
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiphasic aqueous hydroformylation of 1-alkenes with micelle-like polymer particles as phase transfer agents†
Bachir Bibouche a,
Daniel Perala,
Dmitrij Stehlb,
Viktor Söderholmc,
Reinhard Schomäckera,
Regine von Klitzingb and
Dieter Vogt*c
a,
Daniel Perala,
Dmitrij Stehlb,
Viktor Söderholmc,
Reinhard Schomäckera,
Regine von Klitzingb and
Dieter Vogt*c
aDepartment of Chemistry, Technische Universität Berlin, Straße des 17. Juni 124, TC-8, 10623 Berlin, Germany
bDepartment of Physics, Technische Universität Darmstadt, Alarich-Weiss-Straße 10, 64287 Darmstadt, Germany
cDepartment of Biochemical and Chemical Engineering, Technische Universität Dortmund, Emil-Figge-Straße 66, 44227 Dortmund, Germany. E-mail: dieter.vogt@tu-dortmund.de
First published on 27th June 2018
Abstract
Micelle-like polymer particles have been applied in aqueous multiphasic hydroformylation reactions of long chain alkenes. These colloids act as phase transfer agents for the nonpolar substrates and as carriers for the catalyst bearing sulfonated ligands by electrostatic attraction. The catalyst performance and the phase separation were optimized with special focus on the conversion, selectivity and catalyst recovery, as those are key points in multiphasic systems to achieve a feasible industrial process. The effect on the catalyst performance of the number of sulfonate groups and electron withdrawing trifluoromethyl groups in the ligand has been studied. The approach was successfully demonstrated for 1-alkenes from 1-hexene to 1-dodecene. For 1-octene, a TOF of more than 3000 h−1 could be achieved at a substrate to catalyst ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, while keeping the rhodium and phosphorous leaching below 1 ppm. In repetitive batch experiments the catalyst was recycled four times, yielding an accumulated TON of more than 100000 for 1-octene.
000, while keeping the rhodium and phosphorous leaching below 1 ppm. In repetitive batch experiments the catalyst was recycled four times, yielding an accumulated TON of more than 100000 for 1-octene.
Introduction
On an industrial scale, the separation and reuse of homogeneous catalysts is a very important issue. As this is often not easy to achieve, heterogeneous catalysts are being applied in about 80% of industrial catalytic processes.1 However, molecular catalysts often show superior selectivity and activity under milder conditions. One of the biggest homogeneously catalyzed processes is the hydroformylation of olefins with an annual production of 12 Mt. One way to combine easy separation of catalyst and product mixture and the advantages of molecular catalysts is the use of a multiphasic reaction system. This is being applied in the Ruhrchemie–Rhône-Poulenc process for the hydroformylation of short chain alkenes with a molecular rhodium catalyst.2,3 In that process, the substrates and products form a nonpolar phase, while the rhodium catalyst resides in an aqueous phase due to the water soluble, trisulfonated triphenyl-phosphine ligand (TPPTS). This indeed combines most of the advantages of homogeneous catalysis with simple product separation at very low Rh leaching in the ppb range. However, this process is limited to short chain alkenes with sufficient water solubility to enable rapid mass transfer to the aqueous catalyst phase.One strategy to separate and recycle the homogeneous catalyst is to carry out the reaction in a completely homogeneous medium that can be switched into two phases for easy product separation after the reaction. This can be achieved by adding cosolvents4,5 or additives with cosolvents that can be switched in polarity by addition of CO2.6 The homogeneous catalyst itself can also be switched from a nonpolar to a polar state by using special ligands that react with CO2, changing the phase in which the catalyst is dissolved.7,8 In thermomorphic multiphase-systems (TMS) a homogeneous reaction medium is generated at higher (reaction) temperature, while at lower temperature a miscibility gap allows to separate the catalyst phase from the product phase. A mixture of water and 1-butanol has recently been used to carry out the hydroformylation of methyl 10-undecenoate in a continuous miniplant process with efficient catalyst recycling.9,10 Another strategy used for the hydroformylation of long chain alkenes catalyzed by a water soluble molecular catalyst is the use of phase transfer agents (PTA). These additives reduce the mass transfer limitation during reaction and are ideally recycled together with the catalyst. Surfactants11–14 and cyclodextrins15–17 have been used as phase transfer agents for multiphase hydroformylation reactions.
An alternative to a classical phase transfer agent, like a surfactant, is the use of colloidal dispersions with covalently attached ligands. This enables multiphasic reactions with moderate to low leaching of metal to the product phase.18–21 The difference to the before mentioned systems is that the ligands are covalently bound, making the catalyst less similar to a truly homogeneous catalyst. It should be mentioned, that the selectivity does not necessarily suffer from this, and in some cases even increases.18,19
Colloidal, micelle-like polymer particles have been shown to be viable phase transfer agents for multiphasic hydroformylation reactions, although their role in enabling mass transfer has not been fully understood yet.22–24 The particles have a cross-linked polystyrene core, a polyethylene glycol (PEG) shell, making them water soluble, and are functionalized with cationic ammonium salts. This means that any anionic ligand will interact with them, making them flexible in terms of applicability. Although the particles successfully worked as phase transfer agents for the aqueous multiphasic hydroformylation of 1-octene, only moderate turnover frequencies (TOF) could be obtained (350 h−1). Moreover, the formation of stable emulsions after the catalytic reactions prevented easy separation of the aqueous catalyst phase and the organic product phase. The products were extracted with diethylether, which enabled the recycling of the particles and catalyst but still required an undesired cosolvent.22
In this work, the performance of functionalized polymer particles as phase transfer agents and catalyst carriers has been further investigated for the hydroformylation of long chain alkenes. Ligands with different stereoelectronic properties have been evaluated. The focus of this work has been the combination of yield of desired products, catalyst efficiency and ease of recyclability.
