
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe mutual effects between macrophages and cartilage templates in the process of subcutaneous endochondral bone formation
Jing Huang†
a,
Yi Zhou†ab,
Yan Wang a,
Xinjie Cai*ab and
Yining Wang*ab
a,
Xinjie Cai*ab and
Yining Wang*ab
aThe State Key Laboratory Breeding Base of Basic Science of Stomatology (Hubei-MOST), Key Laboratory of Oral Biomedicine Ministry of Education, School and Hospital of Stomatology, Wuhan University, 237 Luoyu Road, Wuhan 430079, China. E-mail: xinjie.cai@whu.edu.cn; wang.yn@whu.edu.cn; Fax: +86 27 87873260; Tel: +86 27 87686318
bDepartment of Prosthodontics, Hospital of Stomatology, Wuhan University, Wuhan 430079, China
First published on 29th June 2018
Abstract
The interplay between implants and the recipient immune environment is key to the long-term effectiveness of bone tissue engineering. In this study, we aimed to investigate the mutual effects between macrophages and cartilage templates in the process of subcutaneous osteogenesis. Primary mice bone marrow derived mesenchymal stem cells (BMSCs) were seeded into gelatin sponge and chondrogenically cultured for 4 weeks in vitro to form cartilage templates. The constructs were then implanted subcutaneously in monocyte-depleted mice or normal C57BL/6 mice. Implants harvested at two months showed inferior osteogenic quality in monocyte-depleted mice compared with that of normal mice. In normal mice, the cartilage templates recruited a high ratio of alternatively activated macrophages (CAM or M2) to classically activated macrophages (AAM or M1), compared with empty sponge. In vitro co-culture assay of macrophages with cartilage templates also showed that the cartilage templates polarized macrophages to the M2 phenotype and that these effects were even stronger than those of primary BMSCs. In turn, the co-culture of polarized macrophages with cartilage templates showed that compared to M0 or M2, M1 significantly increased the expressions of osteogenic and angiogenic markers of cartilage templates. These data suggested that macrophages seem to be indispensable in the osteogenesis of cartilage templates and that cartilage templates have a favorable immunomodulatory ability to polarize macrophages to the M2 phenotype. M1 was the contributing phenotype of macrophages that promoted the osteogenesis and angiogenesis of cartilage templates. Macrophages and cartilage templates cooperate to achieve endochondral bone formation.
Introduction
Bone tissue engineering is a promising approach for bone regeneration.1 Great effort has been made to elevate the functions of three important elements involved in bone tissue engineering, i.e., scaffolds, cells and cytokines.2 For a long time, the osteogenic differentiation of bone marrow derived progenitor/stem cells and the calcification of osteoblasts were considered the optimal approach for bone tissue engineering.3 However, during developmental osteogenesis, bone is formed through two pathways: intramembranous ossification and endochondral ossification.4 Accordingly, the bone tissue engineering approach is designed to resemble developmental bone osteogenesis.5 The term “developmental engineering” has been proposed.6 In an intramembranous ossification approach, bone regeneration is directly formed from progenitor/stem cells.7 In an endochondral ossification approach, a cartilage intermediate is created in vitro before being transformed in vivo for bone regeneration.8 The latter approach has various advantages over the former, i.e., vascularized bone tissue, hypoxia tolerance and angiogenic factor secretion.9 Recently, our group has successfully created cartilage templates with various biomaterials and evaluated their subcutaneous bone generation and periodontal bone regeneration.10,11Currently, the immune environment of the recipient has been considered to have a strong relationship with the effectiveness of bone regeneration.12 Successful bone regeneration highly depends on favourable osteo-immune environments.13 The implants firstly interact with host immune cells before playing a role in bone regeneration, and macrophages are among the first batch of immune cells recruited to implants.14
Macrophages are mononuclear cells of the myeloid lineage derived from hematopoietic stem cells (HSCs).15 Under inflammatory situations, circulating peripheral blood mononuclear cells are recruited into the tissues and differentiate into macrophages.16 Macrophages are traditionally known for their anti-infectious role in innate immunity and have been recently recognized for their contribution to tissue repair and homeostasis.17 Classically activated macrophages (CAM or M1) activated by T helper (Th)1-cytokine interferon-r are the most common and best-known type.18 The concept of alternatively activated macrophages (AAM or M2), which are stimulated by Th2-type cytokines, has gained attention in past decades.19 Different cell markers were used to distinguish between M1 and M2: high levels of CD86 and inducible nitric oxide synthase (iNOS) for M1 and high levels of CD206 and arginase (Arg)-1 for M2.20,21 Different studies show that macrophages play an essential role in autogenous bone healing or bone tissue engineering. In the process of bone fracture healing, a reduction of macrophages via clodronate liposomes resulted in a severely delayed hard callus formation.22 In a transgenic mouse model, macrophage depletion in macrophage Fas-induced apoptosis transgenic mice also showed a complete abolishment of callus formation.23 In bone tissue engineering, modulating the immune environment by targeting the phenotype of macrophages resulted in successful bone regeneration. Local delivery of FTY720, which can activate the sphingosine-1-phosphate (S1P) receptor and subsequently increases the recruitment of M2, enhanced blood vessel growth and bone defect healing.24 The immunomodulatory role of MSC has been identified and applied to MSC-based tissue regeneration. The expression of M2 markers CD206 and Arg1 in bone marrow derived macrophages markedly increased in co-culture with MSCs.25 In vivo implanted BMSCs triggered a functional switch of macrophages from the M1 to M2 phenotype and subsequently resulted in the formation of a bone regenerative niche.26 However, the role of macrophages has not been explored in successful bone regeneration using a cartilage intermediate.
Therefore, it would be of great interest to explore the role of macrophages in the endochondral bone formation process. The aims of this study were to (i) detect whether macrophages were required for the endochondral ossification approach, (ii) determine how the cartilage templates influence the polarization of macrophages, and (iii) detect the effects of M1 or M2 on the expression of osteogenesis and angiogenesis markers in cartilage templates.
