
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfate radical-induced transformation of trimethoprim with CuFe2O4/MWCNTs as a heterogeneous catalyst of peroxymonosulfate: mechanisms and reaction pathways†
Jing Konga,
Ruobai Lia,
Fengliang Wanga,
Ping Chenb,
Haijin Liuc,
Guoguang Liu *a and
Wenying Lva
*a and
Wenying Lva
aSchool of Environmental Science and Engineering, Guangdong University of Technology, Guangzhou 510006, China. E-mail: liugg615@163.com; Fax: +86-20-3932-2548; Tel: +86-13533635690
bSchool of Environment, Tsinghua University, Beijing 100084, China
cSchool of Environment, Henan Normal University, Henan Key Laboratory for Environmental Pollution Control, Xinxiang 453007, China
First published on 10th July 2018
Abstract
Trimethoprim (TMP), a typical antibiotic pharmaceutical, has received extensive attention due to its potential biotoxicity. In this study, CuFe2O4, which was used to decorate MWCNTs via a sol–gel combustion synthesis method, was introduced to generate powerful radicals from peroxymonosulfate (PMS) for TMP degradation in an aqueous solution. The results showed that almost 90% of TMP was degraded within 24 min with the addition of 0.6 mM PMS and 0.2 g L−1 CuFe2O4/MWCNTs. The degradation rate was enhanced with the increase in initial PMS doses, catalyst loading and pH. A fairly low leaching of Cu and Fe was observed during the reaction, indicating the high potential recyclability and stability of CuFe2O4/MWCNTs. Electron paramagnetic resonance analysis confirmed that the CuFe2O4/MWCNT-PMS system had the capacity to generate ·OH and SO4˙−, whereas quenching experiments further confirmed that the catalytic reaction was dominated by SO4˙−. A total of 11 intermediate products of TMP was detected via mass spectrometry, and different transformation pathways were further proposed. Overall, this study showed a systematic evaluation regarding the degradation process of TMP by the CuFe2O4/MWCNT-PMS system.
1 Introduction
Currently, sulfate radical (SO4˙−)-based advanced oxidation processes (AOPs) have garnered increasing research interest as effective methods for the removal of various organic contaminants in aquatic environments.1 The oxidizing potential of SO4˙− is in the range of 2.5–3.1 V under different pH levels in solution, whereas it is only 1.9–2.7 V for the hydroxyl radical (·OH).2 SO4˙− may be generated by the activation of peroxymonosulfate (PMS) with transition metals (e.g., Fe2+, Cu2+, Co2+),3 metal oxides,4 and heating,5 which is very promising for large-scale wastewater treatment. Among these methods, the transition metal activation of PMS has two advantages, i.e., it is easy to operate and energy saving. However, the homogeneous system results in the discharge of dissolved heavy metals into the environment. The toxicity and the potential carcinogenicity of leaching cations limit their practical environmental protection applications. Therefore, it is necessary to prepare a highly stable and recyclable catalyst to produce SO4˙−.CuFe2O4 exhibits effective catalytic activity for PMS-generated SO4˙−.6 In addition to the ease of recycling, another advantage of CuFe2O4 is its magnetic nature, which makes its separation from aqueous systems quite straightforward. Nevertheless, the unique magnetic properties of CuFe2O4 lead to particulate aggregation, which limits the conductivity and the population of active sites.7 The loading of CuFe2O4 onto carbonaceous materials not only limits the aggregation of CuFe2O4 nanoparticles, but also provides additional active sites and facilitates electron transfer within the system.8,9 Compared to other carbon materials (e.g., activated carbon and graphite), multi-walled carbon nanotubes (MWCNTs) have unique properties such as chemical stability and extreme strength.10,11 In a previous study, Zhang et al. used a multi-walled carbon nanotube-loaded CuFe2O4 (CNTs/CuFe2O4)-PMS system for the degradation of 2-phenylbenzimidazole-5-sulfonic acid (PBSA), and they found that this composite exhibited good catalytic activity and stability.12
Pharmaceuticals and personal care products (PPCPs) in ambient aquatic environments have been a growing concern due to their ubiquitous occurrence and potential negative impacts on ecosystems and human health.13 Pharmaceuticals and their bioactive metabolites are being continuously introduced into natural aquatic systems through the incomplete decomposition of urban sewage and excretions from the human body.14,15 Trimethoprim (TMP) as a broad-spectrum antibiotic is employed to treat various bacterial infections including otitis media, gastrointestinal, respiratory, and urinary infections.16,17 The reported concentrations of TMP are 0.1–5 μg L−1, 2.2–10.9 ng L−1, and 0–19.8 ng L−1 in wastewater treatment plant effluents, surface water, and finished drinking water, respectively.18 Therefore, technologies that can effectively remove TMP have been attracting increasing attention. Until now, various methods have been performed for the removal of TMP from water. Zhang et al. demonstrated that the UV/PDS system can efficiently degrade TMP.19 Wang et al. found that the homogenous Fe2+/PS process showed an efficient TMP removal in an aqueous solution.20 However, the degradation of TMP using a CuFe2O4/MWCNT-PMS heterogeneous system has not been attempted yet.
In this study, a CuFe2O4/MWCNT composite was synthesized through a facile method and applied as a heterogeneous activator towards PMS. The catalytic degradation mechanism of TMP as well as the reusability of the CuFe2O4/MWCNT composite was systematically investigated. Furthermore, detailed mechanisms and transformation pathways for TMP degradation by the CuFe2O4/MWCNT-PMS system were proposed based on the intermediate product identification by liquid chromatography–tandem mass spectrometry (HPLC/MS/MS), reactive oxygen species detection, and XPS.
