
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sandwich type polyoxometalates encapsulated into the mesoporous material: synthesis, characterization and catalytic application in the selective oxidation of sulfides
Elham Naseri and
Roushan Khoshnavazi *
*
Department of Chemistry, University of Kurdistan, PO. Box 66135-416, Sanandaj, Iran. E-mail: r.khoshnavazi@uok.ac.ir; Fax: +98 87336224133; Tel: +98 87336224133
First published on 7th August 2018
Abstract
The A-type sandwich polyoxometalates of [(HOSnIVOH)3(PW9O34)2]12− (P2W18Sn3) and [(OCeIVO)3(PW9O34)2]12− (P2W18Ce3) were immobilized for the first time into the porous metal–organic framework MIL-101(Cr). FT-IR, powder X-ray diffraction, SEM-EDX, ICP analysis, N2 adsorption and thermogravimetric analysis collectively confirmed immobilization and good distribution of polyoxometalates into cages of MIL-101(Cr). The catalytic activities of the homogeneous P2W18Sn3 and P2W18Ce3 and the corresponding heterogeneous catalysts were examined in the oxidation of sulfides to sulfones with H2O2 as the oxidant at room temperature. The effects of different dosages of polyoxometalates, type of solvent, reaction time, amount of catalyst and oxidant in this catalytic system were investigated. The new P2W18Sn3@MIL-101 and P2W18Ce3@MIL-101 nanocomposites exhibited good recyclability and reusability in at least five consecutive reaction cycles without significant loss of activity or selectivity.
1. Introduction
Polyoxometalates (POMs) as a class of metal cluster complexes comprising transition metal oxide anions attract attention in light of their significant potential in medicine, structural chemistry, analytical chemistry, surface science, electrochemistry and photochemistry.1–3 Their utility for catalysis applications has been limited, notably due to their tendency toward low surface area (typically ≤ 10 m2 g−1) and porosity (lower than 0.1 cm3 g−1) of bulk POMs which hinder the accessibility of the active sites, together with a high solubility in polar solvents making it inconvenient for recovery and reutilize.4–6 In this sense, the heterogenization or immobilization of POMs onto various solid supports via dative,7 covalent,8 or electrostatic9–11 binding, such as silica, activated carbon, magnetic nanoparticles and titanium dioxide is highly desired to prepare new and robust heterogeneous catalysts, capable of being easily separated from a reaction mixture and recycled.12–14 Recently, metal–organic frameworks (MOFs) materials were also used for immobilization of POMs.15 Because of their structural features, MOFs relative to other porous matrices play an important role in the development of different catalysts, including those for enantioselective chiral reactions, asymmetric epoxidation of alkenes and allyl alcohols, oxidation of alcohols and in a synthesis of porous carbon materials through thermal decomposition of guest-free MOFs.16–20 MIL-101 family of materials (MIL: Materials of the Institute Lavoisier) are a very stable group of MOFs, result from the three-dimensional covalent connection of inorganic clusters and organic linkers. Its open-pore structure with the pores (∼3.5 nm) and pore windows (∼1.5 nm) are large enough to give access to voluminous reactant molecules diffusing into the pores. These properties and also able to be functionalized, accessible cages and very high specific surface area, make it an excellent candidate to support catalytic species.21–25In 2005, Férey et al. reported incorporation of the lacunary polytungstate of PW11O397− within the large cage of MIL-101(Cr).26 Following work by Férey et al., numerous POM@MIL-101 composite materials containing various catalytically active polyoxometalates has been widely studied, and the resulting composites have been applied to H2O2-based alkene epoxidation, knoevenagel condensation, aldehyde–alcohol reactions, oxidative desulfurization, aerobic so on.27–30 Recently, a range of hybrid materials based on various Keggin polyoxometalates building blocks and copper organic frameworks with pyrazine derivatives have been discussed by Haddadi et al. and used as a heterogeneous catalyst for the selective oxidation of sulfides to corresponding sulfoxides