Experimental
Mice
6 week-old male C57BL/6 mice were used in the study. Mice were obtained from Hubei Research Centre of Laboratory Animals (Wuhan, China) and kept in specific pathogen free (SPF) condition. All experimental protocols were approved by the Institutional Animal Care and Use Committee of Wuhan University (Approval Number: S07916090A).Isolation and pre-culture of BMSCs
BMSCs were isolated from 6 week-old male C57BL/6 mice (Hubei Research Centre of Laboratory Animals) using a standard protocol.27 Each gelatin sponge (MS0002, Ethicon, Sweden) was cut into a same size of 10 × 10 × 1 mm3. MSCs were seeded into gelatin sponge and differentiated into cartilage templates as previously described.28In vitro assays of cartilage template
The formation of cartilage template was detected by performing in vitro assays, including Safranin O staining and real-time quantitative polymerase chain reaction (qPCR) (see 2.10).For Safranin O staining, the cartilage template was fixed in 4% paraformaldehyde, dehydrated in graded ethanol and embedded in paraffin. The 5 μm slides were stained in Fast Green (Servicebio, China) for 4 min and rinsed with 1% acetic acid. Then, the slides were stained in Safranin O (Servicebio, China) for 6 min.
Reduction of monocytes/macrophages of mice via clodronate liposome
Liposome-encapsulated dichloromethylene diphosphonate (CL2MDP, clodronate) was used to try to eliminate blood monocytes/macrophages.29 Each mouse received 0.1 ml/10 g clodronate liposome (ClodronateLiposomes.org, Netherlands) via injection in the tail vein twice a week for eight consecutive weeks. Mice received 100 μl of phosphate buffer saline (PBS) liposome in the control group.Subcutaneous implantation in mice
A small incision was made in the back, and a subcutaneous pocket was created lateral to each incision. One implant was placed in each pocket. Afterwards, the skin was sutured with 4-0 absorbable ligature.Micro-CT analysis
The harvested implants were scanned using a μCT system (SkyScan1176). The voxel was set to 9 μm. The X-ray generator was operated at 50 kV and 500 μA. The three-dimensional (3D) volume viewer and analyser software was used for the visualization and quantification of 3D data. A 3D region of interest (ROI) was selected within the whole implants. The following microstructural parameters were characterized: bone volume fraction (BV/TV), trabecular number (Tb.N), bone mineral density (BMD), trabecular bone pattern factor (Tb.Pf), structure model index (SMI), degree of anisotropy (DA).Immunohistochemistry staining
The harvested implants were fixed with 4% paraformaldehyde, decalcified in 10% EDTA, and embedded in paraffin. The implants were cut into 5 μm thick slides. The slides were dewaxed in xylene and re-hydrated in graded ethanol to water. Antigen retrieval was performed in stomach enzyme antigen repair solution for 30 min at 37 °C. The slides were then incubated with 3% hydrogen peroxide for 30 min at 37 °C and blocked in 10% goat serum for 30 min at 37 °C. Immunostaining was performed by incubating with anti-OCN (ab93876, 1:200, Abcam, USA) or anti-CD31 (ab28364, 1:200, Abcam, USA) at 4 °C overnight. Slides were then washed in PBS and incubated with secondary antibody (anti-rabbit system, Maxim Biotechnologies, China) for 30 min at 37 °C. Staining was visualized with 3,3-diaminobenzidine and hematoxylin as a counterstain.Isolation and differentiation of bone marrow derived macrophages
Mouse bone marrow derived macrophages were also isolated from 6 week-old male C57BL/6 mice and cultured with 20 ng ml−1 M−1-CSF (Novus, USA).30 The percentage of F4/80+CD11b+ cells reached more than 90% according to FCM.Macrophages were polarized using a standard protocol.31 The M1 polarization medium was a complete cell culture medium supplemented with 1 μg ml−1 LPS and 10 ng ml−1 IFN-r. The M2 polarization medium was complete cell culture supplemented with 10 ng ml−1 IL-4. M1 or M2 was obtained by culturing macrophages with M1 or M2 polarization medium, respectively, for 48 h.
Co-culture of cartilage templates and macrophages in transwell system
Primary bone marrow derived macrophages were seeded onto a 6-well plate at a density of 106 cells per plate. Empty gelatin sponge, 106 BMSCs or one cartilage template was placed in the insert chambers. The systems were cultured in a humidified incubator for 48 h.Reversely, M0, M1 or M2 were seeded onto a 6-well plate, one cartilage template was placed in the insert chambers. The systems were cultured in a humidified incubator for 48 h.
Flow cytometry analysis
For the identification of reduction of monocytes/macrophages in mice, peripheral blood was collected with an anti-coagulative tube, and red blood cells were removed with RBC lysis buffer (Beyotime Biotechnology, China). The cells suspension was incubated with APC-CyTM 7 anti-CD3 (560590, BD Bioscience, USA), PE anti-CD45RA (553380, BD Bioscience, USA), APC anti-F4/80 (123115, BioLegend, USA) and FITC anti-CD11b (557396, BD Bioscience, USA) for 1 h at 4 °C.The phenotype of infiltrated macrophages in the implants in normal mice were analysed by harvesting the implants at different times and digesting them with collagenase (Sigma, USA) at 37 °C for 1 h. Single cell suspensions were incubated with PE anti-F4/80 (565410, BD Bioscience, USA), APC anti-CD86 (105011, BioLegend, USA) and PerCP/Cy5.5 anti-CD206 (141715, BioLegend, USA) for 1 h at 4 °C.
The macrophages co-cultured in vitro with empty gelatin sponge, BMSCs or cartilage templates were digested with trypsin. Sequential extracellular and intracellular staining was performed using an intracellular staining kit (562574, BD Biosciences, USA) according to the manufacturer's instructions. Briefly, the cell suspensions were incubated with APC anti-CD86 (105011, BioLegend, USA) and PerCP/Cy5.5 anti-CD206 (141715, BioLegend, USA) for 30 min at 4 °C; then the cell suspensions were incubated in 1× fix/permeate work solution for 50 min at 4 °C, followed by incubation with PE-Cy7 anti-iNOS (25-5920-82, eBioscience™, USA) and PE anti-Arg-1 (IC5868P, RnD system, USA) for 50 min at 4 °C.
All samples described above were examined by FACS Aria, and the percentage of targeted cells was analysed with FlowJo (FLOWJO, LLC, USA).