Results and discussion
Colloidal polymer particles
The colloidal polymer particles used in this work as phase transfer agents for the multiphasic hydroformylation of longer chain 1-alkenes were described in a recent publication.22 They are comprised of a nonpolar cross-linked polystyrene core and a hydrophilic shell based on PEG-modified styrene and ammonium salt-modified styrene monomers. The polymer particles were designed to act as microreactors in aqueous liquid–liquid catalytic reactions, as they are similar to micelles and can solubilize non polar solvents. In this approach, the particles are dispersed in the aqueous catalyst phase. Unlike the polymer particles carrying a covalently bound ligand, mentioned in the introductory part, our particles are functionalized with cationic groups. This allows to attach any ligand carrying anionic groups, like the common sulfonated TPPTS-ligand, by electrostatic interaction (ion-exchange). The colloidal suspensions have been synthesized by emulsion polymerization in water. The molar monomer composition and the reaction conditions for the polymer particles used in this work are depicted in Fig. 1.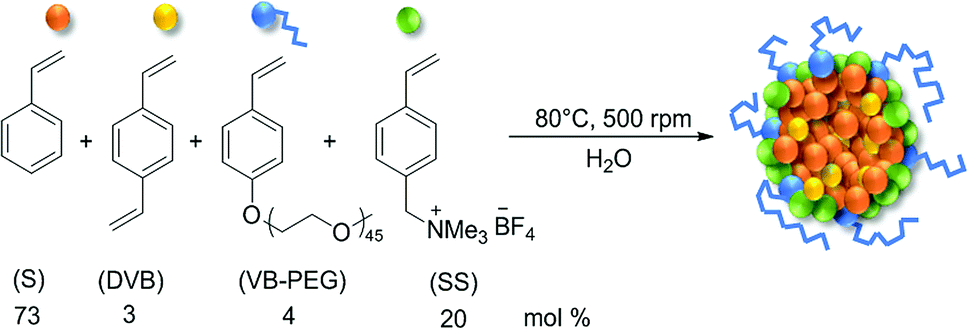 | ||
| Fig. 1 Molar composition and reaction conditions for the synthesis of the polymer particle suspensions by emulsion polymerization. | ||
The polymer particles were characterized by dynamic light scattering (DLS), atomic force microscopy (AFM), transmission electron microscopy (TEM) and scanning electron microscopy (SEM). The cores of the particles have diameters around 30 nm, as shown by TEM (30 nm) and SEM images (32 nm), where only the polystyrene core could be observed. By AFM the particles showed diameters of 72 nm and the core and the shell could be differentiated in the phase mode images. The hydrodynamic diameter of the particles in aqueous suspension found by DLS was 95 nm.
The functionalization of the polymer particles with ammonium salts stabilizes the particles in water, as indicated by the zeta potential of +48 mV. Moreover, the cationic ammonium salts have been proven to electrostatically interact with anionic ligands, rendering the particles catalyst carriers. This was confirmed by membrane filtration experiments with mixtures of polymer particles, anionic ligand (L), and the rhodium-precursor in the concentration regime of the actual catalytic reaction conditions ([Rh]:[L]:[S] = 1:20:40000, 35 mg ml−1 polymer particles, where S stands for substrate). The filtrate did not contain any particles and no detectable amounts of functionalized ligand (TPPTS, TPPMS, TPPDS, Danphos, Dan2phos, see Fig. 5).
Hydroformylation of 1-octene
The multiphasic hydroformylation of 1-octene was performed using [Rh(acac)(CO)2] as catalyst precursor and 6 equivalents of the trisulfonated triphenylphosphine (TPPTS) as water soluble ligand. The reactions were carried out in high pressure autoclaves equipped with a mechanical gas-impeller stirrer to have better control over the mass-transfer processes between the nonpolar substrate phase and the aqueous catalyst phase. As standard reaction conditions 100 bar of syngas (CO:H2 = 1:1), a stirring rate of 700 rpm at 80 °C reaction temperature were chosen (Scheme 1). The concentration of the polymer particles in the aqueous phase was 25 mg ml−1 and n-dodecane was used as internal standard for GC analysis. Hydroformylation of 1-octene was carried out at a scale of 150 mmol, where the amount of catalyst was successively decreased from a substrate to catalyst ratio of 5000 to 80000 (Table 1).
 | ||
| Scheme 1 Multiphasic hydroformylation of 1-octene. | ||
| Entry | [S]:[C] |
Conversion (%) | Chemosel. (%) | l:b |
TOF at 10% conv. (h−1) |
|---|---|---|---|---|---|
| a Reaction conditions: [Rh(acac)(CO)2], TPPTS, [L]:[Rh] = 6, 150 mmol 1-octene, 15 mmol n-dodecane as internal standard. 500 mg of polymer particles, 20 ml H2O. 80 °C, 700 rpm, 100 bar CO:H2 (1:1), 22 h. |
|||||
| 1 | 5000 | 87 | 99 | 2.6 | 347 |
| 2 | 10000 |
86 | 98 | 2.5 | 541 |
| 3 | 20000 |
90 | 96 | 2.4 | 1200 |
| 4 | 40000 |
93 | 90 | 2.3 | 2024 |
| 5 | 80000 |
96 | 85 | 2.1 | 3280 |
Lower amounts of catalyst lead to higher rates. However, as expected, lower amounts of rhodium precursor and ligand, while keeping their ratio constant, lead to lower selectivities, as the lower ligand concentration is expected to lead to less phosphine ligands coordinating to the metal center (Table 1 and Fig. 2). Furthermore, when using half of the amount of catalyst, the conversion stayed similar, but the TOF roughly doubled, indicating a mass transfer limitation of substrate to the catalyst.
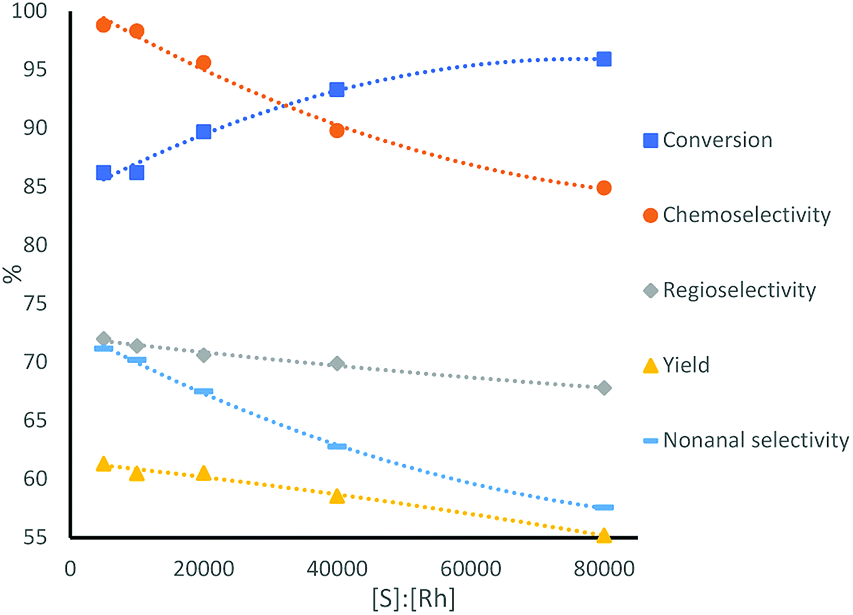 | ||
| Fig. 2 Catalytic performance of the multiphasic hydroformylation of 1-octene using different ratios of substrate to catalyst, conditions see Table 1. | ||
The excellent activity obtained at very high substrate:Rh ratio led us to perform further optimization studies in order to counteract the simultaneous drop in linear selectivity. At the same time, we monitored and strove for efficient and fast phase separation, in order to achieve effective catalyst separation and reuse. The reaction optimization was carried out at a substrate to catalyst ratio of 40000, since the selectivity of the reaction significantly dropped at a ratio of 80000. For the optimization, the amount of ligand and phase transfer agent, as well as the stirring rates were varied systematically (Table 2 and Fig. 2).
| Entry | [L]:[C] |
[PTA] (mg ml−1) | Stirring (rpm) | Conv. (%) | Chemo-sel. (%) | l:b |
TOF at 10% conv. (h−1) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: [Rh(acac)(CO)2], TPPTS, 150 mmol 1-octene, 15 mmol n-dodecane as internal standard, [S]:[Rh] = 40000, 20 ml H2O. 80 °C, 100 bar CO:H2 (1:1), 22 h. |
|||||||
| 1 | 6 | 25 | 700 | 93 | 90 | 2.3 | 2024 |
| 2 | 20 | 25 | 700 | 59 | 98 | 2.5 | 1240 |
| 3 | 20 | 25 | 350 | 50 | 98 | 2.5 | 1076 |
| 4 | 20 | 35 | 350 | 73 | 97 | 2.5 | 1600 |
An increase of the L:Rh ratio from 6 to 20 gave the expected increase of chemo- and regioselectivity towards the linear aldehyde, although the conversion and rate of the reaction dropped significantly (entries 1 and 2, Table 2). Moreover, the formation of a stable Pickering emulsion was observed under these conditions, hampering the phase separation. In order to minimize the formation of stable emulsions, the stirring rate was decreased to 350 rpm. The phase separation improved, although both the conversion and the rate of the reaction further dropped (entry 3, Table 2). To increase the performance of the catalytic reaction while keeping a good phase separation, the amount of particles was increased from 25 mg ml−1 to 35 mg ml−1. Under these conditions, a satisfying combination of phase separation, rate, selectivity and conversion was obtained (entry 4, Table 2). Under these conditions simple decantation of the product phase enabled catalyst recycling without the need for an extraction with a cosolvent (Fig. 3).
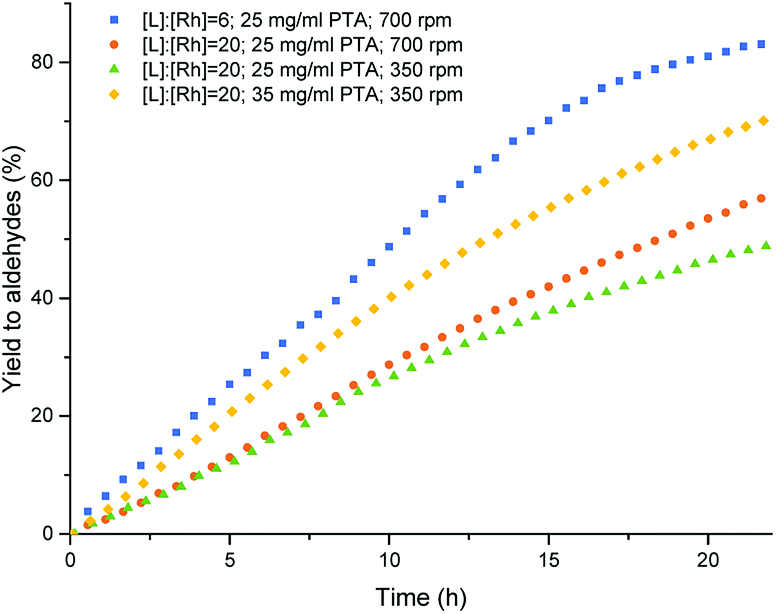 | ||
| Fig. 3 Aldehyde yield vs. time in the multiphasic hydroformylation of 1-octene, end point from GC-results and the graph by gas uptake conditions see Table 2. | ||
An increase of stirring rate or particle concentration leads to faster conversion, as mass transfer is improved. The formation of these Pickering emulsions could suggest that an important feature of the particles is the ability to lead to a locally high catalyst concentration in the interphase between the aqueous phase and the substrate droplets. Therefore, mass transfer limitations are reduced. Under catalytic conditions, both, carrying the catalyst to the interphase and acting as a microreactor might be relevant (Fig. 4).
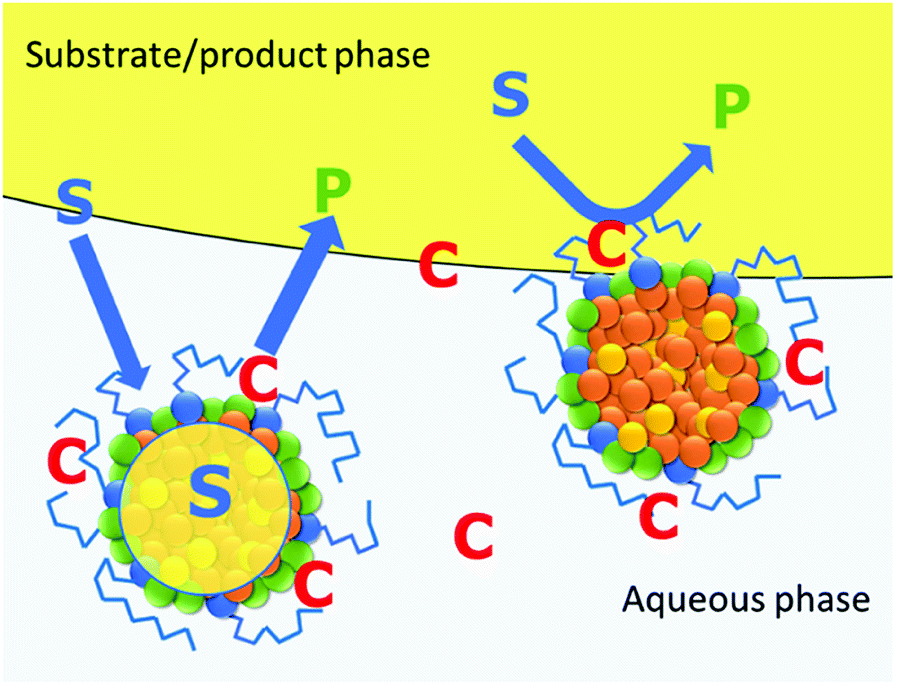 | ||
| Fig. 4 Schematic representation of the role of the polymer particles under catalytic conditions. Left as microreactor and right as catalyst carriers to the interphase (S: substrate, P: product, C: catalyst). | ||
Ligand effect
In order to further improve the catalyst performance, different sulfonated ligands were tested using the optimized conditions previously described: [Rh]:[L]:[S] = 1:20:40000, using 35 mg ml−1 of polymer particles as phase transfer agents and a stirring rate of 350 rpm, to minimize the formation of stable Pickering emulsions.
Triphenylphosphines with fewer sulfonate groups than TPPTS (TPPDS and TPPMS) were tested. Moreover, two different sulfonated phosphines bearing electron withdrawing trifluoromethyl groups were used (Danphos and Dan2phos, Fig. 5). The incorporation of electron withdrawing groups in the phosphine structure both decreases the σ-donating and the π-accepting ability of the ligand, changing the electronic properties of the rhodium catalyst. It has previously been reported that the ligands of the Danphos-family improved the catalyst activity in the biphasic hydroformylation of vinyl acetate and allyl cyanide.25
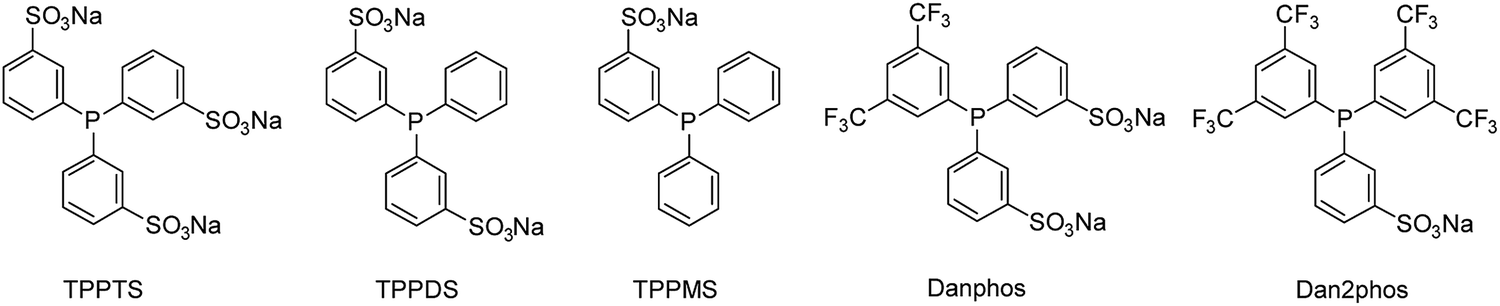 | ||
| Fig. 5 Sulfonated ligands employed in the multiphasic hydroformylation of 1-octene. | ||
The catalyst performance using different sulfonated ligands decreased when decreasing the number of sulfonate groups; TPPTS > TPPDS > TPPMS (entries 1–3, Table 3 and Fig. 6). As mentioned above, the trimethylammonium salt in the shell of the polymer particles interacts with the sulfonated ligands, rendering the particles catalyst carriers. A reasonable explanation for the inferior performance of the lower sulfonated ligands TPPDS and TPPMS is the decreasing number of binding sites of the catalyst at the particles.
| Entry | Ligand | Conversion (%) | l:b |
TOF at 10% conv. (h−1) |
|---|---|---|---|---|
| a [Rh(acac)(CO)2], 150 mmol 1-octene, 15 mmol n-dodecane as internal standard, 700 mg of polymer [S]:[L]:[Rh] = 40000:20:1, 20 ml H2O. 80 °C, 350 rpm, 100 bar CO:H2 (1:1), 22 h. Complete chemoselectivity towards aldehydes was observed for all reactions. |
||||
| 1 | TPPTS | 73 | 2.5 | 1600 |
| 2 | TPPDS | 37 | 1.9 | 732 |
| 3 | TPPMS | 33 | 2.8 | 652 |
| 4 | Danphos | 56 | 2.9 | 1640 |
| 5 | Dan2phos | 46 | 4.6 | 1050 |
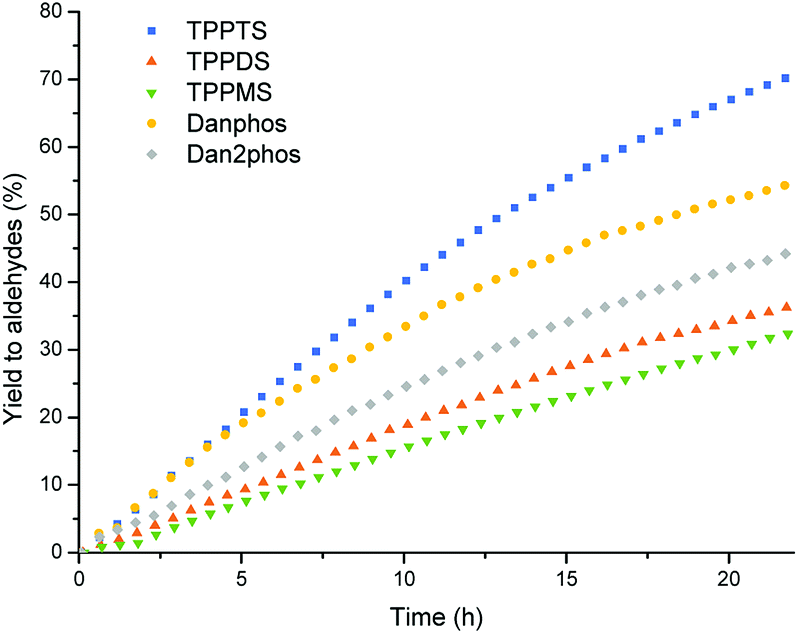 | ||
| Fig. 6 Aldehyde yield vs. time in the multiphasic hydroformylation of 1-octene with different sulfonated ligands, conditions see Table 3. | ||
The trifluoromethylated and sulfonated phosphines, Danphos and Dan2phos increased the activity of the system, compared to TPPDS and TPPMS, respectively (entries 4 and 5, Table 3). It is well-known that trifluoromethyl groups have a very strong electron withdrawing effect on phosphines, leading to a significant decrease of the electron density at the metal center.26,27 This can change the rate determining step of the reaction in certain cases, improving the catalytic activity. It has been previously reported that for the unmodified rhodium carbonyl catalyst and for catalysts modified with electron-poor ligands, the rate determining step of the reaction is the hydrogenolysis of the acyl species,28 in contrast to electron-rich phosphines, where the reaction rate is controlled by the first step of the catalytic cycle, CO dissociation.29
Moreover, the trifluoromethylated phosphines improved the regioselectivity towards the linear aldehyde. Especially with the Dan2phos ligand, which contains two bis(3,5-trifluoromethyl)phenyl rings in its structure, linear to branched ratios of 4.6 could be obtained (Table 3, entry 5). The improvement of regioselectivity by π-accepting ligands like fluorinated phosphines has been reported previously for the hydroformylation of 1-alkenes.27,30
Recycling experiments
The aqueous catalyst phase obtained after the hydroformylation of 1-octene with the ligands TPPTS and Danphos (entries 1, 2, Table 4) were recycled three times for consecutive reactions. Although under those conditions the phase separation after the reaction was satisfactory, the reaction mixtures were centrifuged for 15 min at 3000 rpm in order to quantitatively recover the catalyst together with the polymer particles.| Run | Ligand | Conversion (%) | Chemosel. (%) | l:b |
TOF at 10% conv. (h−1) |
|---|---|---|---|---|---|
| a Reaction conditions: 0.00375 mmol [Rh(acac)(CO)2], 0.075 mmol ligand, 150 mmol 1-octene, [Rh]:[L]:[S] = 1:20:40000, 15 mmol n-dodecane as standard, 700 mg of PTA, 20 ml H2O, 80 °C, 350 rpm, 100 bar CO:H2 (1:1), 22 h. |
|||||
| 1 | TPPTS | 73 | 97 | 2.5 | 1600 |
| 2 | 74 | 95 | 2.4 | 1700 | |
| 3 | 67 | 93 | 2.4 | 1384 | |
| 4 | 74 | 88 | 2.3 | 1400 | |
| 1 | Danphos | 56 | 98 | 2.9 | 1640 |
| 2 | 50 | 97 | 2.8 | 1532 | |
| 3 | 44 | 96 | 2.7 | 1104 | |
| 4 | 44 | 91 | 2.4 | 988 | |
The recycling experiments with TPPTS were successful, and the yield dropped by only 5% from the first to the forth run. The regioselectivity did drop towards the last runs, but this can easily be explained by partial oxidation of the phosphine ligands, since phase separation and centrifugation were carried out under non inert conditions. ICP-MS measurements of the organic phase after the first and the fourth reaction with TPPTS indicated that the loss of rhodium in the oil phase was 1.6 and 1.8% of the initial amount of rhodium, respectively. However, since the substrate to catalyst ratio is 40000, the product phase contains less than 0.3 ppm of rhodium.
Similar results were obtained for the recycling of the Danphos system, but apart from the drop in regioselectivity, the rate dropped more significantly. With the Danphos ligand, the amount of rhodium in the oil phase after the first reaction determined by ICP-MS was 0.2 ppm, which represents 1.4% of the initial rhodium amount.
In our previous work the separation of the organic product phase and the catalyst containing aqueous phase was supported by the use of diethylether for extraction.22 The particles showing improved phase separation in this work, allowed to avoid additional organic solvents. Further improvement of the phase separation kinetics would make centrifugation obsolete and only require simple decantation. Additionally, ultrafiltration can be examined as a means to retain the catalyst-carrying particles in future work.
After the recycling experiments, it was possible to obtain accumulated TONs of 115000 with the TPPTS ligand and 77000 for Danphos, indicating a high catalytic activity and good recyclability.
Different substrates
1-Alkenes of different chain lengths were converted by multiphasic hydroformylation using the polymer particles as phase transfer agents in order to prove the versatility of the approach. The results applying the optimized conditions for 1-octene are summarized in Table 5.| Entry | Substrate | Conversion (%) | Chemosel. (%) | l:b |
TOF at 10% conv. (h−1) |
|---|---|---|---|---|---|
| a Reaction conditions: 0.00375 mmol [Rh(acac)(CO)2], 0.075 mmol TPPTS, 150 mmol of substrate, [Rh]:[L]:[S] = 1:20:40000, 15 mmol of internal standard, 700 mg of PTA, 20 ml H2O, 80 °C, 350 rpm, 100 bar CO:H2 (1:1), 22 h. |
|||||
| 1 | 1-Hexene | 83 | 99 | 2.6 | 1873 |
| 2 | 1-Octene | 73 | 97 | 2.5 | 1600 |
| 3 | 1-Decene | 55 | 91 | 2.3 | 1024 |
| 4 | 1-Dodecene | 36 | 92 | 2.3 | 744 |
With all the substrates tested, conversion towards aldehydes were observed using the optimized conditions found for 1-octene (Table 5, entry 2), showing the versatility of the polymer particles as phase transfer agents and catalyst carriers. However, reaction rates decreased with increasing chain length of the substrate. This is known in literature31 and is caused by the lower concentration of double bonds in the bigger volume of the long chain alkenes. In this case, the lower solubility of the longer chain alkenes in the catalyst phase might also attribute to the difference in rate. The regioselectivity for the different substrates is similar in all cases, but slightly decreases for the longer chains. This might be caused by the increased number of double bond isomers formed in competing isomerization reactions for the longer chain alkenes, which can then lead to more branched aldehydes (Fig. 7).