and sulfones using H2O2.31 A-type sandwich polyoxometalate of K11H[(HOSnIVOH)3(PW9O34)2]·20H2O was immobilized for the first onto Nd-doped TiO2 nanoparticles as support material and these new nanocomposites used for the oxidation of different sulfides and alcohols.32 Herein we report the synthesis and application of new POM@MIL-101 composites materials containing A-type sandwich polyoxometalates in selective oxidation of sulfides. To the best of our knowledge, this is the first report about the immobilization of various potassium salts of [(HOSnIVOH)3(XW9O34)2]12− (X = P and Si), [(OCe)3(XW9O34)2]12− (X= P) and sodium salts [WM3(H2O)2(XW9O34)2]12− (X = M = Zn and Co) into a MIL-101 metal–organic framework resulting in a series of POM@MOF composite materials in selective oxidation of sulfides. The structure of P2W18Sn3 and P2W18Ce3 consists of two A-type PW9O34 anions linked by three tin(IV) or cerium(IV) cations leading to A-type sandwich POMs (Scheme 1).
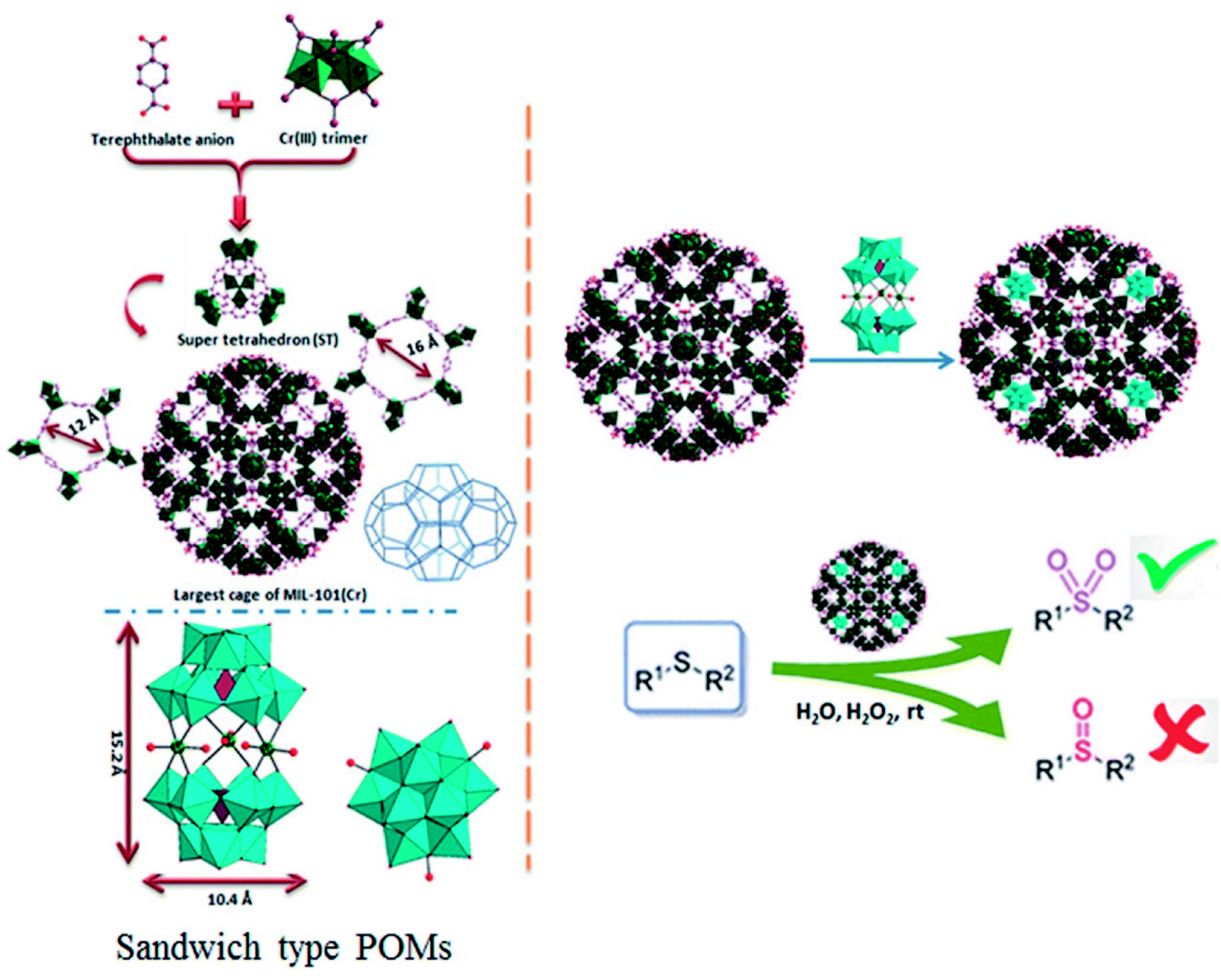 | ||
| Scheme 1 The synthesis pathway for preparation of nanocomposites. | ||
2. Experimental
2.1. Materials
All chemicals were used as received from different commercial sources. All the reagents used in the preparation of the composite materials, namely analytically pure chromium(III) nitrate nonahydrate (Cr(NO3)3·9H2O, Aldrich), benzene-1,4-dicarboxylic acid (C6H4(CO2H)2, Aldrich), fluorhydric acid (HF, Merck) and DMF (N,N-dimethylformamide, Merck) were purchased and used without further purification. The chemicals used for desulfurization experiments, namely diphenyl sulfide, methyl phenyl sulfide, dimethyl sulfide, 2-(methylthio)ethanol, methyl 3-(methylthio)propionate, 2-furfuryl methyl sulfide, allyl sulfide, dibutyl sulfide (Fluka, 95%), acetonitrile (CH3CN, Merck) and hydrogen peroxide (H2O2, Merck 35%) were also used as received. The A-type sandwich polyoxometalates of K11H[(HOSnIVOH)3(PW9O34)2]·20H2O and K9(NH4)H2[(OCeIVO)3(PW9O34)2]·20H2O were prepared according to the previously described procedures.33,342.2. Characterization methods
The synthesized MOF and nanocomposites were characterized using; Infrared absorption spectra were recorded on a Bruker Vector 22 infrared spectrometer using KBr pellet method. Thermogravimetric analysis (TGA) was carried out under N2 flow while gradually increasing the temperature with a rate of 10 °C min−1, using a STA PT-1000 LINSEIS. The morphology of nanocomposites was revealed by a scanning electron microscope (MIRA3 FEG-SEM). The elements in the nanocomposite samples were probed with energy-dispersive X-ray (EDX) spectroscopy accessory to the MIRA3 FEG-SEM scanning electron microscopy. Nitrogen adsorption–desorption isotherms used to obtain the MIL-101(Cr) and POM@MIL 101(Cr) specific surface areas and pore volumes, calculated by the Brunauer–Emmett–Teller (BET) method. All as-prepared samples were degassed at 100 °C in vacuum for overnight prior to adsorption measurements. The experiments took place at −196 °C under variable relative pressure. The equipment was a Belsorp mini II adsorption analyzer (Bel-Japan) at −196 °C. Powder X-ray diffraction (XRD) analyses were collected at ambient temperature by a X'PertPro Panalytical, Holland diffractometer using a CuKα radiation (λ = 1.5418 Å). The accelerating voltage and applied currents were 40 kV and 30 mA, respectively. The C microanalyses was carried out with CHNS–O Elemental Analyzer Vario EL III, ELEMENTARY, Hanau-Germany while the amount of W and Cr were measured by inductively coupled plasma mass spectrometry (ICP-MS).2.3. Preparation of the materials
2.4. General test for the oxidation