Real-time quantitative polymerase chain reaction (qPCR)
Total RNAs were isolated by TRIzol reagent (Takara Bio, Japan), Takara's reverse transcription kit (Takara Bio, Japan) was used to convert RNA into cDNA. SYBR Green Reagent (Takara Bio, Japan) was used to perform qPCR in a 7500 Fast Real-Time PCR system (Applied Biosystems, USA). Table 1 shows the primer sequences used in the study. Experiments were repeated three times. The relative expression levels were calculated by the 2−ΔΔCt method.32| Gene | Primer sequence (5′-3′) |
|---|---|
| a COL, collagen; Acan, aggrecan; Sox, Sry related HMG box; OPN, osteopontin. | |
| COL II | F: CAGGATGCCCGAAAATTAGGG |
| R: ACCACGATCACCTCTGGGT | |
| Acan | F: GCACCATCACAGAGTCCGAG |
| R: ACATTGCTCCTGGTCTGCAA | |
| Sox9 | F: AGTACCCGCATCTGCACAAC |
| R: ACGAAGGGTCTCTTCTCGCT | |
| VEGF | F: GAGTACCCCGACGAGATAGA |
| R: GGCTTTGGTGAGGTTTGAT | |
| COL I | F: TGACTGGAAGAGCGGAGAGT |
| R: AGTAGACCTTGATGGCGTCC | |
| OPN | F: CGGTGAAAGTGACTGATTCTGG |
| R: GGAGATTCTGCTTCTGAGATGG | |
| β-Actin | F: CATCCGTAAAGACCTCTATGCCAAC |
| R: ATGGAGCCACCGATCCACC | |
Statistical analyses
Quantitative data were expressed as means ± standard deviation. Statistical analysis was carried out by one-way analysis of variance (ANOVA) and post hoc testing for the following measurements, for which differences were considered significant at p < 0.05.Results and discussion
The purpose of this study was to observe the role of macrophages in the process of subcutaneous endochondral bone formation and detect the mutual effects between macrophages and cartilage templates. Firstly, the impact of macrophage reduction, using clodronate liposomes, on the subcutaneous bone formation was examined. According to the results of micro-CT and histology, it was implied that the macrophages were needed in the process. Then we backtrack the interplays between macrophages and cartilage templates respectively. The polarization effects of cartilage templates to macrophages were detected via the FCM analyse of in vivo implants and in vitro cell assay. The results consistently showed that cartilage templates had a tendency of polarizing macrophage to M2 phenotype. At last, the effects of macrophages to cartilage templates were detected. The results showed that both M1 and M2 could promote the expressions of osteogenesis and angiogenesis markers, compared to control group. Interestingly, the effects of M1 were even more evident.In vitro preparation and identification of cartilage templates
Safranin O staining visualized the orange-red cartilage matrix deposition distributed inside the gelatin sponge (Fig. 1a), and a number of cells showed a typical chondrocyte morphology embedded in large lacunae (Fig. 1b and c). qPCR analysis of the cartilage templates indicated that the expression of chondrogenic markers COL II, Acan, Sox9 increased significantly compared to that of BMSCs from passage 2 (Fig. 1d). Primary BMSCs had differentiated into hypertrophic chondrocytes, and the cartilage templates were prepared as expected. | ||
| Fig. 1 In vitro identification of cartilage templates. (a) General view of Safranin O staining of cartilage template (10×). Typical chondrocyte morphology embedded in large lacunae in Safranin O staining in (b) (20×) and (c) (40×). (d) qPCR analysis of chondrogenic markers COL II, Acan, Sox9. (yellow arrow: chondrocytes; GS: gelatin sponge; ***p < 0.001). | ||
The priming procedures for the creation of a cartilage template in vitro to induce following bone formation in vivo has been widely researched, especially in human and rat mesenchymal stem cells. In a previous study,10 it has been demonstrated that as short as 2 weeks was enough for the rat MSC-scaffold construct to form a cartilage template in vitro. However, a longer in vitro priming time, up to 4 weeks could lead to a more homogeneous bone formation. Similarly, human MSC pellets that chondrogenically primed for 5 weeks resulted in accelerated bone formation compared to those primed for 2 weeks in a subcutaneously osteogenic model.9 As to murine MSCs,33 Freeman et al.33 cultured murine MSC pellets in chondrogenic medium for different time duration. The results showed that at least 3 week was needed for obtaining the best mineralization capacity. Based on literature, in the present study, mice MSC seed into gelatin sponge were chondrogenically primed for 4 weeks before being implanted in vivo. According to in vitro assay, hypertrophic chondrocytes were detected and the expression of chondrogenic markers significantly increased compared to primary MSCs, which could verify the formation of cartilage templates.
