
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective binding of matrix metalloproteases MMP-9 and MMP-12 to inhibitor-assisted thermolysin-imprinted beads
Nicole Schauer†
a,
Mehmet Dinc†b,
Bastian Raabea,
Tim Hummela,
Marlen Müllera,
Harald Sobeka and
Boris Mizaikoff *b
*b
aLabor Dr Merk & Kollegen GmbH, Beim Braunland 1, 88416 Ochsenhausen, Germany
bInstitute of Analytical and Bioanalytical Chemistry, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: boris.mizaikoff@uni-ulm.de
First published on 18th September 2018
Abstract
Protein-imprinted polymers have been synthesized to recognize and specifically bind selected proteins. However, protein imprinting requires substantial amounts of pure protein to efficiently obtain imprinted polymers for large scale applications, e.g. protein purification by affinity chromatography. In the absence of large quantities of a pure protein of interest, an alternative strategy was developed. In this case study, neutral metalloprotease thermolysin was selected as a commercially available surrogate for imprinting polymer beads. Phosphoramidon-assisted thermolysin-imprinted beads were synthesized. During rebinding experiments, it was shown that these beads specifically bind to thermolysin. In addition, it was shown that these beads also bind in CHO cell culture supernatant to the matrix metalloprotease-9 and -12 (MMP-9, -12). Therefore, these beads can be applied as a selective sorbent for the rare metalloproteases MMP-9 and MMP-12 to remove these proteases from CHO cell culture supernatants. The high selectivity of thermolysin-imprinted beads can be extended to other proteases of the family of metalloproteases, and is not limited to thermolysin. This innovative approach is suitable to address the challenges in the field of protease purification and isolation from biotechnologically relevant media.
1. Introduction
Chinese hamster ovary (CHO) cells are a commonly employed platform for the heterologous production of recombinant biopharmaceutical proteins.1 However, the produced proteins are exposed to proteolytic enzymes originating from the host cell line and present in the cell culture supernatant and the proteolytic degradation of the recombinant proteins is a critical task in biotechnological processes. Proteolytic degradation by proteases from CHO cells has been described for the production of recombinant proteins, e.g., human nerve growth factor,2 IFN-γ,3 factor VIII4 and a Fc-fusion protein.5 Proteases from several protease families such as aspartic proteases,5,6 cysteine proteases,7 metalloproteases, serine proteases3 and acidic proteases8 have been reported to cause proteolytic degradation in CHO cell based expression systems. Elliott et al.9 described the identification of matrix metalloprotease-9 (MMP-9) secreted by a CHO-K1 cell line. Additionally, Sandberg et al.4 isolated a metalloprotease from CHO cell culture supernatant which showed a sequence homology to matrix metalloprotease-12 (MMP-12) and might affect the integrity of the recombinant product. Recently, dipeptidyl peptidase 3, a metalloprotease, and prolyl endopeptidase, a serine peptidase have been identified in the manufacturing process of recombinant acid alpha glucosidase.10Cell cultivation processes and purification strategies were optimized to control protease activities in the production processes of recombinant proteins.11 Several chromatographic procedures and the use of affinity matrices such as benzamidine sepharose are well established to remove contaminating proteases.
Another simple and efficient method for selective separation is the use of molecularly imprinted polymers (MIPs). MIPs as synthetic tailor-made affinity materials offer advantages such as chemical and physical stability, easy synthesis, reusability and cost-efficient mass preparation.12–16 In contrast to large molecules, the imprinting of smaller molecular targets is nowadays a well-established technique for generating artificial receptor materials. Especially the imprinting of biomacromolecules remains a challenge due to their large size, limited solubility, complex structure, poor mass transfer, limited stability, and also thermodynamically caused structural flexibility in solution.17,18 In addition, protein-imprinted polymers are most commonly investigated in individual selectivity studies. However, in complex real-world samples different proteins can influence each other and compete for the binding sites.19–21 Due to the lack of competitive selectivity studies, as well as studies with real-world samples it is understandable that all commercially MIPs are only available for small molecules.22
Pluhar et al. prepared protease-imprinted polymers using mini-emulsion polymerization for the selective binding of a target protease from complex protein matrices.20 Recently, inhibitor-assisted imprinting was used to selectively bind the protease pepsin in multi-protein rebinding studies. In the inhibitor-assisted imprinting technique, an inhibitor is immobilized onto the surface of the supporting material, which pre-organize the template molecules with a defined orientation prior to the polymerization process.23–27 The method combines the advantages of surface imprinting strategies and the inhibitor-based affinity interactions. The strategy of the surface imprinting is to place binding sites at or very close to the surface for unhindered access to the binding sites.28
However, protein-imprinting requires large amounts of pure protein to obtain high efficiently imprinted polymers for large scale applications. In the absence of large quantities of the protease of interest an alternative imprinting strategy was developed. Here, inhibitor-assisted surface-imprinted core–shell particles were synthesized for the selective binding of the matrix metalloprotease-9 and -12 using thermolysin as template, N,N′-methylenbisacrylamide as crosslinker as well as acrylamide (AM) and 2-methacryloyloxyethyl phosphorylcholine (MPC) as functional monomers. Highly porous phosphoramidon-immobilized silica particles were used as core materials, which were coated with a protein-imprinted shell. However, protein-imprinting requires large amounts of pure protein to obtain high efficiently imprinted polymers for large scale applications. In the absence of large quantities of the protease of interest the dummy imprinting strategy was utilized, which offers an alternative route using structural analogue of the target protein.29,30 Therefore, neutral metalloprotease thermolysin – a commercially available protease – was used as surrogate in the imprinting process. Thermolysin is a small thermostable globular metalloprotease produced by Bacillus thermoproteolyticus.31 In this study, we show that thermolysin-imprinted beads specifically bind two other metalloproteases present in CHO cell culture supernatants, i.e., matrix metalloprotease-9 and -12.
