
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An expeditious and efficient bromomethylation of thiols: enabling bromomethyl sulfides as useful building blocks†
Carolina Silva-Cuevasa,
Ehecatl Paleob,
David F. León-Rayoa and
J. Armando Lujan-Montelongo *a
*a
aDepartamento de Química, Centro de Investigación y de Estudios Avanzados (Cinvestav), Av. Instituto Politécnico Nacional 2508, San Pedro Zacatenco, 07360 Ciudad de México, México. E-mail: jalujanm@cinvestav.mx
bFacultad de Ciencias, Universidad Nacional Autónoma de México, Ciudad Universitaria, 04510, Ciudad de México, México
First published on 10th July 2018
Abstract
A facile and highly efficient method for the bromomethylation of thiols, using paraformaldehyde and HBr/AcOH, has been developed, which advantageously minimizes the generation of highly toxic byproducts. The preparation of 22 structurally diverse α-bromomethyl sulfides illustrates the chemo-tolerant applicability while bromo-lithium exchange and functionalization sequences, free radical reductions, and additions of the title compounds demonstrate their synthetic utility.
Heteroatom halomethylations1 have proven to be extremely useful for the generation of valuable synthetic intermediates.2 Halomethylation of thiols provides synthetically valuable chloromethylated intermediates (chloromethyl sulfides), which are typically prepared by condensation with bromochloromethane in basic media,3 or with HCl and a formaldehyde source (paraformaldehyde, polyoxymethylene, etc.).4 While chloromethyl sulfides have been traditionally used as alkylating reagents, the analogous bromomethyl counterparts offer superior electrophilicity, recognized since the earliest report describing their syntheses using hydrogen bromide and paraformaldehyde,5 yet they are often overlooked in this role. Moreover, the reactivity scope of bromomethyl thiol derivatives remains largely unexplored, despite a potentially broader synthetic range (e.g. for the generation of organometallics by metal–halogen exchange).6
Other methods for the generation of bromomethylated thiol derivatives consist of replacing hydrogen bromide gas with concentrated aqueous hydrobromic acid, along with a formaldehyde source (usually paraformaldehyde),7 or by using dibromomethane8 in basic media.9 Two or three-step procedures consisting of hydroxymethylation followed by substitution have also been developed.10 A desilylative rearrangement of α-TMS sulfides has also been used for the generation of bromomethylsulfides.11
As part of our interest in the preparation and application of structurally diverse sulfur-based building blocks,12 we investigated the preparation of benzyl(bromomethyl)sulfane (2a), previously used as an olefination reagent.13 However, several attempts to prepare 2a through exposure of benzylmercaptan (1a) to paraformaldehyde and hydrobromic acid,7b led to a ca. 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 2a (32%) and bis(benzylthio)methane 3a (21%, Scheme 1a). Iterations of the experiment always delivered important and variable amounts of the dithioacetal by-product 3a. On the other hand, an alternate approach to the bromomethylation of a cyclohexanethiol bromomethyl derivative 2b, using dibromomethane and K2CO3, resulted in trace amounts of the dithioacetal derivative 3b only (Scheme 1b).14
:1 mixture of 2a (32%) and bis(benzylthio)methane 3a (21%, Scheme 1a). Iterations of the experiment always delivered important and variable amounts of the dithioacetal by-product 3a. On the other hand, an alternate approach to the bromomethylation of a cyclohexanethiol bromomethyl derivative 2b, using dibromomethane and K2CO3, resulted in trace amounts of the dithioacetal derivative 3b only (Scheme 1b).14
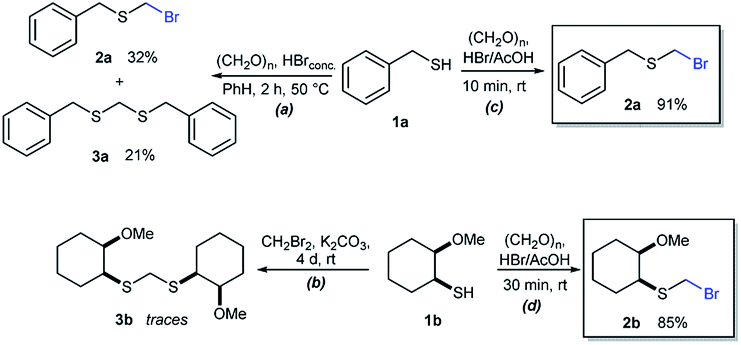 | ||
| Scheme 1 Attempts for the bromomethylation of 1a or 1b under (a) acidic or (b) basic media. (c) and (d) A highly efficient and direct approach for thiol bromomethylation (this work). | ||
HBr/AcOH is a convenient hydrogen bromide source that minimizes exposure to risky set-ups and has been employed as a surrogate to highly corrosive and toxic hydrogen bromide gas in numerous applications.15 Although this reagent has been used previously in the generation of bromomethyl sulfides, installation of the methylene bridge required first a S-pivaloxymethylation of a mercaptan, followed by cleavage by HBr/AcOH.16
Surprisingly, sequential exposure of thiols 1a or 1b to paraformaldehyde and HBr/AcOH,17 rapidly delivered bromomethylated derivatives 2a and 2b with outstanding yields (Scheme 1). The simple experimental setup and straightforward purification procedure offer methodological utility; in most cases extraction with a low-boiling point hydrocarbon such as pentane or hexanes is sufficient to recover the material in high purity (>95%).18 Traces of impurities can be easily discarded through bulb-to-bulb vacuum distillation.
