
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePharmacokinetic and excretion study of three alkaloids in rats using UPLC-MS/MS after oral administration of menispermi rhizoma capsules†
Jia Yu,
Bolin Zhu,
Dan Su and
Zhen Jiang *
*
Department of Pharmaceutical Analysis, School of Pharmacy, Shenyang Pharmaceutical University, 103 Wenhua Road, Shenyang, Liaoning Province, PR China 110016. E-mail: jiangzhen8487@163.com; Fax: +86-24-2398 6285
First published on 10th September 2018
Abstract
A sensitive, specific and rapid ultra-performance liquid chromatography tandem mass spectrometry method (UPLC-MS/MS) was developed for simultaneous determination of three main alkaloids (daucicoline, daurisoline, dauricine) in rat plasma, urine and feces after oral administration of menispermi rhizoma capsules. The chromatographic separation was performed on a Waters ACQUITY UPLC® BEH C18 column (50 mm × 2.1 mm, id, 1.7 μm) with a column temperature of 30 °C and a linear gradient elution using a mobile phase consisting of 0.1% formic acid and acetonitrile. The flow rate was set at 0.3 mL min−1, and the total run time was 10 min. The detection was performed, without interference, using positive electrospray ionization with a multiple reaction monitoring mode. A comprehensive validation of the method was performed. The linearity of the analytical response was good over a wide concentration range with correlation coefficients greater than 0.9903 and the lower limits of quantification were 1.5–5.0 ng mL−1 for all matrices. Both accuracy and precision of the assay were satisfactory. The mean extraction recoveries of analytes and internal standard from rat plasma, urine and feces were all more than 78.3%. The validated methods of the three types of substrates were successfully applied to a pharmacokinetic and excretion study of the three alkaloids in rats after oral administration of menispermi rhizoma capsule. The pharmacokinetics and excretion study of these active components in menispermi rhizoma capsule have not been reported. The results provided a meaningful basis for the clinical application of menispermi rhizoma capsule.
1. Introduction
Menispermi rhizoma (MR), called beidougen in Chinese, is prepared from the dried rhizome of Menispermum dauricum DC (Menispermaceae). MR is one of traditional Chinese medicines and widely used because of its definite therapeutic effect on sore throat, dysentery and rheumatoid arthralgia.1 In order to promote the clinical application, MR capsules have been developed and included in the Chinese Pharmacopoeia.2 With the alkaloid extract of MR as the main material, MR capsules, with the functions of clearing away heat and toxic material, antitussive and expectorant effects, is widely used to treat sore throat, tonsillitis, chronic bronchitis and so on. The previous phytochemical and pharmacological reports on the alkaloid of MR indicated that alkaloids were the major effective constituents responsible for the bioactivities, such as anti-inflammation,3 anti-arrhythmia,4 anti-bacterial,5 anti-tumor,6 platelet aggregation inhibition,7 neuroprotection8 and protection of concurrent myocardial–cerebral ischemia/reperfusion injury.9–11Previous investigations of MR capsules revealed that daucicoline, daurisoline and dauricine were generally considered as the most abundant components responsible which were representative physiologically active alkaloids.12 Having an activity against blood platelet aggregation induced by adenosine 5′-diphosphate,13 these three compounds have been demonstrated to have significant effects on inhibiting blood platelet aggregation, arrhythmias, and tumors. The anti-arrhythmic effects of dauricine and daurisoline have been verified in different experimental arrhythmic models and used in cardiac arrhythmia patients.14 Dauricine blocked the cardiac transmembrane Na+, K+ and Ca2+ ion currents, which exhibited a reverse use-dependent effect.15 Daurisoline has been found to prolong the duration of the cardiac action potential in a normal use dependent manner, and its direct effect was investigated on the human ether-a-go-go-related gene current and the expression of mRNA and protein in human embryonic kidney 293 cells stably expressed the human ether-a-go-go-related gene channel.16 The anti-arrhythmic effect of daurisoline was more potent than that of dauricine.15 Dauricine was also reported to inhibit human breast cancer angiogenesis by suppressing hypoxia-inducible factor-1α protein accumulation and vascular endothelial growth factor expression.17 Up to now, much research on MR capsules has been conducted in the fields of pharmacology and quality control.18,19
However, although the MR capsules demonstrated various pharmacological activities, information regarding the in vivo fate of the MA capsules still remains scarce. This is a great obstacle to its further pharmacological studies, clinically safe medication administration and quality evaluation.20 As is known, pharmacokinetic study is important for illustrating the action mechanism, druggability, target organs and the first-pass effect on gastrointestinal tract/liver of the active compounds.21 Furthermore proper knowledge on the excretion of the drug is essential to investigate and interpret the in vivo dispersion.22 Therefore, evaluation of the pharmacokinetic and excretion research of the alkaloids in MR capsules is required to determine its efficacy and safety considerations in clinical applications.
Several analytical techniques, such as high performance liquid chromatography (HPLC) coupled with ultraviolet detection (UV), diode array detection (DAD) and evaporative light scatting detectors, fluorescence detection, and high-performance capillary electrophoresis, have been used to quantify the active ingredients in biological samples. They were insufficient in terms of sensitivity and specificity for pharmacokinetic and excretion studies.23 Increasing evidence proved that liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) has become the most efficient way for analysing various bio-samples with high sensitivity and selectivity.24 In this study, an ultra-performance liquid chromatography – electrospray ionization – tandem mass spectrometry method (UPLC-ESI-MS/MS) method was developed to investigate the pharmacokinetics and excretion of dauricine, daurisoline, daucicoline of MR capsules in rats, and no one has studied MR capsules with this technique before. It is expected that the results will provide helpful information to further determine the action mechanism, safety evaluation and clinical application of MR capsules.
