharmacoEconomics, 2003, 21, 305–314 CrossRef PubMed.
-
(a) A. L. Schwan, Chem. Soc. Rev., 2004, 33, 218–224 RSC;
(b) F. M. Tappe, V. T. Trepohl and M. Oestreich, Synthesis, 2010, 3037–3062 Search PubMed.
-
(a) Q. Xu and L.-B. Han, J. Organomet. Chem., 2011, 696, 130–140 CrossRef;
(b) Y. H. Budnikova and O. G. d. Sinyashin, Russ. Chem. Rev., 2015, 84, 917–951 CrossRef.
- M. Abdoli and H. Saeidian, J. Sulfur Chem., 2015, 36, 556–582 CrossRef.
- G. J. Perry and I. Larrosa, Eur. J. Org. Chem., 2017, 3517–3527 CrossRef PubMed.
-
(a) S. Arshadi, E. Vessally, M. Sobati, A. Hosseinian and A. Bekhradnia, J. CO2 Util., 2017, 19, 120–129 CrossRef;
(b) E. Vessally, M. Babazadeh, A. Hosseinian, S. Arshadi and L. Edjlali, J. CO2 Util., 2017, 21, 491–502 CrossRef;
(c) K. Didehban, E. Vessally, M. Salary, L. Edjlali and M. Babazadeh, J. CO2 Util., 2018, 23, 42–50 CrossRef;
(d) E. Vessally, A. Hosseinian, M. Babazadeh, L. Edjlali and R. Hosseinzadeh-Khanmiri, Curr. Org. Chem., 2018, 22, 315–322 CrossRef;
(e) A. Hosseinian, S. Farshbaf, R. Mohammadi, A. Monfared and E. Vessally, RSC Adv., 2018, 8, 17976–17988 RSC.
-
(a) K. Didehban, E. Vessally, A. Hosseinian, L. Edjlali and E. S. Khosroshahi, RSC Adv., 2018, 8, 291–301 RSC;
(b) E. Vessally, K. Didehban, R. Mohammadi, A. Hosseinian and M. Babazadeh, J. Sulfur Chem., 2018, 39, 332–349 CrossRef;
(c) E. Vessally, R. Mohammadi, A. Hosseinian, K. Didehban and L. Edjlali, J. Sulfur Chem., 2018, 39, 443–463 CrossRef;
(d) A. Hosseinian, L. Zare Fekri, A. Monfared, E. Vessally and M. Nikpassand, J. Sulfur Chem., 2018 DOI:10.1080/17415993.2018.1436712;
(e) S. Farshbaf, L. Zare Fekri, M. Nikpassand, R. Mohammadi and E. Vessally, J. CO2 Util., 2018, 25, 194–204 CrossRef;
(f) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 23–42 CrossRef PubMed;
(g) E. Vessally, A. Hosseinian, L. Edjlali, E. Ghorbani-Kalhor and R. Hosseinzadeh-Khanmiri, J. Iran. Chem. Soc., 2017, 14, 2339–2353 CrossRef.
- J. Hu, N. Zhao, B. Yang, G. Wang, L. N. Guo, Y. M. Liang and S. D. Yang, Chem.–Eur. J., 2011, 17, 5516–5521 CrossRef PubMed.
- Y. Wu, L. Liu, K. Yan, P. Xu, Y. Gao and Y. Zhao, J. Org. Chem., 2014, 79, 8118–8127 CrossRef PubMed.
- L. Tang, L. Wen, T. Sun, D. Zhang, Z. Yang, C. Feng and Z. Wang, Asian J. Org. Chem., 2017, 6, 1683–1692 CrossRef.
- L. Liu, D. Zhou, J. Dong, Y. Zhou, S.-F. Yin and L.-B. Han, J. Org. Chem., 2018, 83, 4190–4196 CrossRef PubMed.
- X. Li, F. Yang, Y. Wu and Y. Wu, Org. Lett., 2014, 16, 992–995 CrossRef PubMed.
- G. Hu, Y. Gao and Y. Zhao, Org. Lett., 2014, 16, 4464–4467 CrossRef PubMed.
- G. Hu, Y. Zhang, J. Su, Z. Li, Y. Gao and Y. Zhao, Org. Biomol. Chem., 2015, 13, 8221–8231 RSC.
- J. Li, X. Bi, H. Wang and J. Xiao, Asian J. Org. Chem., 2014, 3, 1113–1118 CrossRef.
- D. Yang, D. Zhao, L. Mao, L. Wang and R. Wang, J. Org. Chem., 2011, 76, 6426–6431 CrossRef PubMed.
- B. Kaboudin, L. Karami, J.-y. Kato, H. Aoyama and T. Yokomatsu, Tetrahedron Lett., 2013, 54, 4872–4875 CrossRef.
- M. Zhou, M. Chen, Y. Zhou, K. Yang, J. Su, J. Du and Q. Song, Org. Lett., 2015, 17, 1786–1789 CrossRef PubMed.
- P. Zhang, L. Zhang, Y. Gao, J. Xu, H. Fang, G. Tang and Y. Zhao, Chem. Commun., 2015, 51, 7839–7842 RSC.
- Y. F. Zeng, D. H. Tan, W. X. Lv, Q. Li and H. Wang, Eur. J. Org. Chem., 2015, 2015, 4335–4339 CrossRef.
- M. Zhou, Y. Zhou and Q. Song, Chem.–Eur. J., 2015, 21, 10654–10659 CrossRef PubMed.
- X. Chen, X. Chen, X. Li, C. Qu, L. Qu, W. Bi, K. Sun and Y. Zhao, Tetrahedron, 2017, 73, 2439–2446 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c
c