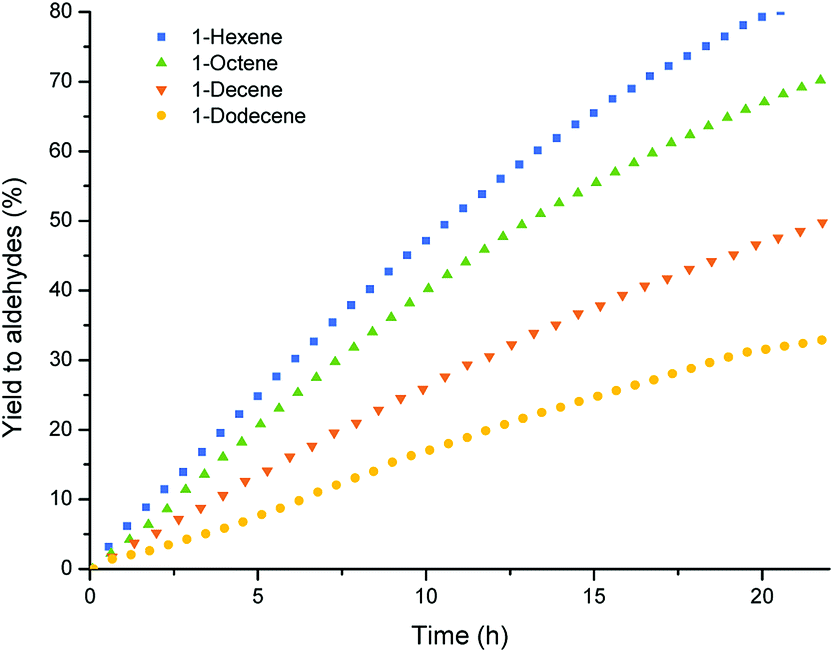 | ||
| Fig. 7 Aldehyde yield vs. time in the multiphasic hydroformylation of different 1-alkenes, conditions see Table 5. | ||
Conclusions
Stable polymer particles suspended in water were successfully applied as phase transfer agents in the multiphasic hydroformylation of long chain alkenes, catalyzed by water soluble rhodium catalysts. For 1-octene, TOFs of 3000 h−1 were achieved using a substrate to catalyst ratio of 80000. However, the chemoselectivity of the reaction towards aldehydes, and particularly the regioselectivity towards the desired linear aldehyde, were quite low. Therefor the [S]:[C] ratio of 40000 was chosen as it yielded a good TOF of 2000 h−1 as well as a standard selectivity for monodentate phosphine ligand.
The optimization of the multiphasic hydroformylation of 1-octene was carried out, focusing on the catalyst performance and the phase separation afterwards. With the resulting system recycling experiments of the aqueous catalyst phase were carried out in four consecutive runs without significant loss of activity and achieving accumulated TONs of 115000. Moreover, ICP-MS measurements of the product phase indicated that the organic phase contained less than 0.3 ppm of rhodium.
The effect of the sulfonated ligand on the catalyst performance was also studied. While TPPTS gives the highest activity, trifluoromethylated phosphine sulfonates improved the regioselectivity of the system towards the desired linear aldehyde.
The polymer particles were also successfully applied as phase transfer agents in the hydroformylation of other long chain 1-alkenes. The rate and regioselectivity of the process decreased with increasing chain length. However, these results prove the versatility of the particles for other multiphasic catalytic reactions.
Experimental section
The commercial substrates used for the hydroformylation experiments, 1-octene (Sigma Aldrich, 98%) 1-hexene (Alfa Aesar, 98%), 1-decene (Alfa Aesar, 96%) and 1-dodecene (Sigma Aldrich, 95%) were filtered through alumina and purged with Ar before use. [Rh(acac)(CO)2] (abcr, 99%), TPPTS (Sigma-Aldrich, ≥95), TPPMS (abcr, 95%), n-dodecane (Sigma-Aldrich, ≥99%), water (VWR, HPLC-grade) and n-decane (Sigma-Aldrich, >95%) were used as received from the commercial sources. Danphos,25 Dan2phos32 and TPPDS33 ligands were prepared according to the literature.Preparation of the monomers and the polymer particles
The monomers and the polymer particles used as phase transfer agents and catalyst carriers in multiphasic hydroformylation reactions were synthesized according to the literature.22Multiphasic hydroformylation reactions
:1) and heated to 80 °C at the corresponding stirring rate. After 1 hour of catalyst preformation, the substrate and standard were added to the catalyst aqueous phase and the gas consumption was recorded. After 22 hours, the temperature was lowered to 65 °C and the autoclave was slowly depressurized, after which it was cooled to room temperature. A drop of the organic phase was diluted with diethyl ether and analyzed by GC to calculate the conversion and the selectivity of the reaction. The TOF was calculated from the gas uptake.Instruments and characterization methods
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support by the German Science Foundation (DFG) (Project SFB/TR 63: “InPROMPT – Integrated Chemical Processes in Multi-Phase Fluid Systems”, Projects A2 EVF, B6). The authors would like to thank the Einstein Foundation Berlin for the financial support and express our gratitude to Umicore AG & Co. KG for a generous loan of noble metal compounds. We would also like to thank Maxi Spiekermann, Jonas Diekmann who conducted experiments as part of their bachelor and master thesis, respectively, Lorna Eades for the ICP analysis and Thomas Seidensticker for correcting the manuscript.References
- A. Behr and P. Neubert, Applied Homogenous Catalysis, Wiley-VCH, 2012 Search PubMed.
- C. W. Kohlpaintner, R. W. Fischer and B. Cornils, Appl. Catal., A, 2001, 221, 219–225 CrossRef.
- L. Obrecht, P. C. J. Kamer and W. Laan, Catal. Sci. Technol., 2013, 541–551 RSC.
- J. P. Hallett, J. W. Ford, R. S. Jones, P. Pollet, C. A. Thomas, C. L. Liotta and C. A. Eckert, Ind. Eng. Chem. Res., 2008, 47, 2585–2589 CrossRef.
- P. Pollet, R. J. Hart, C. A. Eckert and C. L. Liotta, Acc. Chem. Res., 2010, 43, 1237–1245 CrossRef PubMed.
- S. M. Mercer, T. Robert, D. V. Dixon and P. G. Jessop, Catal. Sci. Technol., 2012, 2, 1315 RSC.
- S. L. Desset and D. J. Cole-Hamilton, Angew. Chem., Int. Ed., 2009, 48, 1472–1474 CrossRef PubMed.
- M. Mokhadinyana, S. L. Desset, D. B. G. Williams and D. J. Cole-Hamilton, Angew. Chem., Int. Ed., 2012, 51, 1648–1652 CrossRef PubMed.
- T. Gaide, J. M. Dreimann, A. Behr and A. J. Vorholt, Angew. Chem., Int. Ed., 2016, 55, 2924–2928 CrossRef PubMed.
- A. Rost, Y. Brunsch, A. Behr and R. Schomäcker, Chem. Eng. Technol., 2014, 37, 1055–1064 CrossRef.
- H. Fu, M. Li, H. Chen and X. Li, J. Mol. Catal. A: Chem., 2006, 259, 156–160 CrossRef.
- S. L. Desset, S. W. Reader and D. J. Cole-Hamilton, Green Chem., 2009, 11, 630 RSC.
- T. Pogrzeba, D. Müller, T. Hamerla, E. Esche, N. Paul, G. Wozny and R. Schomäcker, Ind. Eng. Chem. Res., 2015, 54, 11953–11960 CrossRef.
- M. Illner, D. Müller, E. Esche, T. Pogrzeba, M. Schmidt, R. Schomäcker, G. Wozny and J. U. Repke, Ind. Eng. Chem. Res., 2016, 55, 8616–8626 CrossRef.
- E. Monflier, G. Fremy, Y. Castanet and A. Mortreux, Angew. Chem., Int. Ed. Engl., 1995, 34, 2269–2271 CrossRef.
- L. Leclercq, M. Sauthier, Y. Castanet, A. Mortreux, H. Bricout and E. Monflier, Adv. Synth. Catal., 2005, 347, 55–59 CrossRef.
- M. Elard, J. Denis, M. Ferreira, H. Bricout, D. Landy, S. Tilloy and E. Monflier, Catal. Today, 2015, 247, 47–54 CrossRef.
- E. Lobry, A. F. Cardozo, L. Barthe, J. Blanco, H. Delmas, S. Chen, F. Gayet, X. Zhang, M. Lansalot and F. D. Agosto, et al., J. Catal., 2016, 342, 164–172 CrossRef.
- A. F. Cardozo, C. Julcour, L. Barthe, J. Blanco, S. Chen, F. Gayet, E. Manoury, X. Zhang, M. Lansalot and B. Charleux, et al., J. Catal., 2015, 324, 1–8 CrossRef.
- E. Maria Claesson, N. C. Mehendale, R. J. M. Klein Gebbink, G. van Koten and A. P. Philipse, J. Magn. Magn. Mater., 2007, 311, 41–45 CrossRef.
- B. G. P. van Ravensteijn, D.-J. Schild, W. K. Kegel and R. J. M. Klein Gebbink, ChemCatChem, 2017, 9, 440–450 CrossRef.
- D. Peral, D. Stehl, B. Bibouche, H. Yu, J. Mardoukh, R. Schomäcker, R. Von Klitzing and D. Vogt, J. Colloid Interface Sci., 2018, 513, 638–646 CrossRef PubMed.
- K. Kunna, C. Müller, J. Loos and D. Vogt, Angew. Chem., Int. Ed., 2006, 45, 7289–7292 CrossRef PubMed.
- H. Nowothnick, A. Rost, T. Hamerla, R. Schomäcker, C. Müller and D. Vogt, Catal. Sci. Technol., 2013, 3, 600–605 RSC.
- D. Peral, D. Herrera, J. Real, T. Flor and J. C. Bayón, Catal. Sci. Technol., 2015, 6, 800–808 RSC.
- M. Rahman, H. Liu, K. Eriks, A. Prock and W. P. Giering, Organometallics, 1989, 8, 1–7 CrossRef.
- D. R. Palo and C. Erkey, Organometallics, 2000, 38, 81–86 CrossRef.
- M. Garland and P. Pinot, Organometallics, 1991, 10, 1693–1704 CrossRef.
- E. Zuidema, L. Escorihuela, T. Eichelsheim, J. J. Carbó, C. Bo, P. C. J. Kamer and P. W. N. M. Van Leeuwen, Chem. - Eur. J., 2008, 14, 1843–1853 CrossRef PubMed.
- A. C. J. Koeken, M. C. A. Van Vliet, L. J. P. Van Den Broeke, B. Deelman and J. T. F. Keurentjes, Adv. Synth. Catal., 2008, 350, 179–188 CrossRef.
- C. Disser, C. Muennich and G. Luft, Appl. Catal., A, 2005, 296, 201–208 CrossRef.
- D. Peral Crespo and J. C. Bayón Rueda, Triphenylphosphine Derivatives, WO2011045417 A1, 2011.
- T. Thorpe, S. M. Brown, J. Crosby, S. Fitzjohn, J. P. Muxworthy and J. M. J. Williams, Tetrahedron Lett., 2000, 41, 4503–4505 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04022b |
| This journal is © The Royal Society of Chemistry 2018 |