2. Experimental
2.1. Chemicals and materials
Dauricine, daurisoline and daucicoline were isolated and purified from MR in the laboratory. Their structures were identified using MS, proton nuclear magnetic resonance spectrometry (1H-NMR) and 13C-NMR data. The purity of each reference standard was above 97.0% with area normalization by HPLC-DAD. The internal standard carbamazepine was supplied by the National Institutes for Drug and Food Control (100142-201105). The structures of reference standards are shown in Fig. 1. The MR capsules were manufactured by the Yichun Pharmaceutical Co. Ltd (Heilongjiang). The LC-MS grade acetonitrile was purchased from Fisher Scientific (Pittsburgh, PA, USA). The LC-MS grade formic acid was obtained from Sigma-Aldrich (St. Louis, MO, USA). Distilled water was obtained from Wahaha Co. Ltd. (Hangzhou, China). | ||
| Fig. 1 The chemical structures of (A) daucicoline, (B) daurisoline, (C) dauricine and (D) the internal standard, carbamazepine. | ||
2.2. Animals
Male Sprague-Dawley (SD) rats, weighing 260 ± 20 g, were obtained from the Experimental Animal Center of Shenyang Pharmaceutical University (Shenyang, China, SYXK-liao-2014-0004). Animals were bred in a breeding room with temperature of 25 °C, humidity of 55–60%, and a dark–light cycle. All the rats were housed under the conditions mentioned previously for one week for acclimation, and were then were fasted for 12 h, with free access to water, overnight before the experiments.2.3. LC-MS/MS instruments and analytical conditions
The ACQUITY UPLC® system (Waters Corp., Milford, MA, USA) was equipped with a cooling autosampler and a column oven enabling temperature control. Mass spectrometric detection was performed using a Waters Xevo TQ-S tandem quadrupole mass spectrometer (Waters Micromass MS Technologies, Manchester, UK). A UNIFI™ data system (Waters) was used to control the UPLC-MS/MS system and for data acquisition and processing.Chromatographic separation was performed on a Waters ACQUITY UPLC® ethylene bridged hybrid particle (BEH) C18 column (50 mm × 2.1 mm, id, 1.7 μm, Waters, Wexford, Ireland). The column temperature was maintained at 30 °C. The mobile phase consisted of A (0.1% aqueous formic acid, v/v) and B (acetonitrile) and the gradient elution was: 0–2.0 min, isocratic elution with 92% A; 2.0–4.0 min, linear elution changing from 92% to 80% A; 4.0–7.0 min, linear elution changing from 80% to 50% A; 7.0–10.0 min, linear elution changing from 50% to 20% A, then the system was quickly returned to the initial concentration of A (92%, v/v). This was followed by an equilibration period of 4 min prior to the injection of the next sample. The flow rate was set at 0.3 mL min−1. The temperature of the autosampler was kept at 4 °C and the injection volume was 5 μL.
The MS was operated in the positive ESI mode with multiple reaction monitoring (MRM) resolution. High purity nitrogen was used as both a nebulizing and drying gas. The MS/MS setting parameters were as follows: capillary voltage 3.0 kV, source temperature of 150 °C and desolvation temperature of 350 °C, and desolvation and cone gas at a flow rate of 700 L h−1 and 150 L h−1, respectively. Because MRM transitions, cone voltage and collision energy varied with different analytes, the MRM transitions, cone voltage and collision energy were optimized for each analyte (Table 1).
| Analyte | MRM transition (m/z) | Dwell times (ms) | Cone voltage (V) | Collision energy (eV) |
|---|---|---|---|---|
| Daucicoline | 597.2/191.8 | 50 | 5 | 32 |
| Daurisoline | 611.2/191.9 | 50 | 8 | 34 |
| Dauricine | 625.2/205.9 | 50 | 7 | 38 |
| Carbamazepine (internal standard) | 237.4/193.8 | 50 | 20 | 20 |
2.4. Preparation of standards and quality control samples
Stock solutions of daucicoline, dauricine and daurisoline, were prepared in acetonitrile–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at the concentrations of 0.11, 0.18, 0.15 mg mL−1. Then, an appropriate amount of the three stock solutions were mixed and serially diluted with acetonitrile–water (1:1, v/v) into a linear concentration of solutions which were then used as the standard mixture working solutions. Stock solution of the internal standard (carbamazepine) was prepared in methanol at the concentration of 0.10 mg mL−1. Then, an appropriate amount of the internal standard stock solution was diluted with acetonitrile–water (1:1, v/v) to give a concentration of 50 ng mL−1 to be used as the internal standard solution. All solutions were stored at 4 °C and brought to room temperature before use.
:1, v/v) at the concentrations of 0.11, 0.18, 0.15 mg mL−1. Then, an appropriate amount of the three stock solutions were mixed and serially diluted with acetonitrile–water (1:1, v/v) into a linear concentration of solutions which were then used as the standard mixture working solutions. Stock solution of the internal standard (carbamazepine) was prepared in methanol at the concentration of 0.10 mg mL−1. Then, an appropriate amount of the internal standard stock solution was diluted with acetonitrile–water (1:1, v/v) to give a concentration of 50 ng mL−1 to be used as the internal standard solution. All solutions were stored at 4 °C and brought to room temperature before use.
Calibration standards were freshly prepared in a glass tube by evaporating 100 μL of the standard mixture working solutions to dryness with nitrogen (N2) and then thoroughly mixing with a 100 μL of blank plasma, urine or fecal homogenates. The concentration range of the calibration standards are shown in Table 2. Quality control (QC) samples were prepared independently in the same way as the calibration standards, and the concentrations are shown in Table 3.
| Matrix | Compounds | LLOQ (ng mL−1) | Linear range (ng mL−1) | Linear regression equation | Correlation coefficient (R) |
|---|---|---|---|---|---|
| Plasma | Daucicoline | 3.0 | 3.00–1200 | y = 9.85 × 10−3x − 7.67 × 10−4 | 0.9956 |
| Daurisoline | 5.0 | 5.00–2000 | y = 1.59 × 10−2x − 0.288 | 0.9923 | |
| Dauricine | 5.0 | 5.00–2000 | y = 3.30 × 10−2x − 0.209 | 0.9967 | |
| Urine | Daucicoline | 1.5 | 1.50–1500 | y = 7.71 × 10−3x − 2.54 × 10−3 | 0.9975 |
| Daurisoline | 2.5 | 2.50–2500 | y = 1.55 × 10−2x − 0.111 | 0.9929 | |
| Dauricine | 2.5 | 2.50–2500 | y = 1.62 × 10−2x − 3.80 × 10−2 | 0.9976 | |
| Feces | Daucicoline | 1.5 | 1.50–1500 | y = 9.65 × 10−3x + 0.146 | 0.9919 |
| Daurisoline | 2.5 | 2.50–2500 | y = 2.20 × 10−2x − 9.42 × 10−2 | 0.9903 | |
| Dauricine | 2.5 | 2.50–2500 | y = 3.62 × 10−2x − 2.99 × 10−2 | 0.9960 |
| Matrix | Compounds | Low (ng mL−1) | Middle (ng mL−1) | High (ng mL−1) |
|---|---|---|---|---|
| Plasma | Daucicoline | 6.00 | 75.0 | 960 |
| Daurisoline | 10.0 | 125.0 | 1600 | |
| Dauricine | 10.0 | 125.0 | 1600 | |
| Urine | Daucicoline | 3.00 | 60.0 | 1200 |
| Daurisoline | 5.00 | 100.0 | 2000 | |
| Dauricine | 5.00 | 100.0 | 2000 | |
| Feces | Daucicoline | 3.00 | 60.0 | 1200 |
| Daurisoline | 5.00 | 100.0 | 2000 | |
| Dauricine | 5.00 | 100.0 | 2000 |
2.5. Preparation of plasma, urine and feces samples
After thawing the biological samples (plasma, urine and feces) at room temperature, a conventional protein precipitation (PPT) method was applied for the sample preparation.000 rpm, the supernatant was collected and evaporated to dryness under a stream of N2 at 40 °C. The residue was reconstituted in 100 μL of 50% aqueous acetonitrile. Finally, a 5 μL aliquot of supernatant was injected into the UPLC-ESI-MS/MS system in the partial loop mode.2.6. Method validation
The analytical methodology was validated according to the guidelines set by the United States Food and Drug Administration (FDA) for bioanalytical method validation. The method was validated according using linearity, selectivity, precision and accuracy, recovery and stability.25For freeze–thaw stability, the QC samples were stored at −20 °C for 24 h and thawed at room temperature which was one cycle. After three freezing and thawing cycles, the concentrations of the analytes and internal standard were determined. For short-term room temperature stability, the QC samples were thawed at room temperature and kept at room temperature for 12 h prior to analysis. For long-term stability, the QC samples were kept at the storage temperature (−20 °C) for one week. For post-preparation stability, the extracted QC samples were stored in the auto-sampler (4 °C) for 12 h. The stock solutions' (analytes and internal standard) stability was determined at 0, 5, 10, and 15 days.