2 Experimental section
2.1 Preparation and characterization
The CuFe2O4/MWCNT composite was synthesized according to a previously reported method with slight improvement. Detailed information regarding the preparation and characterization methods of the catalyst is provided in Text S1.†2.2 Experimental procedures
Degradation experiments were performed in a 250 mL bath reactor at 27 °C, and the initial pH was set at 7.0. Next, 0.02 mM TMP and 0.2 g L−1 of CuFe2O4/MWCNTs were added to a 100 mL reaction resolution, where the reaction was initiated by the addition of 0.6 mM PMS. Samples (5 mL) of the solution were extracted at 0, 1, 2, 4, 8, 16, and 24 min time intervals, quenched by 0.6 mM Na2S2O3·5H2O, and immediately filtered with a 0.45 μm filter into a 1 mL brown vial for HPLC analysis. Other influencing elements for the catalytic degradation of TMP were evaluated in the same way.2.3 Analysis of TMP degradation
The concentration of TMP was analyzed via a HPLC-UV detector at a wavelength of 239 nm. The column that was used in the experiment was a Chromolith Performance XDB-C18 column (150 × 4.6 mm, 5-micron, Agilent, USA). The oven temperature was set at 35 °C, and the injection volume was 20 μL. The mobile phase was a mixture of acetonitrile and 0.2% methanoic acid in water (85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15, v/v) with a flow rate of 0.2 mL min−1 under isocratic conditions. The total organic carbon (TOC) was measured by a TOC-VCPH analyzer (Japan) to identify the mineralization of the organic contaminants. The analysis of the products is shown in Text S1 of the ESI.†
:15, v/v) with a flow rate of 0.2 mL min−1 under isocratic conditions. The total organic carbon (TOC) was measured by a TOC-VCPH analyzer (Japan) to identify the mineralization of the organic contaminants. The analysis of the products is shown in Text S1 of the ESI.†
3 Results and discussion
3.1 Characterization of catalysts
The morphologies of CuFe2O4/MWCNTs were revealed by TEM observation (Fig. 1a). Furthermore, carbon was detected using the elemental mapping results (Fig. 1b–e) following the synthesis, which indicated the successful loading of CuFe2O4 MNPs onto MWCNTs. The N2 adsorption–desorption isotherms and the pore size distribution are shown in Fig. 1h, indicating the presence of interparticle meso-porosity in this material.21 In addition, the specific surface areas (SSAs) of CuFe2O4/MWCNTs (84.3 m2 g−1) were far higher than that of bulk CuFe2O4 (29.7 m2 g−1). The XRD patterns in Fig. 1f show that both MWCNTs and CuFe2O4/MWCNTs displayed a characteristic peak at 2θ = 26°, which revealed that their carbon skeletons were maintained during the synthesis.22 The diffraction peaks for CuFe2O4/MWCNTs were similar to the standard diffraction data for CuFe2O4 (JCPDS 34-0425).23 The diffraction peaks of CuFe2O4/MWCNTs were narrower than that of bare CuFe2O4, which indicated particle aggregation during annealing.23 As expected in FT-IR spectra (Fig. 1g), the absorption band in the 1700–1600 cm−1 region was assigned to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in both pristine MWCNTs and CuFe2O4/MWCNTs;24 this indicated that the carboxylic groups remained after being calcined, which is critical for the catalyst for preserving the hydrophilicity and high dispersibility. Magnetic hysteresis (M-H) curves for both pristine and used CuFe2O4/MWCNTs (Fig. 1i) demonstrated that the used catalyst still possessed a high level of magnetism.
O in both pristine MWCNTs and CuFe2O4/MWCNTs;24 this indicated that the carboxylic groups remained after being calcined, which is critical for the catalyst for preserving the hydrophilicity and high dispersibility. Magnetic hysteresis (M-H) curves for both pristine and used CuFe2O4/MWCNTs (Fig. 1i) demonstrated that the used catalyst still possessed a high level of magnetism.
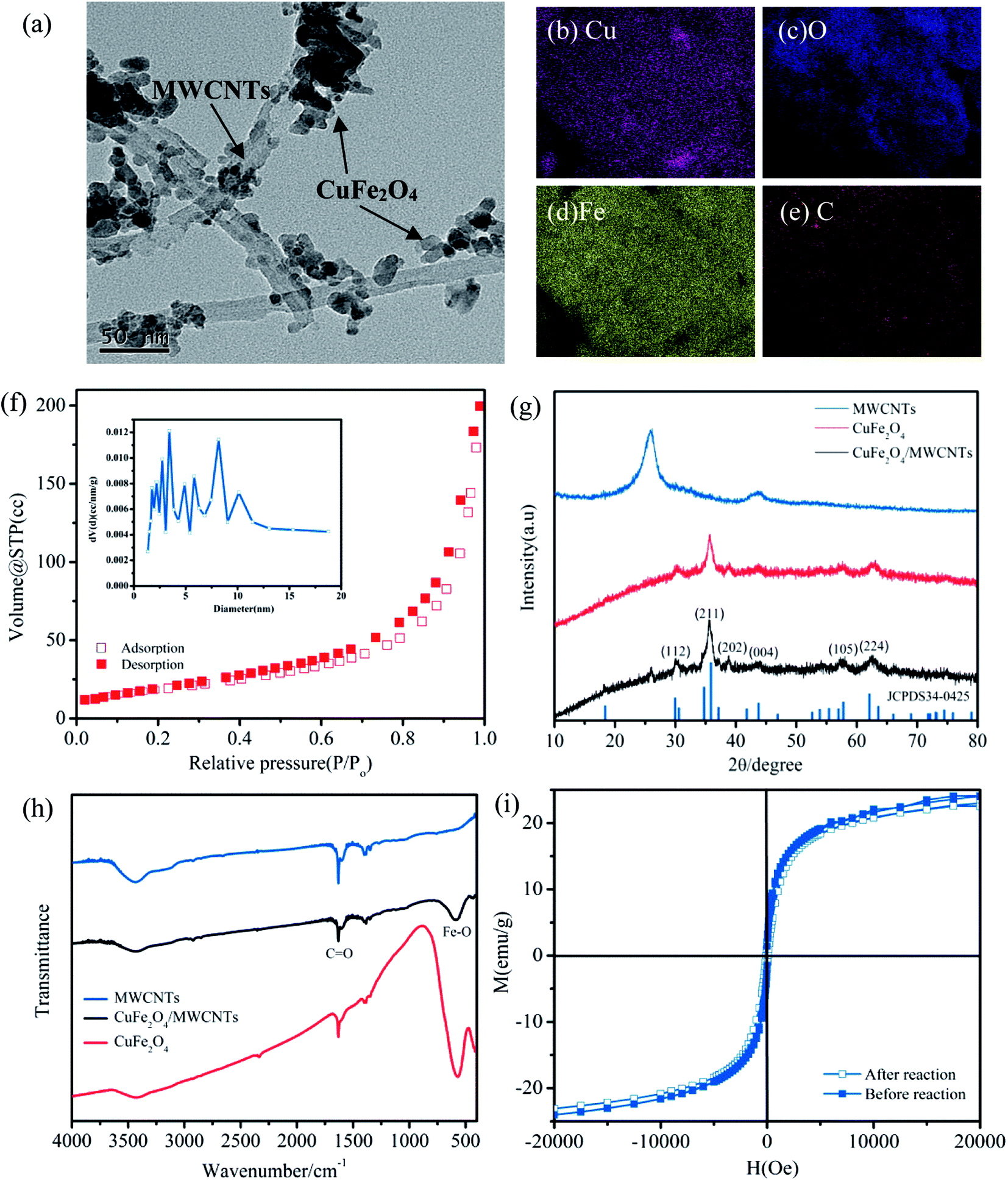 | ||
| Fig. 1 (a) The TEM image and (b–e) elemental mapping of CuFe2O4/MWCNTs; (f) N2 adsorption–desorption isotherms and the corresponding pore size distribution (inset) of CuFe2O4/MWCNTs; (g) XRD patterns of the three samples; (h) FT-IR spectra of the three samples; (i) M-H loops of CuFe2O4/MWCNTs before and after the reaction. | ||
The above-mentioned results illustrated the structure, morphology and physical characteristics of the CuFe2O4/MWCNT composites, which indicated that MWCNTs were successfully coupled with the CuFe2O4 surface. Furthermore, due to the excellent magnetic properties of the catalysts, we could easily separate them under a magnetic field, which is very beneficial for recycling.