The reaction scope was explored with a series of structurally diverse thiols (Table 1). Aliphatic thiols yielded bromomethyl sulfides in excellent yields, although lower yielding 2g is attributed to its high volatility. t-Butyl bromomethyl sulfide (2i), a sterically challenging and volatile material that has been used as a synthetic equivalent of the methylmercaptan group (–CH2–SH),19 was prepared satisfactorily in 76% yield. Fluorinated bromomethyl 2e has been used for the preparation of fluorinated surfactants,10a,20 which some exhibit antimicrobial activity. As a previous method involves a 2-step sequence involving thiol hydroxymethylation and substitution by PBr3, our method directly delivered 2e in 88% yield.







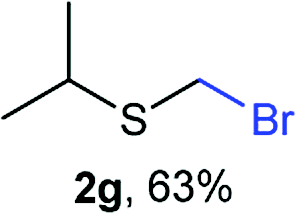



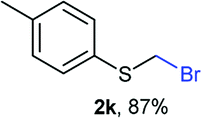
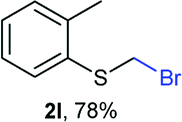



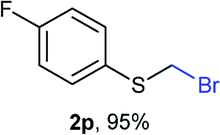


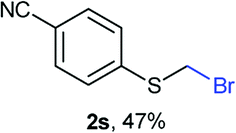

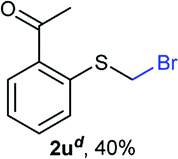




Aromatic substrates (2j–2v) were generally high yielding. For example, (bromomethyl)(phenyl)sulfane (2j), a useful electrophile and precursor to phenylthiomethyl azide21 and diethyl phenylthiomethane phosphonate, an olefination reagent,22 can be prepared in nearly quantitative yield. Aryl derivatives (bromomethyl)(4-methylphenyl)sulfane (2k) and (bromomethyl)(4-chlorophenyl)sulfane (2m), used in the preparation of [(p-phenylphenyl)oxy]methyl (POM) protective group,23 gave 87% and quantitative yields respectively. Comparatively, previously reported methods delivered 2k and 2m in 43% and 75% yield respectively.11 Anisyl thiol 1r was a challenging substrate, as the bromomethylation was highly exothermic and resulted in a near 1:1 mixture of bromomethylsulfide 2r and dithioacetal 3r. The yield of 2r was improved to 4:1 ratio, by cooling the reaction mixture to 0 °C. However, purification of 2r was also problematic as distillation led to partial decomposition. We speculate that integrity of 2r during preparation and purification is influenced by the neighbouring methoxy function. On the other hand, 2s–u modest yields are attributed to a decrease in S-nucleophilicity caused by the EWG groups. Interestingly, although thiol 1v bears an EWG at ortho position, methyl 2-((bromomethyl)thio)benzoate 2v was obtained in excellent yield (85%).
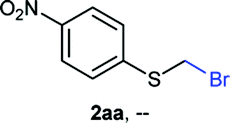
NMR analyses of a fresh mixture of paraformaldehyde and HBr/AcOH24 revealed a mixture consisting mainly of a component with an 1H-NMR 5.8 ppm signal, correlating to a 13C-NMR 68.2 ppm signal (HSQC). This species evolves mainly into two different components: one of them being bis(bromomethyl ether) as determined by a signal at 5.7 ppm (1H-NMR),25 and bis(bromomethoxy) methane (signals at 5.6 ppm and 5.0 ppm).26 The 5.8 ppm signal is presumed to belong to bromomethanol,27 which is consumed promptly by the thiol reagent. This is congruent with our observations, since the best results were obtained when the addition sequence consisted in adding the HBr/AcOH mixture to premixed thiol and paraformaldehyde (Scheme 2). Equimolar ratios of paraformaldehyde are enough for complete transformation, avoiding formation of potentially highly-toxic bis(bromomethyl ether).28 Alkenyl and alkynyl substrates (2w, 2x) were incompatible to this method as the bromomethylation procedure led to complex mixtures. Mercaptans featuring attenuated nucleophilicity such as thioacetic or thiobenzoic acids (2y, 2z), p-nitrothiophenol (2aa), and 2-mercaptopyridine (2bb) were unsuitable for this methodology.