A mixture of bulk polyoxometalates (P2W18Sn3 or P2W18Ce3) or composites (P2W18Sn3@MIL-101 or P2W18Ce3@MIL-101) (50 mg) as catalyst, 35% H2O2 aqueous solution (5.85 mmol) and solvent (2.5 mL) were placed in a 25 mL glass bottle. After 5 min, the substrate (1 mmol) was added under stirring. The reaction time was counted after the addition of sulfide, and then the reaction mixture was stirred at the experiment temperatures for the appropriate time. The sample was collected from the mixture at time intervals and then the progress of the reaction was followed by TLC (eluent: n-hexane/EtOAc, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and stopped when a complete conversion of the substrate was observed. The catalyst was filtered off at the end of reactions, washed several times with ethyl acetate followed by ethanol (4 × 5 mL), heated in an oven at 70 °C overnight and then reused using the same reaction conditions. The starting material and product are insoluble in water and it was used just as an environment for stirring. Therefore, the reaction mixture was transferred to a separating funnel and the product was extracted with CH2Cl2 (3 × 5 mL). After evaporation of organic layer, the crude products were recrystallized from hot ethanol and the pure products were obtained in 94–98% yield. Stability test of the P2W18Sn3@MIL-101 and P2W18Ce3@MIL-101 catalysts were carried out running six consecutive experiments.
:1) and stopped when a complete conversion of the substrate was observed. The catalyst was filtered off at the end of reactions, washed several times with ethyl acetate followed by ethanol (4 × 5 mL), heated in an oven at 70 °C overnight and then reused using the same reaction conditions. The starting material and product are insoluble in water and it was used just as an environment for stirring. Therefore, the reaction mixture was transferred to a separating funnel and the product was extracted with CH2Cl2 (3 × 5 mL). After evaporation of organic layer, the crude products were recrystallized from hot ethanol and the pure products were obtained in 94–98% yield. Stability test of the P2W18Sn3@MIL-101 and P2W18Ce3@MIL-101 catalysts were carried out running six consecutive experiments.
3. Results and discussion
3.1. Synthesis
The MIL-101(Cr) is built up from a corner-sharing of so-called super tetrahedron (hereafter noted ST), which is formed by rigid chromium(III) octahedral trimers (the vertices of the ST) and terephthalate anions (the edges of the ST) (see Scheme 1).The connection between of the corners of the ST building blocks ensures a 3D cubic zeotype structure with two types of mesoporous cages (∅ = 29 and 34 Å) (extended MTN topology), accessible through microporous windows (∅ = 12 and 16 Å). The large cages possess both 12 Å pentagonal and 16 Å hexagonal windows with internal free diameters of 34 Å. Therefore, these cavities are sufficiently spaced to accommodate large guest molecules (such as the successful encapsulation of the sandwich-type anions, P2W18Sn3 or P2W18Ce3 (∼10.4 × 10.4 × 15.2 Å3)), into cages of MIL-101(Cr). The MIL-101(Cr) has been employed as a useful and versatile solid support for preparation of heterogeneous catalysts because of its open-pore structure with large and accessible cages.26 The incorporation of POMs in the mesoporous MIL-101(Cr) has been carried out by the anionic exchange between the counter-ions of the MIL-101(Cr) (nitrate ions coming from the Cr(NO3)3 precursor) and the negatively charged P2W18Sn3 or P2W18Ce3.36 For first time, MIL-101(Cr) with Cr3F(H2O)2O[(O2C)–C6H4–(CO2)]3·nH2O formula was synthesized by the HF–Cr(NO3)3–H2BDC–H2O system.26 The composites of the sandwich-type POM of P2W18Sn3 and P2W18Ce3 with MIL-101 were prepared by impregnation of the synthesized MIL-101 in an autoclave as described above in aqueous solution of P2W18Sn3 or P2W18Ce3.
3.2. Characterization of the material
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups (1514 cm−1), in plane and out–of plane bending modes of COO groups (400–700 cm−1), and the out–of plane deformation vibrations of benzene ring C–H groups at 1164, 1020, 889, and 748 cm−1.38,39
C groups (1514 cm−1), in plane and out–of plane bending modes of COO groups (400–700 cm−1), and the out–of plane deformation vibrations of benzene ring C–H groups at 1164, 1020, 889, and 748 cm−1.38,39
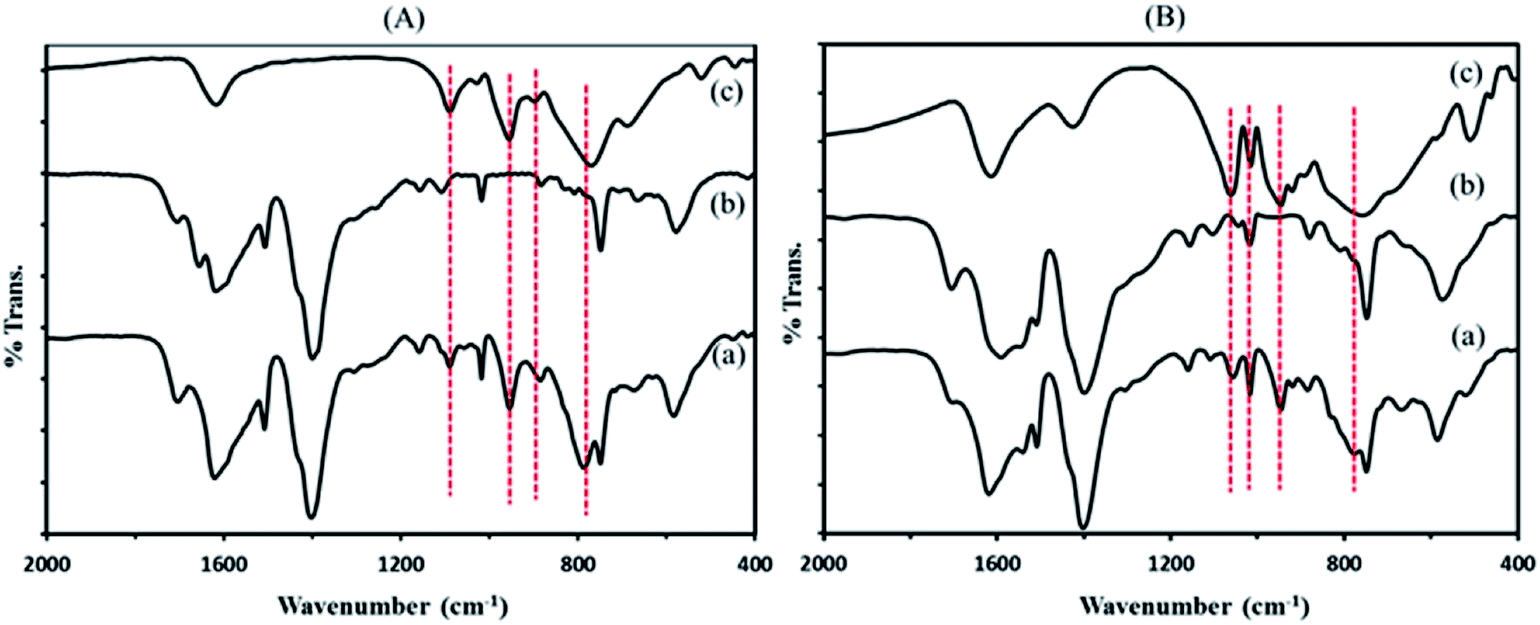 | ||
| Fig. 1 FT-IR spectra of (A) P2W18Sn3@MIL-101 composite (a) the precursors of MIL 101 (b) and P2W18Sn3 (c) and (B) P2W18Ce3@MIL-101 composite (a) the precursors of MIL 101 (b) and P2W18Ce3 (c). | ||
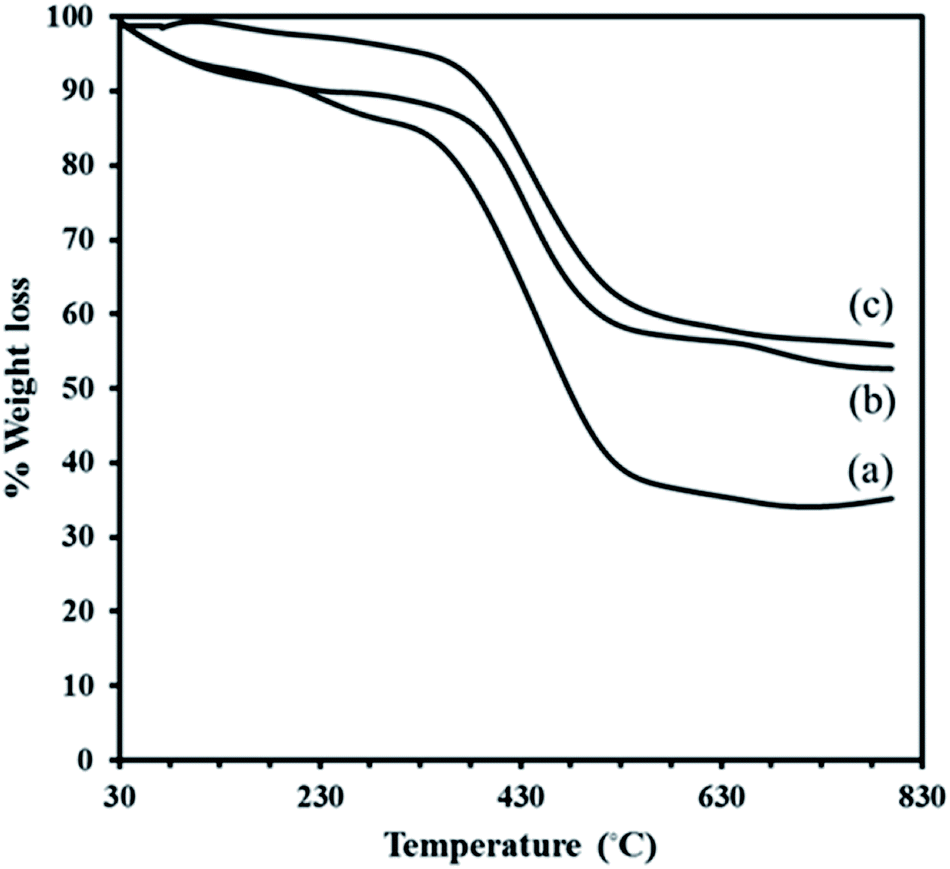 | ||
| Fig. 2 TGA curves of MIL-101 (a), P2W18Ce3@MIL-101 (b) and P2W18Sn3@MIL-101 (c). | ||