3.2 Degradation of TMP by the CuFe2O4/MWCNT-PMS system
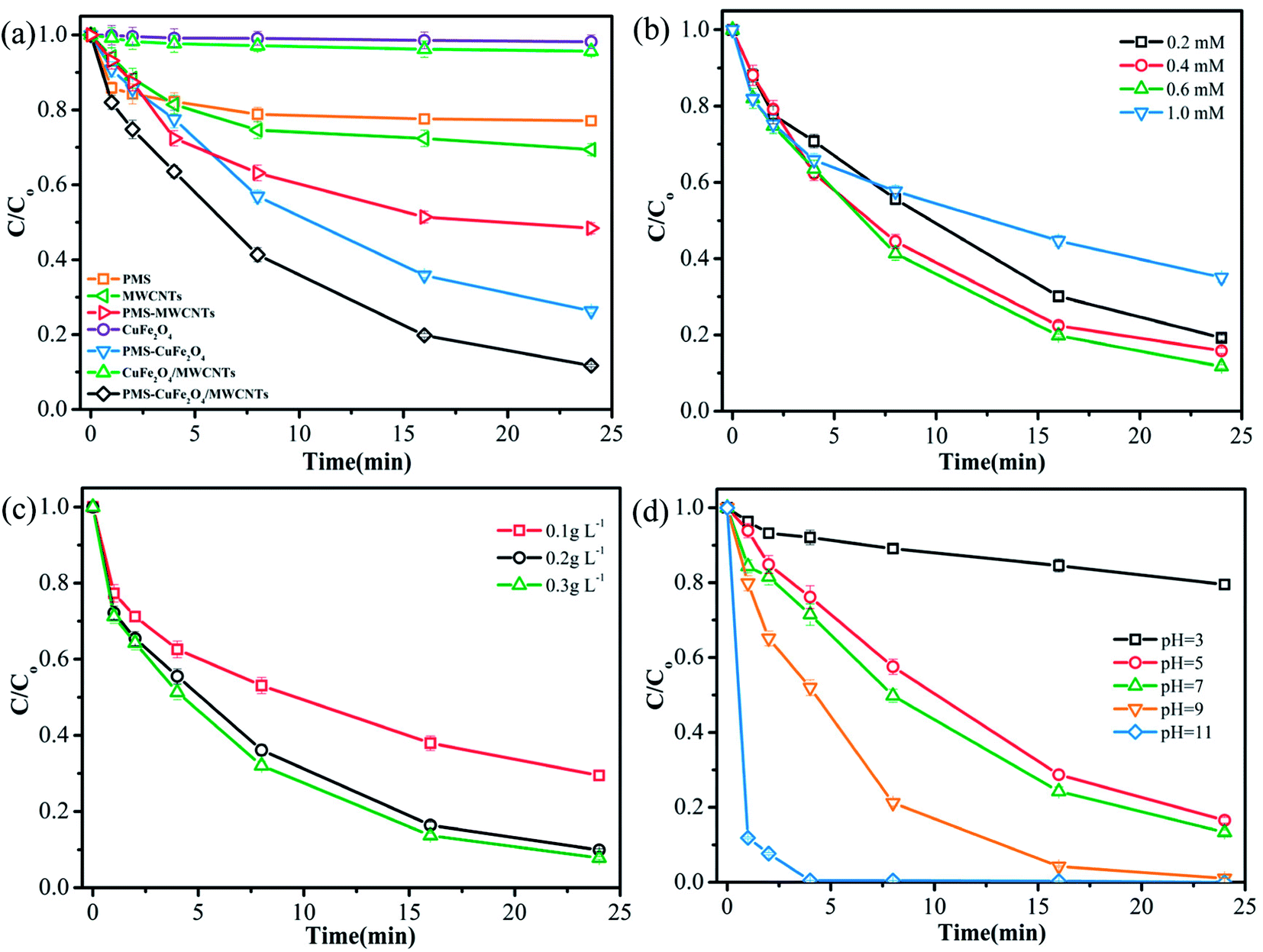 | ||
| Fig. 2 (a) TMP removal efficiencies by various reactions; effects of (b) initial PMS concentration; (c) catalyst dose and (d) initial pH on TMP degradation. Reaction conditions: initial TMP concentration, 0.02 mM; PMS, 0.6 mM; catalyst dose, 0.2 g L−1; pH = 7.0; temperature, 27 °C. | ||
The degradation of TMP under different catalyst doses (0.1, 0.2, and 0.3 g L−1) was investigated, and the results are shown in Fig. 2c. According to Fig. 2c, the increasing catalyst loading could promote the degradation of TMP. At the catalyst doses were varied from 0.1 g L−1 to 0.3 g L−1, the degradation efficiency of TMP improved by about 20%. In a previous study, the decontamination rate attained a maximum and then decreased with the continuously increasing CuFe2O4 catalyst load, which suggested that the detrimental effects of high doses of the catalyst were suppressed by its stimulation effects.6
3.3 Recyclability of CuFe2O4/MWCNTs and cation leaching
The recyclability of the CuFe2O4/MWCNT catalyst is crucial in the practical applications. The degradation experiments were repeated for four cycles to evaluate the catalytic stability of CuFe2O4/MWCNTs. After being used, the CuFe2O4/MWCNT catalyst was magnetically collected and dried. As shown in Fig. 3, the removal efficiency remained at around 82% after four cycles, suggesting the high stability of CuFe2O4/MWCNTs in the PMS oxidation system. Moreover, the leaching of Cu as well as Fe was measured during the time course of TMP degradation with the addition of 0.2 g L−1 catalysts. Under reaction conditions, the atomic absorption spectroscopy (AAS) analysis indicated that the leaching of Cu was about 0.13 mg L−1 over 60 min, which accounted for 0.065% of the total Cu content in the catalyst, whereas the leaching of Fe was less than 0.04 mg L−1.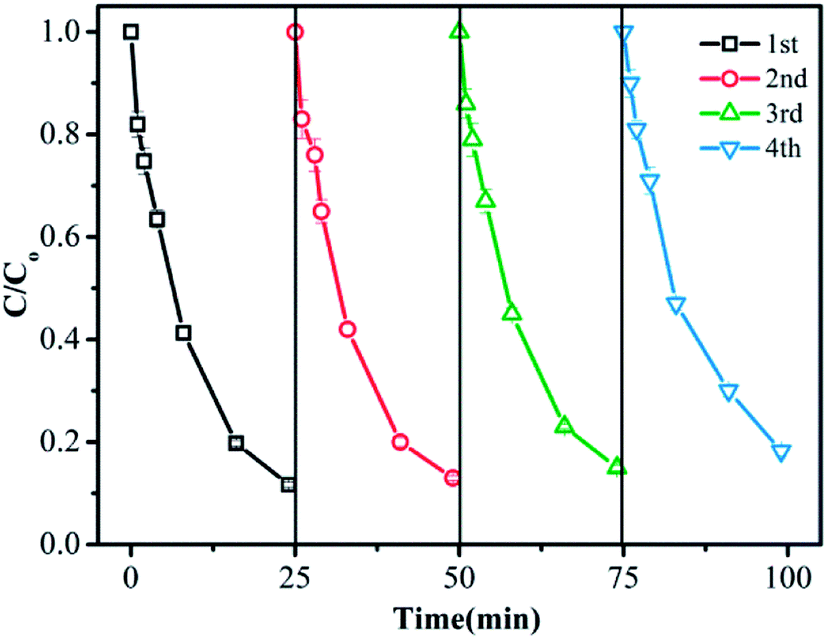 | ||
| Fig. 3 Recyclability of the catalyst for TMP removal in four runs. Reaction conditions: initial TMP and PMS concentrations were 0.02 mM and 0.6 mM, respectively. Catalyst dose, 0.2 g L−1; pH = 7.0; temperature, 27 °C. | ||
3.4 Activation mechanism of the catalyst
PMS has the capacity to generate three primary types of reactive radicals (SO4˙−, SO5˙− and ·OH) through metal-ion activation.29 The species of radicals generated from the oxidation of TMP were probed by the DMPO spin-trapping EPR technique. As shown in the EPR spectrum (Fig. 4a), no distinctive radical was produced by DMPO; the EPR signals represented the presence of DMPO-OH and DMPO-SO4˙− adducts.30,31 Thus, ·OH and SO4˙− were shown to be involved in the catalytic degradation process.32 To further identify the types of major reactive