
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA low-cost electrochemical thio- and selenocyanation strategy for electron-rich arenes under catalyst- and oxidant-free conditions†
Xing Zhang ,
Chenguang Wang,
Hong Jiang* and
Linhao Sun*
,
Chenguang Wang,
Hong Jiang* and
Linhao Sun*
Department of Chemistry, College of Science, Huazhong Agricultural University, Wuhan, 430070, P. R. China. E-mail: jianghong@mail.hzau.edu.cn; sunlinhao@mail.hzau.edu.cn
First published on 14th June 2018
Abstract
A low-cost and efficient thio- and selenocyanation strategy for electron-rich arenes has been developed under constant-current electrolytic conditions in an undivided cell. This strategy is versatile for various (hetero)aromatic compounds such as indole, pyrrole, aniline and anisole under mild conditions without any catalyst or oxidant. Readily available salts NH4SCN and KSeCN are employed respectively as the sole reagent.
Introduction
Organosulfur and organoselenium compounds, possessing broad biological and pharmaceutical activities, have been widely employed as important scaffolds for medicinal chemistry. Some representative examples are shown in Fig. 1, such as arylthioindole,1 albendazole,2 and clioquinol derivatives.3 In synthetic methodology, thio- and selenocyanation have been paid much attention over the past decades because they are facile and efficient ways to incorporate sulfur and selenium elements into organic compounds.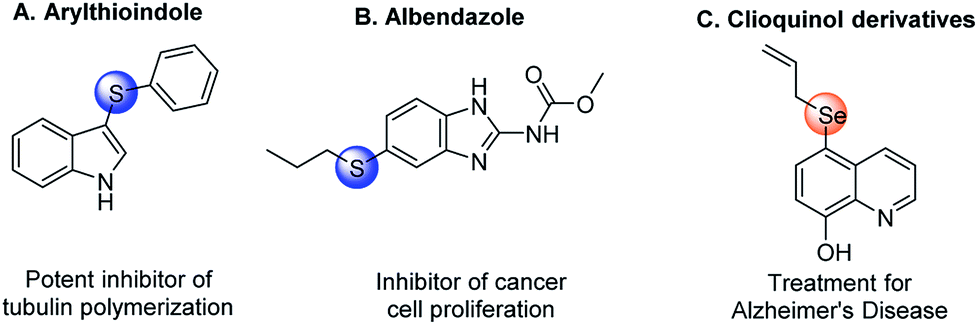 | ||
| Fig. 1 Examples of biologically active organosulfur and organoselenium compounds. | ||
Various methods for thiocyanation and selenocyanation of (hetero)aromatic systems have been established during the past few years. Lots of available methods of thiocyanation have combined thiocyanate salts with chemical oxidants such as CAN,4 oxone,5 DDQ,6 Cu/O2,7 hypervalent iodine reagents,8,9 Mn(OAc)3,10 K2S2O8,11,12 etc. (Scheme 1a) Despite their efficiencies, these approaches suffered from many drawbacks and limitations: harsh oxidizing experimental procedures, the use of stoichiometric oxidants and the bad impact of heavy-metal wastes. Recently photocatalysis13,14 and electrolysis15–17 have been proved to be eco-friendly alternative methods for oxidative aromatic thiocyanation (Scheme 1b and c). Nevertheless, the costly photocatalyst or supporting electrolyte is required and the substrate scope is still narrow in these approaches. As for selenocyanation, only a few orthodox methods have been reported using triselenium dicyanide18,19 or combining KSeCN with a chemical oxidant (CAN,20 TBHP21) as selenocyating reagent (Scheme 1d and e). Hence, the development of an efficient, eco-friendly and versatile strategy for thio- and selenocyanation is still desirable.
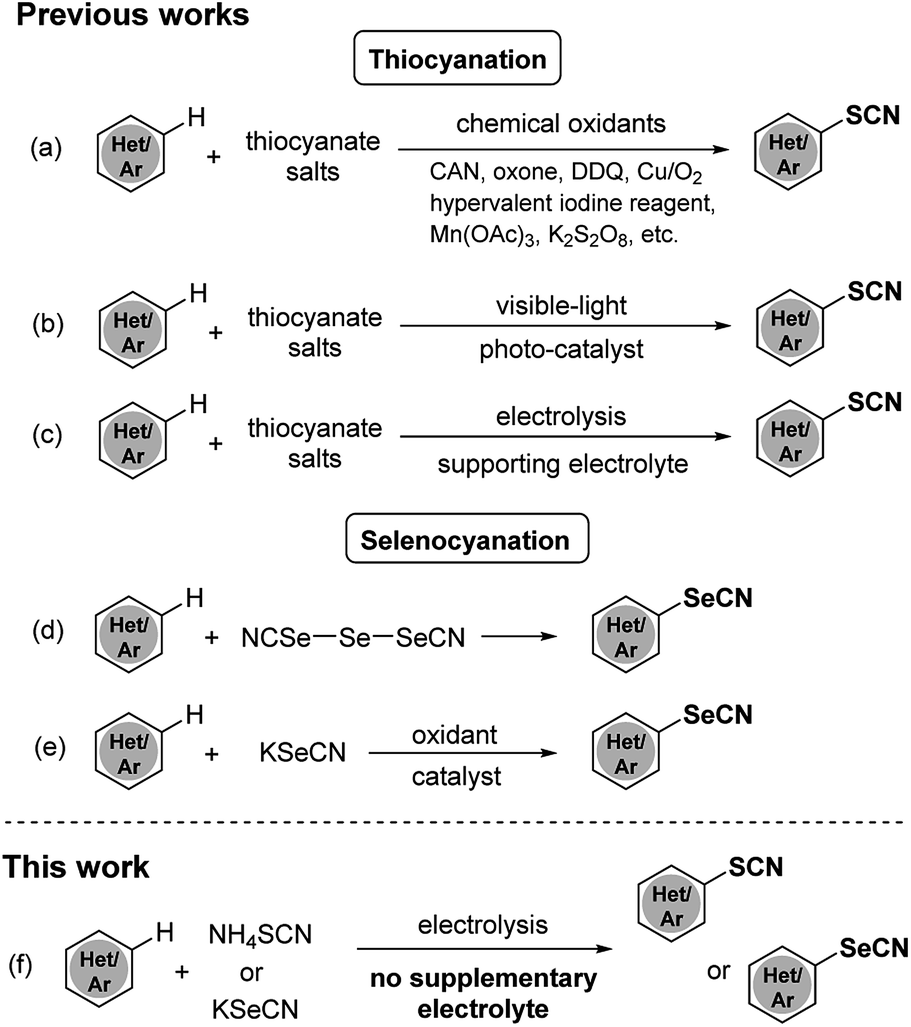 | ||
| Scheme 1 Thio- and selenocyanation strategies for (hetero)aromatic compounds. | ||
Recently, electrochemical functionalization of C–H bonds becomes a hot research topic in organic synthesis. Adopting electron as “reagent” in place of stoichiometric amounts of oxidants or reductants, electrosynthesis has been recognized as an eco-friendly synthetic tool.22,23 As part of our continuous works on electrochemical (pseudo)halogenation reactions,24 herein we present an efficient, low-cost, catalyst- and oxidant- free approach for thio- and selenocyanation of electron-rich arenes using thio- or selenocyanate salts as the sole reagent under mild electrochemical condition (Scheme 1f).
Result and discussion
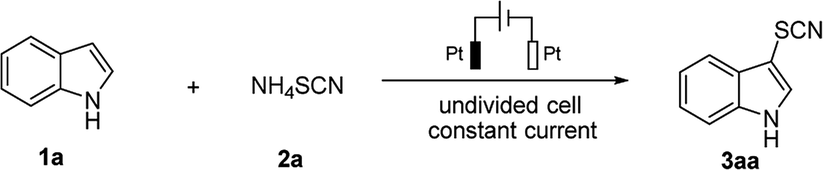
This work is initiated by optimizing the thiocyanation condition between 1H-indole (1a) and ammonium thiocyanate (2a) as a model reaction in an undivided cell. The desired thiocyanation product 3aa was obtained in 96% yield when the electrolysis was performed employing platinum as both anode and cathode under constant-current condition (janode ≈ 5.33 mA cm−2) in an electrolyte solution of CH3CN containing nBu4NPF6 as supporting electrolyte (Table 1, entry 1). Thiocyanation was totally suppressed without electric current (Table 1, entry 2) which led us to rule out the possibility of chemical thiocyanation between 1a and 2a. Atmospheric conditions led slight decrease to the yield of 3aa (Table 1, entry 3). To our delight, electrolysis could be conducted and resulted in an excellent yield without the supporting electrolyte salt nBu4NPF6 (Table 1, entry 4). In this case excessive amount of NH4SCN served as both thiocyanating reagent and electrolyte, so the budget of electrolysis has been remarkably reduced. Moreover the introduction of protonic solvent, such as water, resulted in yield loss (Table 1, entry 5). The highest yield was obtained when electrolysis was performed at higher constant current 18 mA (janode ≈ 8 mA cm−2) within 3 h (Table 1, entry 6). With these optimization studies, further exploration has been carried out using NH4SCN (3 equiv.) as the sole reagent in CH3CN at room temperature under constant current mode (janode ≈ 8 mA cm−2).
|
||
|---|---|---|
| Entry | Electrolysis conditions | Yieldb |
| a Standard conditions: Pt anode, Pt cathode, 1a (0.5 mmol), CH3CN (10 mL), room temperature, argon.b The yield of 3aa was determined by GC. | ||
| 1 | CH3CN, nBu4NPF6 (0.1 M), 2a (1 equiv.), 12 mA, 4.5 h | 96% |
| 2 | Entry 1 but no electric current | 0 |
| 3 | Entry 1 but open to air | 89% |
| 4 | CH3CN, 2a (3 equiv.), 12 mA, 4.5 h | 97% |
| 5 | Entry 4 but CH3CN/H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as solvent :1) as solvent |
78% |
| 6 | Entry 4 but 18 mA, 3 h | 99% |
With the optimized conditions in hand, we have tested the scope of this electrochemical thiocyanation strategy applying indoles derivatives and other electron-rich arenes as substrates (Scheme 2). Generally, electrochemical thiocyanation took place selectively at C3 position of indole derivatives; in the case of electron-rich arenes, the thiocyanation occurs at para or ortho position of arenes. Unsubstituted N–H indoles containing an electron donating group showed better reaction efficiency (products 3ab–3ah 94–99%) than those containing an electron withdrawing group (products 3ai–3ar 43–99%). N-Methyl indoles afforded the thiocyanated products in excellent yield (products 3as–3au 91–99%), however reaction could not occur when indole N–H is protected by “Boc” or “Ts” group, which may due to their strong electron-withdrawing effect. Moreover, 7-azaindole could also lead to product 3ax with 64% yield. The substrate scope of this method is validated: other electron-rich arenes such as pyrrole, aniline and anisole, also showed a great efficiency and afforded the desired product with good to excellent yields (products 3ba–3bj 72–97%).
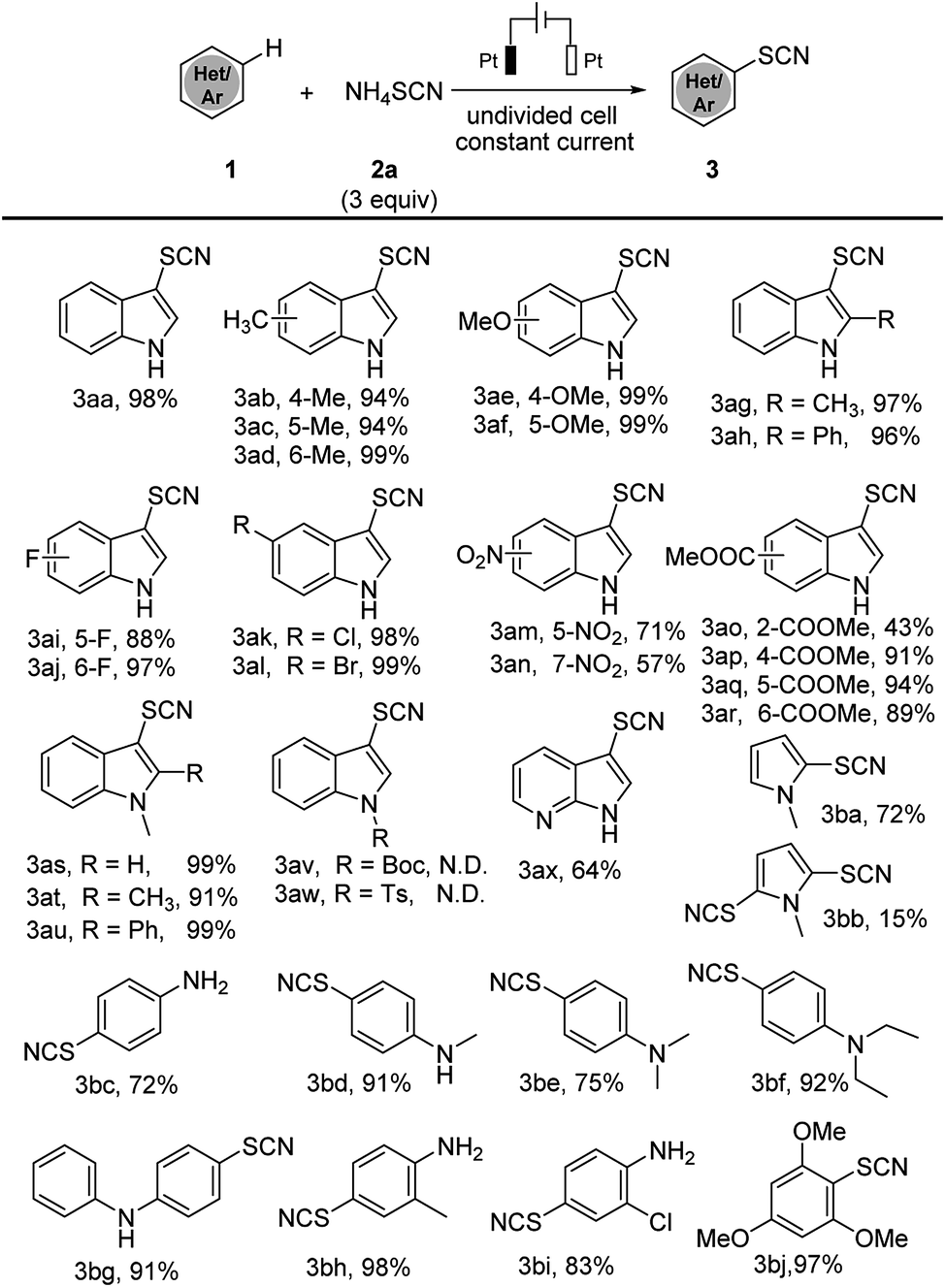 | ||
| Scheme 2 Substrate scope of electrochemical thiocyanation. Standard conditions: Pt anode, Pt cathode, constant current = 18 mA, 1 (0.5 mmol), 2a (1.5 mmol), CH3CN (10 mL), room temperature, argon, 3 h, isolated yields are shown. N.D. = not detected. | ||
The success of electrochemical thiocyanation of electron-rich arenes led us furtherly to explore the electrochemical selenocyanation. We continued our investigations applying the analogous condition as the electrochemical thiocyanation. By treating 1H-indole 1a with KSeCN (2 equiv.) as the sole reagent in CH3CN under constant-current mode, the corresponding selenocyanated product 5aa could be electrogenerated in 94% yield (Scheme 3). We summarized the substrate scope of electrochemical selenocyanation in Scheme 3. Electron-rich indole derivatives furnished the target products in good yields (Scheme 3, products 5aa–5ac). Electron-deficient indole derivatives such as 6-fluoro-, 5-fluoro, 5-chloro and 5-nitroindole could also lead to selenocyanated products in moderate to good yields (products 5ad–5ag 51–91%). When N-methylindole was used as a substrate, the selenocyanation yield was dramatically dropped to 41% (product 5ah). Interestingly, introducing a methyl group at the C-2-position of the N-methylindole afforded the selenocyanated product 5ai in 83% yield, which may due to the electron-donating effect. Unfortunately, the electrochemical selenocyanation could not work in this condition, only the aniline compounds could afford selenocyanated product in moderate yields (product 5ak 52%).
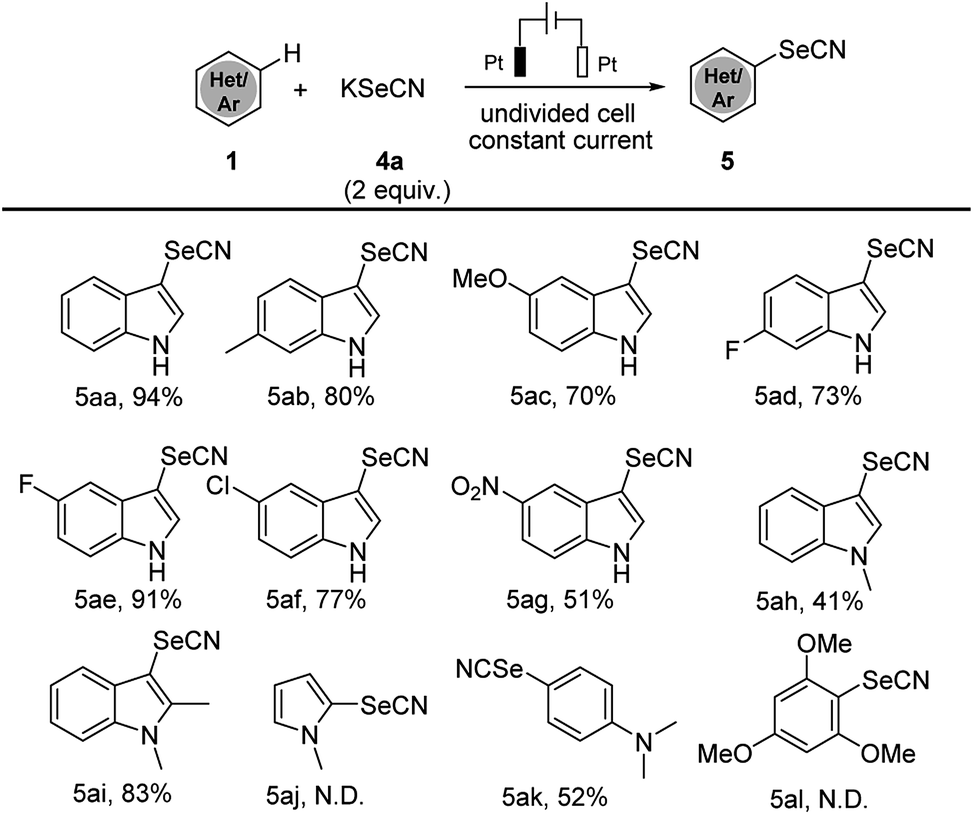 | ||
| Scheme 3 Substrate scope of electrochemical selenocyanation. Standard conditions: Pt anode, Pt cathode, constant current = 18 mA, 1 (0.5 mmol), 4a (1 mmol), CH3CN (10 mL), room temperature, argon, 3 h, isolated yields are shown. N.D. = not detected. | ||
The scalability of this approach was evaluated by performing the electrochemical thio- and selenocyanation of 1H-indole on a gram-scale. The electrolysis of 10 mmol substrate led to 80% isolated yield of 3aa (Scheme 4a) and 77% for 5aa (Scheme 4b). Moreover, compared with the traditional electrochemical approach,16,17,25 the double role of thio- or selenocyanated salt in this approach (as both SCN/SeCN source and supporting electrolyte) has remarkably reduced the budget of electrolysis, showing a great potential on industrial applications.
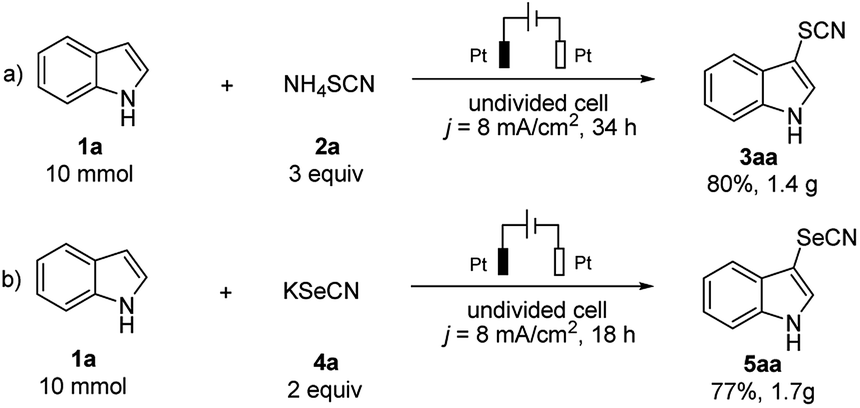 | ||
| Scheme 4 Gram-scale synthesis. | ||
In order to gain insights into the mechanism of the electrolytic process, we performed the radical inhibition experiments26,27 to verify the existence of radical intermediates. Since the oxidation potential of TEMPO is higher than XCN anion (X = S, Se, see ESI† for the voltammograms), it cannot be oxidized before XCN anion, so it could inhibit XCN radical during electrolysis. When an excess amount of TEMPO was added in the reaction between 1a and 2a or 4a under the standard electrolytic conditions, thio- and selenocyanation were totally suppressed (Scheme 5a). These results led us to confirm the presence of radical intermediates during electrolysis. In fact, literature reports16,17,22,25 have already demonstrated a mechanism via electrophilic aromatic substitution for the similar transformation. A putative reaction mechanism was proposed as illustrated in Scheme 5b.
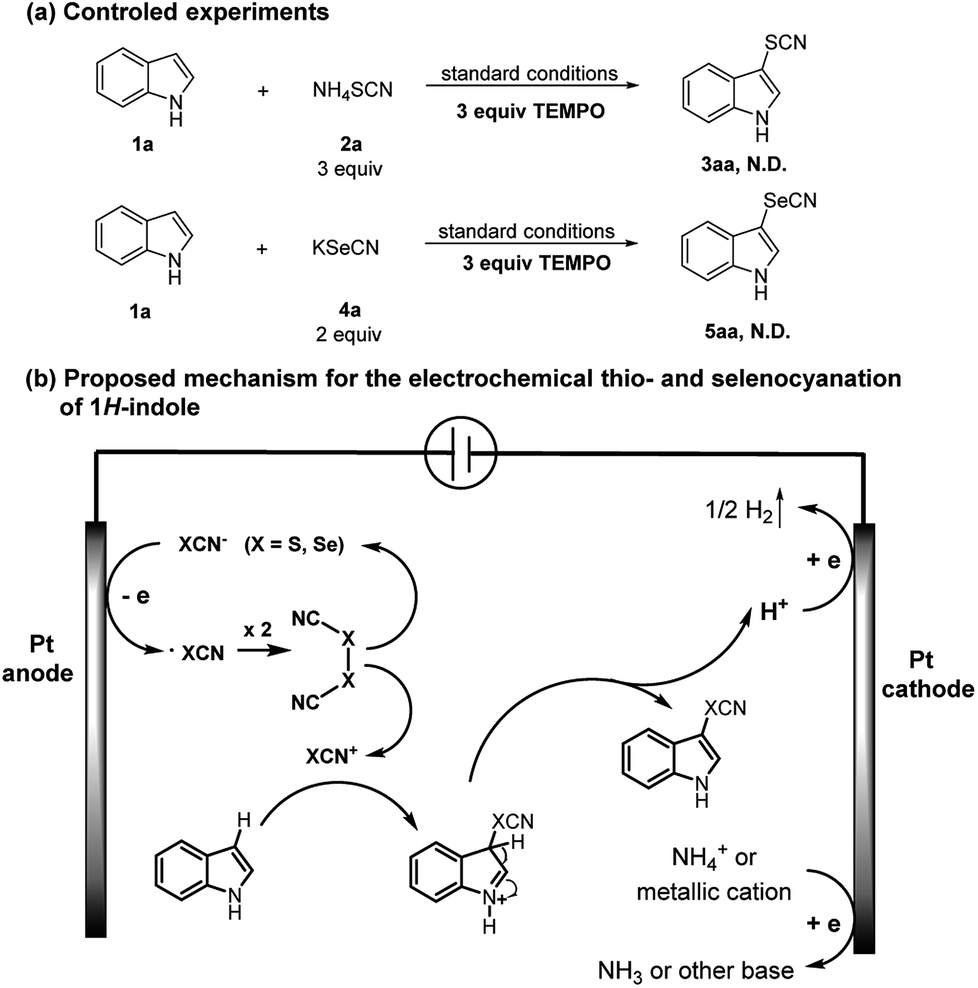 | ||
| Scheme 5 Mechanistic investigations and proposed mechanism. | ||
In the first step, thio- or selenocyanate anion gets oxidized by a single-electron-transfer process at the anode to generate the corresponding radical. (XCN)2 could be formed through radical coupling, which could furtherly generate the electrophile XCN+. After the electrophilic attack, the hydroindole cation intermediate can then release a proton to give the desired product. Concomitant cationic reduction involves the reduction of H+ to H2, which can be confirmed by the observation of gas release at the cathode surface during electrolysis. Another one-electron reduction may concern the cation from electrolyte (NH4+ or K+) leading the accumulation of base during electrolysis.
Conclusions
In summary, a facile, efficient and low-cost electrochemical thio- and selenocyanation strategy has been developed which enables C–S and C–Se bond formation under catalyst- and oxidant-free conditions. Various electron-rich arenes exhibited great efficiencies for this transformation. Importantly, the reaction can be performed on a gram scale with good reaction efficiency and the sole necessary reagent is readily available thio- or selenocyanated salts. These advantages encourage us to have prospect towards industrialization. Further studies on the electrochemical C–H functionalization are underway in our laboratory.Experimental
General procedure for thio- and selenocyanation of electron-rich arenes
In an oven-dried undivided four-necked bottle (25 mL) equipped with a stir bar, electron-rich arene (0.5 mmol), NH4SCN (1.5 mmol) or KSeCN (1.0 mmol) and CH3CN (10 mL) were combined and added. The bottle was equipped with platinum plate (1.5 × 1.5 cm2) as both the anode and cathode and was then charged with argon. The reaction mixture was stirred and electrolyzed at a constant current of 18 mA under room temperature for 3 h. When the reaction was finished, the solvent was removed with a rotary evaporator. The pure product was obtained by column chromatography on a silica gel column using petroleum ether: ethyl acetate = 10:3.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21502058) and the Fundamental Research Funds for the Central Universities (No. 2662015QC031).Notes and references
- G. De Martino, G. La Regina, A. Coluccia, M. C. Edler, M. C. Barbera, A. Brancale, E. Wilcox, E. Hamel, M. Artico and R. Silvestri, J. Med. Chem., 2004, 47, 6120–6123 CrossRef PubMed.
- Q. Guan, C. Han, D. Zuo, M. a. Zhai, Z. Li, Q. Zhang, Y. Zhai, X. Jiang, K. Bao, Y. Wu and W. Zhang, Eur. J. Med. Chem., 2014, 87, 306–315 CrossRef PubMed.
- Z. Wang, Y. Wang, W. Li, F. Mao, Y. Sun, L. Huang and X. Li, ACS Chem. Neurosci., 2014, 5, 952–962 CrossRef PubMed.
- V. Nair, T. G. George, L. G. Nair and S. B. Panicker, Tetrahedron Lett., 1999, 40, 1195–1196 CrossRef.
- G. Wu, Q. Liu, Y. Shen, W. Wu and L. Wu, Tetrahedron Lett., 2005, 46, 5831–5834 CrossRef.
- H. R. Memarian, I. Mohammadpoor-Baltork and K. Nikoofar, Can. J. Chem., 2007, 85, 930–937 CrossRef.
- H. Jiang, W. Yu, X. Tang, J. Li and W. Wu, J. Org. Chem., 2017, 82, 9312–9320 CrossRef PubMed.
- F. Wang, X. Yu, Z. Qi and X. Li, Chem.–Eur. J., 2016, 22, 511–516 CrossRef PubMed.
- J. Yadav, B. Reddy and B. Murali Krishna, Synthesis, 2008, 3779–3782 CrossRef.
- X.-Q. Pan, M.-Y. Lei, J.-P. Zou and W. Zhang, Tetrahedron Lett., 2009, 50, 347–349 CrossRef.
- D. Yang, K. Yan, W. Wei, G. Li, S. Lu, C. Zhao, L. Tian and H. Wang, J. Org. Chem., 2015, 80, 11073–11079 CrossRef PubMed.
- T. B. Mete, T. M. Khopade and R. G. Bhat, Tetrahedron Lett., 2017, 58, 415–418 CrossRef.
- W. Fan, Q. Yang, F. Xu and P. Li, J. Org. Chem., 2014, 79, 10588–10592 CrossRef PubMed.
- S. Mitra, M. Ghosh, S. Mishra and A. Hajra, J. Org. Chem., 2015, 80, 8275–8281 CrossRef PubMed.
- A. Gitkis and J. Y. Becker, Electrochim. Acta, 2010, 55, 5854–5859 CrossRef.
- L. Fotouhi and K. Nikoofar, Tetrahedron Lett., 2013, 54, 2903–2905 CrossRef.
- V. A. Kokorekin, V. L. Sigacheva and V. A. Petrosyan, Tetrahedron Lett., 2014, 55, 4306–4309 CrossRef.
- A. V. Kachanov, O. Y. Slabko, O. V. Baranova, E. V. Shilova and V. A. Kaminskii, Tetrahedron Lett., 2004, 45, 4461–4463 CrossRef.
- S. Redon, A. R. Obah Kosso, J. Broggi and P. Vanelle, Tetrahedron Lett., 2017, 58, 2771–2773 CrossRef.
- N. Vijay, A. Anu and G. G. Tesmol, Eur. J. Org. Chem., 2002, 2363–2366 Search PubMed.
- N. Muniraj, J. Dhineshkumar and K. R. Prabhu, ChemistrySelect, 2016, 1, 1033–1038 CrossRef.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef PubMed.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- L. Sun, X. Zhang, Z. Li, J. Ma, Z. Zeng and H. Jiang, Eur. J. Org. Chem., 2018 DOI:10.1002/ejoc.201800267.
- V. A. Kokorekin, R. R. Yaubasarova, S. V. Neverov and V. A. Petrosyan, Mendeleev Commun., 2016, 26, 413–414 CrossRef.
- T. Vogler and A. Studer, Synthesis, 2008, 2008, 1979–1993 CrossRef.
- P. Wang, S. Tang, P. Huang and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 3009–3013 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04407d |
| This journal is © The Royal Society of Chemistry 2018 |