The residual solid is Cr2O3 for MIL-101. Although, the TGA profile of the composite P2W18Sn3@MIL-101 in the first event, occurs in the range 30–310 °C (2.27%), may be assigned to the removal of water molecules present render in the structure electrostatically neutral or the guest molecules inside the pores. A larger loss of ca. 36.91% is due to relates to the departure of OH/F groups and the decomposition of the framework. The minor weight loss of 2.97% in the temperature range 600–800 °C in TGA profile of the composite P2W18Sn3@MIL-101 can attribute to the loss of oxygen atoms from the residual metal oxides resulted from decomposition of P2W18Sn3. The residual mixture solid is Cr2O3–WO3–P2O5–SnO2–K2O (55.80%).40 Also, the total weight loss of 42.15% for P2W18Sn3@MIL-101 is lower than that for the parent MIL-101 (65.05%), as would be expected from the presence additional residual from partial decomposition of P2W18Sn3 in the cages of composite. The TGA curve of the P2W18Ce3@MIL-101 composite shows a two-stage weight loss below 600 °C, such as the P2W18Sn3@MIL-101 composite, and indicates a lower weight loss than MIL-101. From TGA curves can be recognized that polyoxotungstate anions improve the thermal stability of the neat MIL-101.41
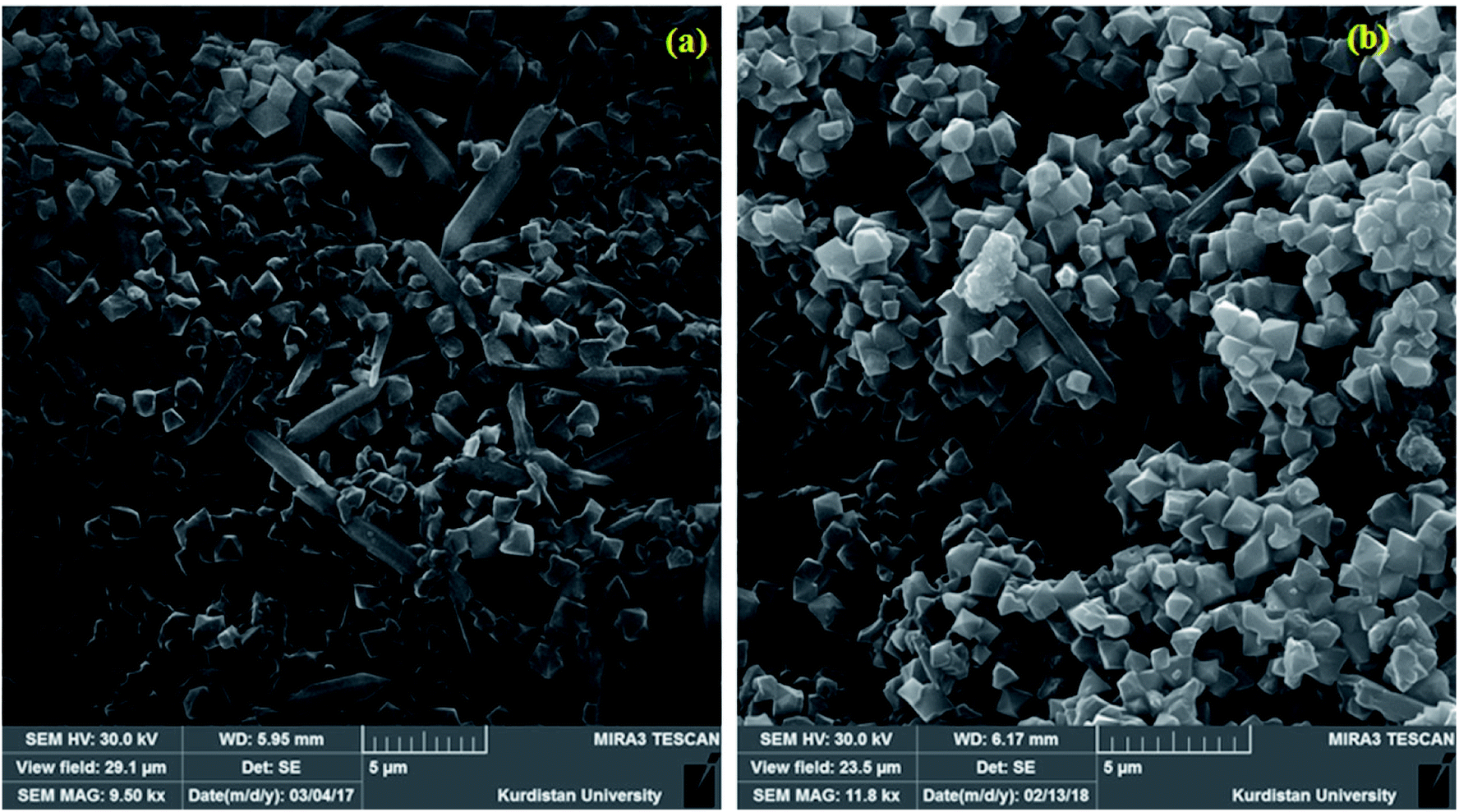 | ||
| Fig. 3 SEM images of MIL-101 (a) and P2W18Ce3@MIL-101 (b). | ||
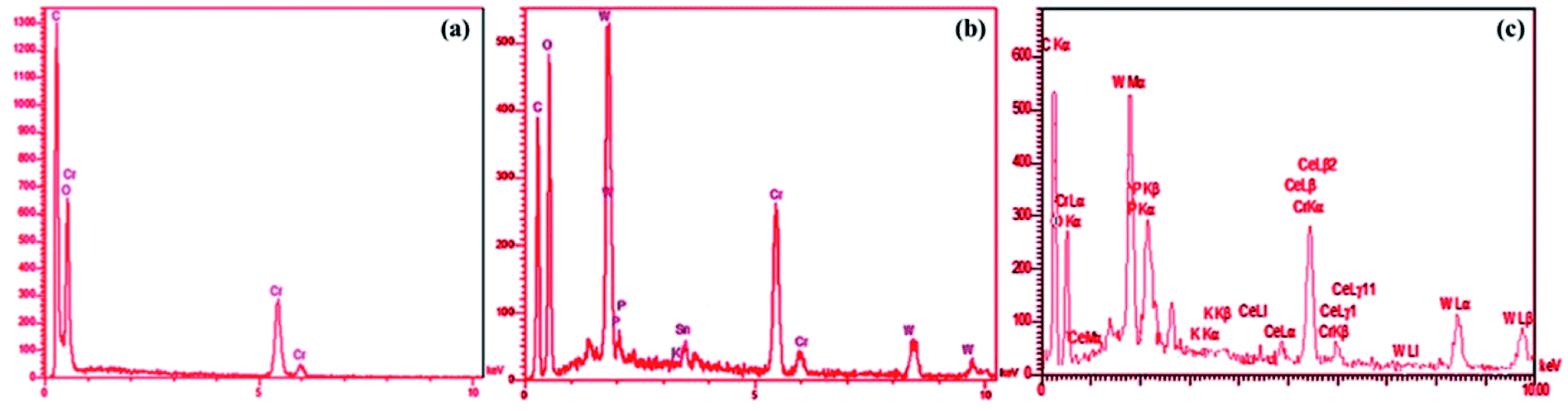 | ||
| Fig. 4 EDX spectra of (a) MIL-101 and the composite of P2W18Sn3@MIL-101 (b) and P2W18Ce3@MIL-101 (c). | ||
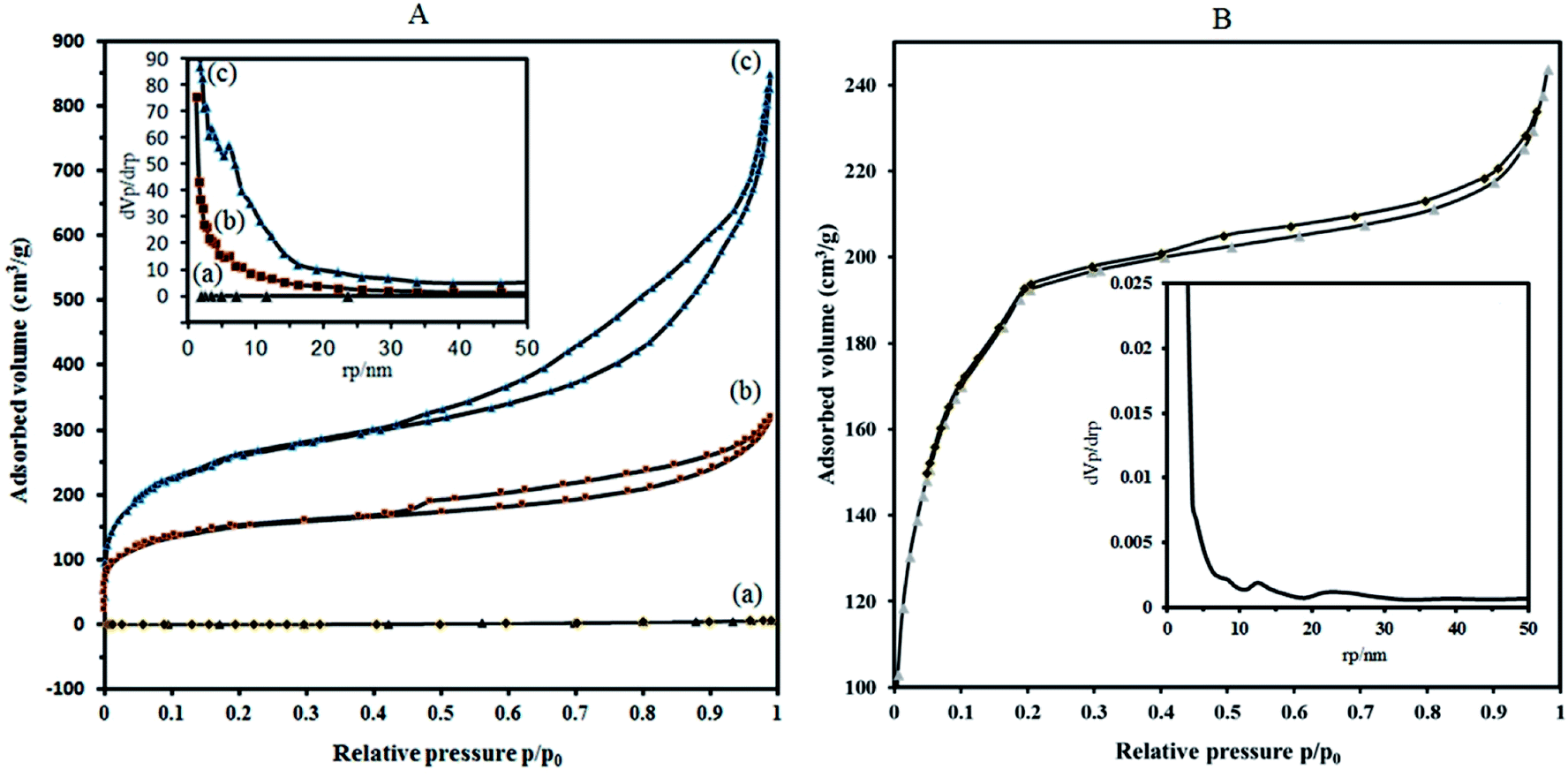 | ||
| Fig. 5 N2 adsorption–desorption isotherms at 77 K of (A) P2W18Sn3 (a), the composite P2W18Sn3@MIL 101 (b) and MIL 101 (c). (B) The composite P2W18Ce3@MIL 101. Inset shows BJH pore-size distribution plots of the samples. | ||
To overcome these problems, it is proposed that increasing the surface area can be achieved by the deposition of sandwich POM into porous solid supports with high surface area. The results of the BET analysis show that the specific surface area of P2W18Sn3 is much lower compared to composite (Fig. 5A(a)).
Nitrogen adsorption isotherms show that the specific surface area of the dehydrated MIL-101 decrease from 946.91 m2 g−1 to 573.08 m2 g−1 for P2W18Sn3@MIL-101. Importantly, compared with MIL-101, P2W18Sn3@MIL-101 shows a smaller pore volume (1.312 cm3 g−1 to 0.491 cm3 g−1), which may help to interpret the lower specific surface area for composite. Combined with N2 sorption isotherms analysis and the pore size distribution (PSD) results, it can be concluded that the doping of the MIL-101 porous structure with POM ions can change morphological characteristics (surface area, pore volume and pore size distributions).43,44
Adsorption isotherms of P2W18Sn3@MIL-101 (Fig. 5A(b)), as well as MIL-101 (Fig. 5A(c)), reveal typical type-I behavior with remarkable H4 hysteresis loop which is coincident with the mesoporous structures. This hysteresis is usually characteristic of solids consisting of aggregates of particles forming slit-shaped pores (plates or edged particles like cubes), with uniform or non-uniform size and/or shape. These results are in good agreement with the results of the pore size distribution (calculated by BJH method based on the adsorption branch) of P2W18Sn3, the MIL-101 and P2W18Sn3@MIL-101 materials. From the distribution curves, the samples have a broad pore-size distribution in the range of 1.2–30 nm (Fig. 5 inset).
The N2 adsorption isotherm and the BJH pore size distribution curve of P2W18Ce3@MIL-101 catalyst shown in Fig. 5B. Importantly, compared with the MIL-101, P2W18Ce3@MIL-101 catalyst shows the surface area and total pore volume is reduced to 690.14 m2 g−1 and 0.376 cm3 g−1, in agreement with the presence of heavy polyoxometalate moieties. According to the results of the Barrett–Joyner–Halenda (BJH) analysis, mesoporous structure are present in both nanocomposites, especially in P2W18Sn3@MIL-101 catalyst, in agreement with its hysteresis loop at relative pressure (P/P0) between 0.4 and 1.0.45 The results of the N2 adsorption–desorption isotherms and pore size distributions of the synthesized nanocomposites show that the type of polyoxometalate loaded in the cavities of the MIL-101 affects the volume and size distribution of the pores in the material.
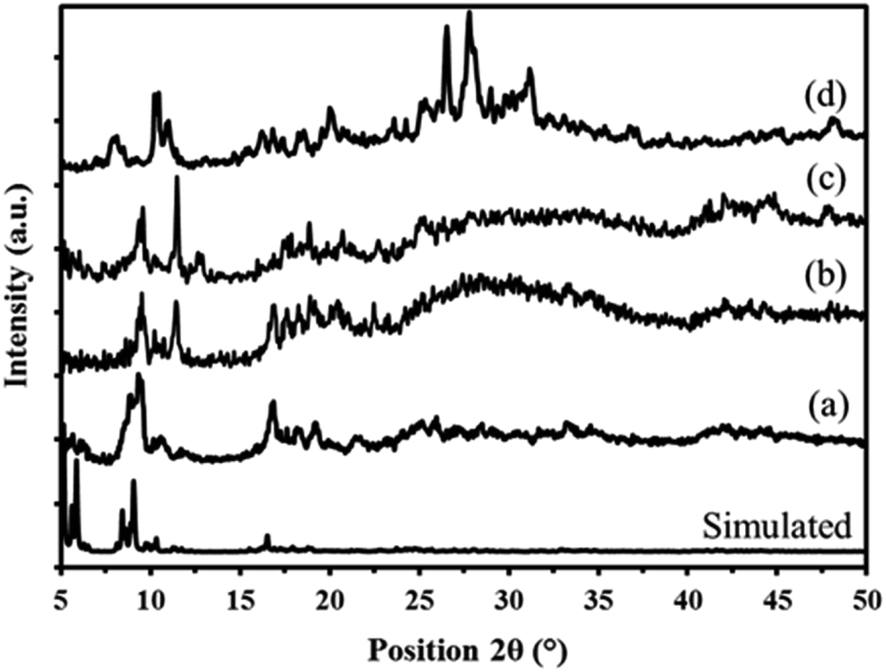 | ||
| Fig. 6 Powder X-ray diffraction patterns of simulated and prepared MIL-101 (a) P2W18Sn3@MIL-101 (b) and P2W18Ce3@MIL-101 (c) sandwich polyoxometalates P2W18M3 (d). | ||
The X-ray diffraction pattern of the P2W18Ce3@MIL-101 nanocomposite is in good agreement with P2W18Sn3@MIL-101, thus confirming both polyoxotungstates have the same diffraction pattern (Fig. 6c).
3.3. Catalytic activity
Organosulfur compounds, such as sulfoxides and sulfones, are useful synthetic intermediates for the construction of various pharmaceutically and biologically active compounds. They are widely utilized as anti-bacterial, anti-fungal, anti-atherosclerotic, anti-ulcer, vasodilators and cardiotonic agents and as well as activation of enzymes. Different synthetic methods for the controlled oxidation of conventional sulfides have been previously reported for fundamental transformation. Traditionally, these transformations take place with stoichiometric amounts of electrophilic reagents, such as peracids, dioxiranes, hypochlorites, periodates and highly toxic oxo metal oxidants (NaIO4, MnO2, CrO3, SeO2, PhIO, NH4MnO4 and so on) but, many of these procedures are accompanied by particular disadvantages such as toxic and corrosive oxidants, long reaction times, inconvenient reaction conditions, environmentally unfavorable catalysts (poor recovery of expensive metal catalysts), low yields and the formation of toxic wastes.49,50 Various catalyst systems of organocatalysts, acid catalysts, enzymes, metal catalysts and organic–inorganic hybrid solid materials have been used for this reaction with H2O2 as an oxygen source. Recently, the use of polyoxometalates was promising not only due to bifunctional redox acid catalysts properties but also due to their unusual properties such as high charges, low cost and low environmental impact.51–53The reaction initially performed in the presence of 5.85 mmol of H2O2 as an oxidant, employing 1 mmol (0.17 mL) diphenyl sulfide and 50 mg of catalyst in CH3CN (2.5 mL) at room temperature. In order to find the optimum reaction conditions, the effect of different reaction parameters such as the amount of catalyst, the stoichiometry of H2O2 respect to the catalyst and substrate, solvent, reaction time, different dosages of POM into MIL-101 and the catalytic activities of the different catalysts on the selectivity and conversion of the oxidation reaction were studied. No product was obtained in the absence of any catalyst (Table 1, entry 1). At first, the catalytic performance was evaluated for the selective oxidation of sulfides using MIL-101 support and the POMs (P2W18Sn3 and P2W18Ce3) as homogeneous catalysts in different reaction conditions (Table 1, entries 2–6).
| Entry | Catalyst (mg) | Solvent | Time (min) | Sulfoxide (%)a | Sulfone (%)a |
|---|---|---|---|---|---|
| a Isolated yield.b Reaction conditions: diphenyl sulfide (1 mmol), 35% H2O2 (5.85 mmol), solvent (2.5 mL),catalyst (50 mg), r.t.c [(HOSnIVOH)3(PW9O34)2]12− @MIL-101 (1), [(HOCeIVOH)3(PW9O34)2]12− @MIL-101 (2) [(HOSnIVOH)3(SiW9O34)2]14− @MIL-101 (3), [WCo3(H2O)2(CoW9O34)2]12− @MIL-101 (4) [WZn3(H2O)2(ZnW9O34)2]12− @MIL-101 (5). | |||||
| 1b | Catalyst free | CH3CN | 1440 | Trace | — |
| 2 | MIL-101 (50) | CH3CN | 1440 | 40 | 20 |
| 3 | P2W18Sn3 (50) | CH3CN | 1440 | 35 | 10 |
| 4 | P2W18Sn3 (50) | H2O | 1440 | Trace | Trace |
| 5 | P2W18Ce3 (50) | H2O | 1440 | Trace | Trace |
| 6 | P2W18Ce3 (50) | EtOH | 180 | — | 98 |
| 7 | Catalyst (1) (50) | CH3CN | 120 | — | 98 |
| 8 | Catalyst (2) (50) | CH3CN | 35 | — | 98 |
| 9 | Catalyst (3) (50) | CH3CN | 210 | — | 98 |
| 10 | Catalyst (4) (50) | CH3CN | 260 | — | 98 |
| 11 | Catalyst (5) (50) | CH3CN | 320 | — | 98 |
| 12 | Catalyst (1) (50) | H2O | 210 | — | 98 |
| 13 | Catalyst (1) (50) | EtOH | 220 | — | 98 |
| 14 | Catalyst (1) (50) | H2O/PEG | 220 | — | 98 |
| 15 | Catalyst (1) (50) | DMF | 220 | 30 | 30 |
| 16 | Catalyst (2) (50) | EtOH | 60 | — | 98 |
| 17 | Catalyst (2) (50) | H2O | 900 | — | 92 |
The P2W18Sn3 as a homogeneous catalyst in the presence of hydrogen peroxide in H2O or CH3CN as solvent unsuitable conversion and selectivity was found even in prolonged reaction time (Table 1 entries 3 and 4). The P2W18Ce3 as a homogeneous catalyst in the presence of hydrogen peroxide, although in H2O do not show suitable activity but, it catalyzes selectively the oxidation of sulfides to sulfone in about 180 min in EtOH (Table 1 entries 5 and 6). Although the conversion and the selectivity of P2W18Ce3 in EtOH were good, but there are two problems about its separation and recovery. We found that like some of POMs, the structure of P2W18Ce3 in the presence of hydrogen peroxide was converted to a mixture of peroxopolytungstophosphate species.54,55 As shown in Table 1, the oxidation of substrate was carried out using the heterogeneous catalysts. Catalytic oxidation of sulfides was carried out in the presence of the MIL-101 composites of various sandwich-type polyoxometalates.
Within this context, [(HOSnIVOH)3(PW9O34)2]12−@MIL-101, [(OCeIVO)3(PW9O34)2]12−@MIL-101, [(HOSnIVOH)3(SiW9O34)2]14−@MIL-101, [WCo3(H2O)2(CoW9O34)2]12−@MIL-101 and [WZn3(H2O)2(ZnW9O34)2]12−@MIL-101 composites were prepared and their catalytic activities investigated for the oxidation of diphenyl sulfide by using H2O2 as an oxidant at room temperature in CH3CN (Table 1, entries 7–11). The results in Table 1 clearly show that P2W18Sn3@MIL-101 and P2W18Ce3@MIL-101 showed the highest selectivity for oxidizing of diphenyl sulfide in short reaction time with 98% yield of the corresponding sulfone. Increasing catalysts over 50 mg and also H2O2 more over than 1 mL doesn't have an appreciable effect on the reaction times. The role of different solvents for the oxidation of diphenyl sulfide was examined. As shown in Table 1; in presence P2W18Sn3@MIL-101 when we used acetonitrile as solvent, the desired product (sulfone) can be obtained in a shorter time (Table 1, entry 7). By using H2O, EtOH or H2O/PEG as solvent similar catalytic conversion and selectivity obtained but at prolonged reaction time (Table 1, entries 12–14). DMF as solvent, the mixture of sulfoxide and sulfone is obtained (Table 1, entry 15). Generally, the solvent type is chosen based on the reaction kinetic and catalyst structure, so choosing the suitable solvents according to the chemical structure, molecular design and its physical and chemical properties is very important in reaction systems. With regard to the porous catalyst in our study, it was not possible to use solvent-free conditions because reactants have to interact with catalytic active species located in the cavities. It is necessary to mention, the reaction in acetonitrile showed a good reactivity (excellent conversion and selectivity), in our this work. On the other hand, in protic solvents such as water, it is possible that Lewis acid sites (CrIII) in the structure of the nanocomposite interact with protons of solvent and thus the cavities are blocked, so it is difficult to insertion–extraction for reactants.
The influence of temperature in the diphenyl sulfide oxidation is illustrated with keeping H2O2 and substrate amount constant. We found similar results at 25 to 50 °C. The reaction was completed selectively in 60 °C at shorter reaction time. Increasing temperature to 85 °C conversion was completed while the selectivity decreased. P2W18Ce3@MIL-101 the best catalytic activity observed in CH3CN and EtOH as solvent (Table 1, entries 8 and 16), while in H2O the same catalytic activity observed only at very prolonged time (Table 1, entry 17).
The used polyoxometalates have similar structure and the difference among them is the metal ions in the belt of the used sandwich type polyoxometalates. Therefore, the difference in their catalytic activity can be attributed to type of metal ions. The nanocomposites of P2W18Sn3@MIL-101 and P2W18Ce3@MIL-101 show the best catalytic activity which it can be attributed to the SnIV or CeIV ions in their structures. A comparison between the latters, show that the best results obtained with P2W18Ce3@MIL-101. Two aligned reasons can be proposed for preference of P2W18Ce3@MIL-101 composite. First, particular ability of cerium in store/release oxygen as an oxygen storage via facile reciprocal transformation of CeIV and CeIII ions under oxidizing and reducing conditions respectively. Due to this feature, activated oxygen species produced from H2O2, may be stored on the composite, which in turn, may be responsible for oxidation of sulfide.56–58 Second, standard electrode potential for reduction of CeIV to CeIII in the P2W18Ce3@MIL-101 is very more positive than that SnIV to SnII in the P2W18Sn3@MIL-101. As an interesting result, in contrast to homogenous condition, the P2W18Ce3 structure was protected in the P2W18Ce3@MIL-101 composite in the presence of hydrogen peroxide.
To investigate the applicability of this procedure, we carried out oxidation of different types of sulfides under the distinctive reaction conditions and the expected products afforded at high yields in CH3CN and H2O solvents for P2W18Sn3@MIL-101 (Table 2) and for P2W18Ce3@MIL-101 in EtOH (Table 3). As a result from this study, all used sulfides were oxidized to the corresponding sulfone and also noteworthy that P2W18Ce3@MIL-101 composite shows better catalytic activity. In other word, substrates have high selectively to oxidation of sulfur singly even with the presence of functional groups such as aromatic and aliphatic sulfides. For example, 2-methylthio ethanol contains the hydroxyl group was transformed to the corresponding sulfone compound in high conversion and selectivity without dehydrogenation of the hydroxyl group.
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Sulfonea,b | Sulfonea,c | ||
| Time (min) | Isolated yield (%) | Time (min) | Isolated yield (%) | ||
| a All the products are known and were characterized by FT-IR and by melting point comparison with those of authentic samples.b Reaction conditions: sulfide (1 mmol), 35% H2O2 (5.85 mmol), catalyst (50 mg), in H2O, r.t.c Reaction conditions: sulfide (1 mmol), 35% H2O2 (5.85 mmol), catalyst (50 mg), in CH3CN, r.t. | |||||
| 1 | 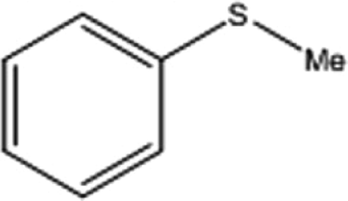 |
150 | 98 | 80 | 98 |
| 2 | 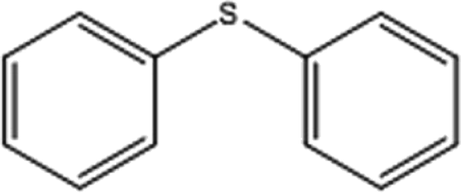 |
210 | 98 | 120 | 98 |
| 3 | 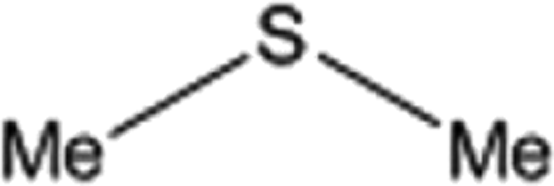 |
10 | 97 | 5 | 98 |
| 4 | 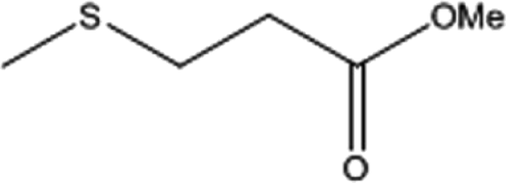 |
15 | 95 | 5 | 96 |
| 5 | 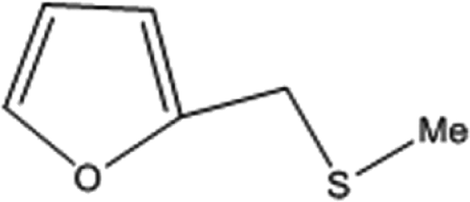 |
15 | 97 | 5 | 97 |
| 6 | 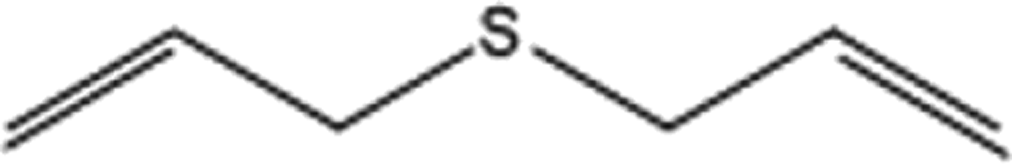 |
200 | 94 | 20 | 98 |
| 7 |  |
240 | 94 | 30 | 95 |
| 8 |  |
15 | 96 | 5 | 97 |

|
|||
|---|---|---|---|
| Entry | Substrate | Sulfonea,b | |
| Time (min) | Isolated yield (%) | ||
| a All the products are known and were characterized by FT-IR and by melting point comparison with those of authentic samples.b Reaction conditions: sulfide (1 mmol), 35% H2O2 (5.85 mmol), catalyst (50 mg), in EtOH, r.t. | |||
| 1 | 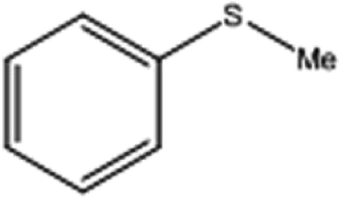 |
30 | 98 |
| 2 | 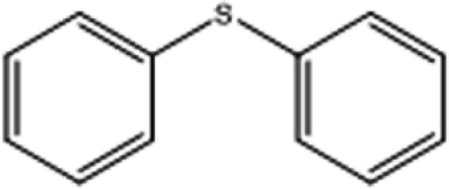 |
60 | 98 |
| 3 | 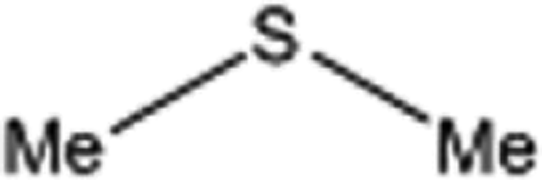 |
5 | 97 |
| 4 | 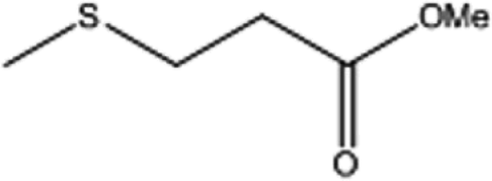 |
10 | 96 |
| 5 | 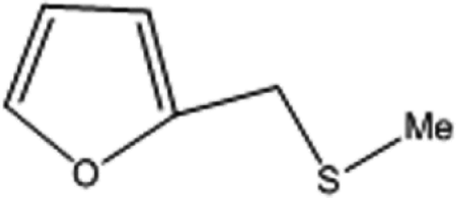 |
20 | 97 |
| 6 | 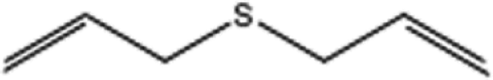 |
30 | 96 |
| 7 | 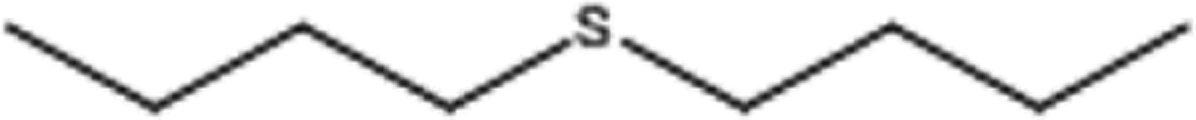 |
70 | 94 |
| 8 |  |
15 | 96 |

3.4. Stability of the catalysts
In our final experimental work, stability tests (recovery and reusability) of the new P2W18Sn3@MIL-101 and P2W18Ce3@MIL-101 catalysts were carried out running six consecutive experiments in the oxidation of diphenyl sulfide at a constant time.At the end of each reaction, the catalyst was recovered by simple centrifuge and was washed with ethyl acetate and ethanol (4 × 3 mL), dried under vacuum then used again as the catalyst. The results show the reactions were completed in run of 1–5 at average time of 210 min (in H2O) and 60 min (in EtOH) for P2W18Sn3@MIL-101 and P2W18Ce3@MIL-101, respectively. At the same reaction times the 6th runs were completed up to 80 and 90% with P2W18Sn3@MIL-101 and P2W18Ce3@MIL-101 respectively (Fig. 7).
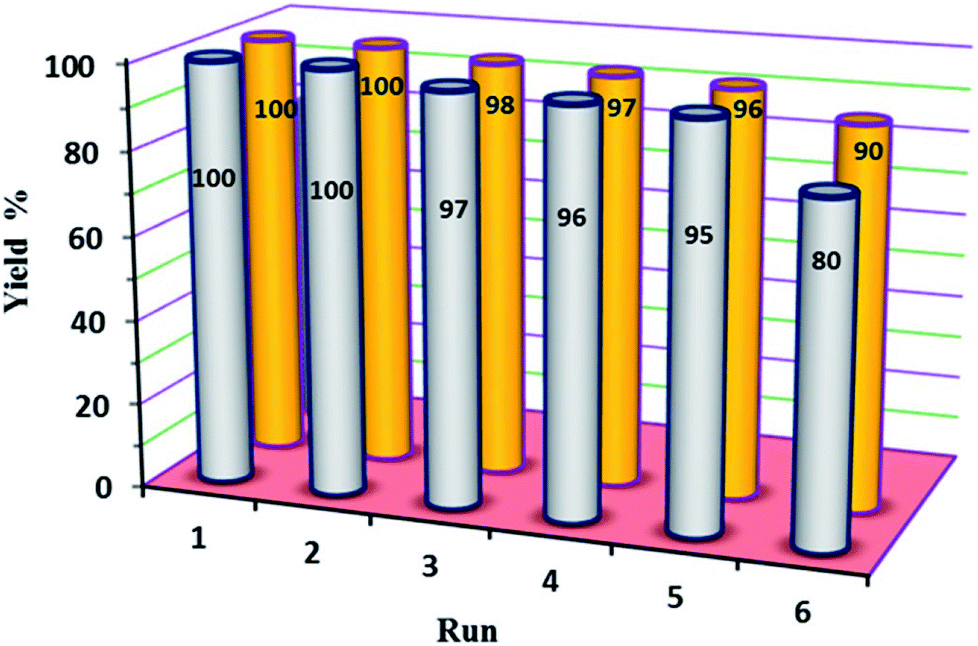 | ||
| Fig. 7 The reusability of P2W18Sn3@MIL-101 (gray) and P2W18Ce3@MIL-101 (orange) catalysts in the oxidation of sulfides. | ||
To investigate the structural stability of catalyst after oxidative reactions, IR spectra fresh and reused nanocomposites were recorded after six catalytic cycles (Fig. 8). The IR spectra of fresh catalyst are identical to those of reused catalyst, confirming the integrity of the support material and also the presence of the sandwich polyoxotungstate anions in the composite material after the catalytic performance.
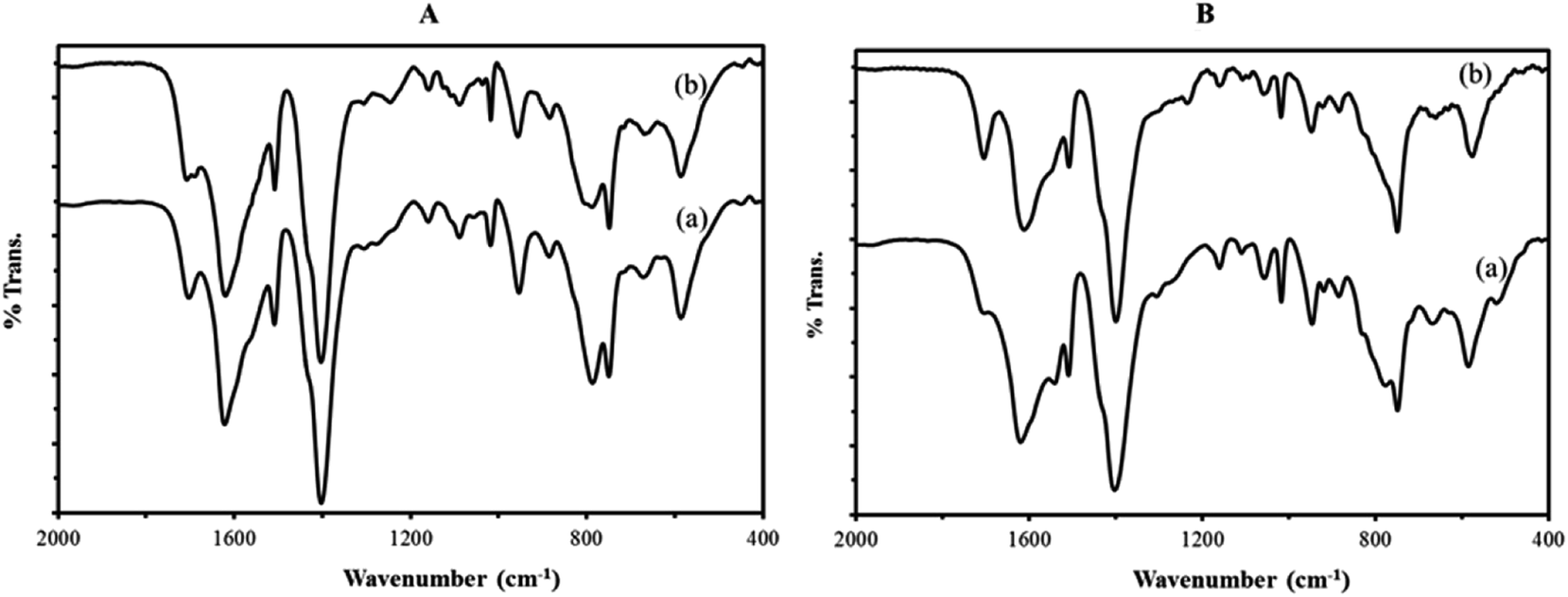 | ||
| Fig. 8 The FT-IR spectra of (A) P2W18Sn3@MIL-101 composite (a) and P2W18Sn3@MIL-101 after six consecutive runs (b) and (B) P2W18Ce3@MIL-101 composite (a) and P2W18Ce3@MIL-101 after six consecutive runs (b). | ||
4. Conclusions
Organic–inorganic frameworks based on different sandwich-type polyoxometalates using commercially and readily available materials were synthesized and their catalytic activities studied in the oxidation of different sulfides with an environmentally benign oxidant in different solvents. The nanocomposites have been achieved by impregnation of the synthesized MIL-101 in aqueous solution of the sandwich-type polyoxometalates. This is important point that by increasing concentration of polyoxometalates in aqueous solution, their loading into cages of MIL-101 is not significantly altered. Even after these relatively large anions signing into the MIL-101, there is still enough space to enter the reactants and get out of the product. The POMs insertion into porous MIL-101 solid was investigated using several methods: XRPD, FT-IR, SEM-EDX, ICP, TGA and N2 adsorption. Catalysts have a useful life and as a result, with time, their activities and their effect on the reaction are reduced. This means that there are active points on the catalyst, which change over time due to different reasons. So today, the ability to recover and recycle catalyst from the environment is very important and significant. In synthesized composites, these active sites include polyoxometalates that are stable in the reaction media and do not suffer from it. The proposed mechanism for sulfides oxidation over polyoxometalates involves formation of peroxo-tungstate species (an electrophilic intermediate) by the interaction of hydrogen peroxide with the polyoxometalate anions which this transformation can oxidize the sulfides into sulfones. Therefore, in composites containing polyoxometalates, these are mainly catalytically active species that act as a catalyst. The best results were obtained by organic–inorganic framework incorporating of [(HOSnIVOH)3(PW9O34)2]12− and [(OCeIVO)3(PW9O34)2]12− among different sandwich polyoxometalates. The results of the BET and TGA analyses show that P2W18Sn3@MIL-101 and P2W18Ce3@MIL-101 catalysts have lower specific surface area than MIL-101, while they show significant higher thermal stability. The advantages of this catalytic synthesis method it can be pointed out to low cost and availability of raw materials, easy synthesis and chemical and thermal stability.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
The authors would like to thank the University of Kurdistan Research Council for their support of this work.References
- S. Zhao, Y. Chen and Y.-F. Song, Appl. Catal., A, 2014, 475, 140–146 CrossRef.
- K. Mijares, Novel hybrid materials: Functionalized polyoxometalates as potential metalloligands, Kansas State University, Germany, 2008 Search PubMed.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605–7622 RSC.
- C. L. Marchenaa, S. Gomeza, C. Sauxa, L. B. Pierellaa and L. R. Pizziob, Quim. Nova, 2015, 38, 518–525 Search PubMed.