radical species generated in the degradation system, ethanol (EtOH) and tert-butyl alcohol (TBA) were added into the reaction solution as radical quenching agents. EtOH (containing α-hydrogen) reacted with ·OH and SO4˙− at high and comparable rates, where the rate constants for the reactions with ·OH and SO4˙− were 1.2–2.8 × 109 mol L−1 s−1 and 1.6–7.7 × 107 mol L−1 s−1, respectively. However, the rate constant of TBA (without α-hydrogen) with ·OH was 1000 times higher compared to that with SO4˙−.22 Therefore, EtOH was employed to quench both ·OH and SO4˙−, and TBA was applied to selectively quench ·OH. As depicted in Fig. 4b, when no quenching agents were added, about 90% of TMP degraded in 24 min. In contrast, the addition of 0.3 mol L−1 of EtOH and TBA to the reaction significantly decreased the degradation efficiencies to 21% and 58%, respectively. The addition of more EtOH and TBA further decreased the TMP removal and when their concentrations were fixed at 0.6 mol L−1, the removal rates of TMP were only 15% and 45%, respectively. The enhanced decrease in the TMP removal by EtOH rather than that by TBA suggested that the primary radical species generated during the activation of PMS was SO4˙−.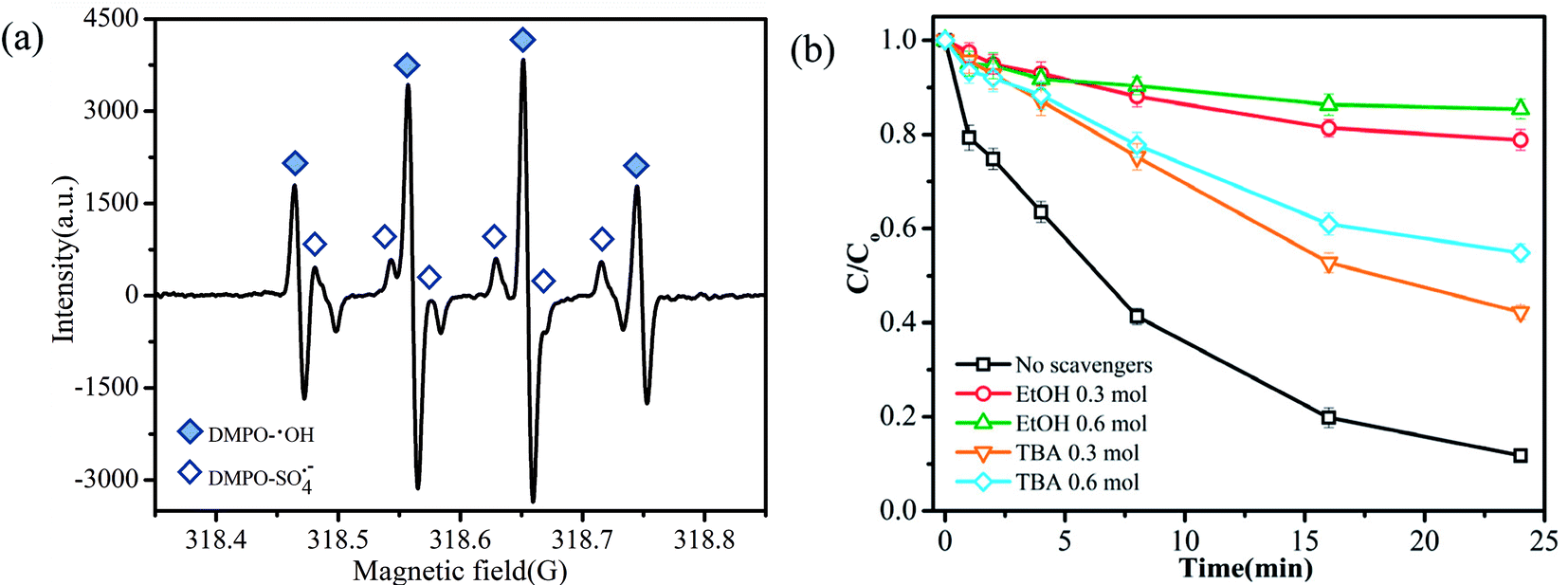 | ||
| Fig. 4 (a) EPR spectrum of the CuFe2O4/MWCNT-PMS system (CDMPO = 50 mM); (b) effects of quenchers on TMP degradation. Reaction conditions: initial TMP and PMS concentrations were 0.02 mM and 0.6 mM, respectively. Catalyst dose 0.2 g L−1; pH = 7.0, temperature was 27 °C. | ||
To better understand the roles of Fe and Cu in the activation of PMS, XPS spectra were obtained prior to and following the catalytic reaction (Fig. 5). The results showed that the valence of Fe3+ on the surface of CuFe2O4/MWCNTs did not change after the reaction. Ren et al. reported a similar conclusion, which stated that only the metals in the A site had a valence change on the surface of MFe2O4.33 Fig. 5c shows a peak at 934.001 eV before the catalytic reaction, which was assigned to Cu 2p3/2 of Cu2+; this value slightly shifted to a lower binding energy value (933.9 eV) following the catalytic reaction, which indicated that the valence of Cu on the surface of the catalyst after the reaction was a combination of the valences of Cu2+ and Cu+. Based on the characteristic peaks, the relative contributions of Cu2+ and Cu3+ accounted for about 40% and 26%, respectively.22 Cu species were mainly present as Cu2+ before the reaction, and they were transformed to Cu3+ and several Cu+ ions following the catalytic reaction, which might have been due to the coexisting oxidation and reduction conditions in the solution.
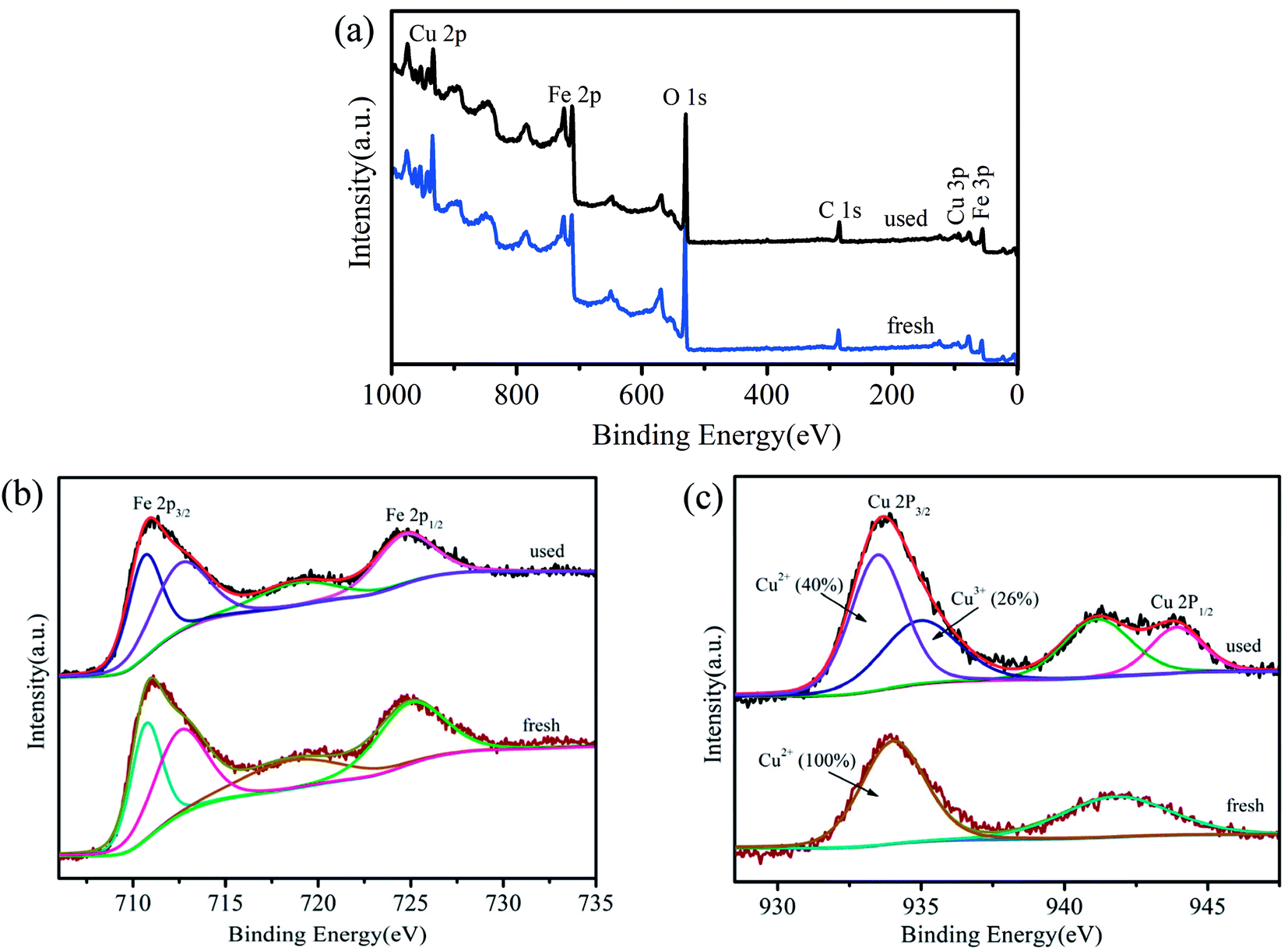 | ||
| Fig. 5 (a) Wide survey XPS spectrum; (b) Fe 2p and (c) Cu 2p XPS envelopes of used and fresh catalysts. | ||
The mechanism for the activation of PMS by CuFe2O4/MWCNTs was proposed as follows (eqn (1)–(7)):
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Cu2+ + HSO−5 → Cu3+ + SO4˙− + OH− Cu2+ + HSO−5 → Cu3+ + SO4˙− + OH−
| (1) |
|
Cu2+ + HSO−5 → Cu+ + SO4˙− + H+
| (2) |
|
Cu+ + HSO−5 → Cu2+ + SO4˙− + OH−
| (3) |
|
Cu3+ + HSO−5 → Cu2+ +SO5˙− + H+
| (4) |
| SO4˙− + TMP → degraded products → H2O + CO2 | (5) |
| SO4˙− + H2O → SO2−4 + H+ + ·OH | (6) |
| ·OH + TMP → degraded products → H2O + CO2 | (7) |
Interestingly, the oxidation of HSO5− by Cu2+ was thermodynamically unfavorable (eqn (2)) (Cu2+/Cu+ (0.17 V) and SO52−/HSO5− (1.1 V) or SO5˙−/SO52− (0.81 V));34 however, the reactions were still possible as SO5˙− was at a low concentration, which rendered the actual reduction potential of SO52−/HSO5− or SO5˙−/SO52− to a relatively low value in the catalyst system. However, due to the weak oxidizing ability of SO5˙− (E(SO5˙−/SO42−) = 1.1 V), it was considered that it did not contribute to TMP degradation.35 Cu+ could be oxidized by HSO5− (eqn (3)), which was an additional pathway for the generation of SO4˙−, and it also returned the metal centers to their initial oxidation states for the next catalytic cycle.
On the basis of the experimental results mentioned above, the potential activation mechanism was concluded. When PMS was added, SO4˙− and ·OH were generated from PMS, which was activated by the unveiled active sites of Cu2+.36 During this process, PMS could also be activated by MWCNTs to generate additional reactive species through non-radical reactions.37,38 In addition, owing to the highly electronic conductive trait of MWCNTs, they facilitated the electron transfer between metal ions.39 To balance the charge on the CuFe2O4/MWCNT surface, Cu2+ not only provided electrons, but also accepted the electrons from the system, which indicated the involvement of Cu2+–Cu3+–Cu2+ and Cu2+–Cu+–Cu2+ redox processes during the reaction.33 Finally, TMP was adsorbed onto the surface of the CuFe2O4/MWCNT catalyst, which was attacked by SO4˙− and ·OH, leading to decomposition and mineralization.
3.5 Degradation transformation products and pathways
TMP was degraded into a variety of transformation products, as identified by the HPLC/MS/MS technique. A positive full scan mass spectrum of SPE enrichment revealed the by-products of TMP that were detected in the CuFe2O4/MWCNT-PMS system. For TMP degradation, the potential transformation pathways are shown in Fig. 6. As seen, hydroxylation, the cleavage of methylene groups, and demethylation were proposed to be existing in this system. Among them, hydroxylation was the most prominent pathway. Moreover, the free radicals reacted relatively easily with –CH2– group-generated carbon centers; the cleavage of the methylene bridge was another major pathway. Furthermore, demethylation was often accompanied with hydroxylation. These proposed TMP pathways likely included different reaction mechanisms. On the basis of previous results, we inferred that TMP was primarily destroyed by SO4˙− and ·OH. The SO4˙− ions preferentially reacted through an electron transfer process, whereas ·OH reacted via hydrogen abstraction or addition reactions.40 Detailed MS2 spectra and the proposed structures of TMP and each by-product are provided in the ESI.†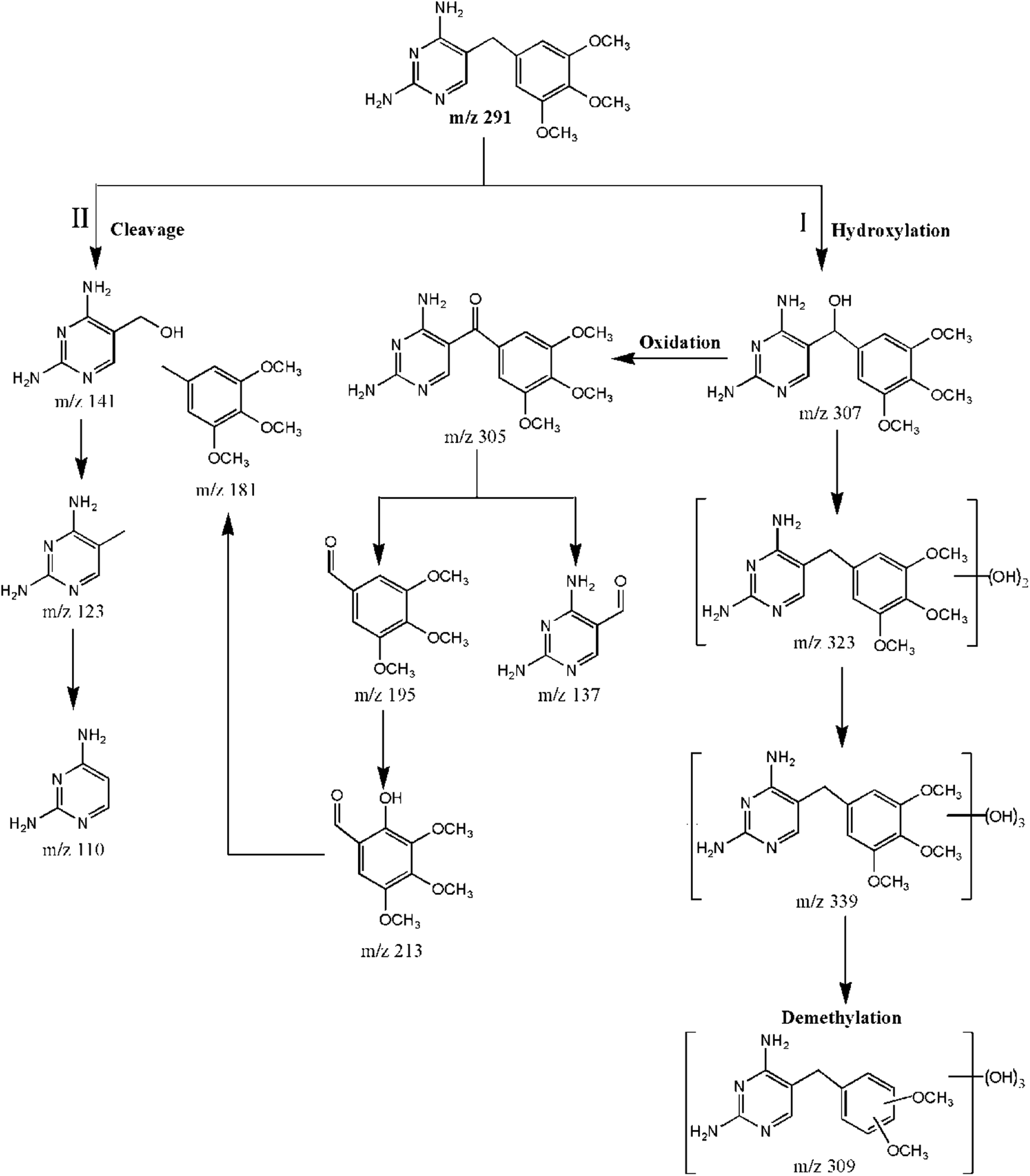 | ||
| Fig. 6 Proposed transformation pathways for TMP degradation. | ||
Pathway I is hydroxylation by which hydroxyl groups can be attached to either the ring of the TMP molecule or to the methylene bridge. The products at m/z 307 (P4) and 305 (P5) were identified as α-hydroxytrimethoprim and α-ketotrimethoprim, respectively. The fragment ions at m/z 289 and m/z 274 of the P4 (Fig. S5f†) implied that a hydroxyl group was added to the C-7 atom; otherwise, the hydroxyl groups on either aromatic ring would lack a hydrogen on the β-atom, which is essential for the formation of a double bond.41 P4 and P5 were likely generated by the electron transfer mechanism and the attack by SO4˙−.42 Furthermore, P4 reacted with SO4˙− and was oxidized to P5. These two compounds were previously identified as transformation products of TMP using the UV-PS system,42 Fe0-PS system43 and permanganate treatment.44 The additional value of 32 amu relative to TMP (m/z 291) suggested that the products at m/z 323 (P2) were dihydroxylated isomers on the basis of TMP;43 thus, they possessed several positional isomers. Fig. S5c† and d shows only the MS2 spectra of two isomers, and m/z 339 (P1) possessed a molecular weight that was 16 amu higher than that of P2, which suggested a transformation pathway of hydroxylation. Compared to P1, P3 (m/z 309) lost one methoxy group. P2, P1, and P3 were the products that were generated by the direct addition of ·OH.
Pathway II included the cleavage of the C–C bond between the two rings in TMP, which was due to a direct attack by SO4˙− or ·OH at the methylene bridge. The cleavage of the methylene group generated m/z 141 (P7) and m/z 181 (P9), and a similar reaction also occurred for P5. The fragmentation pathway of P5 was consistent with those proposed by Ji et al.45 For P7, m/z 123 (P10) could be explained by the loss of water; this ion could be stabilized by mesomeric resonance. The value of m/z 123 or 110 (P11) corresponded to the characteristic fragments of protonated TMP containing pyrimidine groups (Table S1†).10 In addition, P9 was proposed to be a pseudo-molecular ion of the trimethoxytoluene moiety.
During the TMP oxidation process, TOC removal was measured to reflect the mineralization of TMP. After 2 hours, TMP was completely decomposed, but the TOC removal was approximately 35% (Fig. S4†). These results indicated that TOC reduction proceeded much more slowly than the degradation of TMP. Most of the by-products generated during the degradation of TMP maintained the two-ring TMP structure, which could account for this phenomenon.
4 Conclusions
Magnetic porous CuFe2O4/MWCNTs were synthesized by a sol–gel combustion method, and they were fully characterized. The synergistic effects of CuFe2O4 and MWCNTs facilitated the activation of PMS to decompose TMP, showing the notable catalytic performance of CuFe2O4/MWCNTs. In addition, CuFe2O4/MWCNTs exhibited good stability with low levels of metal cation leaching. An investigation of the reaction parameters revealed that the PMS dosage, catalyst loading, and initial pH had influences on the degradation efficiency of TMP to a large extent. The results of EPR and quenching experiments proved that both ·OH and SO4˙− participated in the degradation of TMP, whereas SO4˙− was the primary radical species. The major transformation pathways were proposed to be hydroxylation, cleavage of methylene group and demethylation. This is the first time that the CuFe2O4/MWCNT-PMS system was evaluated by investigating the degradation mechanisms of pharmaceuticals, which provided more insights into a new catalyst for advance oxidation processes. Overall, this systematic investigation presented a promising heterogeneous catalyst for practical applications in the treatment of pharmaceutical wastewater and drinking water.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China [No. 21377031 and 21677040]; and the Innovative Team Program of High Education of Guangdong Province [2015KCXTD007].References
- Y. Yao, H. Chen, J. Qin, G. Wu, C. Lian, J. Zhang and S. Wang, Water Res., 2016, 101, 281–291 CrossRef PubMed.
- P. Neta, R. E. Huie and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 1027–1284 CrossRef.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef PubMed.
- G. D. Fang, D. D. Dionysiou, S. R. Al-Abed and D. M. Zhou, Appl. Catal., B, 2013, 129, 325–332 CrossRef.
- M. Nie, Y. Yang, Z. Zhang, C. Yan, X. Wang, H. Li and W. Dong, Chem. Eng. J., 2014, 246, 373–382 CrossRef.
- Y. H. Guan, J. Ma, Y. M. Ren, Y. L. Liu, J. Y. Xiao, L. Q. Lin and C. Zhang, Water Res., 2013, 47, 5431 CrossRef PubMed.
- Y. Q. Wang, R. M. Cheng, Z. Wen and L. J. Zhao, Eur. J. Inorg. Chem., 2011, 2011, 2942–2947 CrossRef.
- X. M. Chen, Z. J. Lin, T. T. Jia, Z. M. Cai, X. L. Huang, Y. Q. Jiang, X. Chen and G. N. Chen, Anal. Chim. Acta, 2009, 650, 54–58 CrossRef PubMed.
- Y. Yao, Y. Cai, F. Lu, F. Wei, X. Wang and S. Wang, J. Hazard. Mater., 2014, 270, 61–70 CrossRef PubMed.
- X. Hu, B. Liu, Y. Deng, H. Chen, S. Luo, C. Sun, P. Yang and S. Yang, Appl. Catal., B, 2011, 107, 274–283 CrossRef.
- Y. Yao, H. Chen, C. Lian, F. Wei, D. Zhang, G. Wu, B. Chen and S. Wang, J. Hazard. Mater., 2016, 314, 129–139 CrossRef PubMed.
- X. Zhang, M. Feng, L. Wang, R. Qu and Z. Wang, Chem. Eng. J., 2017, 307, 95–104 CrossRef.
- L. P. Padhye, H. Yao, F. T. Kung'U and C. H. Huang, Water Res., 2014, 51, 266 CrossRef PubMed.
- C. Sirtori, A. Agüera, W. Gernjak and S. Malato, Water Res., 2010, 44, 2735–2744 CrossRef PubMed.
- F. Wang, P. Chen, Y. Feng, Z. Xie, Y. Liu, Y. Su, Q. Zhang, Y. Wang, K. Yao and W. Lv, Appl. Catal., B, 2017, 207, 103–113 CrossRef.
- A. Aboueisha, Toxicol. in Vitro, 2006, 20, 601–607 CrossRef PubMed.
- F. Wang, Y. Wang, Y. Li, X. Cui, Q. Zhang, Z. Xie, H. Liu, Y. Feng, W. Lv and G. Liu, Dalton Trans., 2018, 47(20), 6924–6933 RSC.
- Z. Wu, J. Fang, Y. Xiang, C. Shang, X. Li, F. Meng and X. Yang, Water Res., 2016, 104, 272 CrossRef PubMed.
- R. Zhang, Y. Yang, C. H. Huang, N. Li, H. Liu, L. Zhao and P. Sun, Environ. Sci. Technol., 2016, 50, 2573 CrossRef PubMed.
- S. Wang and J. Wang, Chemosphere, 2017, 191, 97–105 CrossRef PubMed.
- M. Q. Yang, B. Weng and Y. J. Xu, Langmuir, 2013, 29, 10549–10558 CrossRef PubMed.
- Y. Ding, L. Zhu, N. Wang and H. Tang, Appl. Catal., B, 2013, 129, 153–162 CrossRef.
- P. Laokul, V. Amornkitbamrung, S. Seraphin and S. Maensiri, Curr. Appl. Phys., 2011, 11, 101–108 CrossRef.
- X. Zhang, M. Feng, R. Qu, H. Liu, L. Wang and Z. Wang, Chem. Eng. J., 2016, 301, 1–11 CrossRef.
- C. Qi, X. Liu, C. Lin, H. Zhang, X. Li and J. Ma, Chem. Eng. J., 2017, 315, 201–209 CrossRef.
- M. C. Dodd, M. O. Buffle and U. V. Gunten, Environ. Sci. Technol., 2006, 40, 1969–1977 CrossRef PubMed.
- O. S. Furman, A. L. Teel and R. J. Watts, Environ. Sci. Technol., 2010, 44, 6423–6428 CrossRef PubMed.
- I. M. Kolthoff and I. K. Miller, J. Am. Chem. Soc., 1951, 73, 1–30 CrossRef.
- M. Pagano, A. Volpe, G. Mascolo, A. Lopez, V. Locaputo and R. Ciannarella, Chemosphere, 2012, 86, 329–334 CrossRef PubMed.
- G. S. Timmins, K. J. Liu, E. J. Bechara, Y. Kotake and H. M. Swartz, Free Radical Biol. Med., 1999, 27, 329 CrossRef PubMed.
- P. Chen, F. Wang, Z. F. Chen, Q. Zhang, Y. Su, L. Shen, K. Yao, Y. Liu, Z. Cai and W. Lv, Appl. Catal., B, 2017, 204, 250–259 CrossRef.
- J. Kang, X. Duan, C. Wang, H. Sun, X. Tan, M. O. Tade and S. Wang, Chem. Eng. J., 2018, 332, 398–408 CrossRef.
- Y. Ren, L. Lin, J. Ma, J. Yang, J. Feng and Z. Fan, Appl. Catal., B, 2015, 165, 572–578 CrossRef.
- T. N. Das, R. E. Huie and P. Neta, J. Phys. Chem. A, 1999, 103, 3581–3588 CrossRef.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2003, 37, 4790 CrossRef PubMed.
- X. Duan, C. Su, J. Miao, Y. Zhong, Z. Shao, S. Wang and H. Sun, Appl. Catal., B, 2018, 220, 626–634 CrossRef.
- X. Duan, H. Sun, Z. Shao and S. Wang, Appl. Catal., B, 2018, 224, 973–982 CrossRef.
- X. Duan, H. Sun and S. Wang, Acc. Chem. Res., 2018, 51, 678–687 CrossRef PubMed.
- H. Lee, H. J. Lee, J. Jeong, J. Lee, N. B. Park and C. Lee, Chem. Eng. J., 2015, 266, 28–33 CrossRef.
- R. Li, J. Kong, H. Liu, P. Chen, G. Liu, F. Li and W. Lv, RSC Adv., 2017, 7, 22802–22809 RSC.
- J. Kuang, J. Huang, B. Wang, Q. Cao, S. Deng and G. Yu, Water Res., 2013, 47, 2863–2872 CrossRef PubMed.
- R. Zhang, P. Sun, T. H. Boyer, L. Zhao and C. H. Huang, Environ. Sci. Technol., 2015, 49, 3056 CrossRef PubMed.
- X. Liu, X. Zhang, K. Shao, C. Lin, C. Li, F. Ge and Y. Dong, RSC Adv., 2016, 6, 20938–20948 RSC.
- L. Hu, A. M. Stemig, K. H. Wammer and T. J. Strathmann, Environ. Sci. Technol., 2011, 45, 3635–3642 CrossRef PubMed.
- Y. Ji, W. Xie, Y. Fan, Y. Shi, D. Kong and J. Lu, Chem. Eng. J., 2016, 286, 16–24 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04103b |
| This journal is © The Royal Society of Chemistry 2018 |