
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegio-selective and stereo-selective hydrosilylation of internal alkynes catalyzed by ruthenium complexes†
Wenhao Dai‡
ab,
Xiaowei Wu‡bc,
Chunpu Lib,
Rui Zhangbc,
Jiang Wangb and
Hong Liu *ab
*ab
aSchool of Pharmacy, China Pharmaceutical University, Nanjing 210009, Jiangsu, China
bKey Laboratory of Receptor Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zu Chong Zhi Road, Shanghai, 201203, China. E-mail: hliu@simm.ac.cn
cUniversity of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing 100049, China
First published on 7th August 2018
Abstract
In this study, ruthenium(II)-catalyzed direct hydrosilylation of internal alkynes with high regio-selectivity and stereo-selectivity is reported. This title transformation led to various vinylsilanes in good to excellent yields. This approach features mild reaction conditions, low catalyst loading, air-stability, and good functional group tolerance. Furthermore, gram-scale preparation and some transformations of vinylsilanes were carried out, which further underscored its synthetic utility and applicability.
Introduction
Vinyl-metal reagents play a key role in organic synthesis. Among the available vinyl-metal reagents, silicon-based reagents are increasingly important. Vinylsilanes are versatile synthetic building blocks, because of their minimal toxicity, low cost, ease of handling, and tendency to undergo different kinds of transformation.1 Among the available methods for preparation of vinylsilanes, the hydrosilylation of alkynes is the most powerful strategy because it is direct and atom-economical, and it offers the potential to control the regio-selectivity and stereo-selectivity.2–4 Although there are many methods for the hydrosilylation of terminal alkynes, particularly for the preparation of cis- and trans-β-vinylsilanes, the regio-selective and stereo-selective hydrosilylation of internal alkynes still remains a great challenge due to their low reactivity and close similarity in terms of electronic and steric properties to the acetylenic substituents.5–7 Non-selective hydrosilylation of internal alkynes would potentially give four isomeric addition products (Fig. 1).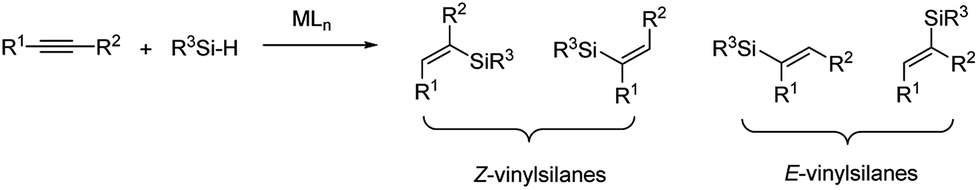 | ||
| Fig. 1 Four isomeric addition products. | ||
To address the difficulty of regiocontrol in the hydrosilylation of unsymmetrical (internal) alkynes, a directing group (DG) was introduced into an unsymmetrical alkyne to control their regio-selectivity of the hydrosilylation. Zakarian6d and co-workers developed the silylation of internal alkyne and sequential iododesilylation with high regioselectivity. The regioselectivity is guided by coordination of the silylcupration reagent to hydroxy or alkoxy groups in the substrate (Scheme 1a). In the early 2000s, Trost8 and co-workers reported the regio-selective trans-hydrosilylation of propargylic alcohols catalyzed by the cationic complex [Cp*Ru(MeCN)3]PF6, which resulted in β-selective silylation at the alkyne carbon (Scheme 1b). The introduction of a hydroxyl group at the propargylic position is crucial to control the regioselectivity of hydrosilylation. In addition, a carbonyl group has been also shown to direct hydrosilylation of internal alkynes to provide α-vinylsilanes. While, the regio-selectivity largely depends on the electronic effect.7d On the contrary, the examples of α-selective hydrosilylation of unbiased unsymmetrical internal alkynes are very limited,7c,7e,9 especially for the propargylic alcohol substrates. To date, selective hydrosilylation on the α-carbon of propargylic alcohols are rarely reported. In 2011, Tomooka's group7c described Pt-catalyzed α-selective hydrosilylation of unsymmetrical alkynes using a dimethylvinylsilyl (DMVS) group as the directing group (Scheme 1c). However, the substrate scope of this method was limited for the primary alcohols. More recently, Fürstner9a,9b,9c and co-workers disclosed that [Cp*RuCl]4 also catalyzed the hydrosilylation of propargylic alcohols to form α-vinylsilanes with a high degree of regioselectivity (Scheme 1d). While, the R1 part of propargylic alcohols was alkyl, alkenyl and H. In view of the importance of the broad synthetic utility of vinylsilanes, we are interested in developing another efficient catalyst for α-selective hydrosilylation of propargylic alcohols. Herein, we describe a new method for the hydrosilylation of differentially propargyl alcohols with a new ruthenium complex under mild conditions (Scheme 1e). The transformation is highly regio-selective and stereo-selective for the preparation of α-vinylsilanes from unbiased and unsymmetrical internal alkynes.