2. Experimental
2.1 Materials
Gelatin (porcine skin) and SDS Gel Preparation Kit were purchased from Fluka. N-Hydroxysulfosuccinimide sodium salt (≥98.5%) (Sulfo-NHS), ammonium persulfate (98%) (APS), thermolysin (from Geobacillus stearothermophilus, 39–175 units per mg), 1-octanol, poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (P123), MES hydrate (>99.5%), Fmoc-8-Aoc-OH (≥98%), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (≥99.0%) (EDC), 2-methacryloyloxyethyl phosphorylcholine (97%) (MPC), N,N′-methylenebis(acrylamide) (99%) (MBA), N,N,N′,N′-tetramethylethylenediamine (99%) (TEMED), acrylamide (≥99%) (AM) and hydroxypropyl cellulose (HPC) were purchased from Sigma Aldrich (Steinheim, Germany). Tetraethyl orthosilicate (TEOS), sorbitan monooleate (Span 80), toluene (max. 0.005% H2O), methanol (>99.89%) and dimethylsulfoxid (DMSO) (≥99.9%) were obtained from Merck (Darmstadt, Germany). (3-aminopropyl)-trimethoxysilane and (3-aminopropyl)triethoxysilane (APTES) were purchased from Alfa Aesar (Karlsruhe, Germany). Phosphoramidon was from Pepta Nova (Sandhausen, Germany). The CHO-S cell line, CD CHO Medium and HT Supplement (100×) were from Gibco Life Technologies. SG-200 (200 mM L-alanyl-L-glutamine) was from GE Healthcare. Anti-rat MMP-9 polyclonal antibodies were obtained from Millipore. Anti-human MMP-12 (C-terminal fragment) polyclonal antibodies were purchased from abcam. Protein samples were concentrated using Amicon Ultra-4 devices (10 and 30 kDa). Porablot PVDF membranes were obtained from Machery-Nagel. Secondary antibody solution alkaline phosphatase conjugated was from Invitrogen.2.2 Cell culture
First cells were propagated in 100 mL of medium. Upon reaching a cell density of >106 cells per mL, two 100 mL flasks were seeded. When a cell density of >106 cells per mL was reached, one 8 L bioreactor containing 4 L medium was seeded. The same serum free medium containing L-glutamine and HT Supplement was used. After 10 days of cultivation, cells were removed by centrifugation (1000 × g) for 10 min followed by a filtration step (Whatman filter) and an ultra-filtration step (0.22 μm) and stored at −20 °C.2.3 SDS gel electrophoresis
Protein samples were separated by SDS-polyacrylamide gel electrophoresis using stacking gels of 8% polyacrylamide and separating gels of 10% polyacrylamide. SDS-PAGE was carried out according to the kit instructions of the manufacturer. Samples were diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 with sample buffer and boiled for 2 min. Samples (15 μL per well) were added and electrophoresis was carried out. Gels were stained with Quick Coomassie™ Stain (Serva Electrophoresis GmbH) for 4 h and then destained in deionized water for 16 h.
:2 with sample buffer and boiled for 2 min. Samples (15 μL per well) were added and electrophoresis was carried out. Gels were stained with Quick Coomassie™ Stain (Serva Electrophoresis GmbH) for 4 h and then destained in deionized water for 16 h.
2.4 Identification of MMP-9 and -12 by western blot analysis
For antibody-based detection of CHO MMP-9 and -12, protein samples were separated by SDS-polyacrylamide gel electrophoresis using stacking gels of 8% polyacrylamide and separating gels of 10% polyacrylamide. After the electrophoresis, the proteins were transferred to a porablot PVDF membrane (Machery-Nagel). Blots were blocked using 5% Difco™ Skim Milk solution (BD), washed with T-TBS buffer and incubated with anti-human MMP-9 and -12 antibodies (0.5 μg mL−1). Signals were detected by 2° antibody solution alkaline phosphatase conjugated as described by the supplier.2.5 Zymography
Zymography activity assays were performed using gelatin and casein as substrates.32 Due to their different substrate specificities MMP-9 and -12 can be detected using gelatin and casein zymography respectively. Thermolysin was detected using gelatin as substrate. Samples were run on non-reducing SDS-polyacrylamide gels (stacking gels of 8% polyacrylamide and separating gels of 10% polyacrylamide) which contained 10% substrate of a 10 mg mL−1 stock solution. After running the gels at 150 V, gels were washed 1 h in renaturation buffer (2.5% Triton X-100). Then the gels were incubated in developing buffer (50 mM Tris, 5 mM CaCl2 × 3H2O) for 16 h. Protease activity was visualized by staining the gels in Coomassie Brilliant Blue R-250 (2.5 g L−1, 40% methanol, 10% acetic acid, 50% water) for 8 min and then destained in destaining solution (40% methanol, 10% acetic acid, 50% water). Zymography revealed the presence of MMP-9, MMP-12 and thermolysin as negatively stained bands due to digestion of the substrate proteins in the gel. A semi-quantitative approach was used to estimate the amount of proteases present in CHO cell culture supernatants and in different fractions. Serial dilutions of purified proteases (MMP-9 and -12) were prepared and used as reference standard in zymography activity assays. By comparing band intensities of the reference standard and of the samples the contents of MMP-9 and -12 were estimated.2.6 Synthesis of porous silica particles (SPs)
SPs were synthesized using the method of co-condensation of TEOS and APTMS in a water-in-oil (W/O) emulsion described by Oh C. et al.33 First the water phase prepared by dissolving 10 wt% of P123 in deionized water. After the dissolution, 2 wt% NH3(aq) (>25%) as a catalyst was added to the phase. For the preparation of the oil phase 1.6 wt% HPC was dissolved in 1-octanol by using a mechanical stirrer at 800 rpm and then kept at 80 °C for 3 h. Then the viscous solution was cooled to 40 °C and Span 80 was added. Afterwards the water phase was mixed with the oil phase under mechanical stirring (600 rpm). The weight ratio of water phase and oil phase was kept as 1:9. After 5 minutes TEOS and APTMS of RW = 9 (RW = the molar ratio of water to TEOS and APTMS) were simultaneously added into the emulsion. The molar ratio of APTMS to TEOS (RAT) was 0.08. The emulsion was stirred at a rate of 600 rpm at 40 °C for 16 hours. After reaction, ethanol was added to solution to decrease the viscosity. The product was centrifuged at 1500 rpm for 15 minutes to obtain the silica particles. The SPs were dispersed in ethanol using an ultrasonic bath for 30 minutes and then centrifuged again at 1000 rpm for 15 minutes. In order to eliminate excess and unreacted reactants the particles were washed several times with ethanol and water. The obtained beads were dried in a vacuum oven at 50 °C overnight. For further use, the amount of amino-groups at the surface was increased by post-grafting method. First the SPs (500 mg) were dispersed in toluene (50 mL) under inert conditions by using a magnetic stirrer. Then, APTES (1 mL) was added to the mixture under moderate stirring. The dispersion was heated to reflux for 5 h. The obtained amino-functionalized silica beads were centrifuged and washed several times with ethanol and dried in vacuum 24 h at 60 °C.