2.7. Plasma pharmacokinetics
The validated UPLC-MS/MS method was applied to a pharmacokinetic study of the three alkaloids in rats after single oral administration of MR capsules. Male SD rats (260 ± 20 g) used for this experiment were supplied by the Experimental Animal Center of Shenyang Pharmaceutical University (Shenyang, China). The rats were housed in a temperature controlled room at ambient temperature (about 25 °C), humidity of 55–60%, with a 12 h light–dark cycle. Animal experiments were carried out in accordance with the Regulations of the Experimental Animal Administration issued by the State Commission of Science and Technology of the People's Republic of China. All the rats were fasted for 12 h but with access to water. The suspension of MR capsules was dissolved in 0.5% carboxymethyl cellulose sodium aqueous solution and was orally administered to the rats at a dose of 2.5 g kg−1 (equivalent to 0.50 mg kg−1 of daucicoline, 13.25 mg kg−1 of dauricine, or 9.17 mg kg−1 of daurisoline). Blood samples of approximately 0.3 mL were collected from the orbital vein into heparin containing tubes before drug administration (0 min) and subsequently at 0.033, 0.083, 0.250, 0.333, 0.75, 2.0, 4.0, 6.0, 8.0, 12.0, 24.0, 48.0, 72.0, 96.0 and 120.0 h after a single oral administration. The blood sample was centrifuged at 15000 rpm for 10 min, and the supernatant was transferred into a clean tube and stored at −20 °C until analysis. The plasma concentration of the three analytes at different times was calculated from the daily calibration curve, which was expressed as mean ± SD. The plasma concentration of the three analytes versus time data for each rat and the pharmacokinetic parameters [time for maximal concentration (Tmax), maximum concentration a drug achieves after dosage (Cmax), half-life (t1/2), area under the curve (AUC(0–t) and AUC(0–∞))] were calculated using non-compartmental analysis using the DAS 2.0 pharmacokinetic program (Chinese Pharmacological Society).
2.8. Excretion study
The previously described UPLC-MS/MS method was also suitable for use for the excretion study of urine and feces. For urinary and fecal excretion study, six rats were given a single oral dose of 2.5 g kg−1 MR suspension (equivalent to 13.25 mg kg−1 of dauricine, 9.17 mg kg−1 daurisoline, 0.50 mg kg−1 daucicoline) using the same preparation method as described previously. Afterwards, the rats were individually placed in stainless-steel metabolic cages, which allowed the separate collection of urine and feces. The urine and feces samples were collected before administration and at time intervals of 0–8, 8–12, 12–24, 24–36, 36–48, 48–72, 72–96 and 96–120 h post dosing. The volumes of urine and the weight of dry feces samples were recorded. The urine and feces samples were stored at −80 °C until analysis. The animal protocol was permitted by the Committee on the Ethics of Animal Experiments of Shenyang Pharmaceutical University (permit number: SYPU-IACUC-C2016-03-15-206).3. Results and discussion
3.1. Method development
To obtain better responses for each analyte and the internal standard, the MS conditions were optimized using the corresponding standard solutions. As they are all nitrogen compounds, the results showed that the ionization of all the analytes and internal standard were more efficient in the positive mode. Thereby, intense and stable protonated molecular ion peaks, [M + H]+, were obtained for daucicoline, daurisoline, dauricine and carbamazepine (internal standard) at m/z 597.2, 611.2, 625.2 and 237.4, respectively. Then these precursor ions were used to obtain product ion spectra. The parameters of cone voltage and collision energy were optimized for the MRM mode and the other parameters were adopted for the recommended value of the instrument. Therefore, the most sensitive response was obtained for MRM transitions from m/z 597.2 → 191.8 for daucicoline, m/z 611.2 → 191.9 for daurisoline, m/z 625.2 → 205.9 for dauricine and m/z 237.4 → 193.8 for carbamazepine (internal standard).