Reduction of blood monocytes influenced subcutaneous osteogenesis and angiogenesis of cartilage templates
Following the verification of cartilage templates, the animal and model were chosen for the study. As firstly stated by Einhorn,34 “appropriate use of animal models in tissue-engineering research begins with careful consideration of the question asked”. Various questions can be answered in the field of bone tissue engineering via different animal models.35 In the present study the question was the role of macrophages in the process of endochondral bone formation via cartilage templates, but not the specific effectiveness of bone defect healing. Therefore, a simple mice subcutaneous osteogenic model without bone defect was used. Moreover, normal C57BL/6 other than nude mice were used for the purpose of mimicking the normal recipient immune environment. For the same reason, the scaffold used in this study was commercial hemostatic gelatin sponge, which is suitable for use as an in vitro model for chondrocyte 3D culture but will not induce host immune inflammatory responses.36,37 The properties of gelatin sponge made it possible to precisely detect the interplay between cartilage templates and macrophages without the influence of scaffold.To detect whether macrophages played a role in the process of bone tissue engineering via cartilage templates, a direct idea is to eliminate monocytes/macrophages. Clodronate encapsulated in liposomes is a widely used agent for reduction of monocytes/macrophages.38 Moreover, the agent had no significant effect on the osteoblasts and osteoclasts.22,39 Therefore, clodronate liposome is an appropriate strategy to specifically target monocytes/macrophages in the present study. However, the reduction is transient and macrophages recover over time, which need to be applied repeatedly to maintain the reduction.40
In the present study, the clodronate injection was repeated twice a week during the subcutaneous osteogenic process for 8 weeks, which was similar with another study that clodronate injection was sustained for 28 days.22 The PBS liposome-treated mice served as a control. Flow cytometry analysis of the peripheral blood showed that clodronate treatment induced a slight reduction of CD3−CD45RA− non-lymphoid cell frequency and achieved a more than 50% reduction of F4/80+CD11b+ monocytes among the CD3−CD45RA− population (Fig. 2), which was slightly lower than the reduction ratio described in other studies.41
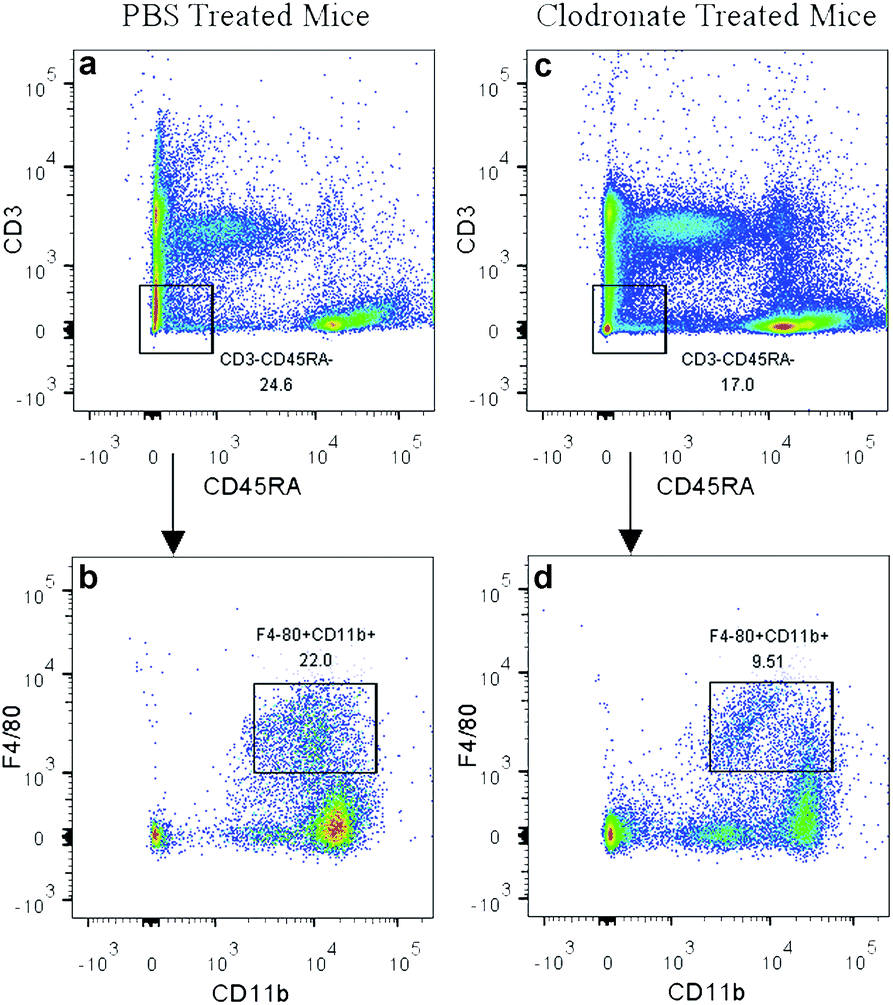 | ||
| Fig. 2 Depletion of monocytes in clodronate liposome-treated mice. CD3−CD45RA− non-lymphoid cell frequency was slightly reduced in clodronate liposome-treated mice (c) compared to PBS liposome-treated mice (a). A more than 50% reduction of F4/80+CD11b+ monocytes was observed among the CD3−CD45RA− population in clodronate liposome-treated mice (d) compared to PBS liposome-treated mice (b). | ||
3D reconstruction of implants after two months showed that the cartilage templates showed less compact bone-like tissues in the clodronate-treated mice compared to in the PBS-treated mice (Fig. 3a). Quantitative μCT analysis showed that the BV/TV, Tb.N and BMD of the implants in the clodronate-treated mice were lower than those of the PBS-treated mice, although the results were not significant. However, the SMI, Tb.Pf and DA of the implants in the clodronate-treated mice were significantly lower than those of the PBS-treated mice (Fig. 3b), which indicated that the quality of bone trabecula in the clodronate-treated mice was much lower.
 | ||
| Fig. 3 Depletion of monocytes/macrophages reduced the osteogenesis and angiogenesis of cartilage templates. (a) General 3D views of implants in clodronate- or PBS liposome-treated mice. (b) Microstructural μCT parameters of implants. (c and e) The number of OCN positive cells was much greater in the PBS liposome-treated mice than in the clodronate-treated mice at one or two months. (d and f) The number of CD31 positive blood vessel-like tissues was much greater in the PBS liposome-treated mice than in the clodronate-treated mice at one or two months. (S: spongostan; black arrow: OCN positive cell; gray arrow: CD31 positive vessel, *p < 0.05, **p < 0.01, ***p < 0.001). | ||
Osteogenesis and angiogenesis were also evaluated via immunohistochemistry analysis of OCN and CD31 of the implant slides at four weeks and eight weeks after implantation (Fig. 3c and d). The OCN positive cells were distinctly higher in the PBS-treated mice compared to in the clodronate-treated mice (Fig. 3e). Similarly, CD31 positive vessel-like tissues were obviously observed in the PBS-treated mice, whereas they were almost not found in the clodronate-treated mice (Fig. 3f). Above all, the major alternations of subcutaneous endochondral bone formation were detected due to the reduction of macrophages.
In the present study, the reduction efficiency of CD3−CD45RA− F4/80+CD11b+ monocytes has achieved more than 50%, which was basically consistent with but less than described in other studies where reduction rates were around 65% and 80%.22,41 However, the influence on the bone generation and expressions of osteogenic and angiogenic markers were still severe at this relative low reduction rate, underling the necessity of the macrophages in the bone generative process via cartilage. Whether macrophages involve in the process directly (macrophages themselves contact with the cartilage templates) or indirectly (macrophages recruit other cells to perform the process) need to be deeply investigated in the future studies.