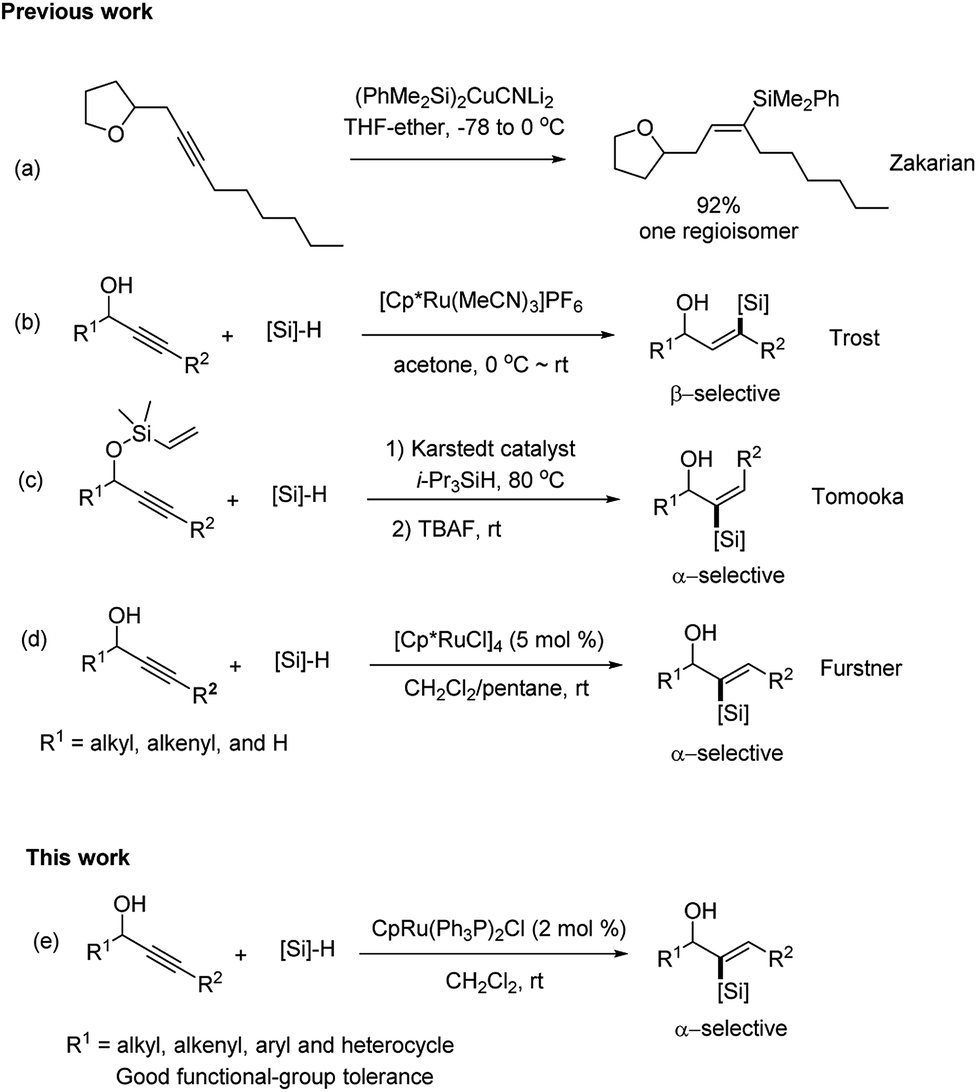 | ||
| Scheme 1 Directed hydrosilylation of internal alkynes. | ||
Results and discussion
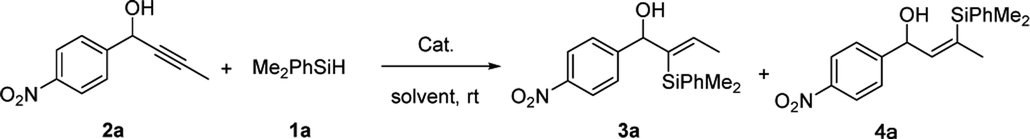
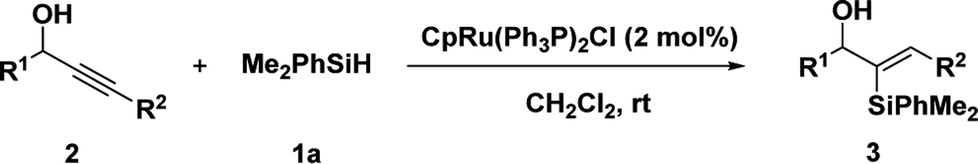
We initiated our studies with the hydrosilylation reaction of 1-(4-nitrophenyl) but-2-yn-1-ol 2a and the easily available PhMe2SiH 1a in the presence of a Ru complex. Interestingly, when the reaction was carried out with CpRu(Ph3P)2Cl (5 mol%) in THF for 12 h, 3a was obtained in 68% yield with good regioselectivity (α/β = 82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18) (Table 1, entry1). Then solvent screening was conducted, when the reaction was conducted in CH2Cl2, the best result was obtained (94% yield with excellent regioselectivity (α/β = 95:5)) (entries 2–4). Other catalysts, such as [{RuCl2(p-cymene)}2], [Cp*RuCl]4, reduced the yield of 3a with poor regioselectivity compared to CpRu(Ph3P)2Cl (entries 5 and 6). To further improve the reaction efficiency, the loading amount of the catalyst was investigated. When we reduced the loading amount of the CpRu(Ph3P)2Cl from 5 mol% to 2 mol%, the reaction also produced 3a in excellent yield (94%) and excellent regioselectivity (α/β = 95:5) (entry 7). However, when 1 mol% CpRu(Ph3P)2Cl was used in the hydrosilylation reaction, the yield of 3a was slightly decreased (entry 8). Then this reaction was carried out in air atmosphere, the yield and regioselectivity slightly decreased (entry 9). When 5 mol% [CpRu(Ph3P)(tht)]BF4 was applied, we got the poor regio-selectivity (α/β = 22:78) (entry 10), it showed the Cl ligand played a crucial role. Briefly, the optimal results could be obtained when 1-(4-nitrophenyl) but-2-yn-1-ol 2a (0.2 mmol) and PhMe2SiH 1a (0.24 mmol, 1.2 equiv.) were treated with 2 mol% CpRu(Ph3P)2Cl in CH2Cl2 at room temperature for 12 h.
:18) (Table 1, entry1). Then solvent screening was conducted, when the reaction was conducted in CH2Cl2, the best result was obtained (94% yield with excellent regioselectivity (α/β = 95:5)) (entries 2–4). Other catalysts, such as [{RuCl2(p-cymene)}2], [Cp*RuCl]4, reduced the yield of 3a with poor regioselectivity compared to CpRu(Ph3P)2Cl (entries 5 and 6). To further improve the reaction efficiency, the loading amount of the catalyst was investigated. When we reduced the loading amount of the CpRu(Ph3P)2Cl from 5 mol% to 2 mol%, the reaction also produced 3a in excellent yield (94%) and excellent regioselectivity (α/β = 95:5) (entry 7). However, when 1 mol% CpRu(Ph3P)2Cl was used in the hydrosilylation reaction, the yield of 3a was slightly decreased (entry 8). Then this reaction was carried out in air atmosphere, the yield and regioselectivity slightly decreased (entry 9). When 5 mol% [CpRu(Ph3P)(tht)]BF4 was applied, we got the poor regio-selectivity (α/β = 22:78) (entry 10), it showed the Cl ligand played a crucial role. Briefly, the optimal results could be obtained when 1-(4-nitrophenyl) but-2-yn-1-ol 2a (0.2 mmol) and PhMe2SiH 1a (0.24 mmol, 1.2 equiv.) were treated with 2 mol% CpRu(Ph3P)2Cl in CH2Cl2 at room temperature for 12 h.