3.2. Method validation
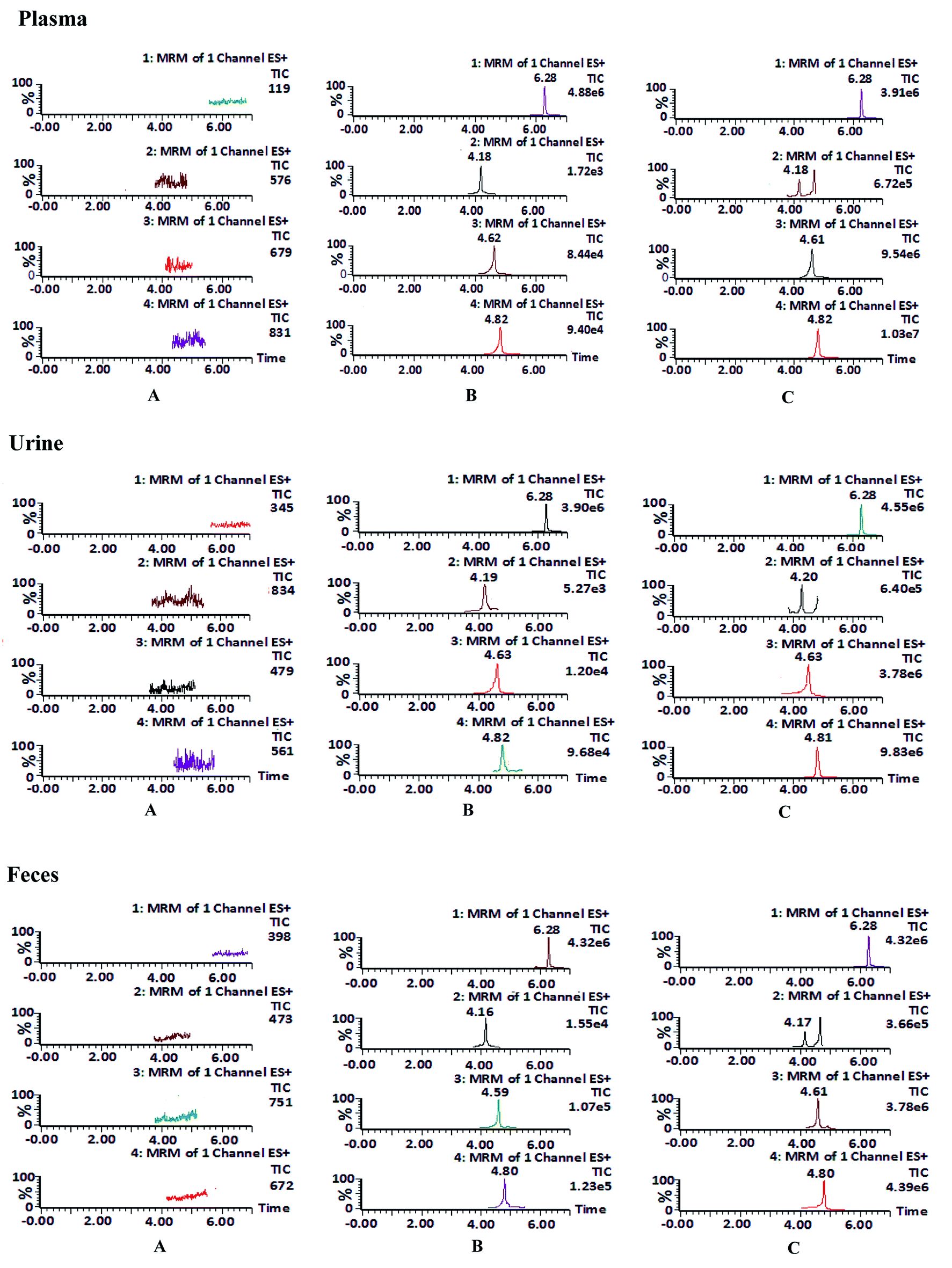 | ||
| Fig. 2 Representative extract ion MRM chromatograms of internal standard, daucicoline, daurisoline and dauricine in plasma, urine and feces. (A) Blank biological samples for carbamazepine (internal standard) (channel 1), daucicoline (channel 2), daurisoline (channel 3) and dauricine (channel 4) in plasma, urine and feces; (B) blank biological samples spiked with internal standard, daucicoline, daurisoline and dauricine; (C) test plasma sample collected at 120 min after oral administration of MR capsules (a dose of 2.5 g kg−1), test urine and feces sample collected at 8–12 h after oral administration of MR capsules. | ||
| Matrix | Compounds | Added concentration (ng mL−1) | Precision | Accuracy (RE, %) | Extract recovery (mean ± SD, %) | Matrix effect (mean ± SD, %) | |
|---|---|---|---|---|---|---|---|
| Intra-day (RSD, %) | Inter-day (RSD, %) | ||||||
| Plasma | Daucicoline | 6.00 | 7.9 | 8.7 | −3.2 | 85.7 ± 8.3 | 106.9 ± 3.9 |
| 75.0 | 6.7 | 7.9 | 4.3 | 85.9 ± 7.9 | 97.3 ± 4.6 | ||
| 960 | 9.0 | 7.1 | 6.4 | 93.2 ± 5.4 | 100.4 ± 5.7 | ||
| Daurisoline | 10.0 | 10.3 | 7.8 | 8.3 | 80.3 ± 6.7 | 103.9 ± 10.7 | |
| 125.0 | 3.6 | 4.9 | 5.7 | 81.3 ± 7.1 | 97.7 ± 9.8 | ||
| 1600 | 4.9 | 3.8 | −4.6 | 79.8 ± 7.9 | 107.2 ± 9.9 | ||
| Dauricine | 10.0 | 8.9 | 8.6 | 4.6 | 85.8 ± 5.4 | 113.0 ± 7.6 | |
| 125.0 | 8.1 | 4.1 | 3.0 | 89.4 ± 8.9 | 110.2 ± 7.2 | ||
| 1600 | 7.8 | 6.8 | 5.3 | 90.3 ± 9.6 | 108.3 ± 8.2 | ||
| Internal standard | 50 | 91.3 ± 4.3 | 104.2 ± 5.4 | ||||
| Urine | Daucicoline | 3.00 | 8.9 | 10.7 | 10.7 | 81.7 ± 7.6 | 103.9 ± 5.9 |
| 60.0 | 6.7 | 6.9 | 8.2 | 81.9 ± 5.9 | 96.3 ± 4.6 | ||
| 1200 | 9.3 | 10.1 | 7.6 | 83.2 ± 6.4 | 106.4 ± 5.7 | ||
| Daurisoline | 5.00 | 4.3 | 6.8 | 8.6 | 78.3 ± 7.7 | 87.9 ± 10.7 | |
| 100.0 | 5.9 | 7.9 | 7.6 | 81.3 ± 7.9 | 93.7 ± 7.8 | ||
| 2000 | 4.9 | 8.6 | 6.5 | 79.8 ± 7.9 | 93.2 ± 9.1 | ||
| Dauricine | 5.00 | 2.9 | 3.7 | −2.2 | 82.8 ± 5.4 | 115.0 ± 4.6 | |
| 100.0 | 6.1 | 6.7 | 9.7 | 83.4 ± 8.9 | 110.2 ± 8.2 | ||
| 2000 | 5.2 | 8.8 | 5.3 | 87.3 ± 9.6 | 107.3 ± 7.2 | ||
| Internal standard | 50 | 89.3 ± 6.3 | 97.2 ± 6.7 | ||||
| Feces | Daucicoline | 3.00 | 5.7 | 8.4 | 7.3 | 80.4 ± 7.2 | 110.9 ± 6.8 |
| 60.0 | 4.7 | 6.9 | 6.2 | 81.3 ± 6.4 | 111.6 ± 7.2 | ||
| 1200 | 3.1 | 4.7 | 3.3 | 79.8 ± 9.1 | 105.4 ± 6.3 | ||
| Daurisoline | 5.00 | 4.3 | 6.8 | −1.6 | 81.2 ± 4.8 | 97.9 ± 10.7 | |
| 100.0 | 5.9 | 7.9 | 7.5 | 84.1 ± 5.3 | 108.5 ± 5.7 | ||
| 2000 | 4.9 | 9.3 | 6.7 | 80.7 ± 7.4 | 96.2 ± 6.9 | ||
| Dauricine | 5.00 | 2.9 | 7.2 | 6.8 | 83.0 ± 4.5 | 113.0 ± 4.6 | |
| 100.0 | 6.1 | 8.1 | 10.3 | 85.4 ± 9.6 | 107.2 ± 7.8 | ||
| 2000 | 5.2 | 5.8 | 3.8 | 81.6 ± 8.3 | 97.9 ± 8.6 | ||
| Internal standard | 50 | 90.0 ± 5.5 | 103.2 ± 6.7 | ||||
| Storage conditions | Compounds | Spike concentration (ng mL−1) | Found concentration (ng mL−1) | RSD (%) | Accuracy (RE; %) |
|---|---|---|---|---|---|
| Plasma | |||||
| Short-term stability (room temperature for 12 h) | Daucicoline | 6.00 | 6.19 | 3.9 | 3.2 |
| 75.0 | 79.6 | 5.6 | 6.1 | ||
| 960 | 987 | 6.5 | 2.8 | ||
| Daurisoline | 10.0 | 9.34 | 7.9 | −6.6 | |
| 125.0 | 135 | 7.1 | 8.0 | ||