2.7 Surface modification
The Spacer Fmoc-8-Aoc-OH was covalently attached onto the bead surface via an amide linkage. 20 mL of 2.0 mg mL−1 Fmoc-8-Aoc-OH prepared in DMSO was mixed with 20 mL of 3.0 mg mL−1 EDC and 15 mL of 2.0 mg mL−1 Sulfo-NHS. EDC and Sulfo-NHS solutions were prepared at pH 4.75 MES buffer, 0.1 M. The resulting mixture was magnetically stirred for 20 min. 0.35 g of amino-functionalized beads – conditioned with 5 mL of pH 4.75 MES buffer solution – were added into the solution. This coupling reaction was proceeded 4 h on a magnetic stirrer at room temperature. To terminate the coupling reaction, the suspension was centrifuged at 1500 rpm for 12 min. After removal of the supernatant, the microbeads were washed several times with water and methanol, respectively. The beads were dried in a vacuum oven at 40 °C overnight. Fmoc deprotection was carried out using 20% piperidine in DMF for 2 h. The deprotected microbeads were washed several times with DMF and ethanol, respectively. After the deprotection, phosphoramidon, a selective inhibitor of the protease thermolysin, was immobilized via an amide bond onto the surface of the beads. 0.3 g of the spacer-modified beads were dispersed in 6 mL of pH 4.75 MES buffer solution using ultrasonication for 10 min. Afterward, EDC (15.00 mL, 3.0 mg mL−1), phosphoramidon (10 mL, 1.0 mg mL−1) and Sulfo-NHS (15 mL, 2.0 mg mL−1) were added to the solution. The resulting mixture was magnetically stirred for 2 h. All solutions were prepared at pH 4.75 MES buffer (0.1 M). The resultant phosphoramidon-immobilized particles were purified with methanol and water and dried in a vacuum oven at 30 °C for 16 h.2.8 Synthesis of the surface-imprinted inhibitor-immobilized beads
The next step is the grafting of an imprinted polymer layer onto the surface of the porous beads in presence (MIP) and absence (NIP) of the template thermolysin. 0.1 g of the inhibitor-immobilized beads were dispersed in 10 mL PBS buffer (pH 7.2, 0.05 M) using ultrasonication for 5 min. Subsequently, 20.0 mg AM (0.28 mmol), 14.0 mg MPC (0.044 mmol), 4.5 mg MBA (0.026 mmol) and 6 mg thermolysin were added to the solution and stirred for 30 min at room temperature and then 5 mg APS and 10 μL TEMED was added for the initiation of the polymerization. The polymerization carried out with gentle stirring at room temperature for 8 h. The surface-imprinted beads were centrifuged at 1500 rpm for 12 min and washed several times with water in order to remove excess reactants. NIPs were prepared by same conditions, except no thermolysin was added to the reaction solution. The template thermolysin was extracted using NaCl (0.5 M) until no free thermolysin was detectable in the supernatant using UV-Vis spectroscopy (detection wavelength: 277 nm). Following the extraction procedure, the beads were washed twice with water and then dried in a vacuum oven at 30 °C for 16 h. NIP microbeads were treated with exactly the same procedure as the MIP particles. In addition, to study the influence of the inhibitor also MIPs and NIPs were prepared without immobilized phosphoramidon as an assistive recognition moiety. The SEM images of the synthesis steps were collected with a Quanta 3D FEG (FEI).2.9 Rebinding experiments
For a typical rebinding assay, MIP- and NIP-particles (10 mg) were washed with 0.5 M NaCl solution for five times. The beads were equilibrated in 3 mL vPBS (1:1 PBS and deionized water, pH 7.0) for three times. Thermolysin was added in vPBS to a final concentration of 10 μg mL−1 and incubated for 20 min at room temperature. After binding the beads were extensively washed using 1.25 mL vPBS for ten times. After each incubation step the beads were centrifuged for 10 min at 1500 × g. Bound thermolysin was eluted from the beads for 20 min at room temperature in 1.0 M NaCl solution. From each step samples were removed from the supernatant and analyzed by zymography for protease activity. For binding studies with MMP-9 and MMP-12 the MIP- and NIP-particles were treated as described above. Then the particles (10 mg) were incubated with concentrated (10×) CHO-S cell culture supernatants (5 mL). The beads were washed, and the matrix metalloproteases were eluted as described for thermolysin.