 | ||
| Scheme 2 (A) 1H-NMR spectra of paraformaldehyde + HBr/AcOH (<1 min). (B) 1H-NMR spectra of paraformaldehyde + HBr/AcOH (after 5 min) (left). Conditions: (a) paraformaldehyde addition to 1k, 5 min, then HBr/AcOH addition, 45 min. (b) HBr/AcOH addition to paraformaldehyde, 5 min, then 1k, 45 min (top). Bromomethanol autocondensation decomposition pathway (bottom). | ||
Attempts to diversify the α-alkyl component, found that exclusively highly reactive aldehydes underwent bromoalkylation with thiols (Table 2). Bromoalkylation yields using aldehydes is evidence that reaction efficiency is strongly dependent on the carbonyl reactivity, as bromo(4-nitrobenzylation) or bromoethylation of 4-methylbenzenethiol (1k) using electrophilic 4-nitrobenzaldehyde or acetaldehyde29 respectively, feature fair yields compared to the corresponding bromomethylation using paraformaldehyde (cf. entries 1, 3 and 4). Interestingly, thiol nucleophilicities have a larger impact in thiol bromoalkylations using aldehydes compared to bromomethylations with paraformaldehyde, as illustrated with superior reaction efficiency when benzyl mercaptan 1a was used instead of 1k (cf. entries 6, 7 and 9). Thiol bromoalkylation using ketones had no practical use as dithioketal 3k4 was the only product when acetone was used as the carbonyl component (entries 10 and 11) and acetophenone yielded a complex mixture (entry 12).
|
||||||
|---|---|---|---|---|---|---|
| Entry | T | R1-SH | R2 | R3 | 2 yield (%) | 3 yield (%) |
| a Reaction time 16 h. | ||||||
| 1 | rt | 1k | H | H | 2k (87%) | 3k — |
| 2 | rt | 1a | H | H | 2a (91%) | 3a — |
| 3 | rt | 1k | 4-(NO2)C6H4 | H | 2k1 (60%) | 3k1 (21%) |
| 4a | rt | 1k | 4-(NO2)C6H4 | H | 2k1 (67%) | 3k1 (9%) |
| 5 | rt | 1k | Me | H | 2k2 (56%) | 3k2 (36%) |
| 6 | rt | 1k | Ph | H | 2k3 (0%) | 3k3 (69%) |
| 7 | 30 °C | 1k | Ph | H | 2k3 (26%) | 2k3 (69%) |
| 8a | 30 °C | 1k | Ph | H | 2k3 (46%) | 2k3 (29%) |
| 9 | 30 °C | 1a | Ph | H | 2a3 (61%) | 3a3 (19%) |
| 10 | rt | 1k | Me | Me | — | 3k4 (52%) |
| 11 | 40 °C | 1k | Me | Me | — | 3k4 (29%) |
| 12 | 40 °C | 1k | Ph | Me | Complex mixture | |

To illustrate the versatility of bromomethyl sulfides as building blocks, we first carried out a polarity reversal through a halogen–metal exchange approach, a relatively rare procedure for the generation of α-sulfanylmethyl organometallics.30–32 This approach is underdeveloped, probably because of difficulties in synthesizing bromomethylsulfides.33 Classically, generation of α-sulfanylmethyl organolithiums has been carried out mainly by deprotonation.34 However, the deprotonation approach has important drawbacks, such as a substitution side-process that generate thiolates or regioselectivity issues when dialkyl sulfides are deprotonated.35 Sequentially exposing (bromomethyl)(cyclohexyl)sulfide (2f) or (bromomethyl)(p-tolyl)sulfide (2k) to nBuLi, generated nucleophilic organolithiums 4f and 4k, that were quenched by benzaldehyde thus assembling alkylated derivatives 5f and 5k in good yields (Scheme 3a). Using (+)-neomenthanethiol bromomethyl sulfide derivative (2h) for the bromo-lithium exchange and benzaldehyde in the electrophilic quench, generated β-hydroxysulfide 5h in good yield albeit low diastereoselectivity (ca. 1.4:1). This constitutes a novel approach for the application of sulfenyl methyllithium organometallics for the access of β-hydroxysulfides, valuable intermediates or fragments of natural products and biologically relevant compounds, usually prepared under acidic media or free radical oxidative conditions.36 On the other hand, preparation methods of mixed or unsymmetrical dithioacetals are scarce,37 some of them displaying selectivity limitations.38 Similar bromo-lithium exchange/functionalization procedures were also carried out on probes 2f and 2k using diphenyldisulfide as electrophile,39 delivering mixed thioacetals 6f and 6k respectively also with good yields (Scheme 3b). The exceptional electrophilicity of bromomethyl sulfides 2f and 2k, also enabled the access to mixed thioacetals 6f and 6k by simple exposure to sodium thiophenolate,40,41 thus demonstrating the versatility of bromomethyl sulfides either as electrophiles or nucleophiles after umpolung.