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | α:β ratiob |
Yieldc (%) |
| a Reaction conditions: 1a (0.24 mmol), 2a (0.2 mmol), in 2 mL solvent at room temperature, Ar atmosphere for 12 h.b Determined by 1H NMR analysis of the crude reaction mixtures.c NMR yields using CH2Br2 as an internal standard, isolated yields in parenthesis.d This entry's major product is α-selective cis- hydrosilylation (see NOE analysis in SI).e 1a (0.24 mmol), 2a (0.2 mmol), in 2 mL solvent at room temperature under air atmosphere for 12 h. | ||||
| 1 | CpRu(Ph3P)2Cl (5) | THF | 82:18 |
68 |
| 2 | CpRu(Ph3P)2Cl (5) | Toluene | 94:6 |
59 |
| 3 | CpRu(Ph3P)2Cl (5) | Acetone | 79:21 |
65 |
| 4 | CpRu(Ph3P)2Cl (5) | CH2Cl2 | 95:5 |
94 |
| 5 | [{RuCl2(p-cymene)}2] (5) | CH2Cl2 | — | —d |
| 6 | [Cp*RuCl]4 (5) | CH2Cl2 | 92:8 |
83 |
| 7 | CpRu(Ph3P)2Cl (2) | CH2Cl2 | 95:5 |
94 (94) |
| 8 | CpRu(Ph3P)2Cl (1) | CH2Cl2 | 95:5 |
89 |
| 9 | CpRu(Ph3P)2Cl (2) | CH2Cl2 | 91:9 |
86e |
| 10 | [CpRu(Ph3P)(tht)]BF4(5) | CH2Cl2 | 22:78 |
18 |
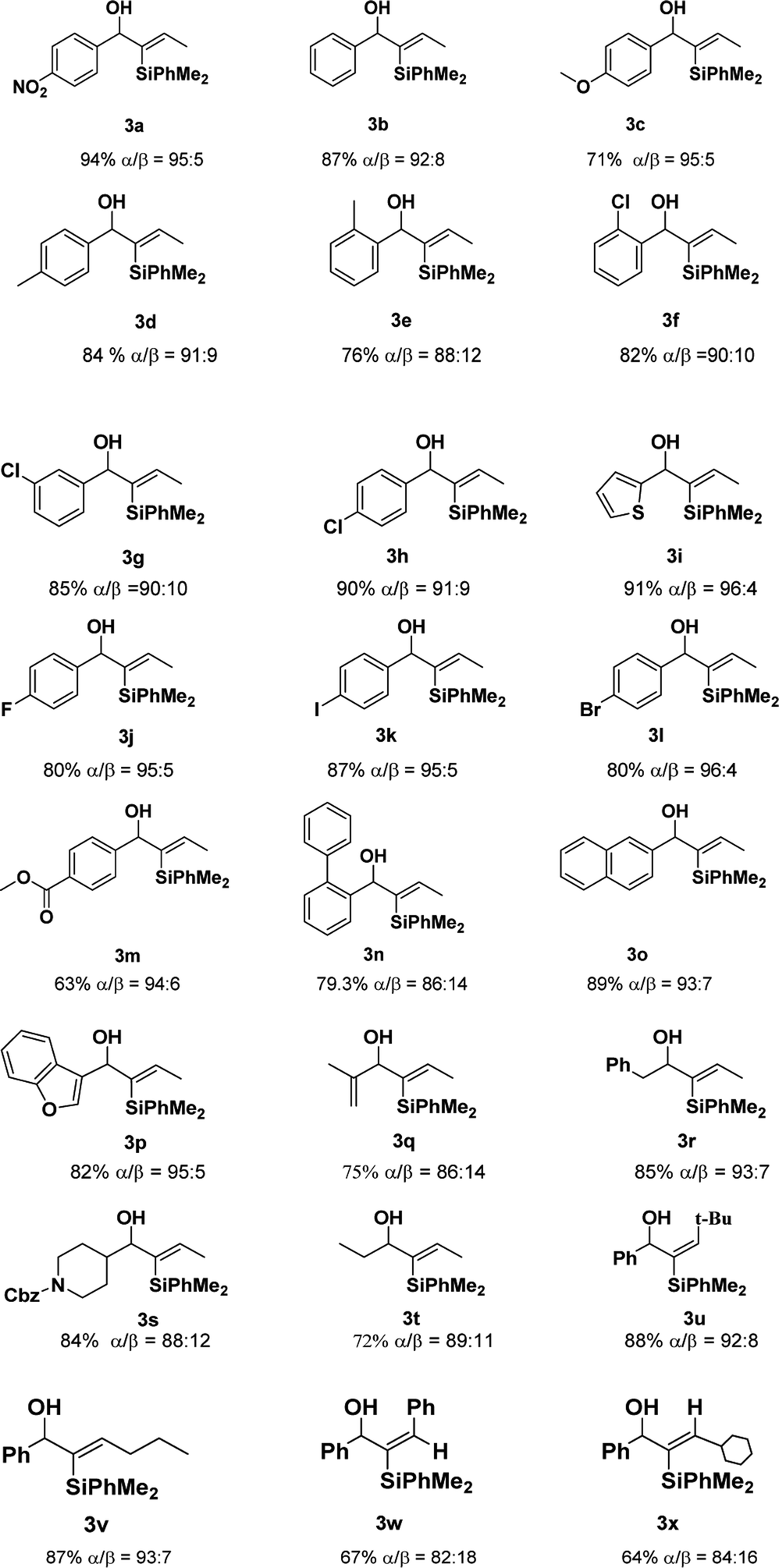
With the optimized conditions in hand, we examined the scope of different propargyl alcohols 2 in the hydrosilylation reaction with PhMe2SiH 1a (Table 2). Substrates containing electron-withdrawing groups, such as nitro and halides, at the phenyl moiety of propargyl alcohols were well tolerated under the standard reaction conditions (3a, 3f–3h, and 3j–3l). A similar level of efficiency was observed when substrates having phenyl, p-tolyl, o-tolyl, and 1,1′-biphenyl groups (3b, 3d, 3e, and 3n), while some other electron-rich substrates provided the desired products in decreased yields (3c). In addition, the desired products were obtained in higher yields and better regio-selectivity when substituents, such as methyl and chloride, were introduced at the para position of the phenyl moiety (3d vs. 3e, and 3h vs. 3f). The tolerance of the reaction with active groups such as halides in the substrates meant that they could be further transformed into other functional groups. When an ester group was placed at the para position of phenyl ring, 3m was obtained in 63% yield, and it seemed that the carbonyl may participate in coordination. Furthermore, substrates bearing other heteroaryl groups, such as thiophene and benzofuran, also could react with 1a to produce the desired product in good yields with excellent regio-selectivity (3i and 3p). Significantly, replacing the phenyl moiety of the propargyl alcohols with vinyl, benzyl, N-protected piperidyl and alkyl, the hydrosilylation reaction also underwent smoothly with satisfactory results (3q–3t). While the introduction of bulkier groups (such as tert-butyl and Ph) in the R2 moiety of the substrates provided the different stereo-selective products in good yields (3u,3w), which was different from Fürstner's work. 9a
Moreover, when Et3SiH was used, the hydrosilylation of 2a proceeded with excellent result (α/β = 95:5, 94% yield) (Scheme 2). Given the poor reactivity of alkylsilanes toward a variety of synthetically transformations, we extended the method to more reactive and operationally convenient alkyoxysilanes. Hydrosilylation of 2a with triethoxylsilane was performed under standard conditions and provided the desired product 4b in 61% yield with excellent regioselectivity. However, hydrosilylation of 2a with t-BuMe2SiH produced desired product 4c in 60% yield with poor regio-selectivity.
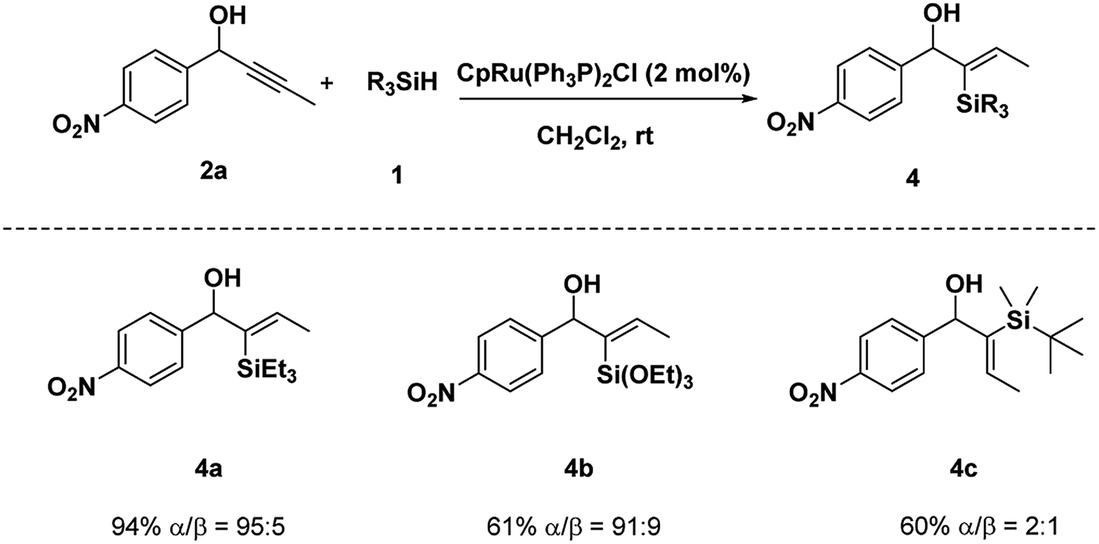 | ||
| Scheme 2 Alternative silane scope. | ||
To assess the efficiency and potential applications of this method, we carried out gram-scale preparation and transformations of the products (Scheme 3). When propargyl alcohol 2a (1.0 g, 5 mmol) was used, the reaction afforded the corresponding product 3a in 75% yield. Besides, there were few works about the transformation of α-hydroxy alkynylsilanes by coupling reactions to date.9d In our work, the coupling of vinylsilane 3za and phenylacetylene could provide the product 5a in good yield.10 The epoxidation of 3a with mCPBA resulted in product 5b. Furthermore, vinylsilane 3a could be converted to trans-olefin 5c by tetrabutylammonium fluoride (TBAF) in 70% yield at room temperature (Scheme 3d).
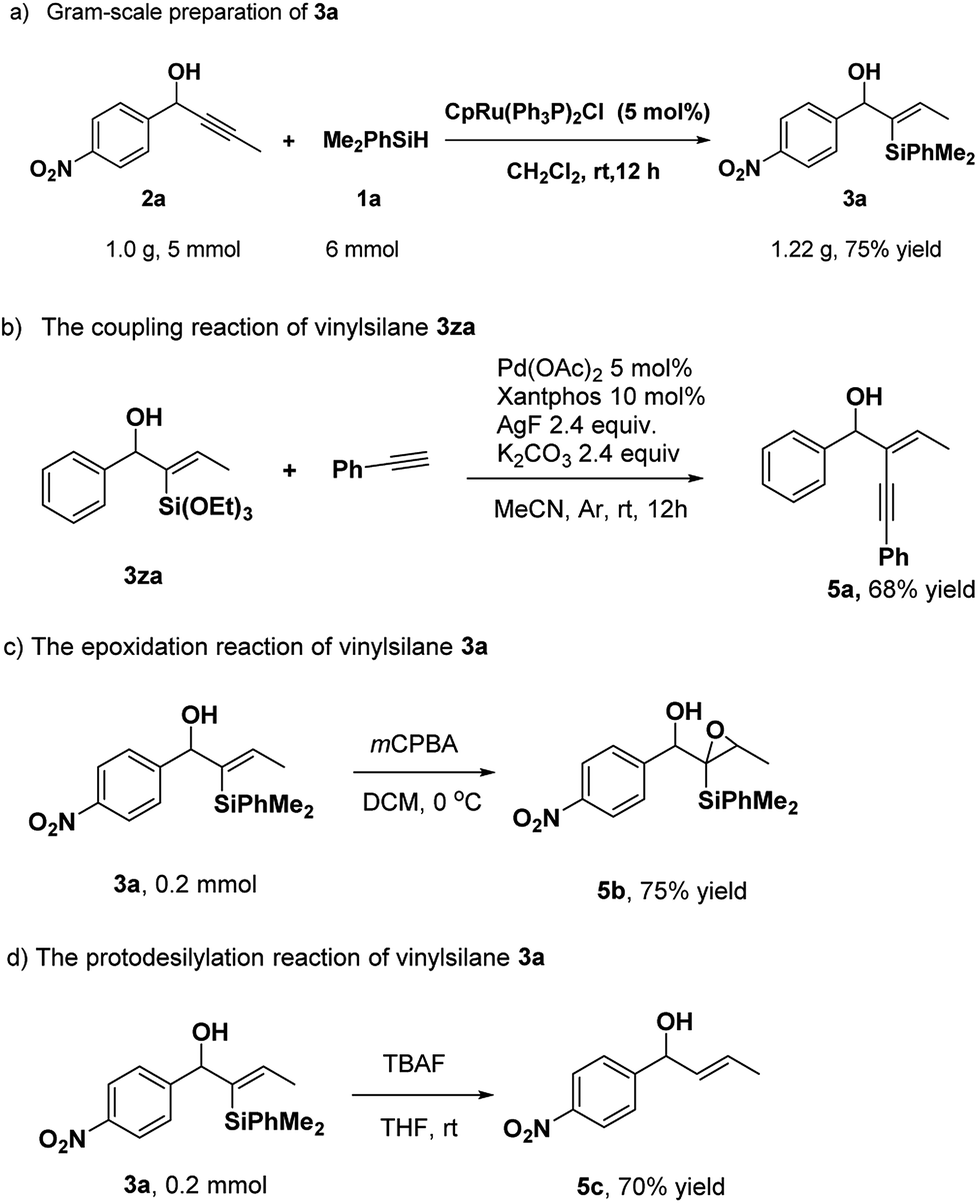 | ||
| Scheme 3 Gram-scale experiment and transformations of the products. | ||
Conclusions
In summary, we reported a mild and efficient ruthenium-catalyzed hydrosilylation of internal alkynes. This method enables the efficient preparation of vinylsilanes in good to excellent yields with highly regio-selectivity and stereo-selectivity. Its mild reaction conditions mean that this approach is also compatible with various functional groups. We also achieved the gram-scale production and some transformations of vinylsilanes, which further underscored the synthetic utility and versatile applicability of this method.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (81620108027, 21632008 and 81602975), the Major Project of Chinese National Programs for Fundamental Research and Development (2015CB910304)Notes and references
- (a) T. A. Blumenkopf and L. E. Overman, Chem. Rev., 1986, 86, 857 CrossRef; (b) E. Langkopf and D. Schinzer, Chem. Rev., 1995, 95, 1375 CrossRef; (c) I. Ojima, Z. Li and J. Zhu, The Chemistry of Organic Silicon Compounds. Wiley, Hoboken, 2003, p. 1687 Search PubMed; (d) I. Beletskaya and C. Moberg, Chem. Rev., 1999, 99, 3435 CrossRef PubMed; (e) M. Suginome and Y. Ito, Chem. Rev., 2000, 100, 3221 CrossRef PubMed; (f) D. S. W. Lim and E. A. Anderson, Synthesis, 2012, 44, 983 CrossRef; (g) M. Zaranek, B. Marciniec and P. Pawluć, Org. Chem. Front., 2016, 3, 1337 RSC.
- (a) H. Brunner, Angew. Chem., Int. Ed., 2004, 43, 2749 CrossRef PubMed; (b) B. M. Trost and Z. T. Ball, Synthesis, 2005, 6, 853 CrossRef; (c) L. Iannazzo and G. A. Molander, Eur. J. Org. Chem., 2012, 2012, 4923 CrossRef PubMed; (d) A. K. Roy, Adv. Organomet. Chem., 2008, 55, 1 Search PubMed; (e) Q. Q. Xuan, C. L. Ren, L. Liu, D. Wang and C. Li, Org. Biomol. Chem., 2015, 13, 5871 RSC.
- (a) S. Schwieger, R. Herzog, C. Wagner and D. Steinborn, J. Organomet. Chem., 2009, 694, 3548 CrossRef; (b) Y. Sumida, T. Kato, S. Yoshida and T. Hosoya, Org. Lett., 2012, 14, 1552 CrossRef PubMed; (c) T. Sanada, T. Kato, M. Mitani and A. Mori, Adv. Synth. Catal., 2006, 348, 51 CrossRef; (d) T. Konno, K. Taku, S. Yamada, K. Moriyasu and T. Ishihara, Org. Biomol. Chem., 2009, 7, 1167 RSC; (e) C. Belger and B. Plietker, Chem. Commun., 2012, 48, 5419 RSC; (f) T. Murai, E. Nagaya, F. Shibahara, T. Maruyama and H. Nakazawa, J. Organomet. Chem., 2015, 794, 76 CrossRef; (g) W. Guo, R. Pleixats, A. Shafir and T. Parella, Adv. Synth. Catal., 2015, 357, 89 CrossRef; (h) M. Planellas, W. Guo, F. Alonso, M. Yus, A. Shafir, R. Pleixats and T. Parella, Adv. Synth. Catal., 2014, 356, 179 CrossRef.