3. Results and discussion
In heterologous protein production in CHO cells, the produced proteins are often degraded by proteases originating from the host cell. Some cellular proteases can degrade intracellular proteins, but once released after cell lysis they can also degrade extracellular proteins during the fermentation or the early stages of the down-stream process. In previous studies proteases of different families including metalloproteases have been identified in cell culture supernatants of CHO cells. Elliott et al.9 reported on the identification of matrix metalloprotease MMP-9 and Sandberg et al.4 identified metalloproteases with sequence homology to MMP-3, -10 and -12. MMP-9 and -12 are multi-domain enzymes and belong to the family of Zn2+-dependent metalloproteases. They degrade extracellular matrix macromolecules and cleave a variety of regulatory proteins, including cytokines and chemokines. All MMPs consist of a signal peptide, a propeptide, and a catalytic domain. MMP-9 and -12 also contain a hemopexin-like domain and additionally, MMP-9 contains three fibronectin type II domains inserted in the catalytic domain.34 MMP-9 and -12 exhibit a broad range of substrate specificity and are therefore potentially critical to the production of recombinant therapeutic proteins in CHO cell lines.Zymography is a sensitive method to detect proteases.32,35 We used this method to detected proteases in cell culture supernatants of a CHO-S cell line. Using gelatin, respectively casein zymography combined with western blot analysis these proteases were identified as MMP-9, respectively MMP-12 (data not shown). Under the conditions applied no other proteases were detected in the cell culture supernatants. To selectively bind and remove MMP-9 and -12 from CHO cell cultures supernatants the selectivity of inhibitor-assisted protease-imprinted beads was exploited. Additionally, in an innovative approach inhibitor-assisted thermolysin-imprinted MIPs were used to selectively bind other metalloproteases than thermolysin. A schematic illustration of the imprinting process is shown in Fig. 1. Macroemulsion synthesis was used to prepare the porous silica beads.
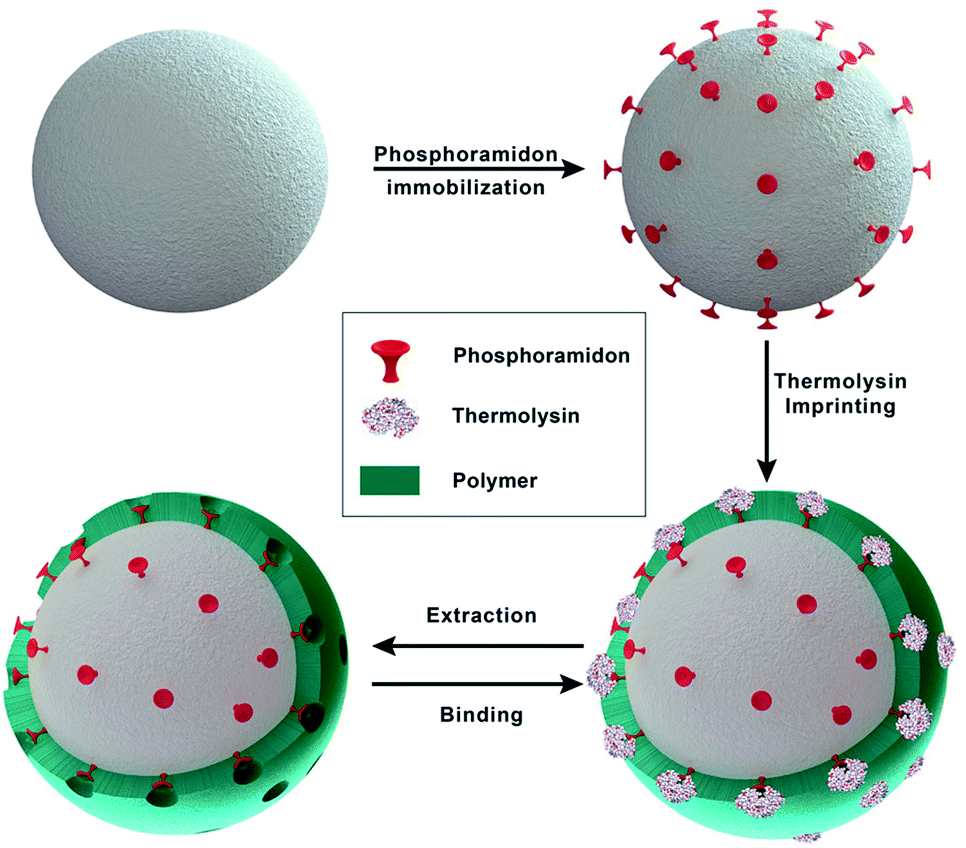 | ||
| Fig. 1 Schematic of the imprinting process. | ||
For the modification of the surface with Fmoc-8-Aoc-OH, the amount of amino-groups on the surface was enhanced by post-grafting technique with APTES.33,36,37 After the FMOC deprotection, the inhibitor phosphoramidon was immobilized onto the surface as a directing agent for the template thermolysin prior to the imprinting process. Fig. 2 shows the representative SEM images of the main synthesis steps. Based on the SEM images, the immobilization of phosphoramidon (b) onto the surface of the spacer-immobilized beads (a) has no visible effect on the surface morphology. After the imprinting process, it is evident that the surface of NIP (c) and MIP (d) appears less porous and homogeneous compared to the initial particles.
 | ||
| Fig. 2 SEM images of (a) spacer-immobilized particles, (b) phosphoramidon-immobilized particles, (c) NIP and (d) MIP. | ||
Thermolysin as template, AM and MPC as functional monomers and MBA as crosslinker were used to graft imprinted films onto the surface of inhibitor-immobilized silica beads. NIPs were prepared using the same procedure, but in the absence of the template. The monomer MPC has a zwitterionic phosphorylcholine group on the side chain. The resultant polymer has an excellent biocompatibility and therefore it can be used to fabricate a wide range of biomaterials.12,38 In addition, on the one hand the phosphorylcholine group prevent protein adsorption and on the other hand it is able to form non-covalent bonds with the calcium ions of thermolysin, MMP9 and MMP12.38,39 Therefore the MPC polymer reduce the non-specific bindings and increase the specific binding of thermolysin, MMP9 and MMP12. The binding properties of the MIPs were compared to the binding properties of non-imprinted polymers (NIPs). Hence, the contributions of non-selective binding processes can be analyzed. The MIP- and NIP-particles were used in a rebinding assay with thermolysin as initial target molecule. Fractions were analyzed using gelatin zymography to detect thermolysin activity. After incubation with MIPs less thermolysin activity was found in the supernatant compared to samples taken from the supernatants obtained after incubation of thermolysin with NIPs (Fig. 3A and B, lanes 2). This indicates that more thermolysin was bound to MIPs than to NIPs. After extended washing steps thermolysin was eluted from the beads and the supernatants from the washing steps were again analyzed by gelatin zymography. Here it was found that thermolysin activity was eluted from the MIPs (Fig. 3A, lane 4). No thermolysin was detected in the elution fractions of the NIPs (Fig. 3B, lane 4). In control experiments the MIP particles did not bind to the serine protease trypsin (data not shown). In a next step the binding studies were extended to study the selective binding of other metalloproteases. Thermolysin-imprinted beads were therefore used to selectively bind metalloproteases present in CHO cell culture supernatants.