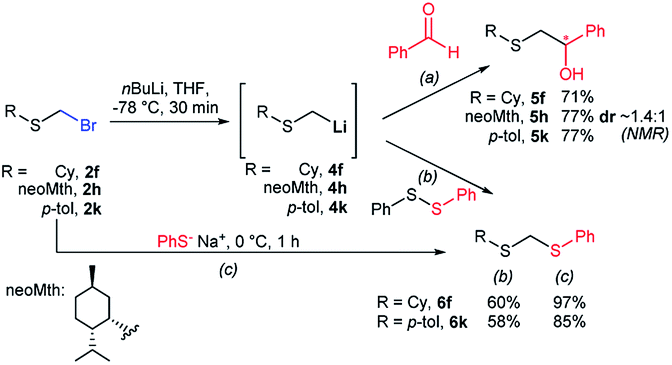 | ||
| Scheme 3 Br–Li exchanges on bromomethyl sulfides for the generation of nucleophilic sulfanylmethyllithiums: (a) β-hydroxysulfide syntheses, (b) unsymmetrical dithioacetal synthesis. (c) Alternate unsymmetrical dithioacetal synthesis by exploiting bromomethyl sulfides (2) electrophilicity. | ||
To our knowledge, bromomethyl sulfides 2 have not been exploited for C–C bond construction through free radical chemistry. As far as we know, there is a single reference to an unrealized effort attempting an intramolecular free radical cyclization of an unavailable alkenyl bromomethylsulfide.14,42 Although our method unfortunately was not compatible with the direct preparation of alkenylsulfide bromomethyl derivatives (see Table 1), we could demonstrate exceptional reactivity of α-thiomethyl radical 7k (generated from 2k), towards (TMS)3SiH reduction,43 thus generating thioether 8k (Scheme 4a). On the other hand, Et3B initiated44 additions of nucleophilic radicals40,45 7f and 7k on radical acceptors acrylonitrile and methyl acrylate led to the generation of γ-functionalized sulfides 9f, 9k, 10f and 10k (Scheme 4b). This constitutes a novel approach for the synthesis of γ-sulfanyl butanenitriles and esters, as an alternative to the thiol-ene reaction approach,46 and establishes bromomethyl sulfides as a new entry on the family of monothiomethyl radical sources.47
 | ||
| Scheme 4 Unprecedented generation of α-thiomethyl free radicals from bromomethyl sulfides and their reduction and addition to acrylonitrile and methyl acrylate. | ||
Conclusions
Development of synthetic methods based on bromomethyl sulfides has been limited mainly by their ambiguous availability, as a consequence of methods lacking selectivity, efficiency, and perilous set-ups. We have developed a simple and high yielding method for thiol bromomethylation, that involves operational simplicity, and minimizes operational risk. The method has broad scope and good functional group tolerance but is unsuitable for low-nucleophilicity mercaptans. The bromomethylating reagent is suspected to be bromomethanol, obtained stoichiometrically and efficiently captured by thiols, thus preventing the formation of undesired toxic species such as bis(bromomethyl)ether. We re-disclosed the applicability of bromomethyl sulfides as precursors of lithiated organometallics and performed unprecedented free radical additions, that support the usefulness of these building blocks. More synthetic applications and derivatizations of bromomethyl sulfides are currently being developed in our laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Conacyt (Mexico) [CB-241455]. C. S.-C. is grateful with Conacyt for a PhD scholarship [340614] and E. P. also thanks Conacyt for a PDF fellowship [217966]. We thank Teresa Cortez Picasso, Victor González Díaz and María Luisa Rodríguez Pérez (Cinvestav), for their assistance with NMR experiments, Géiser Cuellar Rivera (Cinvestav) for his assistance with HRMS measurements, and Ana Lilia Carrasco González (Cinvestav) for her help on performing selected elemental microanalyses. Dr Luis A. Polindara (UNAM) is acknowledged for additional DART-MS and HRMS measurements. We also thank Allen Chao for helpful suggestions on the manuscript and María Martínez-Montoya for assisting C. S. C. on additional experiments.Notes and references
- Selected literature: (a) D. Seyferth, J. K. Heeren, G. Singh, S. O. Grim and W. B. Hughes, J. Organomet. Chem., 1966, 5, 267–274 CrossRef; (b) B. H. Lipshutz and J. J. Pegram, Tetrahedron Lett., 1980, 21, 3343–3346 CrossRef; (c) J. E. Baldwin, R. M. Adlington and J. Robertson, Tetrahedron, 1989, 45, 909–922 CrossRef; (d) R. Hartung, G. Golz, S. Schlaf, G. Silvennoinen, K. Polborn, P. Mayer and H. Pfaendler, Synthesis, 2009, 495–501 Search PubMed.