- (a) C. A. McAdam, M. G. McLaughlin, A. J. Johnston, J. Chen, M. W. Walter and M. Cook, Org. Biomol. Chem., 2013, 11, 4488 RSC; (b) S. Sueki and Y. Kuninobu, Chem. Commun., 2015, 51, 7685 RSC; (c) R. Cano, M. Yus and D. J. Ramón, ACS Catal., 2012, 2, 1070 CrossRef; (d) F. Monteil, I. Matsuda and H. Alper, J. Am. Chem. Soc., 1995, 117, 4419 CrossRef; (e) D. A. Rooke and E. M. Ferreira, Org. Lett., 2012, 14, 3328 CrossRef PubMed; (f) Y. Kim, R. B. Dateer and S. Chang, Org. Lett., 2017, 19, 190 CrossRef PubMed; (g) A. Rivera-Hernandez, B. J. Fallon, S. Ventre, C. Simon, M. H. Tremblay, G. Gontard, E. Derat, M. Amatore, C. Aubert and M. Petit, Org. Lett., 2016, 18, 4242 CrossRef PubMed; (h) P. Gao, W. Zhang and Z. Zhang, Org. Lett., 2016, 18, 5820 CrossRef PubMed.
- (a) B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2001, 123, 12726 CrossRef PubMed; (b) L. N. Lewis, K. G. Sy, G. L. Bryant and P. E. Donahue, Organometallics, 1991, 10, 3750 CrossRef; (c) R. Takeuchi, S. Nitta and D. Watanabe, J. Org. Chem., 1995, 60, 3045 CrossRef; (d) Y. Na and S. Chang, Org. Lett., 2000, 2, 1887 CrossRef PubMed; (e) A. Sato, H. Kinoshita, H. Shinokubo and K. Oshima, Org. Lett., 2004, 6, 2217 CrossRef PubMed; (f) K. H. Huang and M. Isobe, Eur. J. Org. Chem., 2014, 2014, 4733 CrossRef; (g) Z. Mo, J. Xiao, Y. Gao and L. Deng, J. Am. Chem. Soc., 2014, 136, 17414 CrossRef PubMed; (h) C. Z. Wu, W. J. Teo and S. Z. Ge, ACS Catal., 2018, 8, 5896 CrossRef; (i) W. J. Teo, C. Wang, Y. W. Tan and S. Z. Ge, Angew. Chem., Int. Ed., 2017, 56, 4328 CrossRef PubMed; (j) X. Y. Du, W. J. Hou, Y. L. Zhang and Z. Huang, Org. Chem. Front., 2017, 4, 1517 RSC.
- (a) Y. Kawanami, Y. Sonoda, T. Mori and K. Yamamoto, Org. Lett., 2002, 4, 2825 CrossRef PubMed; (b) C. Menozzi, P. I. Dalko and J. Cossy, J. Org. Chem., 2005, 70, 10717 CrossRef PubMed; (c) L. W. Chung, Y. D. Wu, B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2003, 125, 11578 CrossRef PubMed; (d) E. A. Ilardi, C. E. Stivala and A. Zakarian, Org. Lett., 2008, 10, 1727 CrossRef PubMed.
- (a) B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2004, 126, 13942 CrossRef PubMed; (b) B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2005, 127, 17644 CrossRef PubMed; (c) Y. Kawasaki, Y. Ishikawa, K. Igawa and K. Tomooka, J. Am. Chem. Soc., 2011, 133, 20712 CrossRef PubMed; (d) D. A. Rooke and E. M. Ferreira, Angew. Chem., Int. Ed., 2012, 51, 3225 CrossRef PubMed; (e) R. A. García, R. J. A. Romero, P. Mauleón, R. Gómez Arrayás and J. C. Carretero, J. Am. Chem. Soc., 2015, 137, 5506 CrossRef PubMed.
- B. M. Trost, Z. T. Ball and T. Jöge, Angew. Chem., Int. Ed., 2003, 42, 3537 CrossRef.
- (a) S. M. Rummelt, K. Radkowski, D. Roşca and A. Fürstner, J. Am. Chem. Soc., 2015, 137, 5506 CrossRef PubMed; (b) T. G. Frihed and A. Fürstner, Bull. Chem. Soc. Jpn., 2016, 89, 135 CrossRef; (c) D. Roşca, K. Radkowski, L. M. Wolf, M. W. Richard Goddard, W. Thiel and A. Fürstner, J. Am. Chem. Soc., 2017, 139, 2443 CrossRef PubMed; (d) N. Huwyler, K. Radkowski, S. M. Rummelt and A. Ferstner, Chem.–Eur. J., 2017, 23, 12412 CrossRef PubMed.
- Z. S. Ye, M. C. Liu, B. D. Lin, H. Y. Wu, J. C. Ding and J. Cheng, Tetrahedron Lett., 2009, 50, 530 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Data for new compounds and experimental procedures. See DOI: 10.1039/c8ra04083d |
| ‡ Wenhao Dai and Xiaowei Wu contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2018 |