Taken together, the clodronate treatment depleted a majority of blood monocytes, which reduced the number of recruited macrophages into the implanted cartilage templates. μCT results showed that the total quantity of bone generation was reduced in clodronate-treated mice compared to PBS-treated mice; although this difference was not significant, the quality of bone trabecula was significantly reduced. IHC showed that the clodronate-treated mice had fewer cells that were positive for markers of osteogenesis and angiogenesis than the PBS-treated mice. It can be concluded that the monocytes/macrophages seem to be indispensable for perfect osteogenesis and angiogenesis of the cartilage templates.
Infiltrated macrophages showed different phenotypes in empty gelatin sponge and cartilage templates
After confirming the involvement of macrophages, to backtrack the influence of cartilage templates to the recipient macrophages, empty gelatin sponge or cartilage templates were implanted subcutaneously in normal mice to identify infiltrated M1 and M2 subsets. We harvested the implants on seven consecutive days. The implants were collagenase-digested to single cell suspensions for flow cytometry. F4/80 and CD86 were selected as M1 cell markers, and F4/80 and CD206 were selected as M2 cell markers. The results showed a higher ratio of M2 to M1 in the cartilage template implants compared to empty gelatin sponge (Fig. 4).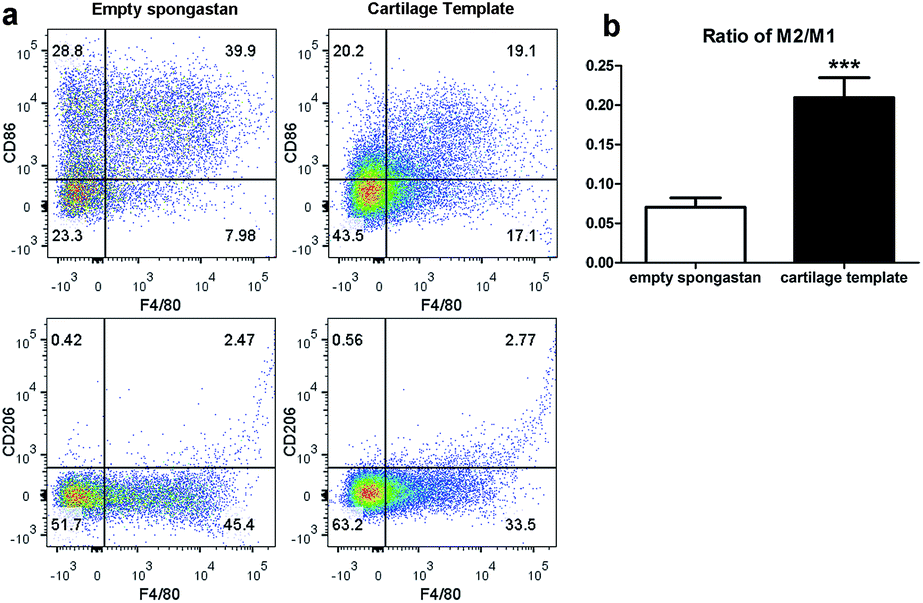 | ||
| Fig. 4 The M2 to M1 ratio of macrophages in the implanted cartilage templates was higher than that of empty spongostan. (a) Representative scatter diagrams of F4/80+CD86+ M1 in empty spongostan (upper left Q2) and cartilage templates (upper right Q2), and F4/80+CD206+ M2 in empty spongostan (lower left Q6) and cartilage templates (lower right Q6). (b) Statistics of M2 to M1 ratio in empty spongostan and cartilage templates. (***p < 0.001). | ||
According to the literature, in a rat model of spinal cord injury (SCI),42 human MSCs were transplanted into the contusion epicenter at 3 days after SCI. At day 7 after SCI, the CD206+Arg-1+ M2 cells significantly increased and the CD16/32+iNOS+ M1 cells decreased in the injury spinal cord of MSC-treated animals compared to control animals. In a mouse model of ectopic bone regeneration using bone marrow mesenchymal stem cells,26 a more elaborate time points were checked. At day 1, 3 and 7, the percentage of CD86+CD40+ M1 cells in MSC-seeded scaffold was less than that of empty scaffold while the percentage of CD206+CD51+ M2 cells was higher in MSC-seeded scaffold. Moreover, the M2/M1 ratio was over 1 in MSC-seeded scaffold at day 1, 3, 7 while it was under 1 all the time. In the present study, the results were partially consistent with above studies. The M2/M1 ratio of infiltrated macrophages in cartilage templates was higher than that of empty gelatin sponge. However, the M2/M1 ratio didn't reach 1 from day 1 to day 7.
Cartilage templates promoted M2 polarization and inhibited M1 polarization of macrophages in vitro
Then in vitro cell assay were conducted to further observe the interplays between cartilage and templates respectively. A transwell model was used for co-cultures of macrophages with empty gelatin sponge, primary BMSCs or cartilage templates, and untreated macrophages were used as a control (Fig. 5a). It is very likely that undifferentiated BMSCs still remained in the cartilage matrix,10 which might have participated in the polarization effect, so undifferentiated BMSCs were served as a control group in the in vitro assay. The results showed that empty gelatin sponge had no effect on the M1 or M2 polarization of macrophages. Primary BMSCs and cartilage templates both down-regulated the percentage of M1 and up-regulated the percentage of M2 (Fig. 5b and c). Furthermore, the effects of cartilage templates on macrophages polarization were stronger than those of primary BMSCs. The results indicated that the cartilage templates polarized macrophages to an M2 phenotype, and this effect was not due to gelatin sponge or any remaining BMSCs.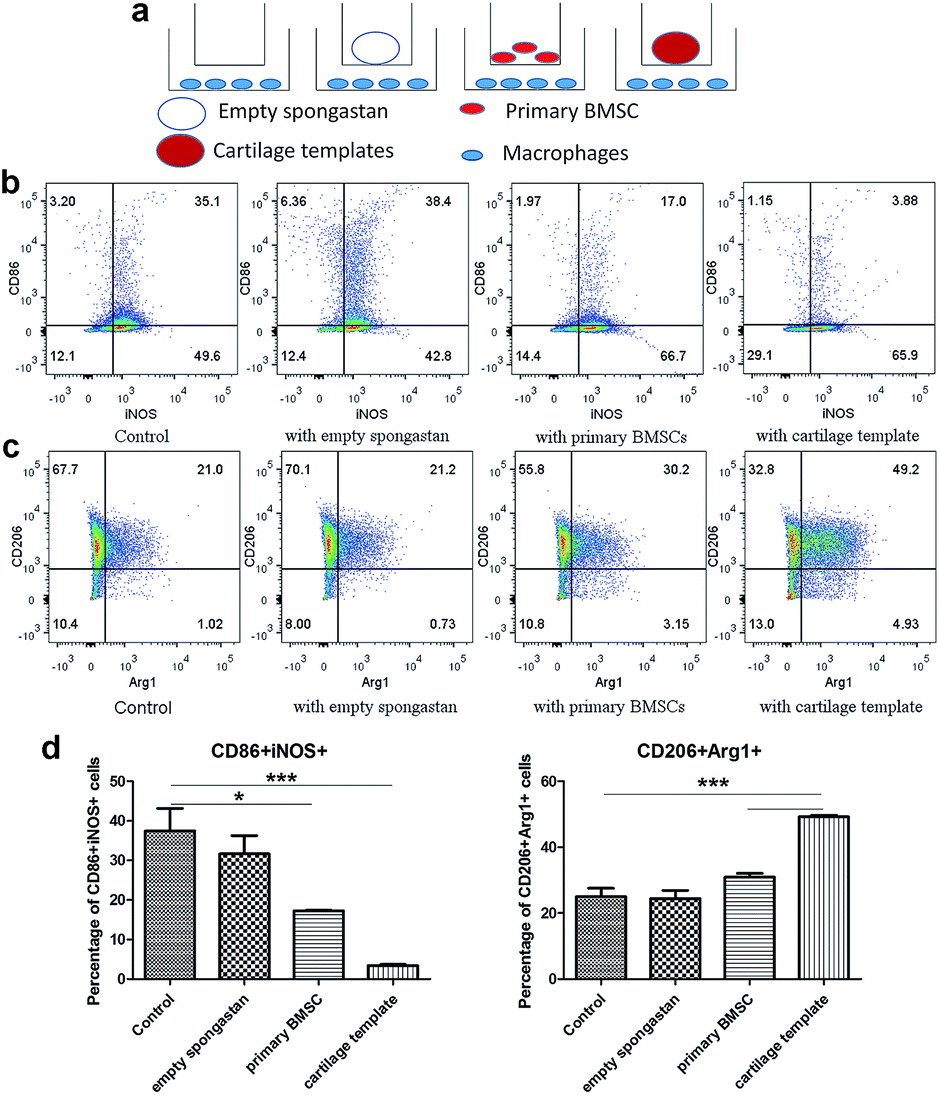 | ||
| Fig. 5 Cartilage templates polarized macrophages to an M2 phenotype. (a) Scheme of co-culture systems. (b) Representative scatter diagrams of CD86+iNOS+ M1 in each group. (c) Representative scatter diagrams of CD206+Arg1+ M2 in each group. (d) Statistics of M1 and M2 percentages in each group. (*p < 0.05, ***p < 0.001). | ||
The present results were consistent with literature that BMSC could promote the M2 activation of macrophages.25 Interestingly, our results showed that chondrogenically primed cartilage templates had an even stronger ability than primary MSCs in promoting M2 activation and inhibiting M1 polarization. The results implied that chondrogenically primed cartilage templates might have a stronger immune-modulatory ability than primary BMSCs, which might explain that cartilage templates performed better than BMSCs in healing bone defects.11 However, the mechanisms involved the phenomenon need future investigations.
M1 promoted the expression of osteogenesis and angiogenesis markers of cartilage templates
At last, a similar transwell model was used to examine the effects of M0, M1 and M2 on the osteogenesis and angiogenesis of cartilage templates in turn, and cartilage templates co-cultured with no cells were used as a control (Fig. 6a). Real-time PCR results showed that the mRNA expression of VEGF in cartilage templates co-cultured with M1 significantly increased compared to those co-cultured with M0, M2 or the control group. The osteogenesis markers COL I and OPN in cartilage templates co-cultured with M1 were significantly increased compared to those of the control group. They were also up-regulated compared to their levels in cartilage templates co-cultured with M0 or M2, but these results were insignificant (Fig. 6b). This observation implied that M1 was the contributing phenotype of macrophages that promoted the osteogenesis and angiogenesis of cartilage templates.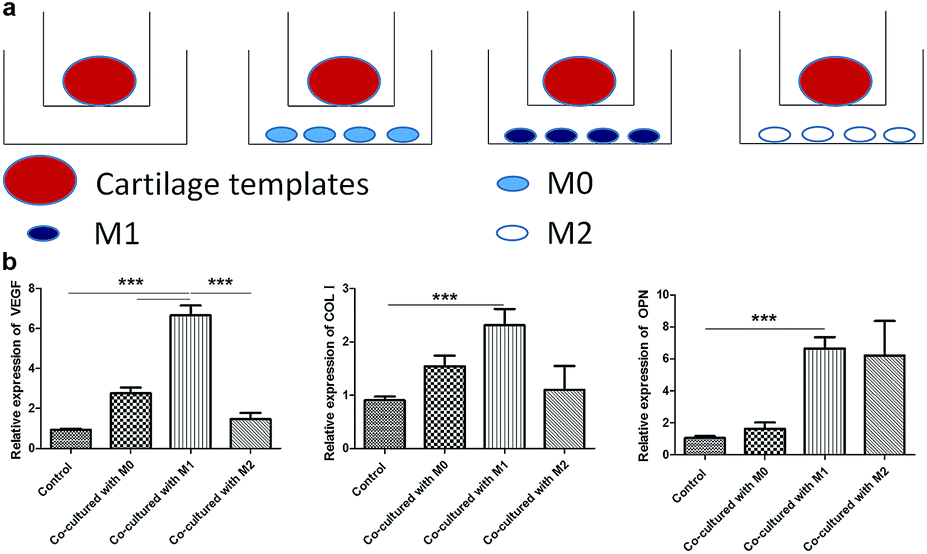 | ||
| Fig. 6 M1 promoted the expression of osteogenesis and angiogenesis markers of cartilage templates. (a) Scheme of co-culture systems. (b) Relative expression of VEGF, COL I and OPN in each group. | ||
Tumultuous literatures demonstrated that advantageous effects of M2 in the process of bone regeneration.22,24 However, more and more attentions were payed to the roles of inflammatory cytokines and pro-inflammatory M1 in bone regenerative biology recently. In a study of the roles of different cytokines to the chondrogenesis of hMSCs, pre-treatment with pro-inflammatory cytokine IL-1β resulted in a higher proteoglycans accumulation compared to hMSCs cultured with chondrogenic medium alone.43 In an osteoclastogenesis assay, addition of M1 macrophages or direct contact with M1, but not M0 or M2, significantly suppressed RANKL-induced osteoclastogenesis in osteoclast precursors.44 The mechanism involved was the secretion of IFN-γ and IL-12. In the present study, the results also showed that co-culture with M1 for 48 h, but not M0 or M2, significantly increased the expressions of osteogenic and angiogenic markers. The result was consistent with another study, in which primary mice BMSCs were co-cultured with M0, M1 or M2 for 4 weeks. It also showed that M1 enhanced the mineralization of BMSCs.45 However, the results need to be verified in vivo in the future studies.
Given the above, the biological performance happened during the early infiltration of macrophages into the cartilage templates is pivotal to the final endochondral bone formation. In a short time after implantation of cartilage templates, M1 is the dominant phenotype in the microenvironment. During this period, M1 was supposed to promote the angiogenesis and osteogenesis of cartilage templates. In turn, to avoid the excessive inflammatory cytokine secretion and damage to the surrounding tissue by M1, cartilage templates were thought to timely polarize M1 to M2. M2 could relieve the inflammatory environment and promote angiogenesis and osteogenesis persistently and slightly until the end. However, although the results highlight the major alternations in subcutaneous endochondral bone formation due to a partial depletion of macrophage population, it cannot be elucidated whether M1 or M2 is a consequence of the disturbed results, unless we could specifically and respectively eliminate M1 or M2.
Conclusions
Based on our findings, and within the limitations of this study, it was implied that macrophages were indispensable in the subcutaneous osteogenesis via cartilage templates and a cascade of interplay between cartilage templates and macrophages lead to the final formation of ectopic bone generative niche. The deficient osteogenesis following reduction of monocytes/macrophages was confirmed by the data of micro-CT and histology.As to the interplay between cartilage templates and macrophages, in vivo implantation of cartilage templates showed a tendency of polarizing infiltrated macrophage to M2 phenotype. In vitro cell assay verified that cartilage templates promote the M2 polarization and inhibit M1 polarization of macrophages. Moreover, the results implied that cartilage templates had a stronger immune-modulatory ability than primary BMSCs. Additionally, the data indicated that the role of M1 to the osteogenesis and angiogenesis should not be neglected and need to be detected in vivo.
The present study offered new insights into the roles of macrophages in tissue engineering using an endochondral ossification approach, which might be a target for achieving optimal bone regeneration. It would be of interest to gain future understanding of (i) the immune-modulatory function of different maturity of cartilage templates, ranging from primary BMSCs to hypertrophy, and the mechanisms involved, (ii) the specific strategies of modulating the recipient phenotypes of macrophages and other immune cells to achieve the best effectiveness of vascularized bone generation.
Conflicts of interest
There are no conflicts of interests to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No. 81571011, 81371170, 81470771 and 81200812), Fundamental Research Funds for the Central Universities (No. 2042017kf0075) and Hubei Provincial Natural Science Foundation of China (No. 2017CFB183).Notes and references
- J. Ma, S. K. Both, F. Yang, F. Z. Cui, J. Pan, G. J. Meijer, J. A. Jansen and J. J. van den Beucken, Stem Cells Transl. Med., 2014, 3, 98–107 CrossRef PubMed.
- E. A. Makris, A. H. Gomoll, K. N. Malizos, J. C. Hu and K. A. Athanasiou, Nat. Rev. Rheumatol., 2015, 11, 21–34 CrossRef PubMed.
- A. M. Yousefi, P. F. James, R. Akbarzadeh, A. Subramanian, C. Flavin and H. Oudadesse, Stem Cells Int., 2016, 2016, 6180487 CrossRef PubMed.
- J. E. Zins and L. A. Whitaker, Plast. Reconstr. Surg., 1983, 72, 778–785 CrossRef.
- R. S. Marcucio, L. Qin, E. Alsberg and J. D. Boerckel, J. Orthop. Res., 2017, 35, 2356–2368 CrossRef PubMed.
- P. Lenas, M. Moos and F. P. Luyten, Tissue Eng., Part B, 2009, 15, 381–394 CrossRef PubMed.
- A. I. Caplan, Tissue Eng., 2005, 11, 1198–1211 CrossRef PubMed.
- D. Gawlitta, E. Farrell, J. Malda, L. B. Creemers, J. Alblas and W. J. Dhert, Tissue Eng., Part B, 2010, 16, 385–395 CrossRef PubMed.
- C. Scotti, B. Tonnarelli, A. Papadimitropoulos, A. Scherberich, S. Schaeren, A. Schauerte, J. Lopez-Rios, R. Zeller, A. Barbero and I. Martin, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7251–7256 CrossRef PubMed.
- W. Yang, S. K. Both, G. J. van Osch, Y. Wang, J. A. Jansen and F. Yang, Acta Biomater., 2015, 13, 254–265 CrossRef PubMed.
- X. Cai, F. Yang, X. Yan, W. Yang, N. Yu, D. A. Oortgiesen, Y. Wang, J. A. Jansen and X. F. Walboomers, J. Clin. Periodontol., 2015, 42, 380–389 CrossRef PubMed.
- Z. Chen, T. Klein, R. Z. Murray, R. Crawford, J. Chang, C. Wu and Y. Xiao, Mater. Today, 2016, 19, 304–321 CrossRef.
- Z. Chen, J. Yuen, R. Crawford, J. Chang, C. Wu and Y. Xiao, Biomaterials, 2015, 61, 126–138 CrossRef PubMed.
- S. Franz, S. Rammelt, D. Scharnweber and J. C. Simon, Biomaterials, 2011, 32, 6692–6709 CrossRef PubMed.
- S. Gordon and P. R. Taylor, Nat. Rev. Immunol., 2005, 5, 953–964 CrossRef PubMed.
- S. Epelman, K. J. Lavine and G. J. Randolph, Immunity, 2014, 41, 21–35 CrossRef PubMed.
- B. Chazaud, Immunobiology, 2014, 219, 172–178 CrossRef PubMed.
- M. Benoit, B. Desnues and J. L. Mege, J. Immunol., 2008, 181, 3733–3739 CrossRef.
- S. Gordon and F. O. Martinez, Immunity, 2010, 32, 593–604 CrossRef PubMed.
- D. M. Mosser and J. P. Edwards, Nat. Rev. Immunol., 2008, 8, 958–969 CrossRef PubMed.
- F. O. Martinez and S. Gordon, F1000Prime Rep., 2014, 6, 1–13 Search PubMed.
- C. Schlundt, T. El Khassawna, A. Serra, A. Dienelt, S. Wendler, H. Schell, N. van Rooijen, A. Radbruch, R. Lucius, S. Hartmann, G. N. Duda and K. Schmidt-Bleek, Bone, 2018, 106, 78–89 CrossRef PubMed.
- L. J. Raggatt, M. E. Wullschleger, K. A. Alexander, A. C. Wu, S. M. Millard, S. Kaur, M. L. Maugham, L. S. Gregory, R. Steck and A. R. Pettit, Am. J. Pathol., 2014, 184, 3192–3204 CrossRef PubMed.
- A. Das, C. E. Segar, B. B. Hughley, D. T. Bowers and E. A. Botchwey, Biomaterials, 2013, 34, 9853–9862 CrossRef PubMed.
- D. I. Cho, M. R. Kim, H. Y. Jeong, H. C. Jeong, M. H. Jeong, S. H. Yoon, Y. S. Kim and Y. Ahn, Exp. Mol. Med., 2014, 46, e70, DOI:10.1038/emm.2013.135.
- R. Tasso, V. Ulivi, D. Reverberi, C. Lo Sicco, F. Descalzi and R. Cancedda, Stem Cells Dev., 2013, 22, 3178–3191 CrossRef PubMed.
- Y. Mabuchi, D. D. Houlihan, C. Akazawa, H. Okano and Y. Matsuzaki, Stem Cells Int., 2013, 2013, 507301 Search PubMed.
- W. Yang, F. Yang, Y. Wang, S. K. Both and J. A. Jansen, Acta Biomater., 2013, 9, 4505–4512 CrossRef PubMed.
- N. van Rooijen and E. van Kesteren-Hendrikx, J. Liposome Res., 2002, 12, 81–94 CrossRef PubMed.
- X. Zhang, R. Goncalves and D. M. Mosser, Curr. Protoc. Immunol., 2008, 1–18 Search PubMed.
- P. J. Murray, J. E. Allen, S. K. Biswas, E. A. Fisher, D. W. Gilroy, S. Goerdt, S. Gordon, J. A. Hamilton, L. B. Ivashkiv, T. Lawrence, M. Locati, A. Mantovani, F. O. Martinez, J. L. Mege, D. M. Mosser, G. Natoli, J. P. Saeij, J. L. Schultze, K. A. Shirey, A. Sica, J. Suttles, I. Udalova, J. A. van Ginderachter, S. N. Vogel and T. A. Wynn, Immunity, 2014, 41, 14–20 CrossRef PubMed.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef PubMed.
- F. E. Freeman, M. G. Haugh and L. M. McNamara, J. Tissue Eng. Regener. Med., 2016, 10, E250–E262 CrossRef PubMed.
- T. A. Einhorn, Clin. Orthop. Relat. Res., 1999,(suppl. 367), S59–S67 CrossRef.
- V. Viateau, D. Logeart-Avramoglou, G. Guillemin and H. Petite, in Sourcebook of Models for Biomedical Research, ed. P. M. Conn, Humana Press, 2008, ch. 74, DOI:10.1007/978-1-59745-285-4_74.
- J. Klangjorhor, P. Nimkingratana, J. Swttakorn, D. Pruksakorn, T. Leerapun, O. Arpornchayanon, S. Rojanasthien, P. Kongtawelert and P. Pothacharoen, J. Orthop. Surg. Res., 2012, 7, 40 CrossRef PubMed.
- M. Cegielski, I. Izykowska, M. Podhorska-Okolow, M. Zebel and P. Dziegiel, In Vivo, 2008, 22, 203–206 Search PubMed.
- S. B. Weisse, N. van Rooijen and L. M. Sly, J. Visualized Exp., 2012, 66, 4105 Search PubMed.
- L. Vi, G. S. Baht, H. Whetstone, A. Ng, Q. Wei, R. Poon, S. Mylvaganam, M. Grynpas and B. A. Alman, J. Bone Miner. Res., 2015, 30, 1090–1102 CrossRef PubMed.
- N. van Rooijen and E. van Kesteren-Hendrikx, J. Liposome Res., 2002, 12, 81–94 CrossRef PubMed.
- C. H. Côté, P. Bouchard, N. van Rooijen, D. Marsolais and E. Duchesne, BMC Musculoskeletal Disord., 2013, 14, 359 CrossRef PubMed.
- H. Nakajima, K. Uchida, A. R. Guerrero, S. Watanabe, D. Suqita, N. Takeura, A. Yoshida, G. Long, K. T. Wright, W. E. Johnson and H. Baba, J. Neurotrauma., 2012, 29, 1612–1625 CrossRef PubMed.
- D. Hingert, H. Barreto-Henriksson and H. Brisby, Tissue Eng., Part A, 2018, 24, 775–785 CrossRef PubMed.
- T. Yamaguchi, A. Movila, S. Kataoka, W. Wisitrasameewong, M. R. Torruella, M. Murakoshi, S. Murakami and T. Kawai, Infect. Immun., 2016, 84, 2802–2812 CrossRef PubMed.
- L. Y. Lu, F. Loi, K. Nathan, T. H. Lin, J. Pajarinen, E. Gibon, A. Nabeshima, L. Cordova, E. Jamsen, Z. Yao and S. B. Goodman, J. Orthop. Res., 2017, 35, 2378–2385 CrossRef PubMed.
Footnote |
| † Both authors contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2018 |