- Recent literature: (a) L. C. Dias and M. A. B. Ferreira, J. Org. Chem., 2012, 77, 4046–4062 CrossRef PubMed; (b) N. Veerasamy, E. C. Carlson, N. D. Collett, M. Saha and R. G. Carter, J. Org. Chem., 2013, 78, 4779–4800 CrossRef PubMed; (c) C. De Monte, S. Carradori, B. Bizzarri, A. Bolasco, F. Caprara, A. Mollica, D. Rivanera, E. Mari, A. Zicari, A. Akdemir and D. Secci, Eur. J. Med. Chem., 2016, 107, 82–96 CrossRef PubMed; (d) A. P. Green, S. Hardy and E. J. Thomas, Org. Biomol. Chem., 2017, 15, 9475–9496 RSC; (e) R.-Y. Liu, Z.-F. Peng and X.-N. Hou, New J. Chem., 2017, 41, 7626–7633 RSC.
- (a) C. T. Goralski and G. A. Burk, J. Org. Chem., 1977, 42, 3094–3096 CrossRef; (b) N. Ono, H. Miyake, T. Saito and A. Kaji, Synthesis, 1980, 952–953 CrossRef.
- Selected literature: (a) H. Böhme, Ber. Dtsch. Chem. Ges. B, 1936, 69, 1610–1615 CrossRef; (b) T. Miyamoto and I. Yamamoto, Agric. Biol. Chem., 1980, 44, 2581–2586 Search PubMed; (c) D. Enders, S. von Berg, B. Jandeleit, J. Cheng and W. R. Roush, Org. Synth., 2002, 78, 169 CrossRef; (d) K. K. Laali, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, Chichester, UK, 2001, pp. 2–4, and references included within Search PubMed.
- (a) H. Böhme, H. Fischer and R. Frank, Justus Liebigs Ann. Chem., 1949, 563, 54–72 CrossRef . See also:; (b) D. A. Evans, D. J. Mathre and W. L. Scott, J. Org. Chem., 1985, 50, 1830–1835 CrossRef.
- (a) D. Seyferth, Organometallics, 2006, 25, 2–24 CrossRef; (b) P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem., Int. Ed., 2003, 42, 4302–4320 CrossRef PubMed.
- (a) L. W. Fancher, Ger. Pat., 1 112 735, 1961Chem. Abstr., 1962, 56, 11499c; (b) H. J. Reich, C. P. Jasperse and J. M. Renga, J. Org. Chem., 1986, 51, 2981–2988 CrossRef.
- Dibromomethane is an ozone depleting substance which use has been discouraged: Y. Yokouchi, T. Saito and H. Mukai, J. Atmos. Chem., 2017, 1–10 Search PubMed , and references included within.
- (a) X. Zhu, K. Pachamuthu and R. R. Schmidt, Org. Lett., 2004, 6, 1083–1085 CrossRef PubMed; (b) Q. Xia, Y. J. Wen, H. Wang, Y. F. Li and H. H. Xu, J. Agric. Food Chem., 2014, 62, 11037–11046 CrossRef PubMed; (c) M. Esseffar, M. El Messaoudi, R. Jalal, L. R. Domingo and M. J. Aurell, J. Phys. Org. Chem., 2008, 21, 457–463 CrossRef.
- (a) F. Marty, E. Bollens, E. Rouvier and A. Cambon, J. Fluorine Chem., 1990, 48, 239–248 CrossRef; (b) L. Massi, F. Guittard, R. Levy and S. Géribaldi, Eur. J. Med. Chem., 2009, 44, 1615–1622 CrossRef PubMed; (c) G. H. Phillipps, E. J. Bailey, B. M. Bain, R. A. Borella, J. B. Buckton, J. C. Clark, A. E. Doherty, A. F. English and H. Fazakerley, J. Med. Chem., 1994, 37, 3717–3729 CrossRef PubMed; (d) H. Trabelsi, M. A. Jouani and A. Cambon, J. Fluorine Chem., 1996, 79, 27–31 CrossRef; (e) H. Trabelsi, F. Szönyi and S. Geribaldi, J. Fluorine Chem., 2001, 107, 177–181 CrossRef.
- H. Ishibashi, K. Maruyama and K. Minami, J. Chem. Soc., Chem. Commun., 1987, 53, 1443–1445 RSC.
- (a) C. Silva-Cuevas, C. Perez-Arrieta, L. A. Polindara-García and J. A. Lujan-Montelongo, Tetrahedron Lett., 2017, 58, 2244–2247 CrossRef; (b) J. A. Lujan-Montelongo, A. O. Estevez and F. F. Fleming, Eur. J. Org. Chem., 2015, 1602–1605 CrossRef PubMed.
- H. J. Reich and J. M. Renga, J. Chem. Soc., Chem. Commun., 1974, 135–136 RSC.
- Dibromomethane condensation with thiols is often problematic, see: E. W. Della and S. D. Graney, J. Org. Chem., 2004, 69, 3824–3835 CrossRef PubMed.
- Selected literature: (a) D. Ben-Ishai, J. Org. Chem., 1954, 19, 62–66 CrossRef; (b) R. C. Desai, R. P. Farrell, G.-H. Kuo and D. J. Hlasta, Synlett, 1994, 933–934 CrossRef; (c) C. Anderson, S. S. Hadida-Ruah, J. M. C. Golec, B. Zhang, B. J. Littler, A. Keshavarz-Shokri, T. E. Alcacio and D. T. Belmont, US Pat. 0376295 A1, 2016.
- J. J. Court, T. A. Lessen and D. J. Hlasta, Synlett, 1995, 423–424 CrossRef.
- A. van der Made, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, Chichester, UK, 2001, pp. 1–2 Search PubMed.
- We believe that our simple isolation procedure is key for optimal performance, since adopting aqueous workups gave us inconsistent results (variable amounts of dithioacetal products)..
- G. Bashiardes and S. G. Davies, Tetrahedron Lett., 1987, 28, 5563–5564 CrossRef.
- L. Massi, F. Guittard, R. Levy and S. Géribaldi, Eur. J. Med. Chem., 2009, 44, 1615–1622 CrossRef PubMed.
- (a) D. Dou, G. He, Y. Li, Z. Lai, L. Wei, K. R. Alliston, G. H. Lushington, D. M. Eichhorn and W. C. Groutas, Bioorg. Med. Chem., 2010, 18, 1093–1102 CrossRef PubMed; (b) K. C. Tiew, D. Dou, T. Teramoto, H. Lai, K. R. Alliston, G. H. Lushington, R. Padmanabhan and W. C. Groutas, Bioorg. Med. Chem., 2012, 20, 1213–1221 CrossRef PubMed; (c) E. J. Hennessy, M. Cornebise, L. Gingipalli, T. Grebe, S. Hande, V. Hoesch, H. Huynh, S. Throner, J. Varnes and Y. Wu, Tetrahedron Lett., 2017, 58, 1709–1713 CrossRef.
- D. J. Fairfax, PhD thesis, University of London, 1991.
- K. S. Fors, J. R. Gage, R. F. Heier, R. G. Kelly, W. R. Perrault and N. Wicnienski, J. Org. Chem., 1998, 63, 7348–7356 CrossRef PubMed.
- Analysed immediately after mixing paraformaldehyde and HBr/AcOH.
- A 13C-NMR signal at 68.9 was assigned to bis(bromomethyl) ether as judged by a HSQC experiment.
- Integrals of signals at 5.6 and 5.0 ppm are congruent with compound identity (2:1 ratio). 5.6 ppm signal correlates to a 13C signal at 69.0 ppm and signal at 5.0 correlates with a 13C signal at 90.6 ppm.
- As judged by its chemical shift compared to the other species (more unshielded protons). No acetyl signals besides AcOH related were found. After extended times 5–24 h in CDCl3, bromomethanol signal in sample decreased substantially, as the others increased.
- Following a strict measurement of the reagents, we couldn't find evidence of formation of bis(bromomethyl)ether by 1H-NMR (see ref. 17). However, extremely caution is mandatory as bis(halomethyl)ethers are highly toxic and carcinogenic: R. T. Drew, S. Laskin, M. Kuschner and N. Nelson, Arch. Environ. Occup. Health, 1975, 30, 61–69 CrossRef.
- Formaldehyde and p-nitrobenzaldehyde are well-known to be superior electrophiles experimentally: (a) M. Regitz and G. Maas in, Diazo Compounds Properties and Synthesis, Academic Press, Inc., Orlando, 1986, ch. 14, p. 472S; and computationally Search PubMed; (b) Private communication with A. U. Orozco-Valencia and A. Vela. See also: A. U. Orozco-Valencia, J. L. Gázquez and A. Vela, J. Phys. Chem. A, 2017, 121, 4019–4029 CrossRef PubMed.
- Selected works by Krief and others cite an unpublished exchange approach for the generation of lithium derivatives from bromomethyl sulfides. However, as far as we know, such lithiation method was never published. See: (a) L. Wartski, M. El Bouz, J. Seyden-Penne, W. Dumont and A. Krief, Tetrahedron Lett., 1979, 20, 1543–1546 CrossRef; (b) J. N. Denis, W. Dumont and A. Krief, Tetrahedron Lett., 1979, 20, 4111–4112 CrossRef; (c) J. J. Zuckerman, in Inorganic Reactions and Methods, ed. J. J. Zuckermam and A. P. Hagen, VCH, Weinheim, 1988, vol. 11, ch. 5.5, pp. 107–129 Search PubMed.
- Formation of thiomethylmanganese organometallics have been reported by a I-Mn exchange protocol: M. Hojo, R. Sakuragi, Y. Murakami, Y. Baba and A. Hosomi, Organometallics, 2000, 19, 4941–4943 CrossRef.
- For alternative methods for the generation of sulfenyl-stabilized carbanions see: C. Ni and J. Hu, in Comprehensive Organic Synthesis II, ed. P. Knochel and G. A. Molander, Elsevier, 2014, vol. 1, pp. 395–423, and references included within Search PubMed.
- A. Krief, Tetrahedron, 1980, 36, 2531–2640 CrossRef.
- Selected literature: (a) H. Gilman and F. J. Webb, J. Am. Chem. Soc., 1940, 62, 987–988 CrossRef; (b) E. J. Corey and D. Seebach, J. Org. Chem., 1966, 31, 4097–4099 CrossRef; (c) E. J. Corey and M. Jautelat, Tetrahedron Lett., 1968, 9, 5787–5788 CrossRef; (d) I. Kuwajima, S. Sato and Y. Kurata, Tetrahedron Lett., 1972, 13, 737–739 CrossRef; (e) A. I. Meyers and M. E. Ford, J. Org. Chem., 1976, 41, 1735–1742 CrossRef; (f) Y. Liu and R. S. Glass, Tetrahedron Lett., 1997, 38, 8615–8618 CrossRef; (g) M. Linnert, C. Bruhn, T. Rüffer, H. Schmidt and D. Steinborn, Organometallics, 2004, 23, 3668–3673 CrossRef.
- D. J. Peterson, J. Org. Chem., 1967, 32, 1717–1720 CrossRef.
- (a) H. Wang, Q. Lu, C. Qian, C. Liu, W. Liu, K. Chen and A. Lei, Angew. Chem., Int. Ed., 2016, 55, 1094–1097 CrossRef PubMed; (b) A. K. Singh, R. Chawla, T. Keshari, V. K. Yadav and L. D. S. Yadav, Org. Biomol. Chem., 2014, 12, 8550–8554 RSC; (c) C. Huo, Y. Wang, Y. Yuan, F. Chen and J. Tang, Chem. Commun., 2016, 52, 7233–7236 RSC; (d) N. Azizi and M. Edrisi, Tetrahedron Lett., 2016, 57, 525–528 CrossRef; (e) S. R. Lanke and B. M. Bhanage, Catal. Commun., 2013, 41, 29–33 CrossRef.
- (a) J. Y. Gauthier, T. Henien, L. Lo, M. Thérien and R. N. Young, Tetrahedron Lett., 1988, 29, 6729–6732 CrossRef; (b) M. Thérien, J. Gauthier and R. Young, Tetrahedron Lett., 1988, 29, 6733–6736 CrossRef; (c) H. E. Morton and Y. Guindon, J. Org. Chem., 1985, 50, 5379–5382 CrossRef; (d) J. M. McNamara, J. L. Leazer, M. Bhupathy, J. S. Amato, R. A. Reamer, P. J. Reider and E. J. J. Grabowski, J. Org. Chem., 1989, 54, 3718–3721 CrossRef.
- (a) B. Roy, D. Sengupta and B. Basu, Tetrahedron Lett., 2014, 55, 6596–6600 CrossRef; (b) X. Ling, S. Zhang, P. Shao, P. Wang, X. Ma and M. Bai, Tetrahedron Lett., 2015, 56, 5242–5244 CrossRef PubMed.
- Although sulfenylation by electrophilic quench of anionic methylene groups using disulfides is well stablished, this is the first time that methylsulfide organometallics are used for this purpose. See: (a) M. Mikolajczyk, J. Drabowicz and P. Kielbasinski, Formation of C-S Bonds, in Houben-Weyl Methods of Organic Chemistry. Stereoselective Synthesis, ed. G. Helmchen, R. W. Hoffmann, J. Mulzer and E. Schaumann, Georg Thieme Verlag, 1995, vol. E 21e, pp. 5040–5041 Search PubMed; (b) T. Takeda and A. Tsubouchi, Product Subclass 1: Acyclic S,S-Acetals, in Science of Synthesis, ed. J. Otera, Thieme, 2007, vol. 30, pp. 149–155, and references included within Search PubMed.
- Related nucleophilic displacements for the preparation of symmetric dithioacetals (a) Y. M. Tsai, F. C. Chang, J. Huang and C. L. Shiu, Tetrahedron Lett., 1989, 30, 2121–2124 CrossRef , and unsymmetric dithioacetals; (b) M. B. Green and W. L. Jenkins, J. Sci. Food Agric., 1958, 9, 536–539 CrossRef.
- R. Jorritsma, H. Steinberg and T. J. de Boer, Recl. Trav. Chim. Pays-Bas, 2010, 101, 288–298 CrossRef.
- For related α-sulfenylalkyl radical generation see: Y. M. Tsai, F. C. Chang, J. Huang, C. L. Shiu, C. L. Kao and J. S. Liu, Tetrahedron, 1997, 53, 4291–4308 CrossRef.
- (a) C. Chatgilialoglu, D. Griller and M. Lesage, J. Org. Chem., 1988, 53, 3641–3642 CrossRef; (b) C. Chatgilialoglu and V. I. Timokhin, in Advances in Organometallic Chemistry, 2008, vol. 57, pp. 117–181 Search PubMed; (c) C. Chatgilialoglu and J. Lalevée, Molecules, 2012, 17, 527–555 CrossRef PubMed.
- (a) C. Ollivier and P. Renaud, Chem. Rev., 2001, 101, 3415–3434 CrossRef PubMed; (b) Y. Yamamoto, T. Yoshimitsu, J. L. Wood and L. N. Schacherer, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, Chichester, UK, 2007, vol. 1, pp. 1–6; and references included within Search PubMed.
- F. De Vleeschouwer, V. Van Speybroeck, M. Waroquier, P. Geerlings and F. De Proft, Org. Lett., 2007, 9, 2720–2724 CrossRef PubMed.
- (a) J. W. Lynn, R. L. Roberts and J. R. Kilsheimer, J. Org. Chem., 1961, 26, 4300–4303 CrossRef; (b) P. Caspari, F. A. Nüesch, A. Neels and D. M. Opris, RSC Adv., 2016, 6, 98059–98065 RSC , and references included within..
- (a) E. Hasegawa, M. A. Brumfield, P. S. Mariano and U. C. Yoon, J. Org. Chem., 1988, 53, 5435–5442 CrossRef; (b) U. Chan Yoon, H.-J. Kim and P. S. Mariano, Heterocycles, 1989, 29, 1041 CrossRef; (c) U. C. Yoon, Y. C. Kim, J. J. Choi, D. U. Kim, P. S. Mariano, I. S. Cho and Y. T. Jeon, J. Org. Chem., 1992, 57, 1422–1428 CrossRef; (d) T. Ikeno, M. Harada, N. Arai and K. Narasaka, Chem. Lett., 1997, 26, 169–170 CrossRef; (e) Y. Li, K. Miyazawa, T. Koike and M. Akita, Org. Chem. Front., 2015, 2, 319–323 RSC; (f) N. El Achi, M. Penhoat, Y. Bakkour, C. Rolando and L. Chausset-Boissarie, Eur. J. Org. Chem., 2016, 4284–4288 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures and NMR data. See DOI: 10.1039/c8ra04002h |
| This journal is © The Royal Society of Chemistry 2018 |