 | ||
| Fig. 3 (A) Analysis of the rebinding experiments using thermolysin-imprinted beads (MIPs) and thermolysin. Gelatin zymography of fractions: samples (15 μL) were separated by a 10% SDS-PAGE containing 1 mg gelatin per mL. The molecular mass of the marker proteins is indicated. The cleared zone represents thermolysin at approx. 35 kDa. Lane 1 supernatant of the rebinding experiment. Lane 2 and 3: aliquots of the washing step. Lane 4: elution of thermolysin (relevant band indicated) lanes marked with x: no samples included. (B) Analysis of the rebinding experiments using non-imprinted beads (NIPs) and thermolysin. Gelatin zymography of fractions: samples (15 μL) were separated by a 10% SDS-PAGE containing 1 mg gelatin per mL. The molecular mass of the marker proteins is indicated. The cleared zone represents thermolysin at approx. 35 kDa. Lane 1: supernatant of the rebinding experiment. Lane 2 and 3: aliquots of the washing step. Lane 4: elution of thermolysin. Lanes marked with x: no samples included. | ||
To bind these contaminating proteases thermolysin-imprinted beads were incubated with aliquots of CHO-S cell culture supernatants. After repeated washing of the beads the bound proteases were eluted from the beads. Gelatin, respectively casein zymography revealed that MMP-9 and MMP-12 could be eluted from the beads (Fig. 4A and 5A, lanes 4). The gelatin zymography also revealed that MMP-9 was eluted from the MIPs but not from the NIPs (Fig. 4A and B, lanes 4). MMP-9 has pro- and active isoforms of 92 and 82 kDa, respectively. A smaller fragment of 68 kDa also shows proteolytic activity.9 Although the 92 kDa form is considered latent, the proteolytic activity observed on gelatin zymograms is a result of the activation of the pro-form under the conditions of this technique. The 68 kDa and the 92 kDa form were detected in the supernatant (Fig. 4A, lane 1). However, the elution fraction showed the only presence of the 82 kDa form of MMP-9. This indicates that the enzyme only the active isoform of MMP-9 binds to the beads (Fig. 4A, lane 4). Based on the intensities of the bands it was estimated that the amount of beads used (10 mg) was sufficient to bind about 0.1 μg of MMP-9. To detect MMP-12 the fractions were also analyzed by casein zymography. Casein zymography showed two protease forms of MMP-12 in the elution fraction of the MIPs (Fig. 5A, lane 4). The apparent molecular weights of 54 and 45 kDa correspond to the latent and the active forms as described for the human matrix metalloprotease MMP-12.40 This indicates that the active isoform and the preform of MMP-12 bind to the beads. The results show that the high selectivity of thermolysin-imprinted beads can be extended to other proteases of the family of metalloproteases and is not limited to the original imprinting molecule thermolysin. The selectivity of MIPs is based on the complementarity of the binding sites to the target molecule. In contrast to conventional imprinting, the technique of inhibitor-assisted imprinting enhances the selectivity of the MIPs by immobilizing an inhibitor prior to the imprinting process.23,24
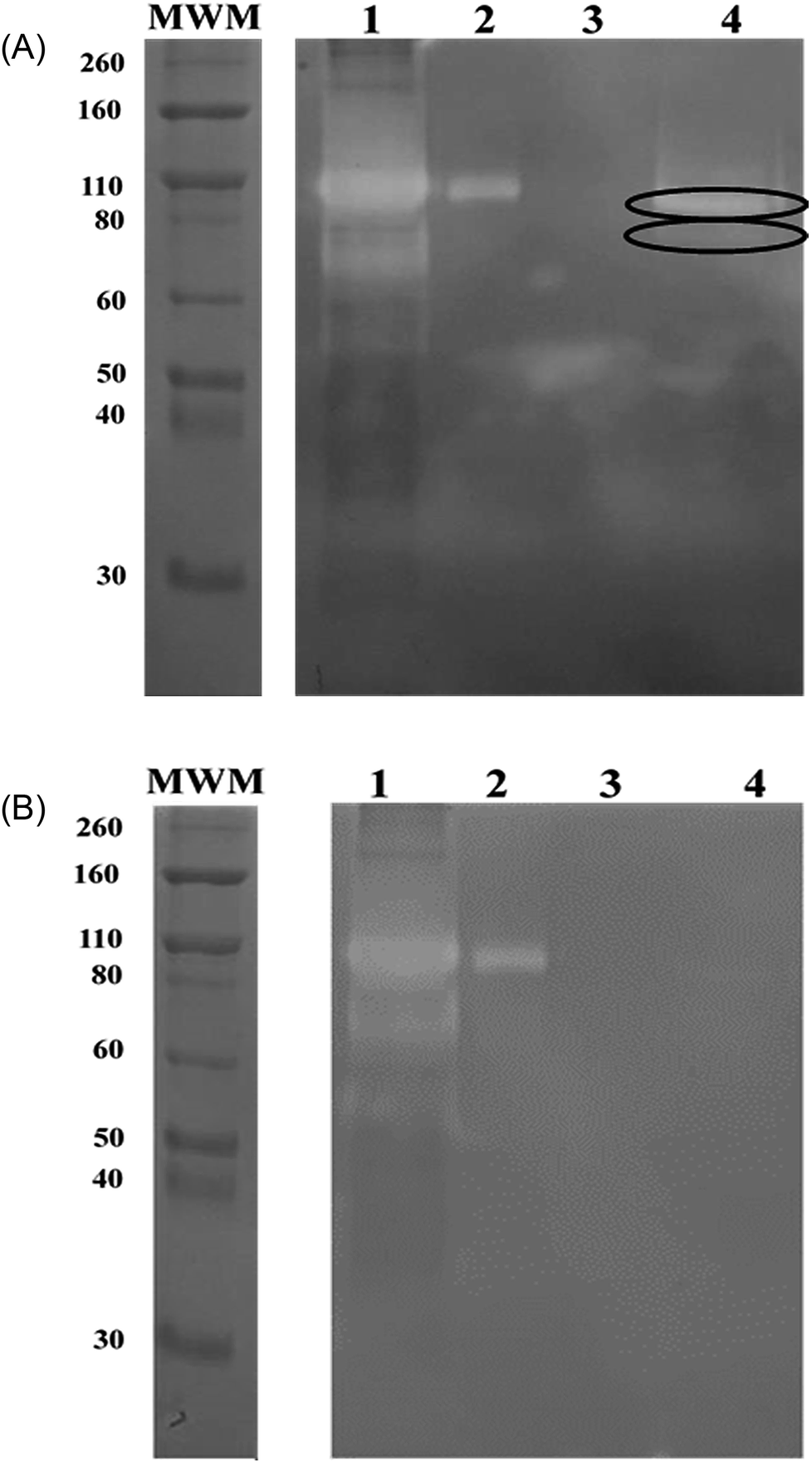 | ||
| Fig. 4 (A) Analysis of the rebinding experiments using thermolysin-imprinted beads (MIPs) and cell culture supernatant. Gelatin zymography of fractions: samples (15 μL) were separated by a 10% SDS-PAGE containing 1 mg gelatin per mL. The molecular mass of the marker proteins is indicated. The cleared zone represents pro-MMP-9 at approx. 92 kDa, active MMP-9 at approx. 82 kDa and a smaller fragment of MMP-9 at approx. 68 kDa. Lane 1: supernatant of the rebinding experiment. Lane 2 and 3: aliquots of the washing step. Lane 4: elution of MMP-9 (relevant bands indicated by circles). (B) Analysis of the rebinding experiments using non-imprinted beads (NIPs) and cell culture supernatant. Gelatin zymography of fractions: samples (15 μL) were separated by a 10% SDS-PAGE containing 1 mg gelatin per mL. The molecular mass of the marker proteins is indicated. The cleared zone represents pro-MMP-9 at approx. 92 kDa and active MMP-9 at approx. 82 kDa. Lane 1: supernatant of the rebinding experiment. Lane 2 and 3: aliquots of the washing step. Lane 4: elution of MMP-9. | ||
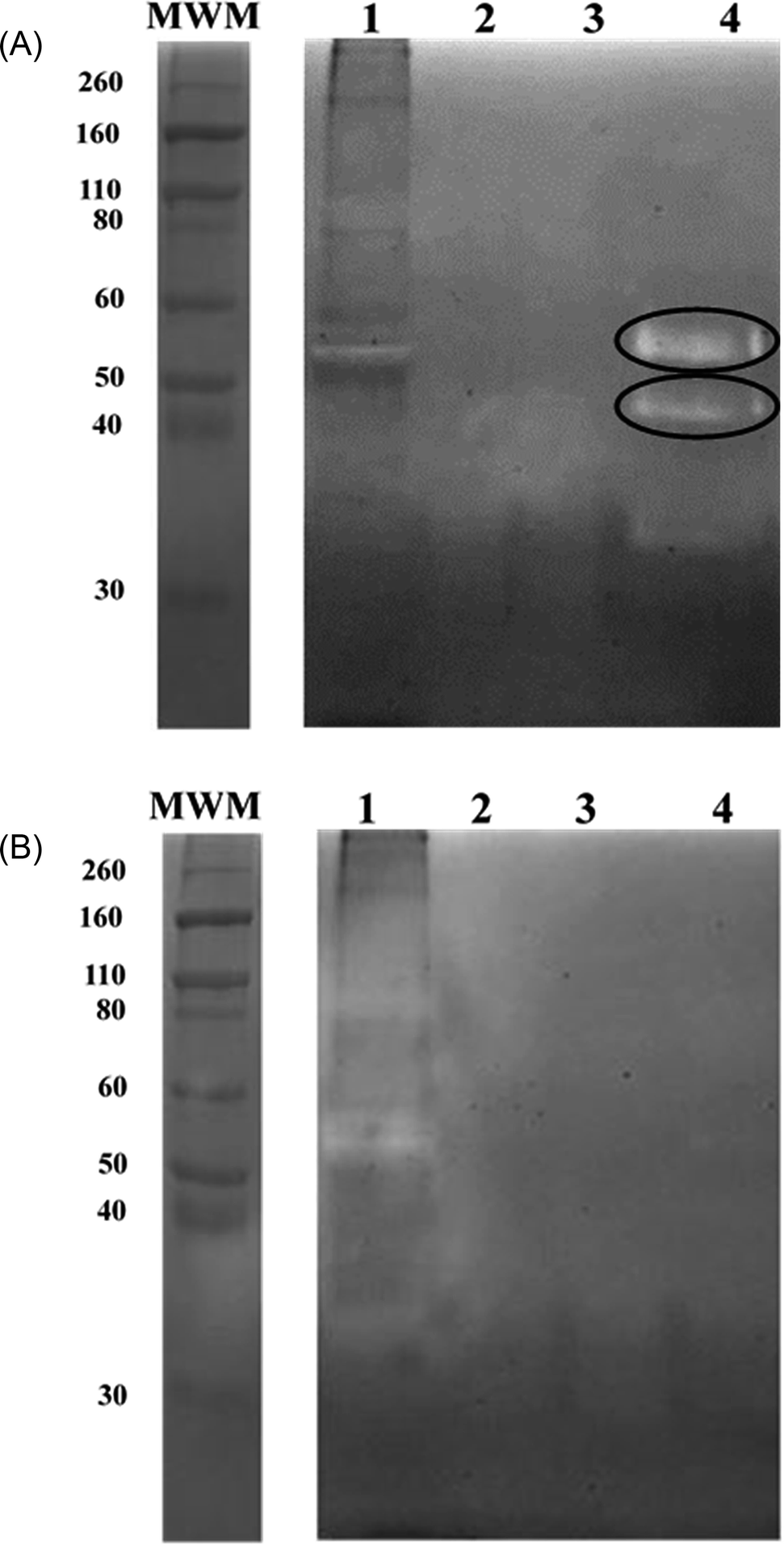 | ||
| Fig. 5 (A) Analysis of the rebinding experiments using thermolysin-imprinted beads (MIPs) and cell culture supernatant. Casein zymography of fractions: samples (15 μL) were separated by a 10% SDS-PAGE containing 1 mg casein per mL. The molecular mass of the marker proteins is indicated. The cleared zone represents MMP-12 at approx. 54 kDa and active MMP-12 at approx. 45 kDa. Lane 1: supernatant of the rebinding experiment. Lane 2 and 3: aliquots of the washing step. Lane 4: elution of MMP-12 (relevant bands indicated by circles). (B) Analysis of the rebinding experiments using non-imprinted beads (NIPs) and cell culture supernatant. Casein zymography of fractions: samples (15 μL) were separated by a 10% SDS-PAGE containing 1 mg casein per mL. The molecular mass of the marker proteins is indicated. The cleared zone represents MMP-12 at approx. 54 kDa and active MMP-12 at approx. 45 kDa. Lane 1: supernatant of the rebinding experiment. Lane 2 and 3: aliquots of the washing step. Lane 4: elution of MMP-12. | ||
The technique of inhibitor-assisted molecularly-imprinting of polymers is based on the copolymerization of functional monomers, an inhibitor and cross-linkers in presence of a template molecule. Thereby binding sites are formed that are complementary in size and shape to the target molecule.
After the removal of the template molecule the obtained binding sites are available for rebinding of target molecules or structural analogues.30 However, to date no structures of CHO MMP-9 and -12 are available. Both the enzymes show high homology to the human enzymes. Therefore, the structures of the human enzymes can be used to discuss the structural features of the hamster enzymes. All MMPs share a number of conserved protein domains. They consist of a signal peptide, a propeptide, and a catalytic domain. Some MMPs contain additional domains such as a hemopexin domain or fibronectin domains.34 The catalytic domains of MMP-9 and -12 show a fold that is similar to the fold of other Zn2+-proteases of the MMP protein family consisting of a five-stranded β-sheet and three α-helices. The catalytic center is composed of the active-site zinc ion and three histidine residues.41,42 Additionally, the catalytic domain of MMP-9 shows three repeats of a type II fibronectin domain inserted in the catalytic domain, which specifically bind to gelatin.34 In contrast, thermolysin is a globular metalloprotease with a molecular weight of 34.6 kDa. The protease consists of a N-terminal domain containing an extended β-sheet and a C-terminal domain containing α-helices. Like the catalytic domain of the MMPs its active center of thermolysin is composed of three histidine residues and an active site zinc-ion. Like MMP-9 and -12 thermolysin also shows flexible surface loops at the active site clefts.31 Despite the low similarity in the overall structure between thermolysin and MMP-9 and -12 the thermolysin-imprinted beads show a high selectivity for other metalloproteases. A common feature of the metalloproteases is the architecture of the active site. The inhibitor phosphoramidon monomer used prior to the grafting of the bead shell may recognize and bind to the Zn2+-ion of the active site, and thus, favor a certain orientation of the thermolysin molecule during the imprinting process. The created complementary binding site may then facilitate rebinding of other metalloproteases in a similar orientation as the original imprinting molecule.
4. Conclusions
In summary, phosphoramidon-assisted surface-imprinted porous silica particles were prepared for MMP-9 and MMP-12 by using the thermolysin as a commercially available surrogate protease. The resulting dummy molecularly imprinted polymers shows a high selectivity for the metalloproteases MMP-9 and MMP-12 in CHO cell culture supernatant. This innovative approach is suitable to address the challenges in the field of protein purification and isolation from biotechnologically relevant media.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by Federal Ministry of Education and Research (BMBF) within the projects PROTSCAV I and II. The authors thank the Electron Microscopy facility and the Focused Ion Beam Center at the University of Ulm for assistance during these studies.Notes and references
- F. Li, N. Vijayasankaran, A. Shen, R. Kiss and A. Amanullah, mAbs, 2010, 2, 466–479 CrossRef PubMed.
- M. Eng, V. Ling, J. a Briggs, K. Souza, E. Canova-Davis, M. F. Powell and L. R. De Young, Anal. Chem., 1997, 69, 4184–4190 CrossRef PubMed.
- M. Clincke, E. Guedon, F. T. Yen, V. Ogier and J. Goergen, BMC Proc., 2011, 5, P115 CrossRef PubMed.
- H. Sandberg, D. Lütkemeyer, S. Kuprin, M. Wrangel, A. Almstedt, P. Persson, V. Ek and M. Mikaelsson, Biotechnol. Bioeng., 2006, 95, 961–971 CrossRef PubMed.
- F. Robert, H. Bierau, M. Rossi, D. Agugiaro, T. Soranzo, H. Broly and C. Mitchell-Logean, Biotechnol. Bioeng., 2009, 104, 1132–1141 CrossRef PubMed.
- J. S. Bee, L. Tie, D. Johnson, M. N. Dimitrova, K. C. Jusino and C. D. Afdahl, Biotechnol. Prog., 2015, 31, 1360–1369 CrossRef PubMed.
- M. Satoh, S. Hosoi and S. Sato, In Vitro Cell. Dev. Biol., 1990, 26, 1101–1104 CrossRef.
- S. X. Gao, Y. Zhang, K. Stansberry-Perkins, A. Buko, S. Bai, V. Nguyen and M. L. Brader, Biotechnol. Bioeng., 2011, 108, 977–982 CrossRef PubMed.
- P. Elliott, A. Hohmann and J. Spanos, Biotechnol. Lett., 2003, 25, 1949–1952 CrossRef PubMed.
- D. Migani, C. M. Smales and D. G. Bracewell, Biotechnol. Prog., 2017, 33, 666–676 CrossRef PubMed.
- H. Dorai and S. Ganguly, Curr. Opin. Biotechnol., 2014, 30, 198–204 CrossRef PubMed.
- X. Li, B. Zhang, L. Tian, W. Li, H. Zhang and Q. Zhang, Sens. Actuators, B, 2015, 208, 559–568 CrossRef.
- K. Haupt and K. Mosbach, Chem. Rev., 2000, 100, 2495–2504 CrossRef PubMed.
- L. Ye and K. Mosbach, Chem. Mater., 2008, 20, 859–868 CrossRef.
- D. S. Janiak and P. Kofinas, Anal. Bioanal. Chem., 2007, 389, 399–404 CrossRef PubMed.
- D. Lakshmi and A. Bossi, Anal. Chem., 2009, 81, 3576–3584 CrossRef PubMed.
- J. O. Mahony, K. Nolan, M. R. Smyth and B. Mizaikoff, Anal. Chim. Acta, 2005, 534, 31–39 CrossRef.
- N. W. Turner, C. W. Jeans, K. R. Brain, C. J. Allender, V. Hlady and D. W. Britt, Biotechnol. Prog., 2006, 22, 1474–1489 CrossRef PubMed.
- B. Pluhar and B. Mizaikoff, Macromol. Biosci., 2015, 15, 1507–1511 CrossRef PubMed.
- B. Pluhar, U. Ziener and B. Mizaikoff, J. Mater. Chem. B, 2015, 3, 6248–6254 RSC.
- R. Gao, Y. Hao, L. Zhang, X. Cui, D. Liu, M. Zhang, Y. Tang and Y. Zheng, Chem. Eng. J., 2016, 284, 139–148 CrossRef.
- J. Wackerlig and R. Schirhagl, Anal. Chem., 2016, 88, 250–261 CrossRef PubMed.
- M. Dinc, H. Basan, T. Diemant, R. J. Behm, M. Linden and B. Mizaikoff, J. Mater. Chem. B, 2016, 4462–4469 RSC.
- M. Dinc, H. Basan, T. Hummel, M. Müller, H. Sobek, M. Lindén, I. Rapp, T. Diemant, R. J. Behm and B. Mizaikoff, ChemistrySelect, 2018, 3, 4277–4282 CrossRef.
- K. Eersels, P. Lieberzeit and P. Wagner, ACS Sens., 2016, 1, 1171–1187 CrossRef.
- H. Zhang, J. Jiang, H. Zhang, Y. Zhang and P. Sun, ACS Macro Lett., 2013, 2, 566–570 CrossRef.
- K. Erol, K. Köse, L. Uzun, R. Say and A. Denizli, Colloids Surf., B, 2016, 146, 567–576 CrossRef PubMed.
- S. Wei, M. Jakusch and B. Mizaikoff, Anal. Chim. Acta, 2006, 578, 50–58 CrossRef PubMed.
- J. Matsui, K. Fujiwara and T. Takeuchi, Anal. Chem., 2000, 72, 1810–1813 CrossRef PubMed.
- S. Lingxin, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- P. M. Colman, J. N. Jansonius and B. W. Matthews, J. Mol. Biol., 1972, 70, 701–724 CrossRef PubMed.
- P. A. M. Snoek-van Beurden and J. W. Von den Hoff, Biotechniques, 2005, 38, 73–83 CrossRef PubMed.
- C. Oh, J.-H. Lee, Y.-G. Lee, Y.-H. Lee, J.-W. Kim, H.-H. Kang and S.-G. Oh, Colloids Surf., B, 2006, 53, 225–232 CrossRef PubMed.
- R. Visse and H. Nagase, Circ. Res., 2003, 92, 827–839 CrossRef PubMed.
- L. Troeberg and H. Nagase, Curr. Protoc. Protein Sci., 2004 Search PubMed , chapter 21, unit 21.15.
- R. S. Fernandes, M. Dinc, I. M. Raimundo and B. Mizaikoff, Microporous Mesoporous Mater., 2018, 264, 28–34 CrossRef.
- R. S. Fernandes, M. Dinc, I. Raimundo Jr and B. Mizaikoff, Anal. Methods, 2017, 8, 1–224 Search PubMed.
- T. Goda, K. Ishihara and Y. Miyahara, J. Appl. Polym. Sci., 2015, 132, 1–10 CrossRef.
- J. Park, S. Kurosawa, J. Watanabe and K. Ishihara, Anal. Chem., 2004, 76, 2649–2655 CrossRef PubMed.
- L. Wu, A. Tanimoto, Y. Murata, T. Sasaguri, J. Fan, Y. Sasaguri and T. Watanabe, Genes Cells, 2003, 8, 225–234 CrossRef PubMed.
- S. Rowsell, P. Hawtin, C. A. Minshull, H. Jepson, S. M. V Brockbank, D. G. Barratt, A. M. Slater, W. L. McPheat, D. Waterson, A. M. Henney and R. A. Pauptit, J. Mol. Biol., 2002, 319, 173–181 CrossRef PubMed.
- H. Nar, K. Werle, M. M. Bauer, H. Dollinger and B. Jung, J. Mol. Biol., 2001, 312, 743–751 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |