
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCooperative effects between π-hole triel and π-hole chalcogen bonds†
Jingru Zhang,
Wenzuo Li*,
Jianbo Cheng,
Zhenbo Liu and
Qingzhong Li *
*
The Laboratory of Theoretical and Computational Chemistry, School of Chemistry and Chemical Engineering, Yantai University, Yantai 264005, People's Republic of China. E-mail: liwenzuo2004@126.com; liqingzhong1990@sina.com; Fax: +86 535 6902063; Tel: +86 535 6902063
First published on 25th July 2018
Abstract
MP2/aug-cc-pVTZ calculations have been performed on π-hole triel- and chalcogen-bonded complexes involving a heteroaromatic compound. These complexes are very stable with large interaction energy up to −47 kcal mol−1. The sp2-hybridized nitrogen atom engages in a stronger π-hole bond than the sp-hybridized species although the former has smaller negative electrostatic potential. The sp2-hybridized oxygen atom in 1,4-benzoquinone is a weaker electron donor in the π-hole bond than the sp2-hybridized nitrogen atom. The π-hole triel bond is stronger than the π-hole chalcogen bond. A clear structural deformation is found for the triel or chalcogen donor molecule in these π-hole-bonded complexes. The triel bond exhibits partially covalent interaction, whereas the chalcogen bond exhibits covalent interaction in the SO3 complexes of pyrazine and pyridine derivatives with a sp2-hybridized nitrogen atom. Intermolecular charge transfer (>0.2e) occurs to a considerable extent in these complexes. In ternary complexes involving an aromatic compound, wherein a triel bond and a chalcogen bond coexist, both the interactions are weakened or strengthened when the central aromatic molecule acts as a double Lewis base or plays a dual role of both a base and an acid. Both electrostatic and charge transfer effects have important contributions toward changes in the strength of both interactions.
1. Introduction
Intermolecular interactions play a crucial role in chemistry, biology, and materials science;1–3 they can lower the activation free energy of a reaction,4 govern the conformational stability and biological activity of proteins and other biological macromolecules,5 and modulate the functions and properties of materials.6 Other than the most important and prevalent intermolecular interaction, i.e., the H-bond, more types of intermolecular interactions have caused considerable interest.7–10 Herein, we focus on chalcogen and triel bonding interactions, wherein the group V and III atoms act as a Lewis acid center to bind with a base.11,12 For most of the cases, the origin of the acidic center of a chalcogen atom can be traced back to the presence of a region with positive electrostatic potentials (σ-hole) in the outer regions of a Ch–R bond.13 For a triel atom, this region is often vertical to the planar molecule or a planar portion of a molecular framework (π-hole).14 Some group V molecules, such as SO3, also have such π-holes.15 It should be pointed out that the π-hole triel bond has a completely different nature from the π-hole in chalcogen bond since in the literature, a triel bond is referred as a dative bond.16Due to their importance in crystalline materials and biological systems, many studies have been conducted on chalcogen bonds by means of experimental and theoretical methods.17–27 Chalcogen-bonded complexes of SOF2, SOFCl, and SOCl2 with a series of N bases are stabilized by coulombic attractions of N to the σ-hole opposite the S atom as well as by the LpN→σ* (S–Z) electron transfer (Z = O, F, Cl).17 Tetravalent SF4 also has one σ-hole along the extension of the S–F bond, and the strength of chalcogen bonding between tetravalent SF4 and amines is similar to that with divalent SF2.18 In realizing its functions in crystalline materials and biological systems, chalcogen bonding often coexists with more than one interaction including that with itself, exhibiting cooperative effects. For instance, bidentate chalcogen bonding increases the association constant by an order of magnitude in binding of perfluoroaryl-substituted tellurophenes with an anion.19 Thus, substantial theoretical attention has been paid to the cooperative effect of chalcogen bonding with itself and other types of interactions,26–33 showing some interesting results. For instance, although SO3⋯NH3⋯NH3 is more stable than NH3–SO3⋯NH3, the latter is identified in argon matrixes, where the two N–S bonds are nonequivalent.33
A trivalent triel atom in TrR3, owing to its electron deficiency, is usually used to bind with Lewis bases such as HCN, CH3CN, and NH3.34–41 Upon complexation, the geometric structure of TrR3 exhibits substantial deviation from the planar one, and it becomes more prominent for a stronger triel bond. There is clear difference between the solid- and gas-phase structures of HCN⋯BF3, which is mainly caused by the cooperative effect in the solid.42 Thus, there is growing interest regarding the cooperative effect of a triel bond with itself and other interactions.43–48 In BF3⋯NCH⋯NCH, where a triel bond and a hydrogen bond coexist, both B⋯N and H⋯N distances are shortened, but the shortening of the B⋯N distance in the stronger triel bond is larger than that of the H⋯N distance in the weaker hydrogen bond.43 This abnormal result is also found in similar complexes composed of a triel bond and a halogen bond47 or a pnicogen bond.47
In this paper, we study the interplay between the triel bond and chalcogen bond in the complexes of pyrazine, 1,4-dicyanobenzene, and 1,4-benzoquinone as well as their derivatives. These three molecules are often used in studying the cooperative effects between an anion–π interaction and other interactions including hydrogen bonding,49 halogen bonding,50 chalcogen bonding,51 pnicogen bonding,52 tetrel bonding,53 triel bonding,54 and metal–Lp interactions.55 It should be noted that there are two important reviews on cooperative effects.56,57 Their derivatives contain 4-BH2-pyrazine, 4-BF2-pyrazine, 1-CN-4-BH2-benzene, and 1-CN-4-BF2-benzene although these molecules are not found in the Cambridge Structural Database. We focus on the comparison of the strengths of triel bonding among these molecules, comparison of the strengths of chalcogen bonding among these molecules, and interplay between triel bonding and chalcogen bonding. The results are estimated by means of binding distances, interaction energies, and electron densities. To reveal the mechanism for the cooperative effects in these systems, charge transfer and molecular electrostatic potentials are analyzed for these complexes.
2. Theoretical methods
All the calculations were performed using the Gaussian 09 program.58 All the complexes were first optimized using the second-order Møller–Plesset perturbation theory (MP2) level with the aug-cc-pVDZ basis set. Frequency analysis at the same computational level was then applied to confirm that the optimized geometries corresponded to stationary points with no imaginary frequencies. Finally, the complexes with MP2/aug-cc-pVDZ geometries were again optimized at the MP2/aug-cc-pVTZ level. The interaction energy (ΔEint) in the binary system was obtained by subtracting the energies of the monomers frozen in the complex from the energy of the complex. If the energies of the optimized monomers are used as the reference, the binding energy (ΔEb) can be calculated. The difference between ΔEb and ΔEint is denoted as the deformation energy (DE). Both ΔEb and ΔEint were corrected from the basis set superposition error (BSSE) by the counterpoise procedure suggested by Boys and Bernardi.59 The total interaction energy (ΔEtotal) in the ternary systems and the interaction energy between the molecule pair were calculated with a similar supramolecular method using the following formulas: ΔEtotal = EABC − EA − EB − EC, ΔEA–BC = EA–BC − EA−EBC, and ΔEAB–C = EAB–C − EAB − EC, where the energies of the monomers and dyads are from the ternary complex. The interplay between the two interactions in the ternary system was estimated with cooperative energy (Ecoop), which was obtained by Ecoop = ΔEtotal − ΔEA–B − ΔEB–C − ΔEA–C; here, ΔEA–B and ΔEB–C are the interaction energies of the optimized binary systems, and ΔEA–C is the interaction energy between the two unbonded molecules in the ternary system.Molecular electrostatic potentials (MEPs) were computed on the 0.001 au electron density contour at the MP2-aug-cc-pVTZ level with the Wave Function Analysis-Surface Analysis-Suite (WFA-SAS) program.60 The topological analysis of all the complexes was carried out using Bader's theory of atoms in molecules (AIM) with the help of the AIM2000 software.61 The contour of the Laplacian of the electron density was plotted by the Multiwfn software.62 Natural bond orbital (NBO) analysis was performed at the HF/aug-cc-pVTZ level via the NBO 3.1 procedure included in Gaussian 09 to analyze charge transfer.63
3. Results and discussion
3.1. MEPs of monomers
Fig. 1 shows the MEP maps of the studied monomers. For BH3 (2), BF3 (3), and SO3 (4), there are two π-holes (red region) along the vertical direction of the molecular plane. The most positive MEP on the π-hole is larger in BF3 than that in BH3, which is consistent with previously reported results.14 The most positive MEP on the π-hole is almost equal for BF3 and SO3. The π-holes in BH3/BF3 and SO3 thus can form a triel bond and a chalcogen bond with a base, respectively. A blue region with negative MEPs is found around the N/O atom for NH3 (1), pyrazine (5), 1,4-dicyanobenzene (6), and 1,4-benzoquinone (7). Due to sp3 hybridization, the N atom of 1 has larger negative MEP than those for 5 and 6. It is thus not difficult to understand the fact that NH3 often acts as the Lewis base in intermolecular interactions. The N atom of 5 has smaller negative MEP than that for 6 due to greater delocalization in the former. In 8–11, a red region (π-hole) and a blue area are found on the B and N atoms, respectively. The π-hole on the B atom in 8–11 has smaller positive MEP than those in 2 and 3, indicating that the aromatic ring is electron-donating. The stronger electron-withdrawing ability of CN in 9/11 results in larger positive MEP on the B atom relative to that in 8/10. The negative MEP on the N atom in 10/11 is smaller than that in 8/9 due to stronger electron-withdrawing ability of BF2.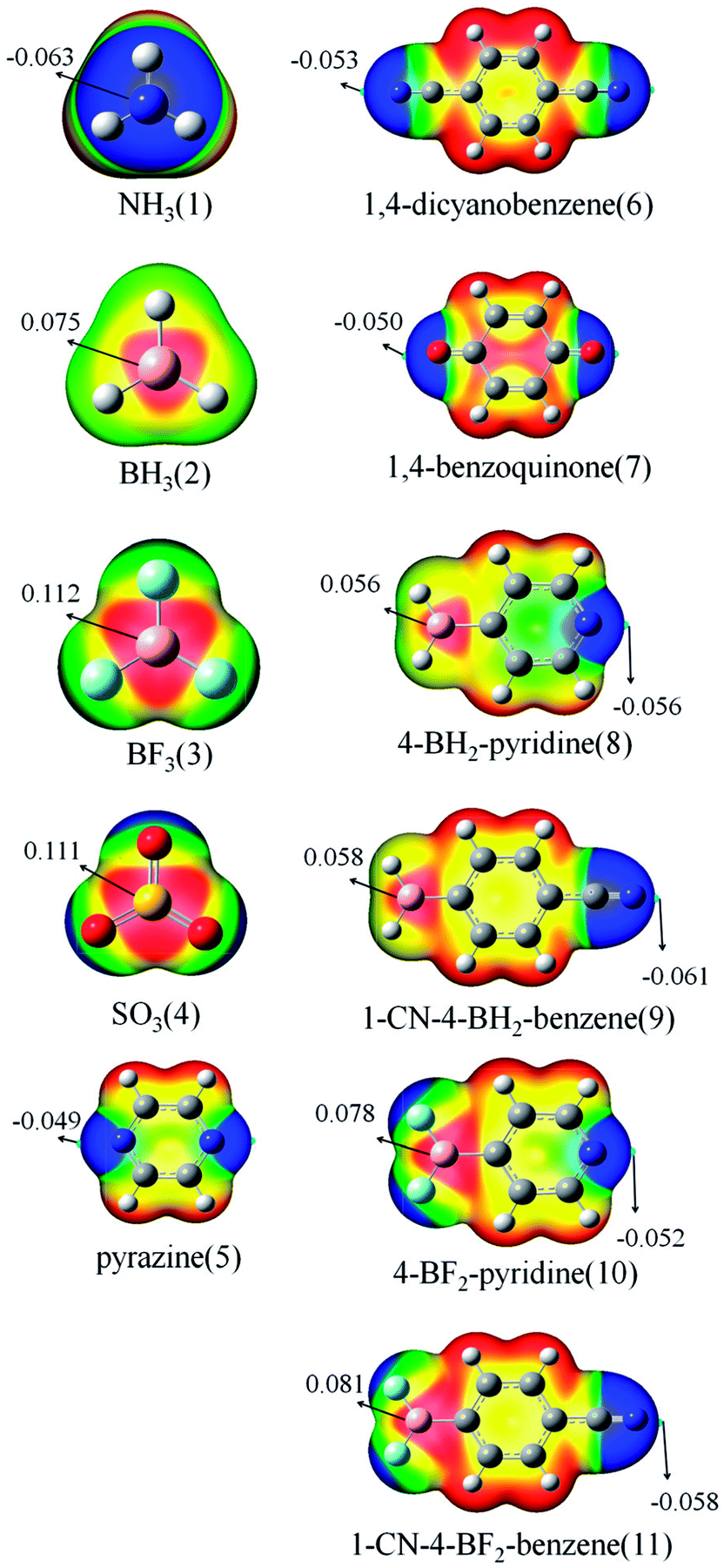 | ||
| Fig. 1 MEP maps of the monomers. Color ranges, in eV, are as follows: red, greater than 0.0210; yellow, between 0.0210 and −0.0028; green, between −0.0028 and −0.0150; and blue, less than −0.0150. | ||
3.2. Triel-bonded dyads
Fig. 2 shows the optimized structures of ten triel-bonded dyads. These structures have approximate Cs symmetry. The plane of symmetry is along the aromatic ring in ZB-2, ZB-3, ZB-5, and ZB-6, whereas it is vertical to the aromatic ring along the B–N–N or B–C–N axis in the other dyads. A clear change in geometry is that the B-containing molecule deviates from the planar structure, and this phenomenon is often observed in triel-bonded complexes.14 The B–C bond exhibits smaller deviation than the B–H/F bond in ZB-8, ZB-9, ZB-10, and ZB-11 due to the stronger B–C bond. To estimate the deformation of the B-containing molecule, we list the average of the three N⋯B–H/F angles in ZB-1 to ZB-6 except the average of the two N⋯B–H/F angles and one N⋯B–C angle in the other dyads (Table 1). The deformation is larger if this average value has larger deviation from 90°; this value is larger than 90° for all the complexes. Thus, the B-containing molecule exhibits deformation. The largest deformation is found in ZB-1, whereas the smallest one is found in ZB-5. It is noted that such an indicator is only an approximate estimation of deformation.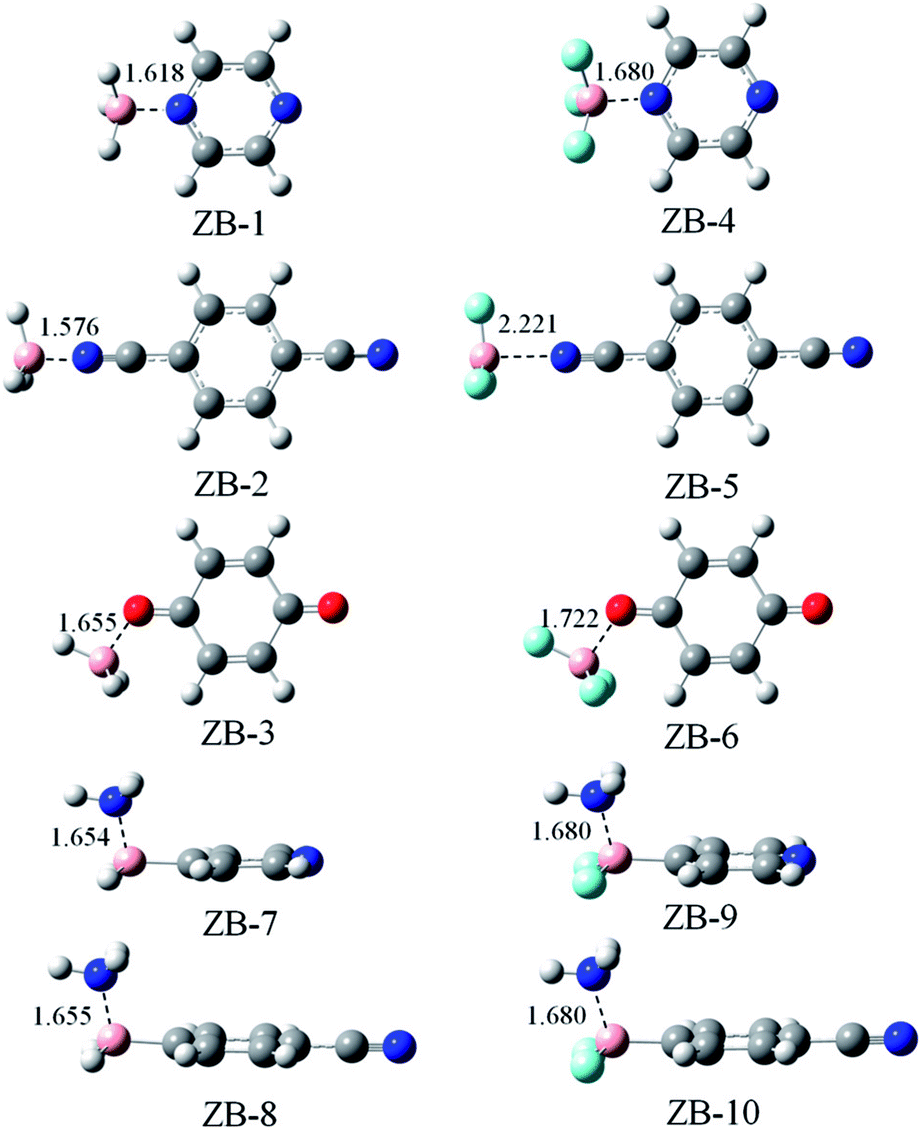 | ||
| Fig. 2 Optimized structures of the triel-bonded binary complexes. Distances are given in Å. | ||
| Dyads | ΔEint | ΔEb | DE | ΔEZPEb | ΔG | α |
|---|---|---|---|---|---|---|
| a ΔEZPEb is the binding energy corrected for zero-point vibrational energy (ZPE). α is the average of the three N⋯B–H/F angles in the former six dyads but the average of the two N⋯B–H/F angles and one N⋯B–C angle in the latter four dyads. | ||||||
| 2 + 5(ZB-1) | −46.81 | −34.42 | 14.03 | −30.51 | −20.67 | 104.8 |
| 2 + 6(ZB-2) | −33.03 | −21.60 | 12.70 | −18.30 | −7.69 | 104.4 |
| 2 + 7(ZB-3) | −27.44 | −17.79 | 11.15 | −13.99 | −3.20 | 103.1 |
| 3 + 5(ZB-4) | −44.52 | −23.08 | 24.47 | −21.24 | −14.28 | 104.2 |
| 3 + 6(ZB-5) | −8.79 | −7.04 | 3.10 | −6.35 | −0.07 | 95.4 |
| 3 + 7(ZB-6) | −25.40 | −10.82 | 17.28 | −9.40 | −0.51 | 102.0 |
| 1 + 8(ZB-7) | −42.06 | −29.50 | 14.17 | −24.86 | −15.29 | 104.4 |
| 1 + 9(ZB-8) | −41.70 | −29.04 | 14.29 | −24.46 | −14.83 | 104.5 |
| 1 + 10(ZB-9) | −37.38 | −19.28 | 20.33 | −15.81 | −9.11 | 103.7 |
| 1 + 11(ZB-10) | −37.53 | −19.22 | 20.53 | −15.77 | −9.00 | 103.8 |
The binding distance is also shown in Fig. 2. The B⋯N distance is in the range of 1.576–2.221 Å, whereas the B⋯O distance varies from 1.655 Å in ZB-3 to 1.722 Å in ZB-6. These distances are much shorter than the sum of the van der Waals (vdW) radii of the corresponding atoms (3.68 Å for B⋯N and 3.66 Å for B⋯O), which indicates that the triel bond is very strong. ZB-2 has shorter distance than ZB-1, whereas ZB-5 has longer distance than ZB-4; the former is consistent with negative MEPs on the N atoms of 5 and 6, whereas the latter shows an inconsistent change. The F-substitution lengthens the binding distance, which is inconsistent with the changes in the positive MEP on the B atom. This implies that electrostatic interaction is not the sole dominant factor in stabilizing the triel-bonded complexes.
Table 1 presents interaction energy and binding energy; their difference is denoted as the deformation energy, which can be used to measure the deformation of both the subsystems in the complex. The deformation energy is relatively large, i.e., from 3.10 kcal mol−1 in ZB-5 to 24.47 kcal mol−1 in ZB-4. The deformation energy amounts to 30–68% of the interaction energy; thus, its contribution is very important. Larger deformation energy corresponds to larger interaction energy although they do not have a linear relationship. In the following discussion, the interaction energy is utilized to estimate the stability of the triel-bonded complex.
The interaction energy varies from −8.79 kcal mol−1 in ZB-5 to −46.81 kcal mol−1 in ZB-1. When BH3 and BF3 are considered, the interaction energy is more negative in the order of ZB-3 < ZB-2 < ZB-1 and ZB-5 < ZB-6 < ZB-4, respectively. Both orders are inconsistent with the negative MEP on the N/O atom; this shows that the N atom of pyrazine has strong affinity toward the B atom, which is similar to that observed for the tetrel atom in tetrel bonds.63 ZB-7 has slightly larger interaction energy than ZB-8, which is inconsistent with the positive MEP on the B atom. However, the interaction energy and deformation energy in ZB-9 are almost equal to those in ZB-10 although the positive MEP on the B atom is larger in 11 than that in 10. The B–F system has smaller interaction energy than the B–H analogue. Interestingly, the interaction energy reduces by 24.24 kcal mol−1 in ZB-5 when compared with that for ZB-2. Although the values of the interaction energy and binding energy have large difference in most systems, their variations are similar, and both are thus appropriate for estimating the relative stability of the complex. The binding energy is reduced by 7–18% if it is corrected for zero-point vibrational energy (ZPE). Nonetheless, its variation tendency remains unchanged. For the triel-bonded complexes, the binding processes are exergonic (ΔG < 0) at 298 K. The entropy change (ΔS) is negative in the formation of complexes; thus, the ΔH term is dominant over the unfavorable TΔS term. Generally, more negative ΔG results in stronger triel bond.
There are some studies on triel bonds between BH3/BF3 and some small compounds containing N. Herein, we compare our results with those of the previous studies. The interaction energies were calculated to be −44.8, −6.7 and −1.8 kcal mol−1 in the BF3 complexes with NH3, HCN, and N2, respectively.64 Thus, the N atoms of pyrazine and NH3 indicated similar affinities toward the B atom of BF3 although the latter exhibited more negative MEP due to sp3 hybridization; moreover, it exhibited stronger affinity toward the B atom of BH3 than the N atom of NH3 since the interaction energy was about −30 kcal mol−1 in the latter.35 For the BF3⋯CH3CN complex, two configurations were found with the interaction energies of −7.7 and −8.7 kcal mol−1.65,66 Clearly, its interaction energy was comparable with that of ZB-5. This comparison indicated that pyrazine and 1,4-dicyanobenzene are good electron donors in triel bonds.
A strong triel bond results in substantial charge transfer from the electron acceptor to the triel donor (Table 2). The charge transfer is larger than 0.2e in most complexes except that for ZB-5. The large charge transfer is chiefly due to the deformation of monomers; Grabowski stated that the deformation energy is strongly related to the above-mentioned charge transfer.67 Although no linear relationship is found between charge transfer and interaction energy, they exhibit a consistent change.
| Dyads | ρBCP | ∇2ρBCP | HBCP | CT |
|---|---|---|---|---|
| a CT is the sum of the NBO charge on all the atoms of the electron donor molecule. | ||||
| ZB-1 | 0.109 | 0.315 | −0.074 | 0.3223 |
| ZB-2 | 0.098 | 0.064 | −0.005 | 0.3133 |
| ZB-3 | 0.072 | 0.564 | −0.018 | 0.2578 |
| ZB-4 | 0.106 | 0.315 | −0.074 | 0.2917 |
| ZB-5 | 0.026 | 0.064 | −0.005 | 0.0488 |
| ZB-6 | 0.073 | 0.317 | −0.038 | 0.2129 |
| ZB-7 | 0.103 | 0.503 | −0.061 | 0.3662 |
| ZB-8 | 0.103 | 0.502 | −0.061 | 0.3659 |
| ZB-9 | 0.104 | 0.351 | −0.070 | 0.3324 |
| ZB-10 | 0.104 | 0.349 | −0.070 | 0.3324 |
The presence of a B⋯N/O bond critical point (BCP) is used to characterize the triel bond (not shown), and its electron density (ρBCP), Laplacian (∇2ρBCP), and total electron energy density (HBCP) are shown in Table 2. The electron density is in the range of 0.026–0.109 au, and this value is large in most complexes. For all the complexes, ∇2ρBCP is positive and HBCP is negative. Therefore, the triel bond belongs to a partially covalent interaction based on the classification of the interaction suggested by Arnold and Oldfield.68 The contour plots of ∇2ρBCP for the triel-bonded complexes are shown in Fig. S1.† Green solid lines represent the region of ∇2ρBCP > 0, and purple dashed lines highlight the area of ∇2ρBCP < 0. For the cases where ∇2ρBCP is positive and HBCP is highly negative, we get a region where electron density is accumulated between the two centers; BCP lies just outside this region in most cases. This phenomenon was reported in the XeBeCN2 cluster69 and B3Ng3+ (Ng = Ar–Rn) complexes.70 Previously, the sign of HBCP was used to characterize the covalent properties of a bond.71 It should be noted that ∇2ρBCP is not sufficient to describe a covalent bond including covalent bonds exhibited by elements other than the first row elements.72–74 In addition, in some cases, HBCP gives a highly negative value even for ionic bonds.75–78
3.3. Chalcogen-bonded dyads
Fig. 3 shows the optimized structures of seven chalcogen-bonded complexes represented from CB-1 to CB-7. These structures also exhibit Cs symmetry similar to the triel-bonded complexes, as shown in Fig. 2. In CB-1, 3, 4, and 6, there are two weak H⋯O interactions other than the chalcogen bond. The geometry of SO3 also deviates from a planar structure, and its deviation is estimated with the average of the three N/O⋯S–O angles (Table 3) since these angles are larger than 90° in chalcogen-bonded complexes. The deformation of SO3 in chalcogen-bonded complexes is smaller than that in a B-containing molecule in triel-bonded complexes due to the smaller N/O⋯S–O angle. The deformation of both the molecules is consistent with the deformation energy (Table 3). This value is larger than 8 kcal mol−1 in the complexes with the sp2-hybridized N atom as the electron donor, whereas it is smaller when the sp-hybridized N atom acts as the electron donor. The deformation energy in the chalcogen-bonded complexes is smaller than that in the triel-bonded complex, that is, the geometrical deformation of the subsystems in the former is smaller than that in the latter.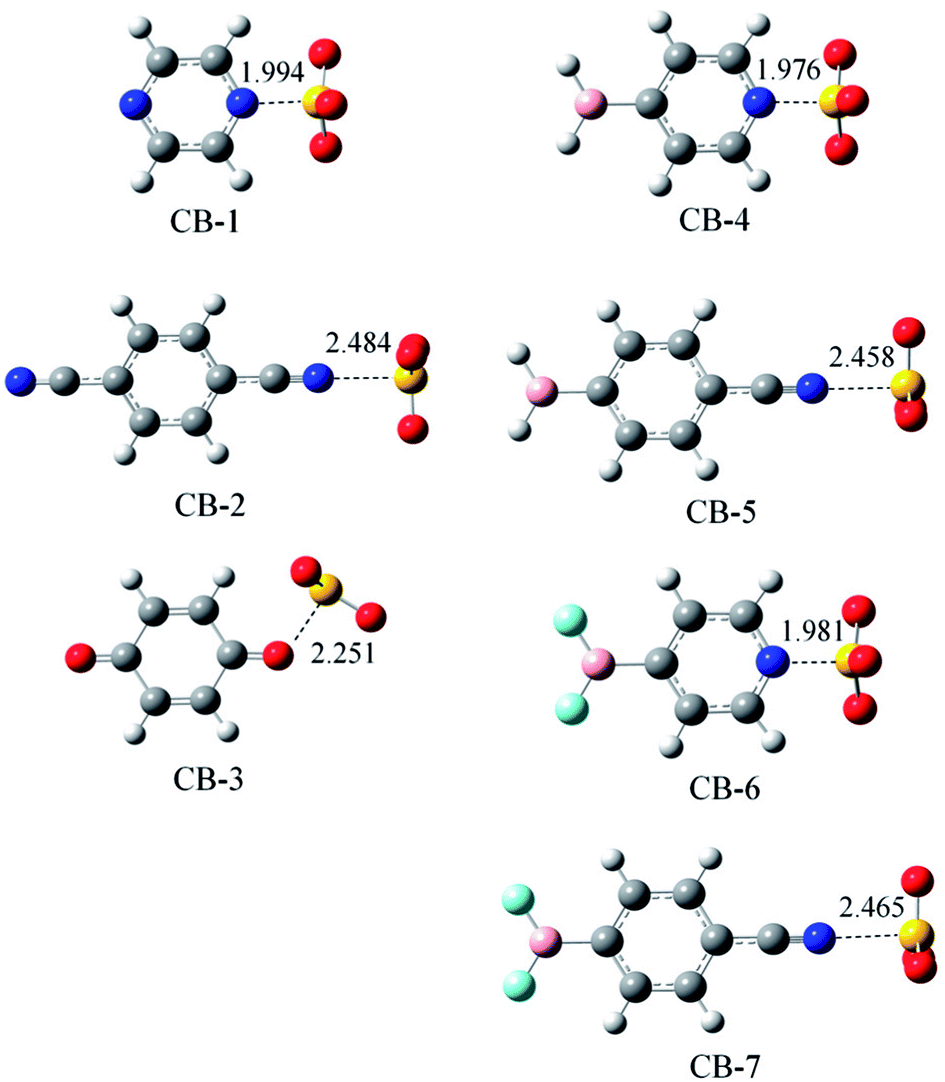 | ||
| Fig. 3 Optimized structures of the chalcogen-bonded binary complexes. Distances are given in Å. | ||
| Dyads | ΔEint | ΔEb | DE | ΔEZPEb | ΔG | α |
|---|---|---|---|---|---|---|
| a α is the average of the three N/O⋯S–O angles. | ||||||
| 4 + 5(CB-1) | −29.93 | −21.81 | 8.12 | −19.96 | −8.04 | 97.4 |
| 4 + 6(CB-2) | −8.67 | −8.11 | 0.56 | −7.31 | −0.28 | 92.1 |
| 4 + 7(CB-3) | −12.25 | −10.04 | 2.21 | −8.91 | 0.28 | 93.8 |
| 4 + 8(CB-4) | −33.48 | −24.47 | 9.01 | −22.55 | −10.39 | 97.8 |
| 4 + 9(CB-5) | −9.26 | −8.57 | 0.69 | −7.74 | −0.33 | 92.3 |
| 4 + 10(CB-6) | −32.61 | −23.77 | 8.84 | −21.90 | −9.71 | 97.7 |
| 4 + 10(CB-7) | −9.12 | −8.47 | 0.65 | −7.65 | −0.27 | 92.2 |
Since the sp2-hybridized N atom can lose electrons more easily than the sp-hybridized N atom, the chalcogen bond is stronger in CB-1 than that in CB-2, as evidenced by the shorter N⋯S distance and larger interaction energy. Similarly, the sp2-hybridized N atom in CB-4/6 forms a stronger chalcogen bond than the sp-hybridized N atom in CB-5/7. Similar to that in the triel bond, the oxygen atom of 1,4-benzoquinone engages in a weaker chalcogen bond than the nitrogen atom of pyrazine. The BF2 substituent decreases the chalcogen bond relative to the BH2 substituent. Although the π-hole of SO3 has more negative MEP than that of BH3, the chalcogen bond is weaker than the corresponding triel bond; the main reason is the larger deformation energy in the latter. For the chalcogen bond, the binding energy also has a consistent change with the interaction energy, and the reduction caused by ZPE (7–11%) is smaller than that in the triel bond. At 298 K, ΔG is negative in most chalcogen-bonded complexes excluding CB-3. The positive ΔG value of CB-3 indicates that the formation of this complex is an endergonic process.
The chalcogen bond leads to smaller charge transfer than the corresponding triel bond. However, the charge transfer in the chalcogen bond has a linear relationship with the interaction energy (Fig. S2†). The electron density at S⋯N/O BCO supports change in the interaction energy (Table 4) since they have a linear relationship (Fig. S3†). HBCP is negative in all chalcogen-bonded complexes, whereas the values of ∇2ρBCP are positive in CB-2, 3, 5, and 7, but they are negative in CB-1, 4, and 6. Therefore, the chalcogen bond exhibits partially covalent interaction in the former and covalent interaction in the latter.
| Dyads | ρBCP | ∇2ρBCP | HBCP | CT |
|---|---|---|---|---|
| a CT is the sum of the NBO charge on all the atoms of the electron donor molecule. | ||||
| CB-1 | 0.1173 | −0.1305 | −0.0810 | 0.2535 |
| CB-2 | 0.0311 | 0.0875 | −0.0011 | 0.0227 |
| CB-3 | 0.0506 | 0.1020 | −0.0086 | 0.0719 |
| CB-4 | 0.1240 | −0.1656 | −0.0905 | 0.2781 |
| CB-5 | 0.0331 | 0.0889 | −0.0016 | 0.0268 |
| CB-6 | 0.1223 | −0.1563 | −0.0880 | 0.2715 |
| CB-7 | 0.0326 | 0.0887 | −0.0016 | 0.0258 |
The charge transfer values were calculated to be 0.0019, 0.010, 0.015, 0.21, and 0.26e in the SO3 complexes with N2, HCN, CH3CN, NH3, and NMe3, respectively.16 The larger charge transfer corresponded to a stronger chalcogen bond; thus, we can infer that NMe3 engages in a stronger chalcogen bond than the other four nitrogenated bases. The charge transfer in CB-1, 4, and 6 is almost equal to that in SO3⋯NMe3; thus, we can infer that the sp2-hybridized N atom in pyrazine, 4-BH2-pyridine, and 4-BF2-pyridine is a good electron donor in the chalcogen bond. The interaction energy is larger than −30 kcal mol−1 in CB-1, 4, and 6. It should be noted that the magnitude of charge transfer is related to the calculation methods used.79
3.4. Interplay between triel bond and chalcogen bond
Fig. 4 shows the optimized structures of ten ternary complexes, where a triel bond and a chalcogen bond coexist. The conformation of the triad is similar to that in a dyad. In the former six triads, the binding distances of both the triel bond and chalcogen bond are elongated relative to that in the corresponding dyad. Interestingly, the B⋯N distance is reduced in ZB-CB-2. Moreover, the lengthening of the S⋯N/O distance is much larger than that of the B⋯N/O distance in ZB-CB-1, 3, 4, and 6; in the latter four triads, the binding distances of both the triel bond and chalcogen bond are shortened, and the shortening is much larger for the chalcogen bond.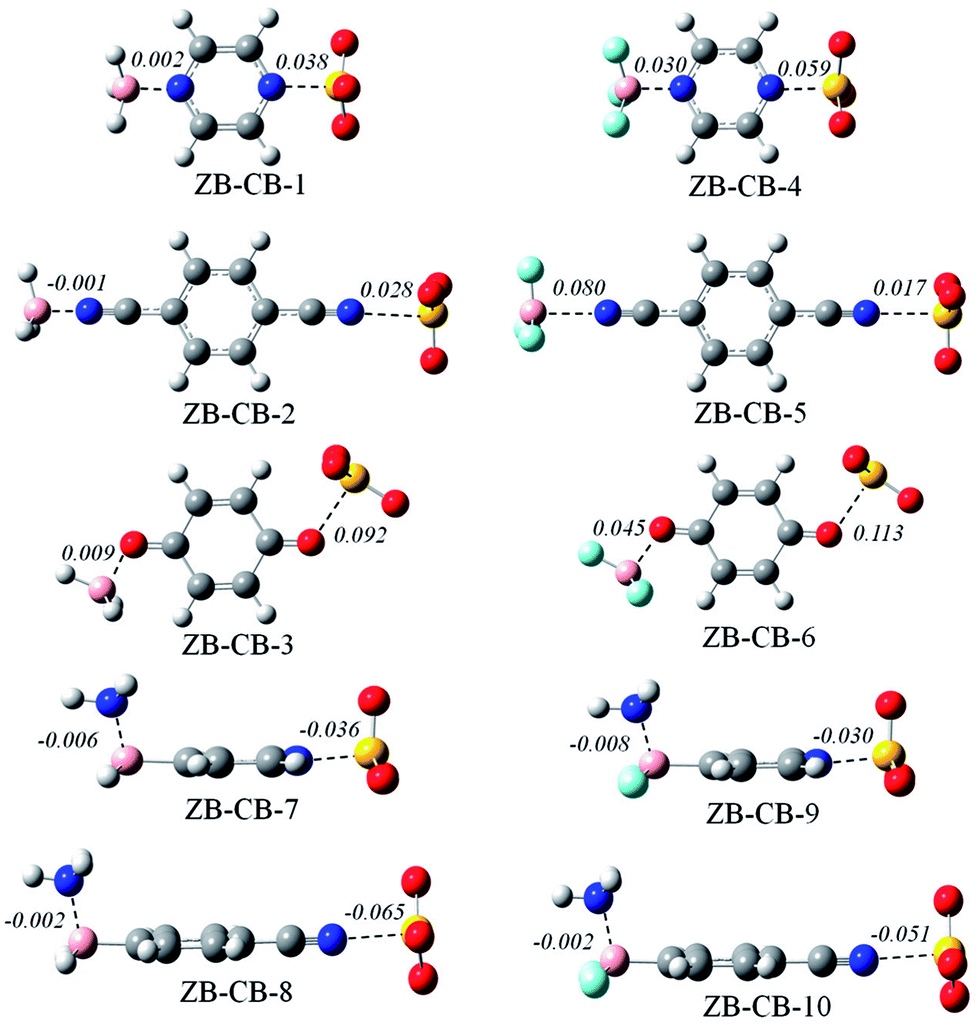 | ||
| Fig. 4 Optimized structures of the ternary complexes. Distances are given in Å. | ||
Table 5 presents the total interaction energy and the interaction energies of both the triel bond and chalcogen bond. The total interaction energy is dependent on the strength of both the interactions. The change (ΔΔE) in the interaction energies of both the triel bond and chalcogen bond is also listed in Table 5. ΔΔE is positive for both the interactions in the former six triads, but it is negative for both the interactions in the latter four triads. This indicates that both the interactions weaken from ZB-CB-1 to 6, but they are strengthened from ZB-CB-7 to 10. Generally, the interaction energies exhibit larger change when both types of interactions have comparable strengths. From ZB-CB-1 to 6, both N/O atoms in the central molecule are taken as a double Lewis base to form a triel bond and a chalcogen bond simultaneously; thus, they weaken each other. However, from ZB-CB-7 to 10, the central molecule plays the dual role of a Lewis acid and base to engage in the triel and chalcogen bonding interactions, respectively; thus, they are enhanced. The interaction energy between the two distant molecules is positive in ZB-CB-1 to 6, but it is negative in ZB-CB-7 to 10. Nonetheless, it is small in all the ternary systems.
| Triads | ΔEtotal | ΔEZB | ΔECB | ΔEfar | ΔΔEZB | ΔΔECB | Ecoop |
|---|---|---|---|---|---|---|---|
| a ΔΔE is the difference of ΔE in the triad relative to the corresponding dyad. ΔEfar is the interaction energy between two unbonded molecules in the ternary system. Data in parentheses denote the percentage of Ecoop to ΔEtotal. | |||||||
| ZB-CB-1 | −71.56 | −42.05 | −24.56 | 0.25 | 4.76 | 5.37 | 4.93(6.9) |
| ZB-CB-2 | −40.93 | −32.36 | −8.05 | 0.01 | 0.67 | 0.62 | 0.76(1.9) |
| ZB-CB-3 | −35.58 | −25.28 | −9.77 | 0.01 | 2.16 | 2.48 | 4.10(11.5) |
| ZB-CB-4 | −63.16 | −35.97 | −22.61 | 0.43 | 8.55 | 7.32 | 10.86(17.2) |
| ZB-CB-5 | −17.24 | −7.53 | −8.28 | 0.01 | 1.26 | 0.39 | 0.21(1.2) |
| ZB-CB-6 | −31.97 | −20.93 | −9.32 | 0.02 | 4.47 | 2.93 | 5.66(17.7) |
| ZB-CB-7 | −82.60 | −47.52 | −39.69 | −0.20 | −5.46 | −6.21 | −6.86(8.3) |
| ZB-CB-8 | −52.77 | −43.21 | −10.49 | −0.03 | −1.51 | −1.23 | −1.78(3.3) |
| ZB-CB-9 | −76.32 | −42.30 | −37.70 | −0.21 | −4.92 | −5.09 | −6.12(8.0) |
| ZB-CB-10 | −48.13 | −38.79 | −10.29 | −0.03 | −1.26 | −1.17 | −1.45(3.0) |
The interplay between both the interactions can be further estimated with cooperative energy. This term is positive in ZB-CB-1 to 6, but it is negative in ZB-CB-7 to 10, which indicates that there is negative synergistic effect in the former, but positive synergistic effect in the latter. In most cases, this effect is relatively prominent when the strength is sufficiently large for both the interactions. For instance, the cooperative energy amounts to 10.86 kcal mol−1 in ZB-CB-4, where the interaction energies of both the interactions are larger than −20 kcal mol−1. The cooperative energy corresponds to 1.2–17.7% of the total interaction energy, and it varies in a large range depending on the strength of both the interactions. This percentage in ZB-CB-1 to 10 is smaller than 17–55% in F3B⋯NCX⋯NCM (X, M = halogen), where a triel bond and a halogen bond coexist.47
The cooperative effect between both the interactions can also be evidenced by the change in the electron density at the intermolecular BCP in the ternary complex relative to that in the corresponding dyad (Table 6). The electron density decreases for both the types of BCPs in ZB-CB-1 to 6, but a reverse result is found in ZB-CB-7 to 10. The change in the electron density supports change in the strength of the corresponding interaction since electron density can be used to estimate the interaction strength. Moreover, the electron density exhibits larger change for the chalcogen bond.
| Triads | ρZB | ΔρZB | ρCB | ΔρCB |
|---|---|---|---|---|
| ZB-CB-1 | 0.1064 | −0.0028 | 0.1060 | −0.0113 |
| ZB-CB-2 | 0.1040 | −0.0001 | 0.0290 | −0.0021 |
| ZB-CB-3 | 0.0746 | −0.0027 | 0.0401 | −0.0105 |
| ZB-CB-4 | 0.0959 | −0.0097 | 0.1004 | −0.0169 |
| ZB-CB-5 | 0.0221 | −0.0041 | 0.0298 | −0.0013 |
| ZB-CB-6 | 0.0651 | −0.0084 | 0.0381 | −0.0126 |
| ZB-CB-7 | 0.1054 | 0.0026 | 0.1356 | 0.0116 |
| ZB-CB-8 | 0.1035 | 0.0007 | 0.0388 | 0.0057 |
| ZB-CB-9 | 0.1070 | 0.0032 | 0.1320 | 0.0098 |
| ZB-CB-10 | 0.1046 | 0.0008 | 0.0369 | 0.0043 |
Although deformation energy is very important in the triel bond and chalcogen bond, it was demonstrated that electrostatic interaction is still the main driving force in the formation of both the interactions.13,14 To explain the interplay between both the interactions, MEPs in the dyads are shown in Table 7. The most negative MEP on the free N/O atom decreases in ZB-1 to 6, but it increases in ZB-7 to 10 and thus, the former is a weaker Lewis base, and the latter is a stronger base; the former forms a weaker chalcogen bond in ZB-CB-1 to 6, whereas the latter engages in a stronger chalcogen bond in ZB-CB-7 to 10. The most negative MEP on the free N/O atom also decreases in CB-1 to 3, corresponding to a weaker base, and it forms a weaker triel bond in ZB-CB-1 to 6. The more positive MEP on the B atom increases in CB-4 to 7; thus, it is a stronger acid and forms a stronger triel bond.
| Dyads | Vmin | ΔVmin | Dyads | Vmax | ΔVmax |
|---|---|---|---|---|---|
| ZB-1 | −0.031 | 0.018 | CB-4 | 0.0917 | 0.0352 |
| ZB-2 | −0.045 | 0.008 | CB-5 | 0.0680 | 0.0097 |
| ZB-3 | −0.036 | 0.014 | CB-6 | 0.1091 | 0.0313 |
| ZB-4 | −0.024 | 0.025 | CB-7 | 0.0892 | 0.0086 |
| ZB-5 | −0.048 | 0.005 | |||
| ZB-6 | −0.032 | 0.018 | |||
| ZB-7 | −0.071 | −0.015 | |||
| ZB-8 | −0.072 | −0.011 | |||
| ZB-9 | −0.065 | −0.013 | |||
| ZB-10 | −0.068 | −0.010 | |||
| CB-1 | −0.020 | 0.029 | |||
| CB-2 | −0.048 | 0.005 | |||
| CB-3 | −0.038 | 0.012 |
The interplay between both the interactions can also be understood with a change in the charge transfer (Table 8). The charge transfer is reduced for both the interactions in ZB-CB-1 to 6, where it moves from the central molecule to BH3/BF3/SO3. In ZB-CB-1 to 6, the direction of charge transfer is reverse for both the interactions. On the other hand, the charge transfer increases for both the interactions in ZB-CB-7 to 10, where the direction of charge transfer is from left to right for both the interactions. ΔΔE has a linear relationship with ΔCT for both the interactions, but a better relationship is found for the chalcogen bond (Fig. S4†).
| Triads | CTZB | ΔCTZB | CTCB | ΔCTCB |
|---|---|---|---|---|
| ZB-CB-1 | 0.2995 | −0.0228 | 0.2150 | −0.0385 |
| ZB-CB-2 | 0.3095 | −0.0038 | 0.0187 | −0.0040 |
| ZB-CB-3 | 0.2492 | −0.0086 | 0.0451 | −0.0268 |
| ZB-CB-4 | 0.2640 | −0.0277 | 0.1976 | −0.0559 |
| ZB-CB-5 | 0.0343 | −0.0145 | 0.0202 | −0.0025 |
| ZB-CB-6 | 0.1882 | −0.0247 | 0.0405 | −0.0314 |
| ZB-CB-7 | 0.3797 | 0.0135 | 0.3161 | 0.0380 |
| ZB-CB-8 | 0.3695 | 0.0036 | 0.0391 | 0.0123 |
| ZB-CB-9 | 0.3454 | 0.0130 | 0.3044 | 0.0329 |
| ZB-CB-10 | 0.3356 | 0.0032 | 0.0348 | 0.0090 |
4. Conclusions
Ab initio calculations have been performed for ternary complexes, where both a π-hole triel bond and a π-hole chalcogen bond coexist, along with the respective binary complexes. These π-hole bonds are very strong, possessing numerous characteristics of at least partially covalent interactions. The stability of these complexes is mainly due to charge transfer from the electron donor to the triel or chalcogen donor, caused by electrostatic, polarization, and deformation effects. HBCP is negative for all the complexes; furthermore, ∇2ρBCP is also negative for the chalcogen-bonded complexes of pyrazine and pyridine derivatives. A comparison of the different hybridized nitrogen atoms indicates that the sp2-hybridized nitrogen atom is favorable to bind with the π-hole at the triel or chalcogen center. The triel molecule is more easily deformed to bind with the electron donor than the chalcogen molecule, thus forming a stronger π-hole bond.Both triel and chalcogen bonds can coexist in the same ternary complex where a heteroaromatic compound is located in the central position. If the central molecule acts as a double Lewis base to bind with another two molecules, both the interactions are weakened, and a larger weakening effect is found for the chalcogen bond. If the central molecule plays a dual role of both a base in the triel bond and an acid in the chalcogen bond, both interactions are strengthened, and larger strengthening effect is found for the chalcogen bond. The cooperative mechanism of both the interactions is mainly governed by electrostatic and charge transfer effects.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21573188).References
- S. Scheiner, Hydrogen Bonding: A Theoretical Perspective, Oxford University Press, Oxford, 1997 Search PubMed.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond, Oxford University Press, Oxford, United Kingdom, 1999 Search PubMed.
- M. Breugst, E. Detmar and D. v. d. Heiden, ACS Catal., 2016, 6, 3203–3212 CrossRef.
- M. Iwaoka, S. Takemoto and S. Tomoda, J. Am. Chem. Soc., 2002, 124, 10613–10620 CrossRef PubMed.
- M. Bai, S. P. Thomas, R. Kottokkaran, S. K. Nayak, P. C. Ramamurthy and T. N. G. Row, Cryst. Growth Des., 2014, 14, 459–466 CrossRef.
- M. X. Liu, Q. Z. Li and S. Scheiner, Phys. Chem. Chem. Phys., 2017, 19, 5550–5559 RSC.
- S. Zahn, R. Frank, E. Hey-Hawkins and B. Kirchner, Chem.–Eur. J., 2011, 17, 6034–6038 CrossRef PubMed.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2013, 52, 12317–12321 CrossRef PubMed.
- Q. Z. Li, R. Li, X. F. Liu, W. Z. Li and J. B. Cheng, ChemPhysChem, 2012, 13, 1205–1212 CrossRef PubMed.
- S. J. Grabowski, ChemPhysChem, 2015, 16, 1470–1479 CrossRef PubMed.
- W. Z. Wang, B. M. Ji and Y. Zhang, J. Phys. Chem. A, 2009, 113, 8132–8135 CrossRef PubMed.
- J. S. Murray, P. Lane, T. Clark and P. Politzer, J. Mol. Model., 2007, 13, 1033–1038 CrossRef PubMed.
- S. J. Grabowski, ChemPhysChem, 2014, 15, 2985–2993 CrossRef PubMed.
- M. D. Esrafili and R. Nurazar, Mol. Phys., 2016, 114, 276–282 CrossRef.
- D. L. Fiacco, Y. Mo, S. W. Hunt, M. E. Ott, A. Roberts and K. R. Leopold, J. Phys. Chem. A, 2001, 105, 484–493 CrossRef.
- L. M. Azofra, I. Alkorta and S. Scheiner, J. Phys. Chem. A, 2015, 119, 535–541 CrossRef PubMed.
- V. d. P. N. Nziko and S. Scheiner, J. Phys. Chem. A, 2014, 118, 10849–10856 CrossRef PubMed.
- G. E. Garrett, E. I. Carrera, D. S. Seferos and M. S. Taylor, Chem. Commun., 2016, 52, 9881–9884 RSC.
- Q. Z. Li, H. Qi, R. Li, X. F. Liu, W. Z. Li and J. B. Cheng, Phys. Chem. Chem. Phys., 2012, 14, 3025–3030 RSC.
- M. D. Esrafili and F. Mohammadian-Sabet, Chem. Phys. Lett., 2015, 628, 71–75 CrossRef.
- W. A. Burns, J. A. Phillips, M. Canagaratna, H. Goodfriend and K. R. Leopold, J. Phys. Chem. A, 1999, 103, 7445–7453 CrossRef.
- M. Pyziak, J. Pyziak, M. Hoffmann and M. Kubicki, Cryst. Growth Des., 2015, 15, 5223–5232 CrossRef.
- H. T. Huynh, O. Jeannin and M. Fourmigué, Chem. Commun., 2017, 53, 8467–8469 RSC.
- K. T. Mahmudov, M. N. Kopylovich, M. F. C. G. d. Silva and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 10121–10138 RSC.
- T. T. Wang, W. Z. Li, Q. Z. Li and J. B. Cheng, Russ. J. Phys. Chem. A, 2015, 89, 812–817 CrossRef.
- X. Guo, X. L. An and Q. Z. Li, J. Phys. Chem. A, 2015, 119, 3518–3527 CrossRef PubMed.
- M. D. Esrafili, F. Mohammadian-Sabet and M. Solimannejad, Struct. Chem., 2014, 25, 1197–1205 CrossRef.
- M. D. Esrafili and M. Vakili, Mol. Phys., 2014, 112, 2746–2752 CrossRef.
- J. George, V. L. Deringer and R. Dronskowski, J. Phys. Chem. A, 2014, 118, 3193–3200 CrossRef PubMed.
- M. D. Esrafili and F. Mohammadian-Sabet, Struct. Chem., 2015, 26, 199–206 CrossRef.
- M. D. Esrafili and M. Vakili, J. Mol. Model., 2014, 20, 2291 CrossRef PubMed.
- K. Haupa, A. Bil and Z. Mielke, J. Phys. Chem. A, 2015, 119, 10724–10734 CrossRef PubMed.
- I. R. Beattie and P. J. Jones, Angew. Chem., Int. Ed., 1996, 35, 1527–1529 CrossRef.
- V. Jonas, G. Frenking and M. T. Reetz, J. Am. Chem. Soc., 1994, 116, 8741–8753 CrossRef.
- W. A. Burns and K. R. Leopold, J. Am. Chem. Soc., 1993, 115, 11622–11623 CrossRef.
- H. J. Jiao and P. v. R. Schleyer, J. Am. Chem. Soc., 1994, 116, 7429–7430 CrossRef.
- S. W. Reeve, W. A. Burns, F. J. Lovas, R. D. Suenram and K. R. Leopold, J. Phys. Chem., 1993, 97, 10630–10637 CrossRef.
- G. Venter and J. Dillen, J. Phys. Chem. A, 2004, 108, 8378–8384 CrossRef.
- J. A. Phillips, D. J. Giesen, N. P. Wells, J. A. Halfen, C. C. Knutson and J. P. Wrass, J. Phys. Chem. A, 2005, 109, 8199–8208 CrossRef PubMed.
- H. Hirao, K. Omoto and H. Fujimoto, J. Phys. Chem. A, 1999, 103, 5807–5811 CrossRef.
- E. Iglesias, T. L. Sordo and J. A. Sordo, Chem. Phys. Lett., 1996, 248, 179–181 CrossRef.
- D. L. Fiacco and K. R. Leopold, J. Phys. Chem. A, 2003, 107, 2808–2814 CrossRef.
- S. Yourdkhani, T. Korona and N. L. Hadipour, J. Comput. Chem., 2015, 36, 2412–2428 CrossRef PubMed.
- M. D. Esrafili and P. Mousavian, Mol. Phys., 2017, 115, 2999–3010 CrossRef.
- M. X. Liu, H. Y. Zhuo, Q. Z. Li, W. Z. Li and J. B. Cheng, J. Mol. Model., 2016, 22, 10 CrossRef PubMed.
- Q. J. Tang and Q. Z. Li, Mol. Phys., 2015, 113, 3809–3814 CrossRef.
- M. D. Esrafili and P. Mousavian, Chem. Phys. Lett., 2017, 678, 275–282 CrossRef.
- D. Escudero, A. Frontera, D. Quiñonero and P. M. Deyà, J. Comput. Chem., 2009, 30, 75–82 CrossRef PubMed.
- C. Estarellas, A. Frontera, D. Quiñonero and P. M. Deyà, ChemPhysChem, 2011, 12, 2742–2750 CrossRef PubMed.
- H. A. Samimi, M. D. Esrafili, F. Mohammadian-Sabet and H. Haddadi, Mol. Phys., 2015, 113, 1442–1450 CrossRef.
- M. D. Esrafili, F. Mohammadian-Sabet and M. Solimannejad, J. Mol. Graphics Modell., 2015, 57, 99–105 CrossRef PubMed.
- H. L. Xu, J. B. Cheng, X. Yang, Z. B. Liu, W. Z. Li and Q. Z. Li, ChemPhysChem, 2017, 18, 2442–2450 CrossRef PubMed.
- M. D. Esrafili and P. Mousavian, Mol. Phys., 2018, 116, 388–398 CrossRef.
- M. Gao, G. Q. Gao, Q. Z. Li, X. Yang, W. Z. Li and J. B. Cheng, RSC Adv., 2015, 5, 76912–76918 RSC.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef PubMed.
- I. Alkorta, F. Blanco, P. M. Deya, J. Elguero, C. Estarellas, A. Frontera and D. Quiñonero, Theor. Chem. Acc., 2010, 126, 1–14 Search PubMed.
- M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision A02. Gaussian Inc., Wallingford, 2009 Search PubMed.
- S. B. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef.
- F. A. Bulat, A. Toro-Labbé, T. Brinck, J. S. Murray and P. Politzer, J. Mol. Model., 2010, 16, 1679–1691 CrossRef PubMed.
- R. F. W. Bader, AIM2000 program, version 2.0, McMasterUniversity, Hamilton, Canada, 2000 Search PubMed.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef PubMed.
- A. E. Reed, L. A. Curtiss and F. A. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef.
- S. J. Grabowski, J. Comput. Chem., 2018, 39, 472–480 CrossRef PubMed.
- D. J. Giesen and J. A. Phillips, J. Phys. Chem. A, 2003, 107, 4009–4018 CrossRef.
- J. A. Phillips and C. J. Cramer, J. Phys. Chem. B, 2007, 111, 1408–1415 CrossRef PubMed.
- S. J. Grabowski, Phys. Chem. Chem. Phys., 2017, 19, 29742–29759 RSC.
- W. D. Arnold and E. Oldfield, J. Am. Chem. Soc., 2000, 122, 12835–12841 CrossRef.
- S. Pan, D. Moreno, J. L. Cabellos, J. Romero, A. Reyes, G. Merino and P. K. Chattaraj, J. Phys. Chem. A, 2014, 118, 487–494 CrossRef PubMed.
- R. Saha, S. Pan, S. Mandal, M. Orozco, G. Merino and P. K. Chattaraj, RSC Adv., 2016, 6, 78611–78620 RSC.
- D. Cremer and E. Kraka, Angew. Chem., Int. Ed., 1984, 23, 627–628 CrossRef.
- I. V. Novozhilova, A. V. Volkov and P. Coppens, J. Am. Chem. Soc., 2003, 125, 1079–1087 CrossRef PubMed.
- P. Macchi, L. S. Garlaschelli, S. Martinengo and A. Sironi, J. Am. Chem. Soc., 1999, 121, 10428–10429 CrossRef.
- P. Macchi, D. M. Proserpio and A. Sironi, J. Am. Chem. Soc., 1998, 120, 13429–13435 CrossRef.
- S. Pan, A. Gupta, S. Mandal, D. Moreno, G. Merino and P. K. Chattaraj, Phys. Chem. Chem. Phys., 2015, 17, 972–982 RSC.
- S. Pan, R. Saha, S. Mandal and P. K. Chattaraj, Phys. Chem. Chem. Phys., 2016, 18, 11661–11676 RSC.
- G. Jana, S. Pan, G. Merino and P. K. Chattaraj, J. Phys. Chem. A, 2017, 121, 6491–6499 CrossRef PubMed.
- S. Pan, A. Gupta, R. Saha, G. Merino and P. K. Chattaraj, J. Comput. Chem., 2015, 36, 2168–2176 CrossRef PubMed.
- B. Szefczyk, W. A. Sokalski and J. Leszczynski, J. Chem. Phys., 2002, 117, 6952–6958 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04106g |
| This journal is © The Royal Society of Chemistry 2018 |