- R. Ghanbaripour, I. Mohammadpoor-Baltork, M. Moghadam, A. R. Khosropour, S. Tangestaninejad and V. Mirkhani, Polyhedron, 2012, 31, 721–728 CrossRef.
- X. Zheng, L. Zhang, J. Li, S. Luo and J.-P. Cheng, Chem. Commun., 2011, 47, 12325–12327 RSC.
- Y. Guo, C. Hu, C. Jiang, Y. Yang, S. Jiang, X. Li and E. Wang, J. Catal., 2003, 217, 141–151 Search PubMed.
- R. Villanneau, A. Marzouk, Y. Wang, A. B. Djamaa, G. Laugel, A. Proust and F. Launay, Inorg. Chem., 2013, 52, 2958–2965 CrossRef PubMed.
- X. Duan, Y. Liu, Q. Zhao, X. Wang and S. Li, RSC Adv., 2013, 3, 13748–13755 RSC.
- M. Kooti and M. Afshari, Mater. Res. Bull., 2012, 47, 3473–3478 CrossRef.
- M. Afshari, M. Gorjizadeh and G. Afshar, Orient. J. Chem., 2014, 29, 1675–1681 CrossRef.
- R. Tan, C. Liu, N. Feng, J. Xiao, W. Zheng, A. Zheng and D. Yin, Microporous Mesoporous Mater., 2012, 158, 77–87 CrossRef.
- E. Rafiee and S. Eavani, Green Chem., 2011, 13, 2116–2122 RSC.
- M. Rahimizadeh, G. Rajabzadeh, S.-M. Khatami, H. Eshghi and A. Shiri, J. Mol. Catal. A: Chem., 2010, 323, 59–64 CrossRef.
- J. Han, D. Wang, Y. Du, S. Xi, Z. Chen, S. Yin, T. Zhou and R. Xu, Appl. Catal., A, 2016, 521, 83–89 CrossRef.
- S. Kaskel, The chemistry of metal–organic frameworks: synthesis, characterization, and applications, John Wiley & Sons, Germany, 2016 Search PubMed.
- V. V. e. Butova, M. A. Soldatov, A. A. Guda, K. A. Lomachenko and C. Lamberti, Russ. Chem. Rev., 2016, 85, 280 CrossRef.
- C.-D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940–8941 CrossRef PubMed.
- S. Lim, K. Suh, Y. Kim, M. Yoon, H. Park, D. N. Dybtsev and K. Kim, Chem. Commun., 2012, 48, 7447–7449 RSC.
- S. Abednatanzi, A. Abbasi and M. Masteri-Farahani, J. Mol. Catal. A: Chem., 2015, 399, 10–17 CrossRef.
- K. M. Taylor-Pashow, J. D. Rocca, Z. Xie, S. Tran and W. Lin, J. Am. Chem. Soc., 2009, 131, 14261–14263 CrossRef PubMed.
- Y.-F. Huang, M. Liu, Y.-Q. Wang, Y. Li, J.-M. Zhang and S.-H. Huo, RSC Adv., 2016, 6, 15362–15369 RSC.
- D. Y. Hong, Y. K. Hwang, C. Serre, G. Ferey and J. S. Chang, Adv. Funct. Mater., 2009, 19, 1537–1552 CrossRef.
- S. H. Jhung, J. H. Lee, J. W. Yoon, C. Serre, G. Férey and J. S. Chang, Adv. Mater., 2007, 19, 121–124 CrossRef.
- S. Biswas, S. Couck, M. Grzywa, J. F. Denayer, D. Volkmer and P. Van Der Voort, Eur. J. Inorg. Chem., 2012, 2012, 2481–2486 CrossRef.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- L. Bromberg and T. A. Hatton, ACS Appl. Mater. Interfaces, 2011, 3, 4756–4764 CrossRef PubMed.
- L. Bromberg, Y. Diao, H. Wu, S. A. Speakman and T. A. Hatton, Chem. Mater., 2012, 24, 1664–1675 CrossRef.
- A. K. Babahydari, R. Fareghi-Alamdari, S. M. Hafshejani, H. A. Rudbari and M. R. Farsani, J. Iran. Chem. Soc., 2016, 13, 1463–1470 CrossRef.
- D. Julião, A. C. Gomes, M. Pillinger, L. Cunha-Silva, B. de Castro, I. S. Gonçalves and S. S. Balula, Fuel Process. Technol., 2015, 131, 78–86 CrossRef.
- H. Haddadi, S. M. Hafshejani and M. R. Farsani, Catal. Lett., 2015, 145, 1984–1990 CrossRef.
- L. Bahrami, R. Khoshnavazi and A. Rostami, J. Coord. Chem., 2015, 68, 4143–4158 CrossRef.
- F. Xin and M. T. Pope, J. Am. Chem. Soc., 1996, 118, 7731–7736 CrossRef.
- W. Knoth, P. Domaille and R. Harlow, Inorg. Chem., 1986, 25, 1577–1584 CrossRef.
- S. Ribeiro, C. M. Granadeiro, P. Silva, F. A. A. Paz, F. F. de Biani, L. Cunha-Silva and S. S. Balula, Catal. Sci. Technol., 2013, 3, 2404–2414 RSC.
- W. Salomon, F.-J. Yazigi, C. Roch-Marchal, P. Mialane, P. Horcajada, C. Serre, M. Haouas, F. Taulelle and A. Dolbecq, Dalton Trans., 2014, 43, 12698–12705 RSC.
- A. Ghiasi Moaser and R. Khoshnavazi, New J. Chem., 2017, 41, 9472–9481 RSC.
- F. Farzaneh and Y. Sadeghi, J. Mol. Catal. A: Chem., 2015, 398, 275–281 CrossRef.
- Q. Liu, L. Ning, S. Zheng, M. Tao, Y. Shi and Y. He, Sci. Rep., 2013, 3, 2916–2921 CrossRef PubMed.
- L. E. Briand, H. J. Thomas and G. T. Baronetti, Appl. Catal., A, 2000, 201, 191–202 CrossRef.
- S. Abednatanzi, K. Leus, P. G. Derakhshandeh, F. Nahra, K. De Keukeleere, K. Van Hecke, I. Van Driessche, A. Abbasi, S. P. Nolan and P. V. Der Voort, Catal. Sci. Technol., 2017, 7, 1478–1487 RSC.
- J. E. Mondloch, O. Karagiaridi, O. K. Farha and J. T. Hupp, CrystEngComm, 2013, 15, 9258–9264 RSC.
- X. Hu, Y. Lu, F. Dai, C. Liu and Y. Liu, Microporous Mesoporous Mater., 2013, 170, 36–44 CrossRef.
- G. Leofanti, M. Padovan, G. Tozzola and B. Venturelli, Catal. Today, 1998, 41, 207–219 CrossRef.
- N. D. Martínez, R. B. Venturini, H. S. Silva, J. E. González and A. M. Rodríguez, Mater. Res., 2009, 12, 45–50 CrossRef.
- Y. Zhang, J. Wan, Y. Wang and Y. Ma, RSC Adv., 2015, 5, 97589–97597 RSC.
- O. Lebedev, F. Millange, C. Serre, G. Van Tendeloo and G. Férey, Chem. Mater., 2005, 17, 6525–6527 CrossRef.
- D. A. Islam, A. Chakraborty and H. Acharya, New J. Chem., 2016, 40, 6745–6751 RSC.
- C. Saux, C. L. Marchena, L. R. Pizzio and L. B. Pierella, J. Porous Mater., 2016, 23, 947–956 CrossRef.
- T.-H. Chen, K. W. Kwong, N. F. Lee, D. Ranburger and R. Zhang, Inorg. Chim. Acta, 2016, 451, 65–72 CrossRef.
- H. Haddadi and M. R. Farsani, J. Cluster Sci., 2016, 27, 373–386 CrossRef.
- M. Dabiri, M. Koohshari, F. Shafipour, M. Kasmaei, P. Salari and D. MaGee, J. Iran. Chem. Soc., 2016, 13, 1265–1272 CrossRef.
- I. C. Santos, J. A. Gamelas, T. A. Duarte, M. M. Simões, M. G. P. Neves, J. A. Cavaleiro and A. M. Cavaleiro, J. Mol. Catal. A: Chem., 2017, 426, 593–599 CrossRef.
- W. Zhao and C. Yang, New J. Chem., 2013, 37, 1867–1870 RSC.
- X. Xue, W. Zhao, B. Ma and Y. Ding, Catal. Commun., 2012, 29, 73–76 CrossRef.
- H. Veisi, F. H. Eshbala, S. Hemmati and M. Baghayeri, RSC Adv., 2015, 5, 10152–10158 RSC.
- Y. Jin, N. Li, H. Liu, X. Hua, Q. Zhang, M. Chen and F. Teng, Dalton Trans., 2014, 43, 12860–12870 RSC.
- L. Zou, Q. Wang, Z. Wang, L. Jin, R. Liu and X. Shen, Ind. Eng. Chem. Res., 2013, 53, 658–663 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |