
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo/Cu-MFF derived mesoporous ternary metal oxide microcubes for enhancing the catalytic activity of the CO oxidation reaction†
Huijun Songabc,
Li Zhang *abcd,
Guancheng Xuabc,
Chi Zhangabc,
Xin Maabc,
Lu Zhangabc and
Dianzeng Jia*abc
*abcd,
Guancheng Xuabc,
Chi Zhangabc,
Xin Maabc,
Lu Zhangabc and
Dianzeng Jia*abc
aKey Laboratory of Energy Materials Chemistry (Xinjiang University), Ministry of Education, Urumqi, Xinjiang 830046, P. R. China. E-mail: zhanglixju@163.com; jdz0991@gmail.com; Fax: +86-991-8580586; Tel: +86-991-8580586
bKey Laboratory of Advanced Functional Materials, Autonomous Region, Urumqi, Xinjiang 830046, P. R. China
cInstitute of Applied Chemistry, Urumqi, Xinjiang 830046, P. R. China
dPhysics and Chemistry Detecting Center, Xinjiang University, Urumqi, Xinjiang 830046, P. R. China
First published on 10th July 2018
Abstract
Metal–organic framework (MOF)-based derivatives with uniform micro/mesoporous structures have attracted a great deal of interest in various research fields. Herein, we report a simple strategy to design functional mesoporous ternary metal oxides with controlled composition through direct pyrolysis of Co/Cu bimetal-formate frameworks (Co/Cu-MFFs), which were prepared by a facile one-step liquid-phase precipitation method, exhibiting uniform distribution of two different metal species and good structural integrity. The obtained mesoporous ternary metal oxide CuxCo3−xO4 (x = 0.5, 1) microcubes exhibit much better performance for CO oxidation than pure Co3O4, which can be mainly attributed to their larger specific surface areas, stronger reducibility, and the synergistic effect of two active metal oxide components.
1. Introduction
CO oxidation over solid catalysts has been extensively studied, not only with regard to its value as a classical probe reaction for fundamental studies but also due to its great importance in practical applications, such as air cleanup, gas sensors for detection, automotive exhaust gas treatment and CO elimination from the hydrogen feed for proton-exchange membrane fuel cells.1–5 It has been reported that noble metals such as Pt, Au, and Rh are excellent CO oxidation catalysts.2,6–11 Platinum group metals (PGMs) are some of the earliest catalysts and are still widely employed in automotive emission control and cleaning of gas originating from the petrochemical industry since Langmuir's pioneering work.6,7,12 Haruta et al. discovered that oxide-supported Au catalysts prepared through a wet chemistry method exhibit an outstanding high activity for CO oxidation even at cryogenic temperatures.13 Recently, a sub-nano Rh/TiO2 catalyst with an unexpectedly high performance of total CO conversion at −50 °C was discovered by Guan et al.10 The excellent properties of this catalyst can be ascribed to the facile formation of Rh–O–O–Ti superoxide easily reacting with the adsorbed CO species on TiO2 sites.Although these noble metals exhibit high activities for CO oxidation, the high cost and limited supply constrain their commercial applications.14 Thus attention has been focused on searching for “noble metal-free” catalysts with excellent catalytic properties.15–21 Co3O4, one kind of the less-expensive alternatives to the noble metal-based catalysts, has been reported to be one of the most efficient catalysts for carbon monoxide oxidation due to its excellent CO adsorption strength, low barrier of CO reacting with lattice O, and remarkable redox properties.14,15,18,22 A pioneering work on Co3O4 for CO oxidation reported by Xie et al. revealed that the Co3O4 nanorods demonstrate a catalytic activity towards CO oxidation at a temperature of −77 °C.23 The superior performance of the catalyst can be attributed to the predominantly exposed (110) planes with richness of active Co3+ sites. A recent theoretical work found that Co3+ has a higher activity than Co2+ in the Co3O4 for CO oxidation because: (i) Co3+ binds CO molecule strongly compared to Co2+, (ii) a high barrier exists between lattice O linked to Co2+ and CO adsorbed on Co2+.22 Therefore, many works are focusing on preparing special nanostructural Co3O4 predominately exposing active faces with more Co3+ cations to enhance their catalytic activities for CO oxidation. However, these active faces still contain many Co2+ cations which have lower activities for CO oxidation.24–26 So, replacing the inactive Co2+ with other active divalent cations to prepare Co-based ternary metal oxides provides an efficient way to further enhance the catalytic performance of Co3O4. On this point, Cu2+ can be a promising candidate for replacing Co2+ in the Co3O4 not only due to its high activities for CO oxidation, but also owning to the similar ionic radius with Co2+ which allows the easy formation of Co/Cu ternary metal oxides.27–29
MOFs are a class of porous functional materials constructed by metal ions and bridging ligands.30–33 Owing to its adjustable cavities and flexible structures, various metal oxides with specific morphologies and interconnected pores have been fabricated by thermal decomposition of MOFs under suitable calcination conditions, expecting to improve their performance in specific applications.34–42 Among them, ternary metal oxides have gained increasing considerations due to the structure merits and synergetic effect of multiple components. Therefore, bimetallic organic frameworks are considered highly desirable to facile synthesis of the ternary metal oxides. However such effort is still scarce because of the challenge in morphology and composition control during the incorporation of different secondary metal nodes into MOFs.39 Divalent metal formate frameworks (MFFs) of [CH3NH3][M(HCOO)3] (M = Mn, Fe, Ni, Co, Zn) could be promising MOFs to tackle this challenge due to the same six-connected (412·63) nodes of the octahedral (MO6) metal ions within the framework.43 Herein, we propose a facial one-step synthesis of bimetallic formate frameworks [CH3NH3][M(HCOO)3] (M = Co/Cu) (Co/Cu-MFFs) and its derivation of ternary oxides CuxCo3−xO4 (x = 0, 0.5, 1, 1.5) microcubes. The CuxCo3−xO4 microcubes are constructed by numerous interconnected nanoparticles with uniform distribution of cobalt and copper species. In addition, the Cu/Co atomic ratio can be controlled exactly through adjusting the compositions of the growth precursor solutions. Owing to its larger specific surface area, stronger reducibility and the synergistic effect of two metal oxide components, the mesoporous CuxCo3−xO4 with moderate Cu/Co atomic ratio exhibit better performance for CO oxidation reaction than pure Co3O4.
2. Experimental
2.1. Synthesis of Co/Cu-MFFs precursors
25 mL of ethanol was mixed with 12 mmol HCOOH, 8 mmol CH3NH2 (30–33% in methanol) and 0.5 g polyvinylpyrrolidone (PVP K-30) to get a colorless solution. Then a transparent solution containing 1 mmol mixed salts with different Cu/Co atomic ratios (0, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, 1:2, 1:1) and 0.5 g PVP K-30 were dropped into the above solution in 10 minutes to obtain a pink colloidal suspension. The whole reaction process was maintained at room temperature with magnetic stirring. One hour later, the colloidal suspension was aged for one day at ambient temperature without any interruption. The resulting pink precipitates were centrifuged and washed several times with ethanol, finally dried at 60 °C for 6 hours in a vacuum oven and noted as [CH3NH3][Co(HCOO)3], [CH3NH3][Cu1/6Co5/6(HCOO)3], [CH3NH3][Cu1/3Co2/3(HCOO)3], [CH3NH3][Cu1/2Co1/2(HCOO)3] respectively.
:5, 1:2, 1:1) and 0.5 g PVP K-30 were dropped into the above solution in 10 minutes to obtain a pink colloidal suspension. The whole reaction process was maintained at room temperature with magnetic stirring. One hour later, the colloidal suspension was aged for one day at ambient temperature without any interruption. The resulting pink precipitates were centrifuged and washed several times with ethanol, finally dried at 60 °C for 6 hours in a vacuum oven and noted as [CH3NH3][Co(HCOO)3], [CH3NH3][Cu1/6Co5/6(HCOO)3], [CH3NH3][Cu1/3Co2/3(HCOO)3], [CH3NH3][Cu1/2Co1/2(HCOO)3] respectively.
2.2. Synthesis of CuxCo3−xO4 (x = 0, 0.5, 1, 1.5) microcubes
The porous ternary metal oxides were fabricated in muffle burner by annealing these prepared Co/Cu-MFFs precursors at 350 °C for 2 hours, at a heating rate of 1 °C min−1 and noted as Co3O4, Cu0.5Co2.5O4, CuCo2O4, Cu1.5Co1.5O4, respectively.2.3. Characterizations
Powder XRD patterns were recorded on a Bruker D8 advance diffractometer with Cu Kα radiation over the 2θ range of 10–80°. Thermogravimetric analyses (TGA) were carried out in a Netzsch SDT449F3 thermal analyzer in air atmosphere with a heating rate of 5 °C min−1. The SEM and TEM images of the prepared samples were obtained through field emission scanning electron microscopy (FESEM, Hitachi S-4800 microscope) and transmission electron microscopy (TEM, JEOL, JEM-2100F). The chemical composition and elemental distribution of the prepared catalysts were examined by Energy-dispersive X-ray spectroscope (EDX) attached to the FESEM instrument. ICP-OES was used to determine the Cu/Co atomic ratio of CuxCo3−xO4. The X-ray photoelectron spectroscopy (XPS) spectra were collected on Thermo ESCALAB 250 Xi spectrometer. N2 adsorption–desorption isotherm were obtained by a Micromeritics ASAP 2020 analyzer at 77 K. H2-temperature-programmed reduction (H2-TPR) experiments were performed under a 10 vol% H2/Ar mixture with a flow rate of 50 mL min−1 over 60 mg of catalyst by a Micromeritics Chemisorb 2920 apparatus and the temperature was increased from ambient temperature to 800 °C at a ramp rate of 1 °C min−1. Prior to each analysis, the catalysts were purged in a flow of pure argon at 200 °C for 2 hours to remove traces water.2.4. Catalytic performance measurements
The catalytic tests of the CuxCo3−xO4 microcubes for CO oxidation reaction were performed in a fixed quartz tubular reactor. The reactor was charged with 50 mg of the catalysts without pre-treatment. The reaction temperature was detected by a thermocouple under catalyst bed. The mixed feed gas consisted of 1 vol% CO, 20 vol% O2, 79 vol% N2 was passed through the reactor at a flow rate of 50 mL min−1, corresponding to a gas hourly space velocity (GHSV) of 60000 mL g−1 h−1. The components of the gas were examined by a gas chromatograph (Agilent GC7890). Two catalytic test data points at each temperature were collected and the average values of them were reported.
3. Results and discussion
The synthesis process of mesoporous CuxCo3−xO4 microcubes is shown in Scheme 1. Firstly, the Co/Cu-MFFs precursors with different Cu/Co atomic ratios were synthesized through a one-step liquid-phase precipitation method under room temperature. Then, the CuxCo3−xO4 microcubes with porous structure were obtained through thermal decomposition of the precursors under air. The phase and purity of the Co/Cu-MFFs precursors and CuxCo3−xO4 were examined by PXRD (Fig. 1a). All strong peaks in the XRD patterns of each Co/Cu-MFFs precursor are well matched with the simulated single crystal structure of [CH3NH3][Co(HCOO)3] according to the reported crystal structure data,44,45 suggesting the successful incorporation of Cu2+ into Co/Cu-MFFs. Fig. S1† shows the TGA curves of as-prepared precursors, indicating that the weight residual of all samples becomes constant when the temperature rises to 300 °C. During the calcination, all samples exhibit similar two-step weight loss. The first weight loss could be relate to the removal of amine and one formate molecule per formula unit. The second weight loss could be due to the decomposition of residual organic components. Fig. 1b shows that the corresponding diffraction profiles of prepared ternary oxides CuxCo3−xO4 are in agreement with the standard CuCo2O4 (JCPDS no. 01-1155) or pure Co3O4 (JCPDS no. 42-1467). CuCo2O4 can be treated as Co3O4 with Co2+ replaced by Cu2+ and therefore these two oxides have almost same XRD patterns. Meanwhile, two weak peaks corresponding to CuO (002), (111) (JCPDS no. 48-1548) can be seen in the XRD patterns of CuCo2O4 and Cu1.5Co1.5O4, suggesting that a few Cu2+ cations did not enter into the lattice of Co3O4 successfully. These two peaks become obvious with the increase of Cu-doping amount, indicating that the excess amounts of Cu2+ cations in the Co/Cu-MFFs precursors lead to a significant phase segregation of the oxides. | ||
| Scheme 1 Schematic illustration of the synthesis of Co/Cu-MFFs precursors and its derived mesoporous CuxCo3−xO4 microcubes for CO oxidation reaction. | ||
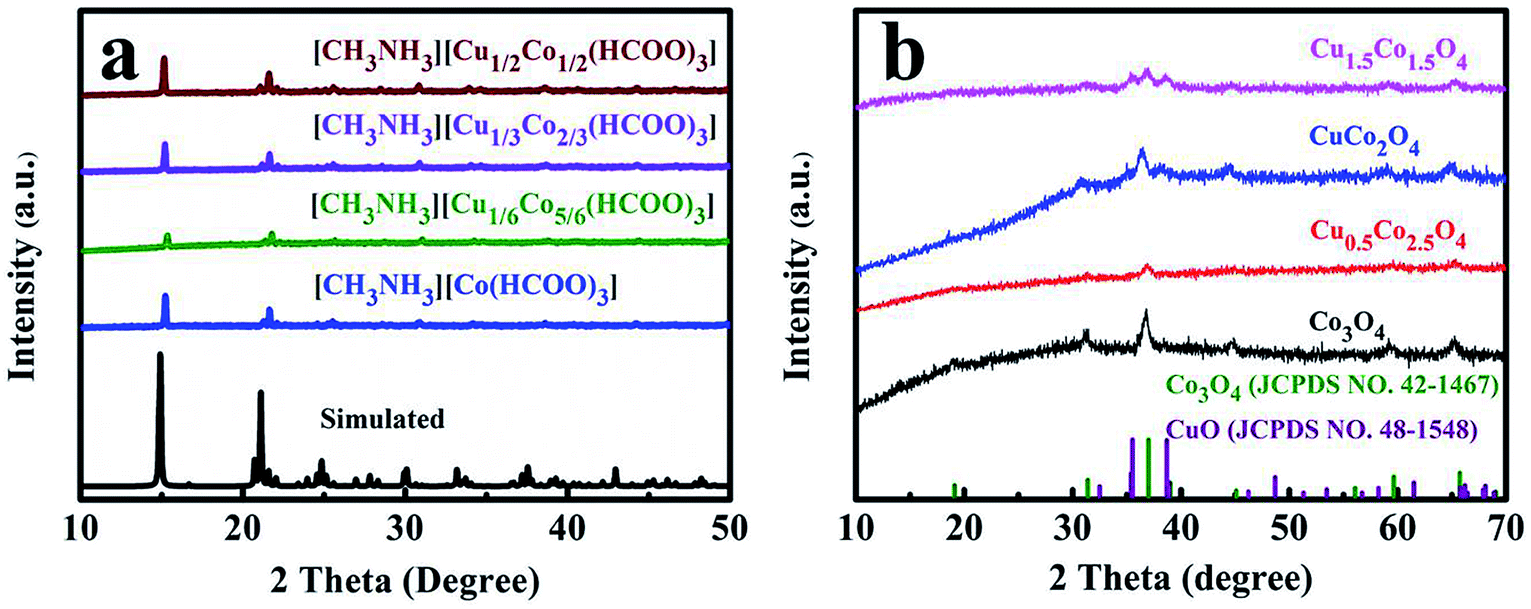 | ||
| Fig. 1 XRD patterns of Co/Cu-MFFs precursors (a) and CuxCo3−xO4 microcubes (b). | ||
The SEM images in Fig. S2a and S2b† reveal that the as-prepared [CH3NH3][Co(HCOO)3] and [CH3NH3][Cu1/3Co2/3(HCOO)3] have similar cubic shapes with smooth surfaces and some macropores about 0.6 μm can be observed clearly on the partial surfaces of these two precursors. Meanwhile, the size of [CH3NH3][Cu1/3Co2/3(HCOO)3] microcubes is about 1–2 μm, which is smaller than that of [CH3NH3][Co(HCOO)3], mainly about 2–3 μm. We can infer accordingly that the introduced Cu2+ does not have great influences on the morphology of precursors. In order to study the effect of PVP K-30 in the preparation of Co/Cu-MFFs precursors, the morphology of [CH3NH3][Co(HCOO)3] prepared in the absence of PVP K-30 was also characterized. As shown in Fig. S3,† the [CH3NH3][Co(HCOO)3] without adding PVP K-30 still retains the cubic morphology but many microbes agglomerate together. So we can infer that PVP K-30 functions here mainly as stabilizing agent to prevent agglomeration of the microcubes. The similar functions have been reported by many other reports.46,47 The SEM images of CuxCo3−xO4 easily prepared by annealing precursors are shown in Fig. 2a–d. The cubic morphology of CuxCo3−xO4 reveals that all the CuxCo3−xO4 almost preserved the original cubic morphology of as-prepared precursors. Meanwhile, a great number of small nanoparticles on the rough surface of cubic CuxCo3−xO4 can be seen clearly in the SEM images, which confirms the successful formation of porous structure. The chemical composition and elemental distribution of the microcubes were further characterized by SEM-EDX. The elemental mapping images (Fig. 2e and S4†) further confirm that all the ternary metal oxides are with similar microcubic morphologies and the uniform distribution of O, Cu and Co. The metal element ratios of Cu and Co in the oxides are similar with those ratios in the growth precursor solution as shown in Table S1 and Fig. S5,† which shows that the compositions of CuxCo3−xO4 can be controlled exactly by adjusting the synthesis of [CH3NH3][Cu1/3Co2/3(HCOO)3]. The high-resolution TEM image of CuCo2O4 microcube is shown in Fig. 2f, the interplanar distances of 0.23 nm can be indexed to the (111) plane of CuO, whereas the interplanar distance of 0.24 nm is correspondence to the (311) plane of CuCo2O4. The interlaced boundaries marked with white circle demonstrate the high interdispersion of the CuO and CuCo2O4.
 | ||
| Fig. 2 SEM images of Co3O4 (a), Cu0.5Co2.5O4 (b), CuCo2O4 (c), Cu1.5Co1.5O4 (d), SEM–EDX mapping images of CuCo2O4 (e), TEM images of CuCo2O4 (f). | ||
Some related mechanisms at the molecular level of Co3O4 for CO oxidation suggest that gas phase CO chemisorbs preferably on the exposed Co3+, then reacts with an oxygen atom linked to the active Co3+ site resulting in CO2 and an oxygen vacancy formation.24,48 Therefore, the amount of the exposed Co3+ cations on the Co3O4 surfaces is responsible for catalytic activity.49 Herein, the XPS analysis was further carried out to examine the surface chemical compositions and elemental states of CuxCo3−xO4 microcubes. The Co2p XPS spectra profiles (Fig. 3a) constructed with two main peaks centering at about 779.5 and 794.6 eV, corresponding to the Co2p3/2 and Co2p1/2 respectively.37,50,51 The relative percentage of Co3+ and Co2+ was calculated through the fitted curves of CuxCo3−xO4, and was plotted as functions of the Cu/Co atomic ratio in the solution of preparing precursors (Fig. 3b). We can find that the ratio of Co2+/Co3+ on the surface of CuxCo3−xO4 first drastically decreases and then almost remains unchanged with the increasing Cu/Co atomic ratio. That decline of Co2+/Co3+ atomic ratio should be originated from the Cu2+ substitution for Co2+ in CuxCo3−xO4. The further steadiness might be ascribed to many Cu2+ leading to the formation of CuO rather than substitution for Co2+, which can also be inferred from the XRD results.
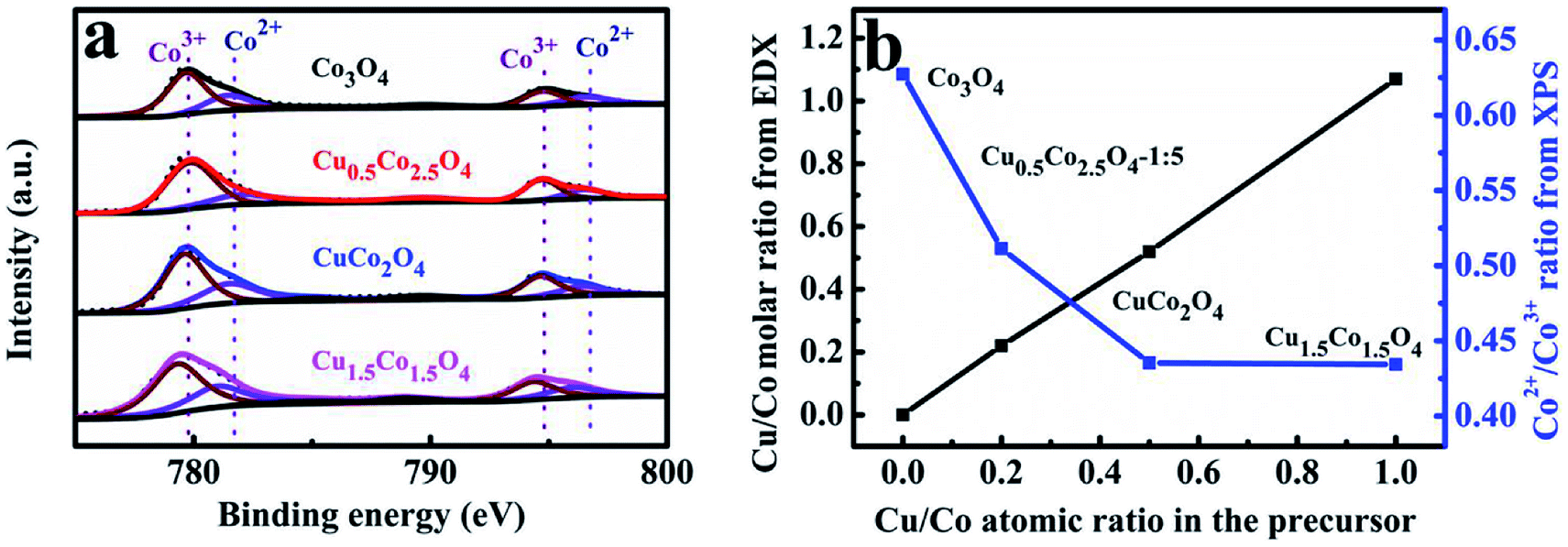 | ||
| Fig. 3 XPS spectra of the CuxCo3−xO4 microcubes (a), the measured Cu/Co atomic ratio and Co2+/Co3+ atomic ratio on the surface of the CuxCo3–xO4 microcubes as functions of the Cu/Co atomic ratio in the solution of preparing precursors (b). | ||
The porous structures of Co/Cu-MFFs derived CuCo2O4 and pure Co3O4 were further examined by measuring sorption isotherms of nitrogen at 77 K. It can be observed in Fig. 4a that both of the oxides present a type-IV adsorption isotherm with a significant hysteresis loop, denoting that they are mesoporous solids. The specific surface areas of Co3O4 and CuCo2O4 microcubes were calculated to be 25.59 and 29.47 m2 g−1 through Brunauer–Emmett–Teller (BET) method. In addition, the corresponding Barrett–Joyner–Halenda (BJH) pore size distribution plots (Fig. S6†) show the main pore size distribution of Co3O4 and CuCo2O4 are 0.71–1.58 nm and 16.15–20.73 nm, respectively.
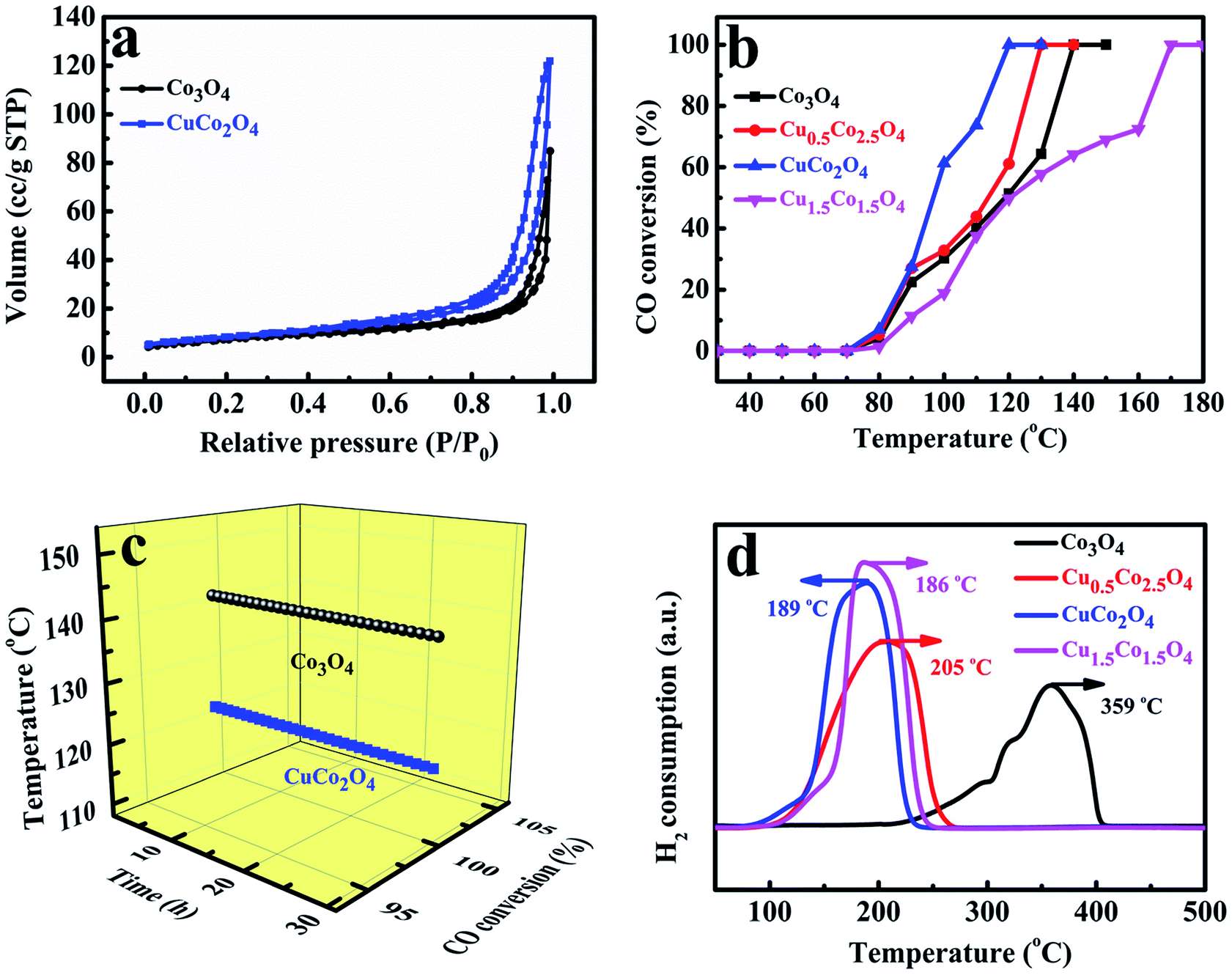 | ||
| Fig. 4 Nitrogen adsorption–desorption isotherms of Co3O4 and CuCo2O4 (a), the catalytic activities of CuxCo3−xO4 for CO oxidation (b), stability of Co3O4 at 140 °C and CuCo2O4 at 120 °C (c), H2-TPR profiles of CuxCo3−xO4 (d). | ||
To directly evaluate the catalytic performance of the Co/Cu-MFFs derived ternary oxides for CO oxidation reaction, 50 mg of the mesoporous CuxCo3−xO4 microcubes without any pre-treatments were put into a fixed quartz glass reactor respectively. As shown in Fig. 4b, with the increasing amount of Cu/Co atomic ratio in the catalysts, the catalytic activities of CuxCo3−xO4 for CO oxidation reaction firstly increase and then decrease. Among them, CuCo2O4 exhibits the highest activity with 100% CO conversion rate at 120 °C, superior or comparable to other transition oxides as CO oxidation catalysts (Table S2†). These results indicate that using more active Cu2+ to replace the inactive Co2+ in the lattice of Co3O4 could be a novel way to enhance the catalytic performance for CO oxidation. Meanwhile, the Cu1.5Co1.5O4 has the lowest activity and the temperature of CO complete conversion is as high as 170 °C, which might be due to the richness of less-active CuO phase. The CO oxidation conversions of without materials and with 50 mg of precursors performed under different temperatures were shown in Fig. S7,† the results indicated that there was no catalytic activity in the absence of catalyst and the Co/Cu-MFFs exhibited extremely low catalytic activity of CO oxidation. We also investigated the stabilities of pure Co3O4 and CuCo2O4 under the similar conditions at 140 and 130 °C, respectively (Fig. 4c). Both samples exhibit good stable performance that retains CO complete conversion within 30 hours. Based on these results, we can infer that the enhanced catalytic activity of CuxCo3−xO4 can be attributed to the structure and component merits achieved through the incorporation of new secondary copper nodes in the Co/Cu-MFFs precursors. In addition, pure Co3O4 presents a good activity for CO oxidation with a CO complete conversion temperature of 140 °C, which is 30 °C lower than that reported by Zhang et al.52 The higher catalytic activity might be ascribed to the larger specific surface area and the lower inactive Co2+ concentration in the surface of Co3O4 according to the BET and XPS results (Table S3†).52
To further investigate the synergetic effects of Cu and Co species, the temperature-programmed reductions were performed using a stream of diluted H2 (H2/Ar2, 1:9 v/v). As shown in Fig. 4d, only a single peak at 359 °C is observed in pure Co3O4 microcubes reduction profile, which can be explained by the direct reduction of cobalt ions into metallic Co in one step for the large particles of Co3O4.53 With the amount of Cu2+ increasing in catalysts, the prominent peaks of Cu0.5Co2.5O4, CuCo2O4 and Cu1.5Co1.5O4 shift towards lower temperature to 205, 189 and 186 °C, respectively, which means that the reducibility of the catalysts was markedly promoted when the Cu/Co atomic ratio increased. Such enhanced reduction may be attributed to the strong Co–Cu interaction originated from the intimate contact and the good interdispersion of the CuO and Co3O4, which possibly leads to a junction of their band levels in the solid solution or the mixed oxidations.54 Such strong interactions also have been found for copper–zinc chromite catalysts and CuO–ZnO system.55,56
4. Conclusion
In this work, we have proposed a novel and simple method to prepare Co-based ternary metal oxides CuxCo3−xO4 with cube-like morphology and studied the effect of replacing Co2+ with Cu2+ on the catalytic activity of Co3O4 towards CO oxidation. The CuxCo3−xO4 microcubes with controlled composition were prepared by one step pyrolysis of Co/Cu-MFFs, in which the Cu/Co atomic ratio can be tuned through adjusting the compositions of growth precursor solution. The obtained mesoporous CuxCo3−xO4 have enhanced catalytic properties for CO oxidation compared to the pure porous Co3O4 when the atomic ratio of Cu/Co is no more than 1/2 in the solution of preparing precursors. The larger specific surface areas and stronger reducibility resulted from the introduced new secondary Cu metal nodes into Co/Cu-MFFs which may play important roles in improving catalytic performance. We believe that the synthetic strategy can also be extended to obtain other ternary metal oxides with different structures by controlling the morphology and the compositions of the bimetal-formate frameworks, which might have a higher catalytic performance or exhibit excellent properties in other applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial supports for this work have been received from Key Laboratory Open Research Foundation of Xinjiang Autonomous Region (No. 2016D03008) and National Natural Science Foundation of China (No. 21661029, 21771157, 21663029).References
- H.-J. Freund, G. Meijer, M. Scheffler, R. Schlögl and M. Wolf, Angew. Chem., Int. Ed., 2011, 50, 10064–10094 CrossRef CAS PubMed.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175–192 CrossRef CAS.
- J.-C. Ding, H.-Y. Li, T.-C. Cao, Z.-X. Cai, X.-X. Wang and X. Guo, Solid State Ionics, 2017, 303, 97–102 CrossRef CAS.
- K. M. Adams and G. W. Graham, Appl. Catal., B, 2008, 80, 343–352 CrossRef CAS.
- P. Landon, J. Ferguson, B. E. Solsona, T. Garcia, A. F. Carley, A. A. Herzing, C. J. Kiely, S. E. Golunski and G. J. Hutchings, Chem. Commun., 2005, 3385–3387 RSC.
- J. Lin, X. Wang and T. Zhang, Chin. J. Catal., 2016, 37, 1805–1813 CrossRef CAS.
- B. Liu, Y. Liu, H. Hou, Y. Liu, Q. Wang and J. Zhang, Catal. Sci. Technol., 2015, 5, 5139–5152 RSC.
- L. W. Guo, P. P. Du, X. P. Fu, C. Ma, J. Zeng, R. Si, Y. Y. Huang, C. J. Jia, Y. W. Zhang and C. H. Yan, Nat. Commun., 2016, 7, 13481 CrossRef CAS PubMed.
- B. Qiao, J. Lin, A. Wang, Y. Chen, T. Zhang and J. Liu, Chin. J. Catal., 2015, 36, 1505–1511 CrossRef CAS.
- H. Guan, J. Lin, B. Qiao, X. Yang, L. Li, S. Miao, J. Liu, A. Wang, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2016, 55, 2820–2824 CrossRef CAS PubMed.
- H. Guan, J. Lin, L. Li, X. Wang and T. Zhang, Appl. Catal., B, 2016, 184, 299–308 CrossRef CAS.
- I. Langmuir, Trans. Faraday Soc., 1992, 17, 621–654 RSC.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS.
- L. F. Liotta, H. Wu, G. Pantaleo and A. M. Venezia, Catal. Sci. Technol., 2013, 3, 3085–3102 RSC.
- X. Wang, W. Zhong and Y. Li, Catal. Sci. Technol., 2015, 5, 1014–1020 RSC.
- Z. Zhao, X. Lin, R. Jin, G. Wang and T. Muhammad, Appl. Catal., B, 2015, 115–116, 53–62 Search PubMed.
- Y. Su, Z. Tang, W. Han, Y. Song and G. Lu, Catal. Surv. Asia, 2015, 19, 68–77 CrossRef CAS.
- C. J. Jia, M. Schwickardi, C. Weidenthaler, W. Schmidt, S. Korhonen, B. M. Weckhuysen and F. Schuth, J. Am. Chem. Soc., 2011, 133, 11279–11288 CrossRef CAS PubMed.
- A. Biabani-Ravandi and M. Rezaei, Chem. Eng. J., 2012, 184, 141–146 CrossRef CAS.
- T. Cwele, N. Mahadevaiah, S. Singh and H. B. Friedrich, Appl. Catal., B, 2016, 182, 1–14 CrossRef CAS.
- Y. Yu, T. Takei, H. Ohashi, H. He, X. Zhang and M. Haruta, J. Catal., 2009, 267, 121–128 CrossRef CAS.
- H.-F. Wang, R. Kavanagh, Y.-L. Guo, Y. Guo, G. Lu and P. Hu, J. Catal., 2012, 296, 110–119 CrossRef CAS.
- X. Xie, Y. Li, Z. Q. Liu, M. Haruta and W. Shen, Nature, 2009, 458, 746–749 CrossRef CAS PubMed.
- J. Jansson, A. E. C. Palmqvist, E. Fridell, M. Skoglundh, L. Österlund, P. Thormählen and V. Langer, J. Catal., 2002, 211, 387–397 CrossRef CAS.
- P. Broqvist, I. Panas and H. Persson, J. Catal., 2002, 210, 198–206 CrossRef CAS.
- F. Grillo, M. M. Natile and A. Glisenti, Appl. Catal., B, 2004, 48, 267–274 CrossRef CAS.
- M. Y. Guo, F. Liu, J. Tsui, A. A. Voskanyan, A. M. C. Ng, A. B. Djurišić, W. K. Chan, K.-Y. Chan, C. Liao, K. Shih and C. Surya, J. Mater. Chem. A, 2015, 3, 3627–3632 RSC.
- R. Zhang, L. Hu, S. Bao, R. Li, L. Gao, R. Li and Q. Chen, J. Mater. Chem. A, 2016, 4, 8412–8420 RSC.
- M. Zhou, L. Cai, M. Bajdich, M. García-Melchor, H. Li, J. He, J. Wilcox, W. Wu, A. Vojvodic and X. Zheng, ACS Catal., 2015, 5, 4485–4491 CrossRef CAS.