| 1600 | 1639 | 8.4 | 2.4 | ||
| Dauricine | 10.0 | 11.1 | 9.6 | 11.0 | |
| 125.0 | 131 | 5.9 | 4.8 | ||
| 1600 | 1699 | 5.3 | 6.2 | ||
| Long-term stability (−20 °C for 20 days) | Daucicoline | 6.00 | 6.19 | 4.6 | 3.2 |
| 75.0 | 77.9 | 6.9 | 3.9 | ||
| 960 | 980 | 6.7 | 2.1 | ||
| Daurisoline | 10.0 | 10.5 | 8.1 | 5.0 | |
| 125 | 130.6 | 3.1 | 4.5 | ||
| 1600 | 1559 | 8.5 | −2.6 | ||
| Dauricine | 10.0 | 10.3 | 3.9 | 3.0 | |
| 125 | 132 | 5.1 | 5.6 | ||
| 1600 | 1750 | 7.3 | 9.4 | ||
| Freeze–thaw stability | Daucicoline | 6.00 | 6.47 | 6.7 | 7.8 |
| 75.0 | 80.9 | 3.8 | 7.9 | ||
| 960 | 986 | 5.4 | 2.7 | ||
| Daurisoline | 10.0 | 11.09 | 2.9 | 10.9 | |
| 125 | 120.5 | 10.7 | −3.6 | ||
| 1600 | 1672 | 4.8 | 4.5 | ||
| Dauricine | 10.0 | 10.39 | 6.3 | 3.9 | |
| 125 | 134 | 8.2 | 7.2 | ||
| 1600 | 1658 | 3.9 | 3.6 | ||
| Post-preparation stability (4 °C for 12 h) | Daucicoline | 6.00 | 6.57 | 11 | 9.5 |
| 75.0 | 69.8 | 7.2 | −6.9 | ||
| 960 | 977 | 6.9 | 1.8 | ||
| Daurisoline | 10.0 | 10.79 | 4.2 | 7.9 | |
| 125 | 134.5 | 6.7 | 7.6 | ||
| 1600 | 1499 | 3.3 | −6.3 | ||
| Dauricine | 10.0 | 11.01 | 10.4 | 10.1 | |
| 125 | 132.3 | 8.7 | 5.8 | ||
| 1600 | 1664 | 4.8 | 4.0 | ||
|
|||||
| Urine | |||||
| Short-term stability (room temperature for 12 h) | Daucicoline | 3.00 | 3.22 | 4.8 | 7.3 |
| 60.0 | 61.6 | 5.2 | 2.7 | ||
| 1200 | 1247 | 6.8 | 3.9 | ||
| Daurisoline | 5.00 | 5.46 | 6.6 | 9.2 | |
| 100.0 | 107 | 7.5 | 7.0 | ||
| 2000 | 2207 | 6.5 | 10.4 | ||
| Dauricine | 5.00 | 4.85 | 7.2 | −3.0 | |
| 100.0 | 95.9 | 6.9 | −4.1 | ||
| 2000 | 2190 | 6.6 | 9.5 | ||
| Long-term stability (−20 °C for 20 days) | Daucicoline | 3.00 | 3.23 | 4.3 | 7.7 |
| 60.0 | 62.1 | 2.8 | 3.5 | ||
| 1200 | 1308 | 5.7 | 9.0 | ||
| Daurisoline | 5.00 | 5.51 | 5.6 | 10.2 | |
| 100.0 | 99.5 | 7.1 | −0.5 | ||
| 2000 | 2168 | 2.8 | 8.4 | ||
| Dauricine | 5.00 | 5.35 | 10.6 | 7.0 | |
| 100.0 | 106.7 | 6.4 | 6.7 | ||
| 2000 | 1886 | 7.9 | −5.7 | ||
| Freeze–thaw stability | Daucicoline | 3.00 | 3.22 | 7.7 | 7.3 |
| 60.0 | 58.4 | 7.1 | −2.7 | ||
| 1200 | 1265 | 5.7 | 5.4 | ||
| Daurisoline | 5.00 | 5.38 | 5.3 | 7.6 | |
| 100.0 | 104.5 | 6.7 | 4.5 | ||
| 2000 | 2128 | 9.6 | 6.4 | ||
| Dauricine | 5.00 | 5.21 | 7.5 | 4.2 | |
| 100.0 | 103.7 | 6.6 | 3.7 | ||
| 2000 | 2104 | 5.9 | 5.2 | ||
| Post-preparation stability (4 °C for 12 h) | Daucicoline | 3.00 | 2.78 | 10.7 | −7.3 |
| 60.0 | 62.3 | 2.1 | 3.8 | ||
| 1200 | 1117 | 6.7 | −6.9 | ||
| Daurisoline | 5.00 | 5.54 | 5.2 | 10.8 | |
| 100.0 | 98.1 | 6.2 | −1.9 | ||
| 2000 | 2106 | 7.2 | 5.3 | ||
| Dauricine | 5.00 | 5.39 | 4.8 | 7.8 | |
| 100.0 | 103.2 | 5.9 | 3.2 | ||
| 2000 | 2136 | 2.8 | 6.8 | ||
|
|||||
| Feces | |||||
| Short-term stability (room temperature for 12 h) | Daucicoline | 3.00 | 3.32 | 10.3 | 10.7 |
| 60.0 | 61.9 | 11.2 | 3.2 | ||
| 1200 | 1256 | 10.3 | 4.7 | ||
| Daurisoline | 5.00 | 5.36 | 8.1 | 7.2 | |
| 100.0 | 108.3 | 11.1 | 8.3 | ||
| 2000 | 2206 | 8.7 | 10.3 | ||
| Dauricine | 5.00 | 4.85 | 5.7 | −3.0 | |
| 100.0 | 95.9 | 4.1 | −4.1 | ||
| 2000 | 2212 | 4.6 | 10.6 | ||
| Long-term stability (−20 °C for 20 days) | Daucicoline | 3.00 | 3.27 | 8.7 | 9.0 |
| 60.0 | 61.4 | 3.5 | 2.3 | ||
| 1200 | 1308 | 10.8 | 9.0 | ||
| Daurisoline | 5.00 | 5.54 | 5.4 | 10.8 | |
| 100.0 | 103.5 | 7.6 | 3.5 | ||
| 2000 | 2168 | 9.1 | 8.4 | ||
| Dauricine | 5.00 | 5.35 | 7.8 | 7.0 | |
| 100.0 | 107.3 | 9.6 | 7.3 | ||
| 2000 | 2164 | 5.9 | 8.2 | ||
| Freeze–thaw stability | Daucicoline | 3.00 | 3.26 | 6.7 | 8.7 |
| 60.0 | 62.9 | 9.1 | 4.8 | ||
| 1200 | 1264 | 6.7 | 5.3 | ||
| Daurisoline | 5.00 | 5.46 | 5.2 | 9.2 | |
| 100.0 | 106.5 | 6.2 | 6.5 | ||
| 2000 | 2178 | 6.9 | 8.9 | ||
| Dauricine | 5.00 | 5.29 | 8.4 | 5.8 | |
| 100.0 | 108.4 | 10.5 | 8.4 | ||
| 2000 | 2052 | 8.1 | 2.6 | ||
| Post-preparation stability (4 °C for 12 h) | Daucicoline | 3.00 | 2.88 | 9.6 | −4.0 |
| 60.0 | 63.54 | 10.3 | 5.9 | ||
| 1200 | 1121 | 7.5 | −6.6 | ||
| Daurisoline | 5.00 | 5.21 | 4.6 | 4.2 | |
| 100.0 | 107.1 | 4.8 | 7.1 | ||
| 2000 | 2104 | 1.5 | 5.2 | ||
| Dauricine | 5.00 | 5.39 | 1.9 | 7.8 | |
| 100.0 | 105.2 | 6.4 | 5.2 | ||
| 2000 | 2136 | 5.2 | 6.8 | ||
3.3. Pharmacokinetics
The plasma concentrations of three active components following oral administration of MR capsules at a dose of 2.5 g kg−1 in rats were successfully determined by the validated method. The mean plasma concentration–time profiles of the three analytes are presented in Fig. 3 and the pharmacokinetic parameters were calculated using DAS 2.0 software with non-compartmental analysis and the results are listed in Table 6.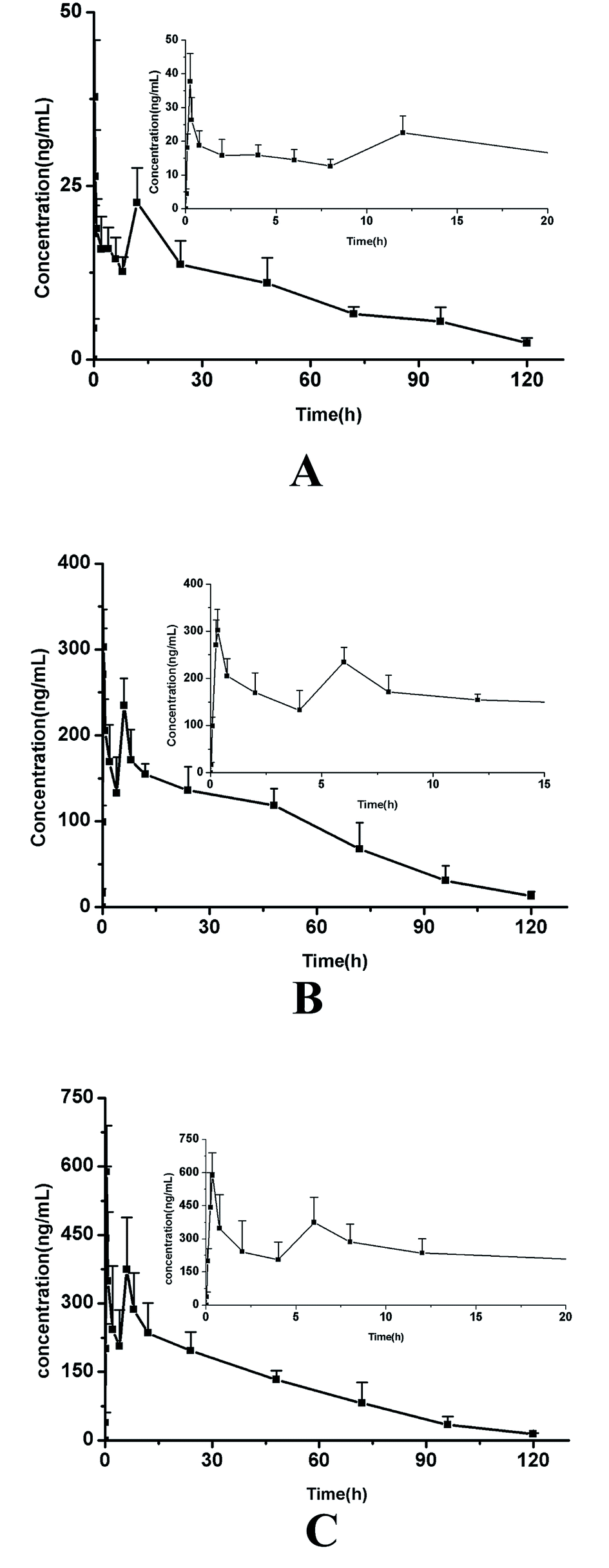 | ||
| Fig. 3 The mean plasma concentration–time curves of daucicoline (A), daurisoline (B) and dauricine (C) in rat plasma after oral administration of MR capsules. | ||
| Parameter | Unit | Daucicoline mean ± SD | Daurisoline mean ± SD | Dauricine mean ± SD |
|---|---|---|---|---|
| a Clz/F: apparent oral clearance, MRT: mean residence time, Vz/F: apparent volume of distribution. | ||||
| AUC(0–t) | ng h mL−1 | 1161.8 ± 233.5 | 10863.0 ± 1748.1 |
14461.9 ± 3279.0 |
| AUC(0–∞) | ng h mL−1 | 1400.3 ± 295.2 | 11844.1 ± 2578.8 |
15223.7 ± 3909.8 |
| MRT(0–t) | h | 41.844 ± 2.54 | 38.845 ± 3.411 | 35.241 ± 2.762 |
| MRT(0–∞) | h | 54.611 ± 10.648 | 43.488 ± 5.239 | 38.839 ± 2.575 |
| t1/2 | h | 27.26 ± 10.10 | 28.84 ± 12.90 | 24.89 ± 8.67 |
| Tmax1 | h | 0.25 ± 0.00 | 0.37 ± 0.19 | 0.32 ± 0.03 |
| Tmax2 | h | 11.33 ± 1.63 | 6.67 ± 1.03 | 6.33 ± 1.51 |
| Cmax1 | ng mL−1 | 37.80 ± 9.20 | 311.99 ± 55.66 | 591.01 ± 102.01 |
| Cmax2 | ng mL−1 | 23.55 ± 7.37 | 241.97 ± 67.05 | 422.48 ± 182.77 |
| Clz/F | L h−1 kg−1 | 0.004 ± 0.001 | 0.008 ± 0.002 | 0.009 ± 0.002 |
| Vz/F | L kg−1 | 0.199 ± 0.053 | 0.315 ± 0.101 | 0.32 ± 0.011 |
As shown in the results, the three alkaloids (daucicoline, dauricine, daurisoline) exhibited similar plasma concentration–time plots because of their similar chemical structures, where the basic parent structure was dibenzyl tetrahydroiso-quinoline. A double-peak phenomenon of the three alkaloids was observed in the rats' specimens. They all achieved the first maximum plasma concentration between 15–30 min post-dose. But the larger difference occurs at Tmax of the second peak. The second peak appears around 11 h for daucicoline, and less than 7 h for daurisoline and dauricine. Some researchers have previously conducted pharmacokinetic studies on daurisoline, and the results described in this paper are basically consistent with those found in the literature.26 In the study of pharmacokinetics of traditional Chinese medicine, the double peak phenomenon was a common phenomenon. It has been reported that the main reason for abnormal double-peak phenomenon of dauricine in vivo was the stomach–intestine circle.27 The emergence of the second peak of the plasma concentration–time curve was the equivalent of an “autogenous drug” process, which can affect the intensity of drug action and the elimination process.28 The retention time of the three compounds in rats was longer, which may be related to the occurrence of the double-peak phenomenon. The study of the double-peak or multimodal phenomenon can provide the basis for the formulation of a more careful drug delivery program.
3.4. Excretion
After oral administration to rats of MR capsules at a dose of 2.5 g kg−1, the cumulative excretion results of the analytes in rat urine and feces are displayed in Fig. 4. In urine, it could be concluded that the three analytes were mainly excreted during 120 h after oral administration. Compared with the dosing, total recoveries of the three compounds in urine within 120 h were about 0.71% for daucicoline, 0.20% for daurisoline, 0.18% for dauricine. Low recoveries of the three analytes in urine indicated that few unconverted forms were excreted in the urine. The cumulative excretion of the three compounds in feces within 120 h were about 29.5% for daucicoline, 19.8% for daurisoline, 14.9% for dauricine. It could be concluded that there was significant increase for the three alkaloids in fecal excretion after 12 h, and the speed of excretion continued to accelerate within 12 h to 36 h. Then the fecal cumulative excretion of the analytes reached a plateau at 48 h after dosing. These results indicated that fecal excretion was the dominant excretion pathway. It has been reported previously26 that dauricine was excreted mainly in feces. The results of this study were consistent with the results of the report in the literature. As far as is known, the excretion characteristics of daucicoline and daurisoline were investigated for the first time in this research.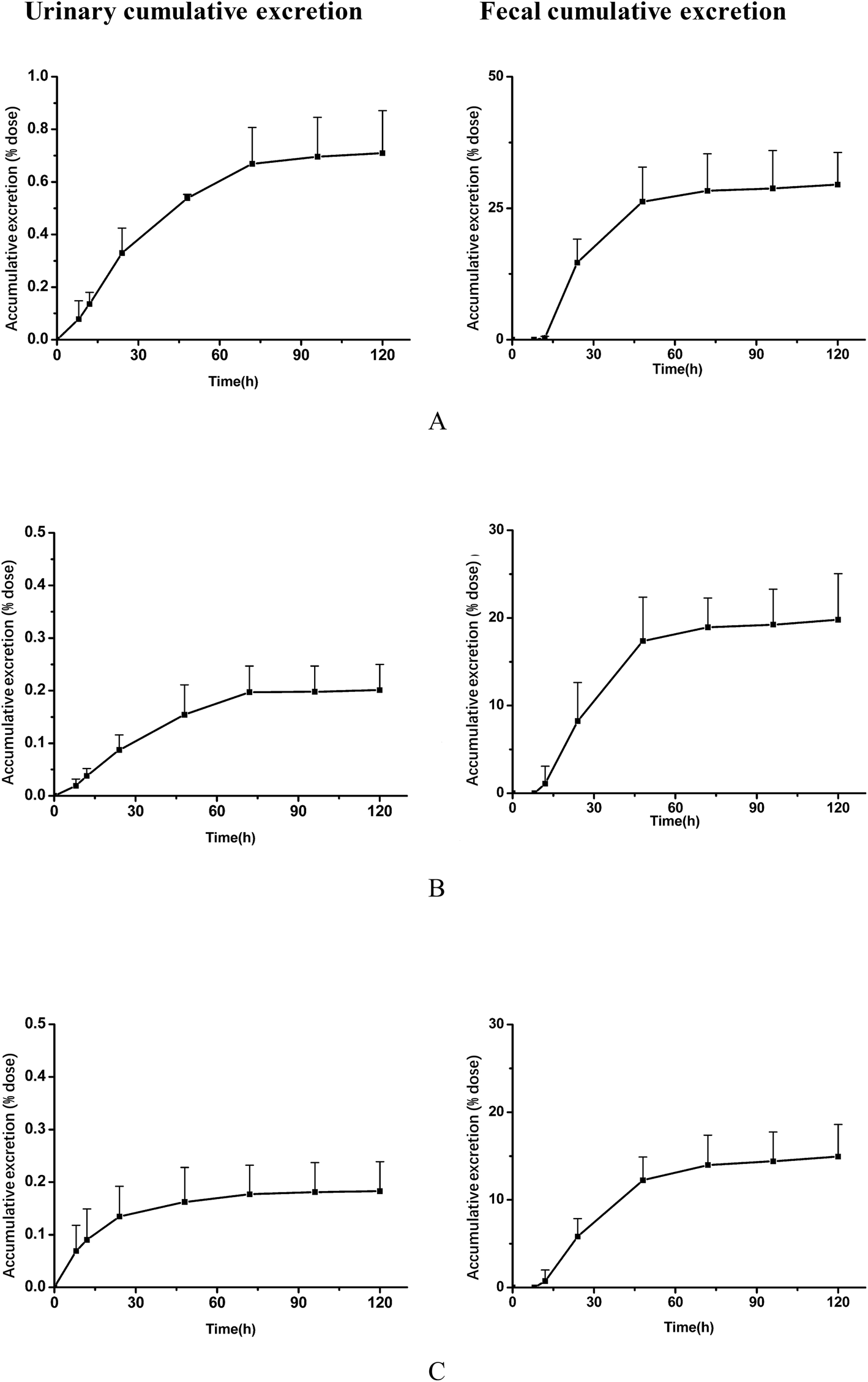 | ||
| Fig. 4 Mean urinary and fecal cumulative excretion curves of daucicoline (A), daurisoline (B) and dauricine (C) in rats after oral administration of MR capsules (2.5 g kg−1; n = 6 for male rats; each point represents mean ± SD). | ||
4. Conclusions
This research is the first demonstration of a UPLC-MS/MS method that was validated for the simultaneous determination of three main alkaloids (daucicoline, dauricine, and daurisoline) of MR capsules in rat plasma, urine and feces. The analytical procedure was successfully applied to pharmacokinetic and excretion studies of the analytes after oral administration of MR capsules. The pharmacokinetic data showed that the plasma concentration–time plots of the three alkaloids exhibited a similar double-peak phenomenon. The excretion data suggested that the three analytes were mainly excreted by feces. The previously described results will be helpful for the further investigation of the mechanism of action of the active ingredients in MR capsules and may provide a useful tool for the clinical application of the formulation of MR capsules.Conflicts of interest
The authors declare no conflict of interest.References
- Chinese Pharmacopoeia Committee, Chinese Pharmacopeia, China Medical Science Press, Beijing, 2015, vol. 1, p. 99 Search PubMed
.
- Chinese Pharmacopoeia Committee, Chinese Pharmacopeia, China Medical Science Press, Beijing, 2015, vol. 1, p. 775 Search PubMed
- Y. Zhang, C. Sun, Q. C. Guan, J. Yang and J. Zhao, Research of anti-inflammatory and analgesic effect of rhizoma of Menispermum dauricum, Mod. Pharm. Clin., 2009, 06, 367–368 Search PubMed
- X. H. Liu and F. L. Han, Antagonistic effect of the total alkaloids of Menispermum dauricum injection on experimental arrhythmias, Heilongjiang Med. J., 2000, 13, 160–162 Search PubMed
- J. Li, H. Bi, B. Du, Y. Sun and H. Zhang, Study on the bacteriostatic action of Menispermi alkaloid by microcalorimetry, J. Qufu Norm. Univ., Nat. Sci., 2010, 04, 96–102 Search PubMed
- Y. Su, C. Zhang and J. Xiao, Effects of PAMD on the proliferation of human tumour cells of PC-3 and BT5637, J. Harbin Med. Univ., 2007, 41, 129–131 Search PubMed
- X. Kong, X. Yang and P. Xi, Effects of phenolic alkaloids of Menispermum dauricum on platelet aggregation and its mechanism, Chin. Pharm. J., 2006, 41, 910–913 Search PubMed
- Z. Wang, J. Wang, Y. Guo, T. Lu, X. Wang and Q. Duan, Neuroprotection of phenolic alkaloids from Menispermum dauricum versus melatonin against oxygen-glucose-serum-deprivation- mediated injury, Neural Regener. Res., 2009, 4, 15–19 Search PubMed
- F. Wang, L. Qu, Q. Lv and L. J. Guo, Effect of phenolic alkaloids from Menispermum dauricum on myocardial-cerebral ischemia-reperfusion injury in rabbits, Acta Pharmacol. Sin., 2001, 22, 1130–1134 Search PubMed
- Y. M. Su, H. Su and B. Sheng, Effect of phenolic alkaloids from Menispermum dauricum on hemodynamics in experimental myocardial ischemia, China Pharm., 2004, 07, 83–85 Search PubMed
- D. Z. Zhao, Y. Q. Du and B. Jiang, Effect of phenolic alkaloids from Menispermum dauricum (PAMD) on apoptosis protein cfos expression in rats with cerebral ischemia, China Pharm., 2009, 12, 1005–1008 Search PubMed
- D. Su, Z. K. Wang, T. T. Sun and X. J. Guo, Simultaneous determination of content of 5 alkaloids in Menispermi Rhizoma capsule by HPLC, J. Shenyang Pharm. Univ., 2016, 01, 46–50 Search PubMed
- S. M. Hu, S. X. Xu, X. S. Yao, C. B. Cui, Y. Tezuka and T. Kikuchi, Dauricoside, a new glycosidal alkaloid having an inhibitory activity against blood-platelet aggregation, Chem. Pharm. Bull., 1993, 10, 1866–1868 CrossRef
- S. Xia, Z. Li, J. W. Dong, H. Tu and F. D. Zeng, Dauricine-induced changes in monophasic action potentials and effective refractory period of rabbit left ventricle in situ, Acta Pharmacol. Sin., 2002, 23, 371–375 Search PubMed
- J. Q. Qian, Cardiovascular pharmacological effects of bisbenzylisoquinoline alkaloid derivatives, Acta Pharmacol. Sin., 2002, 23, 1086–1092 Search PubMed
- Q. Liu, X. Mao, F. Zeng, S. Jin and X. Yang, Effect of Daurisoline on hERG Channel Electrophysiological Function and Protein Expression, J. Nat. Prod., 2012, 75, 1539–1545 CrossRef PubMed
- X. D. Tang, X. Zhou and K. Y. Zhou, Dauricine inhibits insulin-like growth factor-I-induced hypoxia inducible factor 1alpha protein accumulation and vascular endothelial growth factor expression in human breast cancer cells, Acta Pharmacol. Sin., 2009, 30, 605–616 CrossRef PubMed
- Y. Liu, X. Song, R. Yan, T. Li, X. Chai, A. Qi, Y. Wang and Z. Jiang, Development and validation of a UPLC-DAD-MS method for characterization and quantification of alkaloids in Menispermi Rhizoma and its preparations, J. Food Drug Anal., 2013, 21, 206–218 CrossRef
- Y. Zhang, K. Li, P. Zhang, L. Feng and Z. Zhang, Determination of
Dauricine and Daurisoline in Rhizoma Menispermi and Its Preparations by HPLC, Chin. Arch. Tradit. Chin. Med., 2016, 04, 921–923 Search PubMed
- J. L. Geng, Y. Dai, Z. H. Yao, Z. F. Qin, X. L. Wang, L. Qin and X. S. Yao, Metabolites prole of Xian-Ling-Gu-Bao capsule, a traditional Chinese prescription, in rats by ultra performance liquid medicine coupled with quadrupole time-of-fight tandem chromatography spectrometry analysis mass, J. Pharm. Biomed. Anal., 2014, 96, 90–103 CrossRef PubMed
- H. Hao, X. Zheng and G. Wang, Insights into drug discovery from natural medicines using reverse pharmacokinetics, Trends Pharmacol. Sci., 2014, 04, 168–177 CrossRef PubMed
- Y. Xie, G. Zhong, H. He, G. Fan and Y. Wu, Pharmacokinetics, tissue distribution and excretion of porcine fibrinogen after intraperitoneal injection of a porcine-derived fibrin glue to rats, J. Pharm. Biomed. Anal., 2014, 54, 148–153 CrossRef PubMed
- J. Wang, Q. Pang, W. Cen, P. Zhu and Y. Xu, Simultaneous determination of ten active constituents of Yankening in rat plasma by ultra high performance liquid chromatography with tandem mass spectrometry, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 43, 978–979 Search PubMed
- Y. Wu, L. Ou, D. Han, Y. Tong, M. Zhang, X. Xu and C. Zhang, Pharmacokinetics, biodistribution and excretion studies of neotuberostemonine, a major bioactive alkaloid of Stemona tuberose, Fitoterapia, 2016, 112, 22–29 CrossRef PubMed
- Food and Drug Administration, Guidance for Industry: Bioanalytical Method Validation, US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research, 2018 Search PubMed
- Y. X. Jin, C. C. Song, L. J. Shao, L. T. Wang, X. T. Tu, B. B. Chen, J. Y. Chen, Y. H. Zhi, C. C. Wen and W. Z. Zhu, Determination and Pharmacokinetic Study of Daurisoline in Rat Plasma by UPLC-MS/MS, Lat. Am. J. Pharm., 2017, 36, 1204–1209 Search PubMed
- S. Chen, Y. Yang, Y. Liu, B. Zhang, X. Pang and F. Zeng, The pharmacokinetics of dauricine in rats, Chin. Pharmacol. Bull., 2001, 02, 225–229 Search PubMed
- J. X. Wei, L. L. Fang, X. L. Liang, D. Su and X. J. Guo, A sensitive and selective UPLC–MS/MS method for simultaneous determination of 10 alkaloids from Rhizoma Menispermi in rat plasma and its application to a pharmacokinetic study, Talanta, 2015, 144, 662–670 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04084b |
| This journal is © The Royal Society of Chemistry 2018 |