- E. A. Dolgopolova, A. J. Brandt, O. A. Ejegbavwo, A. S. Duke, T. D. Maddumapatabandi, R. P. Galhenage, B. W. Larson, O. G. Reid, S. C. Ammal, A. Heyden, M. Chandrashekhar, V. Stavila, D. A. Chen and N. B. Shustova, J. Am. Chem. Soc., 2017, 139, 5201–5209 CrossRef CAS PubMed.
- L. Chen, R. Luque and Y. Li, Chem. Soc. Rev., 2017, 46, 4614–4630 RSC.
- X. Yang and Q. Xu, Cryst. Growth Des., 2017, 17, 1450–1455 CrossRef CAS.
- L. Zhu, X. Q. Liu, H. L. Jiang and L. B. Sun, Chem. Rev., 2017, 117, 8129–8176 CrossRef CAS PubMed.
- F. Wang, X. Wang, D. Liu, J. Zhen, J. Li, Y. Wang and H. Zhang, ACS Appl. Mater. Interfaces, 2017, 6, 22216–22223 CrossRef PubMed.
- Y. Zhu, C. Cao, J. Zhang and X. Xu, J. Mater. Chem. A, 2015, 3, 9556–9564 RSC.
- L. Shen, L. Yu, X.-Y. Yu, X. Zhang and X. W. D. Lou, Angew. Chem., Int. Ed., 2015, 54, 1868–1872 CrossRef CAS PubMed.
- J. Wei, Y. Feng, Y. Liu and Y. Ding, J. Mater. Chem. A, 2015, 3, 22300–22310 RSC.
- L. Han, X.-Y. Yu and X. W. D. Lou, Adv. Mater., 2016, 28, 4601–4605 CrossRef CAS PubMed.
- H. Li, M. Liang, W. Sun and Y. Wang, Adv. Funct. Mater., 2016, 26, 1098–1103 CrossRef CAS.
- R. R. Salunkhe, Y. V. Kaneti and Y. Yamauchi, ACS Nano, 2017, 11, 5293–5308 CrossRef CAS PubMed.
- Z. Yu, Y. Bai, Y. Liu, S. Zhang, D. Chen, N. Zhang and K. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 31777–31785 CrossRef CAS PubMed.
- S. Zheng, X. Li, B. Yan, Q. Hu, Y. Xu, X. Xiao, H. Xue and H. Pang, Adv. Energy Mater., 2017, 7, 1602733 CrossRef.
- Z. Wang, K. Hu, S. Gao and H. Kobayashi, Adv. Mater., 2010, 22, 1526–1533 CrossRef CAS PubMed.
- L. C. Gomez-Aguirre, B. Pato-Doldan, J. Mira, S. Castro-Garcia, M. A. Senaris-Rodriguez, M. Sanchez-Andujar, J. Singleton and V. S. Zapf, J. Am. Chem. Soc., 2016, 138, 1122–1125 CrossRef CAS PubMed.
- B. Pato-Doldan, L. C. Gomez-Aguirre, A. P. Hansen, J. Mira, S. Castro-Garcia, M. Sanchez-Andujar, M. A. Senaris-Rodriguez, V. S. Zapf and J. Singleton, J. Mater. Chem. C, 2016, 4, 11164–11172 RSC.
- X. Huang and N. Zheng, J. Am. Chem. Soc., 2009, 131, 4602–4603 CrossRef CAS PubMed.
- J. Yin, J. Wang, M. Li, C. Jin and T. Zhang, Chem. Mater., 2012, 24, 2645–2654 CrossRef CAS.
- Y. Xie, F. Dong, S. Heinbuch, J. J. Rocca and E. R. Bernstein, Phys. Chem. Chem. Phys., 2010, 12, 947–959 RSC.
- A. Alvarez, S. Ivanova, M. A. Centeno and J. A. Odriozola, Appl. Catal., A, 2012, 431–432, 9–17 CrossRef CAS.
- G. Li, L. Li, Y. Li and J. Shi, New J. Chem., 2015, 39, 1742–1748 RSC.
- J. Zhu and Q. Gao, Microporous Mesoporous Mater., 2009, 124, 144–152 CrossRef CAS.
- C. Zhang, L. Zhang, G.-C. Xu, X. Ma, Y.-H. Li, C.-Y. Zhang and D.-Z. Jia, New J. Chem., 2017, 41, 1631–1636 RSC.
- J.-Y. Luo, M. Meng, X. Li, X.-G. Li, Y.-Q. Zha, T.-D. Hu, Y.-N. Xie and J. Zhang, J. Catal., 2008, 254, 310–324 CrossRef CAS.
- G. Fierro, M. Lo Jacono, M. Inversi, R. Dragone and P. Porta, Top. Catal., 2000, 10, 39–48 CrossRef CAS.
- G. L. Castiglioni, A. Vaccari, G. Fierro, M. Inversi, M. Lo Jacono, G. Minelli, I. Pettiti, P. Porta and M. Gazzano, Appl. Catal., A, 1995, 123, 123–144 CrossRef CAS.
- G. Fierro, M. Lo Jacono, M. Inversi, P. Porta, F. Cioci and R. Lavecchia, Appl. Catal., A, 1996, 137, 327–348 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04081h |
| This journal is © The Royal Society of Chemistry 2018 |