
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA model study on the photodecarbonyl reaction of (η5-C5H5)M(CO)2 (M = Co, Rh, Ir)†
Zheng-Feng Zhanga and
Ming-Der Su *ab
*ab
aDepartment of Applied Chemistry, National Chiayi University, Chiayi 60004, Taiwan
bDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung 80708, Taiwan. E-mail: midesu@mail.ncyu.edu.tw
First published on 10th July 2018
Abstract
The group 9 organometallic complexes η5-CpM(CO)2 (M = Co, Rh, and Ir) and Si(CH3)3(H) have been considered as a model system to study their photochemical decarbonyl reactions as well as the Si–H bond activation reactions using the CASSCF and MP2-CAS computational methods. For the cobalt complex, three kinds of reaction pathways, which result in the same oxidative addition product, are investigated. Our theoretical finding demonstrated that after the photoirradiation, η5-CpCo(CO)2 loses one CO ligand without any difficulty to form either the triplet ([η5-CpCo(CO)]3) or singlet ([η5-CpCo(CO)]1) species. The former plays a decisive role in the formation of the final oxidative addition product. On the other hand, the latter plays no role in the production of the final product molecule, but its singlet cobalt center interacts weakly with solvent molecules ((Me3)SiH) to produce an alkyl-solvated organometallic complex, which is experimentally detectable. The present works reveal that both η5-CpRh(CO)2 and η5-CpIr(CO)2 should adopt the conical intersection mechanism after they are irradiated by light. Moreover, our theoretical examinations strongly suggest that for the 16-electron monocarbonyl η5-CpM(CO) (M = Rh and Ir) species, the insertion into a Si–H bond by the Ir system is much more facile and more exothermic than that for the Rh counterpart.
I. Introduction
The photochemical oxidative addition reaction of one E–H (E = H or group 14 elements) bonding to the transition metal complex is of essential interest in different fields of chemical research, such as inorganic chemistry, organometallic chemistry, biochemistry, and most importantly, catalytic experimentation,1–6 ever since the groundbreaking discovery by Schuster-Woldan and Basolo.7 The basic approach is to break a metal–ligand bond upon coordination by using light to enable subsequent oxidative addition reactions. In this process, one particularly exciting reaction is the photochemical activation of an E–H bond by one unsaturated intermediate CpML (Cp = η5-C5H5) with a 16 valence electron count, which is formed by the two-legged piano stool (half-sandwich) complex CpML2 upon light irradiation.8–49 Since the CpML complexes can be easily prepared for experiments,11–14 it is thus not surprising that many experimental and theoretical chemists have investigated either the chemical synthesis or the energy sequences of these intermolecular E–H bond activation reactions.8–49 Nevertheless, most of the E–H bond insertion reactions by CpML2 organometallic compounds are heavily focused on their thermodynamical aspects, with a few discussions of their initially photochemical aspects. Indeed, to our knowledge, there has been little mechanistic study regarding the photolysis of the CpML2 complex,24–36 much less related theoretical research. In addition to the limitations of the spectroscopic instruments, there are many fundamental questions about the photolysis of CpML2 in hydrosilane solution that need to be answered. For instance, how does a photo-excited CpML2 molecule go from the excited state to the ground singlet state? And, how does a spin state of the CpML intermediate play a role in its photochemical oxidative addition reactions?.Theoretical considerations should thus help experimental chemists to gain a better understanding for the mechanisms of the photochemical decarbonylation reactions of the CpML2 complexes. Until now, however, there have been no publications on theoretical interpretations for such photochemical mechanisms.8–49 The lack of theoretical investigations could be because the sophisticated computational methods for such photochemical reactions studies have not been suitable until recently. In particular, the photochemical activated systems involve transition metal atoms (concerning the spin–orbit coupling). The current work thus undertakes an examination of the mechanisms for the photochemical CO-extrusion reaction of the group 9 d8 transition metal complex, η5-CpM(CO)2 (M = Co, Rh, and Ir) as well as its oxidative addition of a Si–H bond in trimethylsilane, eqn (1).
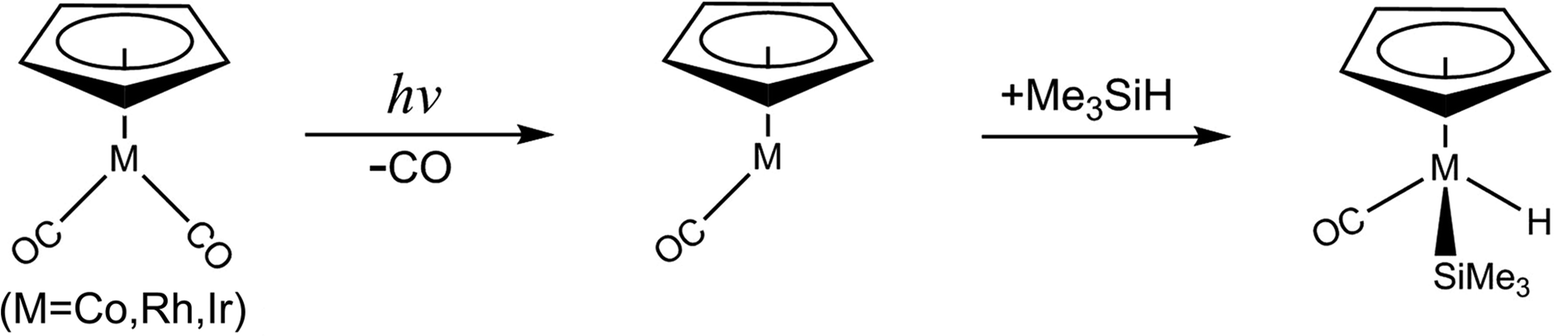 | (1) |
II. Methodology
Since the photochemical reaction paths involve the singlet ground state as well as the exited singlet and triplet states, the ab initio CASSCF (the complete-active-space SCF) program released in GAUSSIAN 0950 is utilized in the presence of this study. That is, the stationary point structures on the S0 and S1 (for Rea-Rh-S0 and Rea-Ir-S0 molecules) and the S0 and T1 (for Rea-Co-S0 molecule) potential energy surfaces are investigated at the CASSCF/Def2-SVPD level of computation.51The electronic structure of an organometallic “half-sandwich” complex of the type CpML2 having only 18 electrons in their valence shell has already been discussed elsewhere,52–54 and is shown in detail in Fig. 1. The active space that needed to properly represent the ground and the lowest excited states of Rea-Co-S0, Rea-Rh-S0, and Rea-Ir-S0 contains twelve electrons in eleven (bonding and antibonding metal d) orbitals. That is to say, according to the valence orbitals given in Fig. 1, the active space for the η5-CpM(CO)2 (M = Co, Rh, and Ir) complex at the singlet ground state can be described as (πCp/dx2−y2)2(πCp/dxz)2(πCp/dyz+py)2(dxy)2(dz2)2(dx2−y2)2 configuration with five low-lying virtual orbitals  As a consequence, the state-averaged CAS(12,11) mode for the η5-CpM(CO)2 molecule is applied in the present study to resolve the stationary points on both ground and excited space. In addition, for the oxidative addition reactions, the active space of trimethylsilane is chosen as two electrons in two orbitals, i.e., one bonding σ(Si–H) orbital and one antibonding σ*(Si–H) orbital. As a consequence, the active space for the whole molecular system (η5-CpM(CO)2 + Si(CH3)3(H)) is referred to as CAS(14,13). For simplicity, in this work, [Si]1 stands for trimethylsilane at the singlet state (see below).
As a consequence, the state-averaged CAS(12,11) mode for the η5-CpM(CO)2 molecule is applied in the present study to resolve the stationary points on both ground and excited space. In addition, for the oxidative addition reactions, the active space of trimethylsilane is chosen as two electrons in two orbitals, i.e., one bonding σ(Si–H) orbital and one antibonding σ*(Si–H) orbital. As a consequence, the active space for the whole molecular system (η5-CpM(CO)2 + Si(CH3)3(H)) is referred to as CAS(14,13). For simplicity, in this work, [Si]1 stands for trimethylsilane at the singlet state (see below).
 | ||
| Fig. 1 Valence molecular orbitals of the group 9 η5-CpM(CO)2 (M = Co, Rh, and Ir) complex. See ref. 45–47. | ||
Frequency computations based on the CASSCF level of theory for their oxidative addition reactions at the singlet ground state are utilized to examine all points calculated being either minima (the number of imaginary frequencies (NIMAG) = 0) or transition states (NIMAG = 1). The standards for the geometrical optimizations are limited to the maximum force and its root mean square, which are less than 0.00065 and 0.00005 hartree/bohr, respectively. Nevertheless, the computed energies rather than zero-point corrected energies and Gibbs free energies are shown in this work.
Considering the dynamic electron correlations, the multireference Møller–Plesset (MP2-CAS) algorithm,55–65 contained in the GAUSSIAN 09 program package,43 is carried out. As a result, the relative energies mentioned in this paper are those calculated at the MP2-CAS/Def2-TZVPD level, using the CAS/Def2-SVPD geometry. The active space for each point used in this study is stated above. In consequence, they are abbreviated as MP2-CAS and CAS, respectively. The Cartesian coordinates (CAS) as well as the CAS and MP2-CAS energetics are given in ESI.†
In order to compare the MP2-CAS MP2-CAS singlet-triplet energy difference for the η5-CpCo(CO)2 complex at different levels of theory, two kinds of density functional theory (DFT) and basis sets are used in this work. That is, B3PW91/Def2-TZVPPD,66,67 and B3LYP/DZP-DKH.68,69 The reason for choosing them is because they have been proved to be reliable for examining the relative energetics.70 Their Cartesian coordinates and energetics are also collected in ESI.†
III. Results and discussion
(1) Mechanism for the photoactivation reaction of η5-CpCo(CO)2 in the triplet state channel
We first study the photodecarbonyl reaction and the Si–H bond activation reaction of the half-sandwich two-legged-piano-stool CpCo(CO)2 complex with trimethylsilane. When the CpCo(CO)2 molecule absorbs light, it may jump directly from the singlet ground state to the excited states, which is called the Frank–Condon (FC) region and still has the geometry of its singlet ground state. The MP2-CAS computations are thus used in this work to compute its vertical excitation energies. As shown in the left-hand side of Fig. 2, the MP2-CAS results display eight electronic states that are achievable around the energies of a 325 nm photon: four triplet states (kcal mol−1); Co-T1-FC (42.8), Co-T2-FC (47.1), Co-T3-FC (69.4), and Co-T4-FC (122.7), and four singlet states (kcal mol−1); Co-S1-FC (74.5), Co-S2-FC (84.4), Co-S3-FC (93.4), and Co-S4-FC (95.5). Since the spin-allowed absorption cross-sections are well accepted to be larger than those for spin-forbidden excitations, photoirradiation thus advances CpCo(CO)2 from the singlet ground state to an excited singlet state (S2). Then, this dicarbonyl complex relaxes to the T1 state (Co-T1-Min). Our calculations estimate the energy of Co-T1-Min is about 2.6 kcal mol−1 above the ground-state minimum (Co-S0-Rea). Also, we have used density functional theory to test the energy difference between the singlet and triplet η5-CpCo(CO)2 species, i.e., B3PW91/Def2-TZVPPD, and B3LYP/DZP-DKH. On the basis of these computations (ESI†), the free energy of the singlet η5-CpCo(CO)2 compound is computed to be lower than that of the triplet η5-CpCo(CO)2 species by about 15 (B3PW91/Def2-TZVPPD)66,67 and 8.6 (B3LYP/DZP-DKH)68,69 kcal mol−1, respectively.70 Again, this reveals that its CO-photoextrusion reaction starts on the triplet state energy surface. Indeed, according to the available experiments,24–28 it was found that excitation of this molecule at 325 nm (= 88.0 kcal mol−1) leads to the generation of a η5-CpCo(CO) intermediate in the triplet electronic state.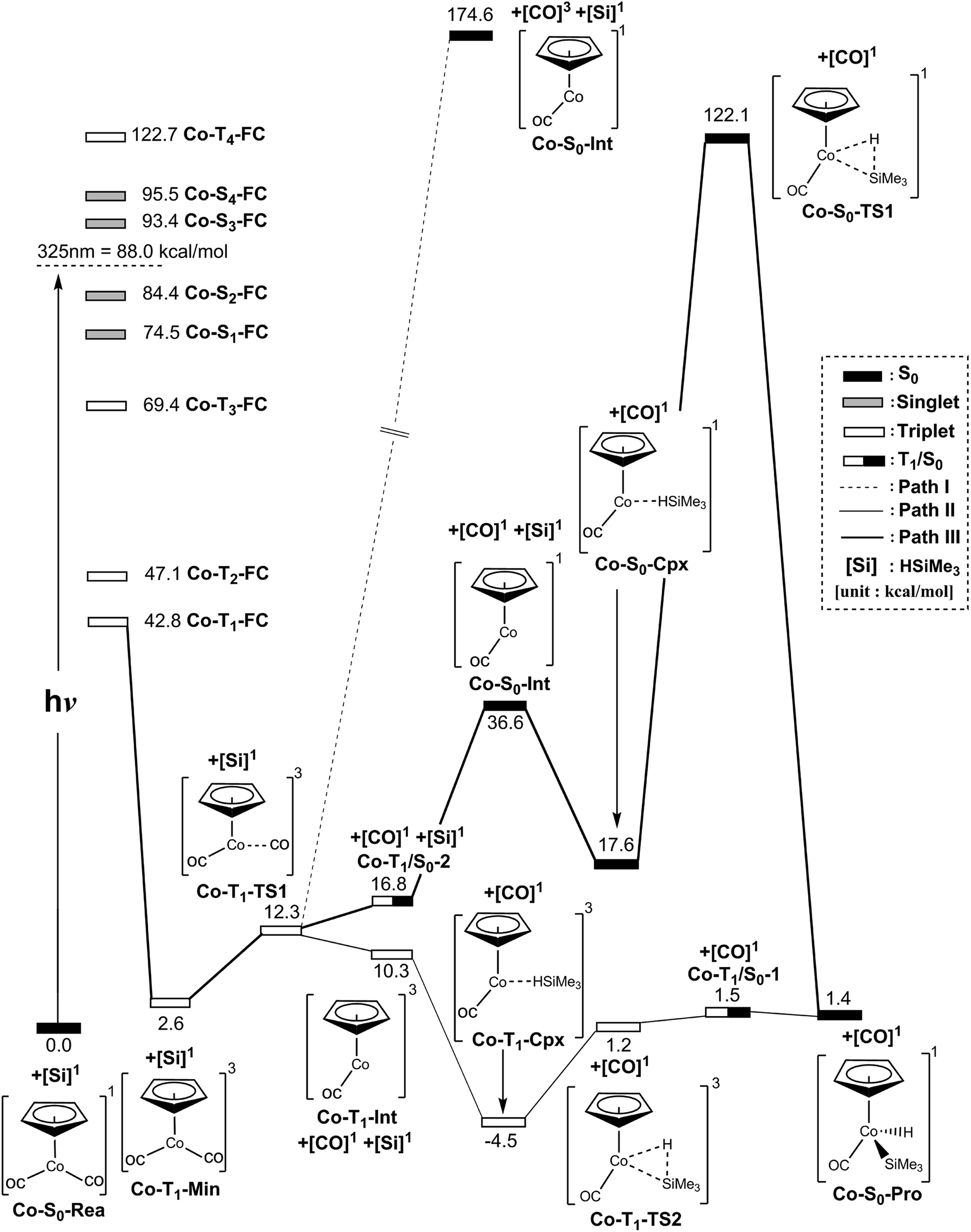 | ||
| Fig. 2 Energy profiles for the photo-oxidative-addition reactions for η5-CpCo(CO)2 (Co-S0-Rea). The computational active spaces used in this work are described in Section II. All energies (in kcal mol−1) are given with respect to the reactant (Co-S0-Rea). For the crucial points of the CASSCF optimized structures, see Fig. 3. For more information, see the text. | ||
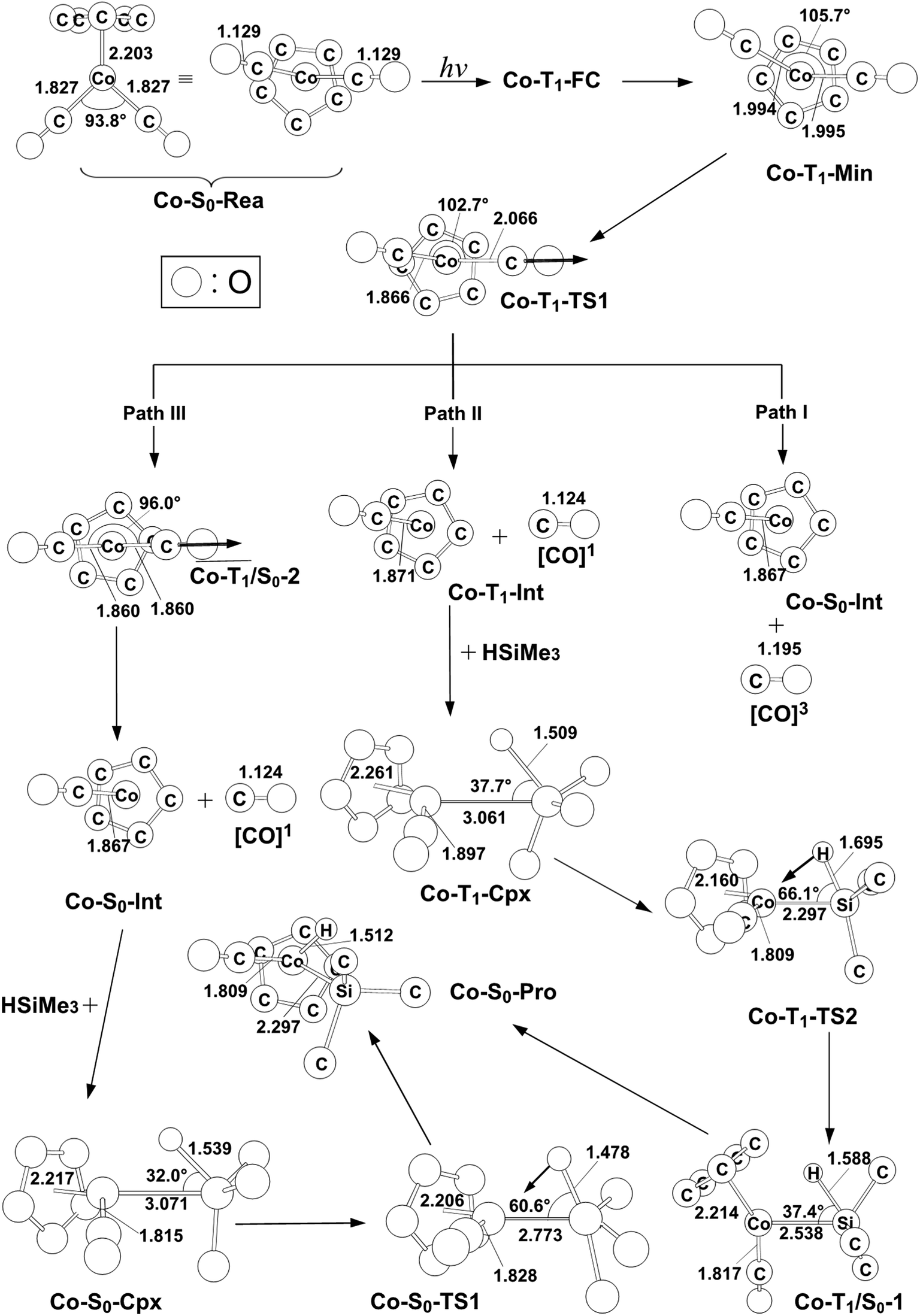 | ||
| Fig. 3 The CASSCF geometries (in Å and deg) for photo-oxidative-addition reactions of η5-CpCo(CO)2 (Co-S0-Rea). The computational active spaces used in this work are described in Section II. Their relative energies are given in Fig. 4. Some hydrogen atoms are omitted for clarity. | ||
As seen in Fig. 2, the Co-T1-Min complex then undergoes a Co–CO bond cleavage (2.066 Å) via a transition state (Co-T1-TS1). From this transition state point, the intermediate complex may go forward three possible reaction pathways (i.e., path I, path II, and path III in Fig. 2) in order to undergo an oxidative addition reaction with trimethylsilane to produce the final insertion product (Co-S0-Pro).
In path I, as seen in Fig. 2, the triplet Co-T1-Min molecule can dissociate one CO ligand to produce two products: one triplet CO molecule and one singlet η5-CpCo(CO) intermediate (Co-S0-Int). The relative energy of the final products ([CO]3 + Co-S0-Int) is, however, estimated to be 87 kcal mol−1 above the irradiated energy (325 nm = 88 kcal mol−1).24–28 As a result, the present computational evidence reveals that the photo-excited energy of Co-S0-Rea at 325 nm is unable to generate one triplet CO and one singlet Co-S0-Int. Therefore, our theoretical finding anticipates that no such photoproducts can be detected in the photochemical activation reactions for η5-CpCo(CO)2, which has been confirmed by the available experimental reports.24–28 For convenience, the mechanism for path I of the Co-S0-Rea complex can be expressed as follows:
| Co system-path I: Co-S0-Rea + [Si]1 + hν → Co-T1-FC + [Si]1 → Co-T1-Min + [Si]1 → Co-T1-TS1 + [Si]1 → Co-S0-Int + [CO]3 + [Si]1 |
In the second pathway, path II, the triplet Co-T1-Min complex can undergo one CO ligand cleavage to produce one singlet CO and one triplet Co-T1-Int intermediate through the transition state (Co-T1-TS1). The MP2-CAS data indicate that the Co-T1-Int + [CO]1 point is computed to be about 10 kcal mol−1 higher than that of Co-S0-Rea. Then, the triplet Co-T1-Int interacts with (CH3)3Si–H to generate the precursor complex (Co-T1-Cpx) in the triplet state. Subsequently, this triplet complex encounters a triplet transition state (Co-T1-TS1) through the Co-T1/S0-1 intersystem crossing to form the final singlet photoproduct (Co-S0-Pro). On the basis of the MP2-CAS computations, path II is anticipated to be energetically accessible. The reason for this is once the initial reactants (Co-S0-Rea + [Si]1) absorb light of 325 nm (= 88 kcal mol−1) wavelength, they can obtain enough energy to overcome the barrier height (5.7 kcal mol−1) from Co-T1-Cpx to Co-T1-TS2 in path II. In consequence, the mechanism for path II for both photochemical decarbonyl reaction and the Si–H bond activation reaction of η5-CpCo(CO)2 is given as follows:
| Co system-path II: Co-S0-Rea + [Si]1 + hν → Co-T1-FC + [Si]1 → Co-T1-Min + [Si]1 → Co-T1-TS1 + [Si]1 → Co-T1-Int + [CO]1 + [Si]1 → Co-T1-Cpx + [CO]1 → Co-T1-TS2 + [CO]1 → Co-T1/S0-1 + [CO]1 → Co-S0-Pro + [CO]1 |
In path III, on the other hand, after the Co–CO bond breaking (Co-T1-TS1), the intersystem crossing from the triplet state to the singlet state comes about in the region of the T1/S0 intersection (Co-T1/S0-2), as depicted in Fig. 2. By way of this crossing point, the triplet η5-CpCo(CO)2 complex separates into one singlet η5-CpCo(CO) (Co-S0-Int) intermediate and one singlet CO molecule. Trimethylsilane is subsequently coordinated to the central cobalt atom of the Co-S0-Int species in the pattern of an η2 to generate a singlet precursor complex (Co-S0-Cpx). As a result, the Si–H bond of (CH3)3Si(H) is broken through a singlet transition state (Co-S0-TS1) to produce the final oxidative addition product (Co-S0-Pro). The MP2-CAS data reveal that the energies of intermediates (Co-S0-Int + CO + (CH3)3Si(H)), the precursor complex (Co-S0-Cpx + CO), the transition state (Co-S0-TS1 + CO) and the final insertion product (Co-S0-Pro + CO) relative to that of the starting materials (Co-S0-Rea + (CH3)3Si(H)) are computed to be 37, 18, 122, and 1.4 kcal mol−1, respectively. That is to say, the theoretical calculations indicate that the energy of Co-S0-Cpx (19 kcal mol−1) is much lower than that of its further activation barrier (105 kcal mol−1) from Co-S0-Cpx to Co-S0-TS1 in path III. Our computational evidence, therefore, predicts that this singlet cobalt precursor complex (Co-S0-Cpx) should be easily located, which has already been verified by the available experimental observations.24–28 Furthermore, the theoretical data given in Fig. 2 show that the activation energy of Co-S0-TS1 is 105 kcal mol−1 much higher than the irradiation energy (325 nm = 88 kcal mol−1) of the corresponding reactants (Co-S0-Rea + (CH3)3Si(H)). That is to say, compared to the excited promotion energy, the barrier height from Co-S0-Cpx to Co-S0-TS1 represented in path III cannot be surmounted. Accordingly, path III is energetically unfavorable for such photochemical oxidative additions of the η5-CpCo(CO)2 compound. Thus, the present theoretical investigations suggest that the mechanism of path III should proceed as follows:
| Co system-path III: Co-S0-Rea + [Si]1 + hν → Co-T1-FC + [Si]1 → Co-T1-Min + [Si]1 → Co-T1-TS1 + [Si]1 → Co-T1/S0-2 + [CO]1 + [Si]1 → Co-S0-Int + [CO]1 + [Si]1 → Co-S0-Cpx + [CO]1 → Co-S0-TS1 + [CO]1 → Co-S0-Pro + [CO]1 |
In brief, the above computations reveal that once the η5-CpCo(CO)2 (Co-S0-Rea) molecule absorbs light, it can easily generate a coordinatively unsaturated 16-electron η5-CpCo(CO) fragment in either the triplet or the singlet excited state, via the loss of a CO molecule upon photolysis. In a similar way, this dynamic half-sandwich intermediate can easily undergo oxidative addition to the Si–H bond of H–Si(Me)3. Three reaction routes (paths I, II, and III) can be used for interpreting the mechanisms of such CO-photoextrusion reaction as well as the Si–H bond activation reaction. The present theoretical evidence demonstrate that only path II, which can form the triplet Co-T1-Int fragment, is the most energetically favorable reaction path for the photochemical reactions. On the other hand, both path I and path III, which can produce the singlet Co-S0-Int species, are energetically unfeasible for the generation of Co-S0-Pro. In other words, our theoretical investigations strongly indicate that it is only the triplet species, [η5-CpCo(CO)]3 (Co-T1-Int), that play a decisive role in the photochemical Si–H bond activation reaction by the organometallic compound, η5-CpCo(CO)2. These theoretical findings are in excellent agreement with the experimental observations reported by the team led by Harris.24–28
(2) Mechanism for the photoactivation reaction of η5-CpRh(CO)2 in the singlet state channel
For comparison with the case of η5-CpCo(CO)2, we next examine the mechanism of the photoactivation reaction of η5-CpRh(CO)2 (Rh-S0-Rea) towards trimethylsilane. The same theoretical method (MP2-CAS) is utilized to explore the potential energy surfaces of Rh-S0-Rea for its Si–H bond activation reactions, which are all shown in Fig. 4. The selected geometrical parameters for the photochemical activation reaction of Rh-S0-Rea with trimethylsilane are given in Fig. 5. According to some availably experimental observations,23,29,30 Harris and co-workers reported that the irradiation of light to Rh-S0-Rea is 295 nm (= 96.9 kcal mol−1).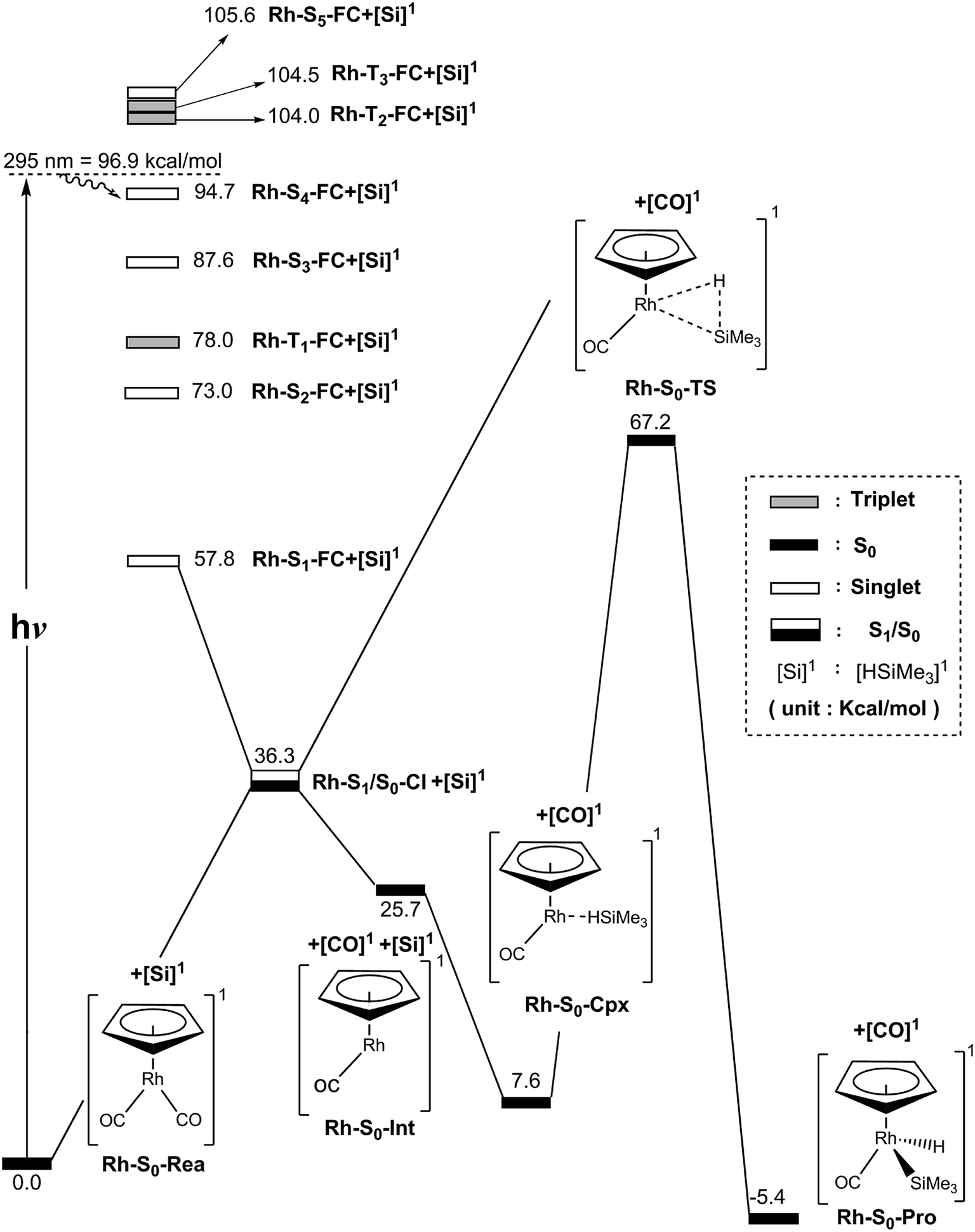 | ||
| Fig. 4 Energy profiles for the photo-oxidative-addition reactions for η5-CpRh(CO)2 (Rh-S0-Rea). The computational active spaces used in this work are described in Section II. All energies (in kcal mol−1) are given with respect to the reactant (Rh-S0-Rea). For the crucial points of the CASSCF optimized structures, see Fig. 5. For more information, see the text. | ||
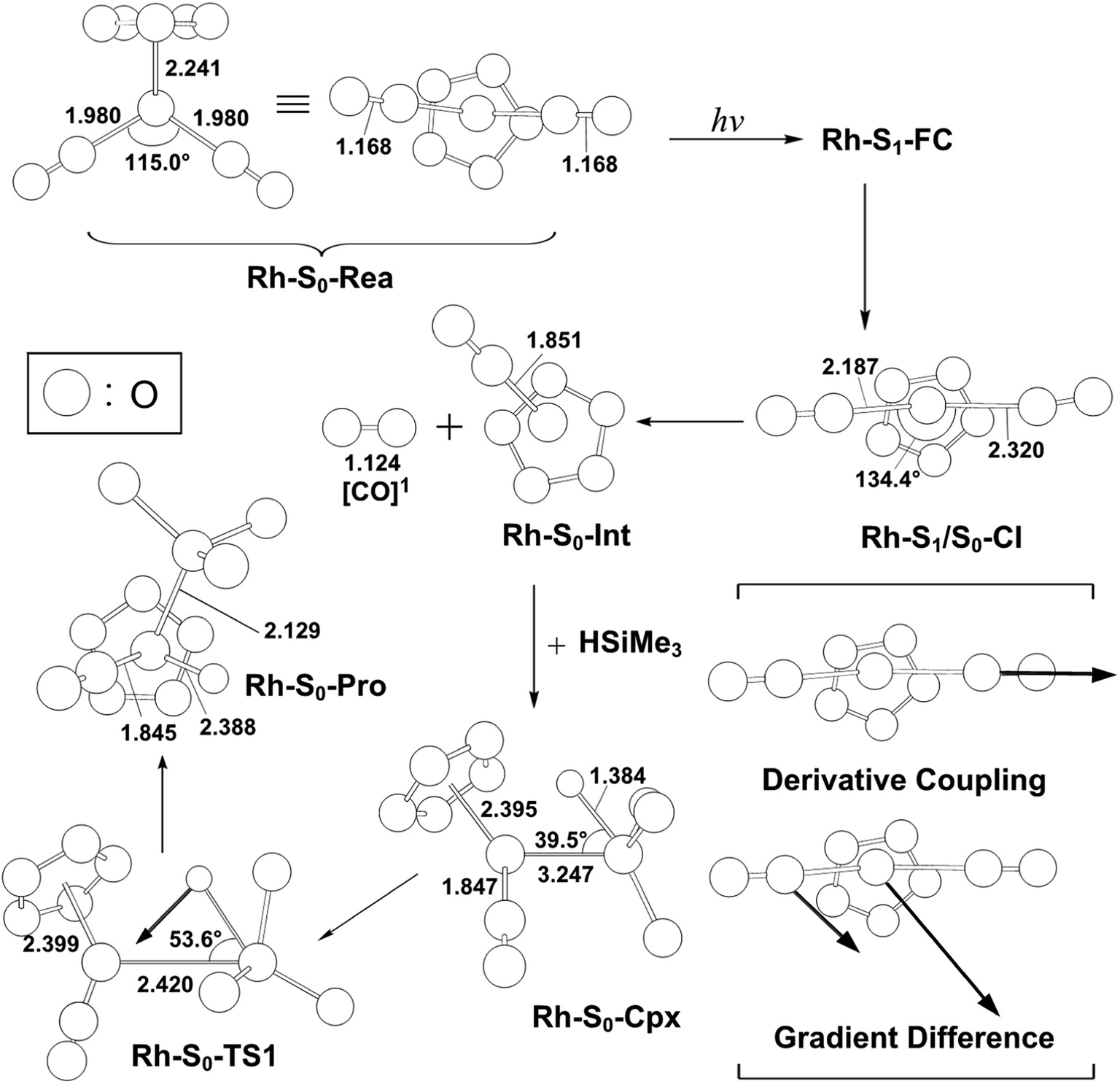 | ||
| Fig. 5 The CASSCF geometries (in Å and deg) for photo-oxidative-addition reactions of η5-CpRh(CO)2 (Rh-S0-Rea). The computational active spaces used in this work are described in Section II. Their relative energies are given in Fig. 4. Some hydrogen atoms are omitted for clarity. | ||
As seen in the left-hand side of Fig. 4, the MP2-CAS computational data indicates that the relative FC energies (kcal mol−1) of the electronic states increase in the order: Rh-S1-FC (57.8) < Rh-S2-FC (73.0) < Rh-T1-FC (78.0) < Rh-S3-FC (87.6) < Rh-S4-FC (94.7) < Rh-T2-FC (104.0) < Rh-T3-FC (104.5) < Rh-S5-FC (105.6). The MP2-CAS results reveal that only the energy of the first excited triplet state (i.e., Rh-T1-FC) is lower than that of its photoirradiated light (295 nm). In addition, we did not calculate the triplet energy surface for the photodecarbonyl reaction of the η5-CpRh(CO)2 (Rh-S0-Rea) complex in the present work. First, on the basis of the available experimental results,23,29,30 the photolysis of η5-CpRh(CO)2 generates only one singlet η5-CpRh(CO) fragment and one singlet CO molecule, i.e., [η5-CpRh(CO)2]1 + hν → [η5-CpRh(CO)]1 + [CO]1. Second, it is well understood that spin-allowed (e.g., singlet ground state → singlet excited state) absorption cross-sections are basically larger than those for spin-forbidden excitations (e.g. singlet ground state → triplet excited state). Due to the above two reasons, photoexcitation can promote the η5-CpRh(CO)2 complex from a singlet ground state a singlet excited state (such as, S4 or S3 in Fig. 4). Then, this excited complex relaxes to its singlet ground state. As a consequence, we focus the on the singlet state surface of the photodecarbonyl reaction of η5-CpRh(CO)2 from now on.
It has to be mentioned here that this work starting on the singlet surface of η5-CpRh(CO)2 does not imply its triplet excited states do not play a role in such a photochemical CO-extrusion reaction. In contrast, we understand the complexity of the photochemical behaviors of the η5-CpRh(CO)2 complex. In order to make the present study simpler and clear, we thus consider the singlet energy surface aspects separately. The photochemical behavior of the triplet excited state is considered later as an effect, which may or may not affect the photochemistry of the η5-CpRh(CO)2 complex.
On the basis of the above information, our computations therefore suggest that once the Rh-S0-Rea complex has absorbed the light, it will jump from the singlet ground state to the more highly excited singlet state (Rh-S4-FC) and then relaxes to the lowest excited singlet state (Rh-S1-FC). Accordingly, in this work we use the conical intersection (CI) concept,57–67 which considers the crossing between states of the same multiplicity (mostly commonly singlet–singlet), to explore the mechanism of the photochemical activation reactions of the Rh-S0-Rea system.
As shown in Fig. 4, after the vertical excitation procedure, the Rh-S0-Rea complex finally decays to the excited singlet Rh-S1-FC point but still maintains the ground-state geometry. Subsequently, this species relaxes to arrive at a Rh-S1/S0-CI point, from which the photoexcited rhodium complex decays nonradiatively to the singlet ground state.57–67 Our MP2-CAS findings suggest that Rh-S1/S0-CI is about 22 kcal mol−1 lower in energy than Rh-S1-FC. In Fig. 4, the directions of the derivative coupling and gradient difference vectors for the Rh-S1/S0-CI point are given. That is to say, funneling through Rh-S1/S0-CI via either the gradient difference vector or the derivative coupling vector can result in two different reaction routes on the singlet ground state surface. As shown in Fig. 4, the major contribution of the derivative coupling vector concerns the Rh–CO bond breaking, whereas the gradient difference vector involves vibrationally hot motion at the ground state configuration. As a result, the former allows the formation of the coordinatively unsaturated 16-electron η5-CpRh(CO) (Rh-S0-Int) intermediate and one CO molecule, both of which are in the singlet ground states. Indeed, this finding has been confirmed by the experimental observations, in which the photodecarbonyl reaction of Rh-S0-Rea finally occurs a singlet 16-electron organometallic η5-CpRh(CO) species.23,29,30
Subsequently, Rh-S0-Int interacts with Si(H)Me3 to form a singlet precursor complex (Rh-S0-Cpx), whose energy is predicted to be 7.6 kcal mol−1 above the energy of the corresponding reactants (Rh-S0-Rea + (CH3)3Si(H)). Then, Rh-S0-Cpx undergoes the oxidative addition into the Si–H bond of trimethylsilane via the transition state, Rh-S0-TS. Our computational findings suggest that the relative energies (kcal mol−1) of Rh-S0-Cpx, Rh-S0-TS and Rh-S0-Pro with respect to the corresponding reactants are 7.6, 67.2, and −5.4, respectively. As a result, from the reaction profiles given in Fig. 4, it is clear that the photoexcitation energy at 295 nm of the η5-CpRh(CO)2 reactant has sufficient internal energy to overcome the energy barrier from Rh-S0-Cpx to Rh-S0-TS. In brief, the theoretical observations demonstrate that the conical intersection mechanism plays a decisive role in the mechanism of the Si–H bond-activation reaction of photogenerated singlet η5-CpRh(CO) with trimethylsilane. Therefore, the mechanism of singlet photochemical oxidative addition reaction of η5-CpRh(CO)2 can be represented as follows:
| Rh system: Rh-S0-Rea + [Si]1 + hν → Rh-S1-FC + [Si]1 → Rh-S1/S0-CI + [Si]1 → Rh-S0-Int + [CO]1 + [Si]1 → Rh-S0-Cpx + [CO]1 → Rh-S0-TS + [CO]1 → Rh-S0-Pro + [CO]1 |
3(3) Mechanism for the photoactivation reaction of η5-CpIr(CO)2 in the singlet state channel
In spite of the fact that the photochemical behavior of η5-CpIr(CO)2 (Ir-S0-Rea) has been experimentally reported elsewhere, unlike its cobalt and rhodium counterparts,24–29 the photodecarbonyl mechanism of Ir-S0-Rea has not been investigated neither experimentally nor theoretically.30–34In this work, the same computational method (MP2-CAS) is used to theoretically investigate the potential energy surfaces of η5-CpIr(CO)2 for its photochemical oxidative reaction. The computational reaction routes and the related geometrical parameters for the photoactivation reaction of Ir-S0-Rea with trimethylsilane are schematically represented in Fig. 6 and 7, respectively. To enable comparisons with the previous η5-CpCo(CO)2 (Co-S0-Rea) and η5-CpRh(CO)2 (Rh-S0-Rea) model systems, the irradiation of light to Ir-S0-Rea is assumed to be 295 nm (= 96.9 kcal mol−1), as shown on the left-hand side of Fig. 6.
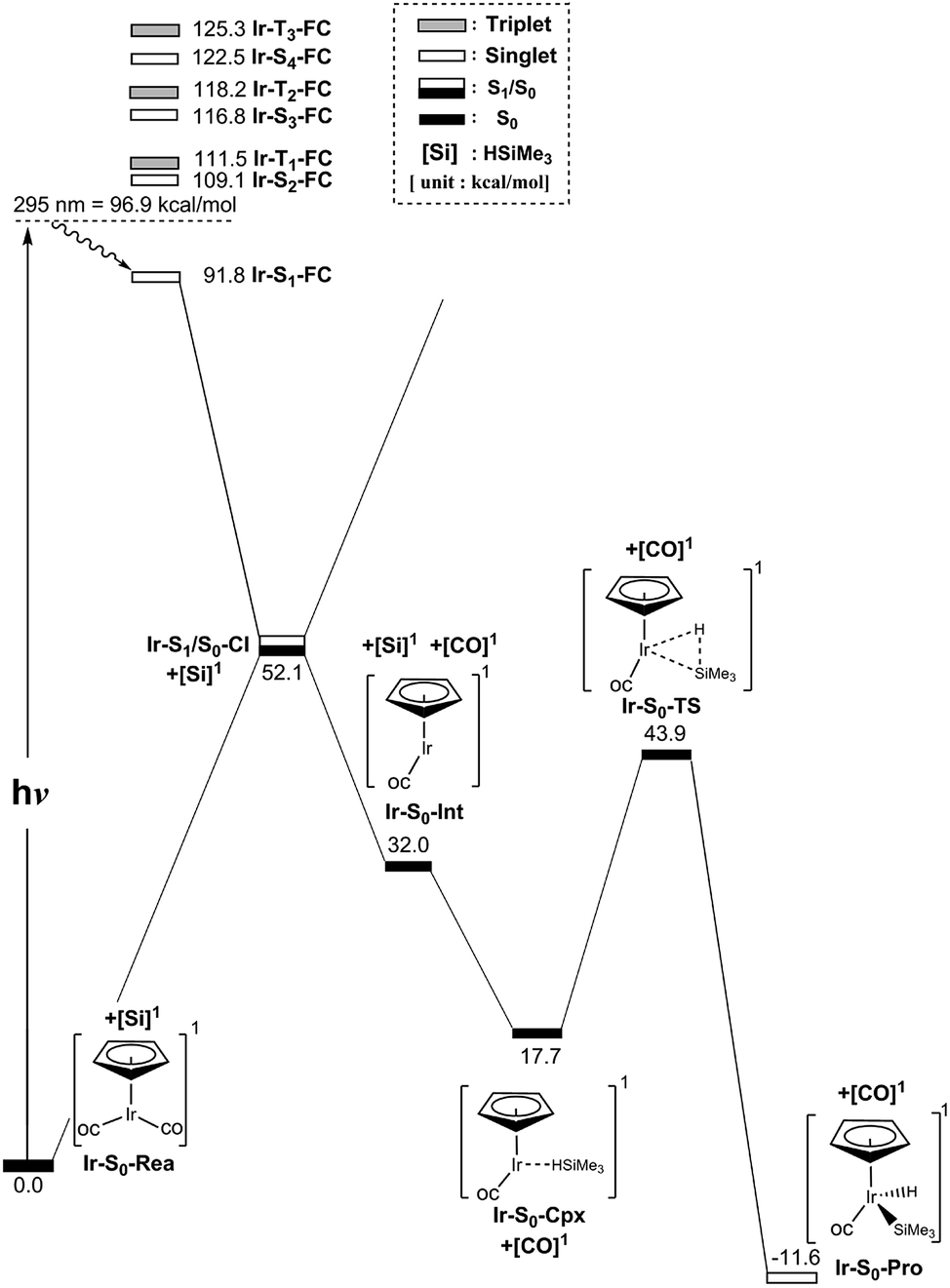 | ||
| Fig. 6 Energy profiles for the photo-oxidative-addition reactions for η5-CpIr(CO)2 (Ir-S0-Rea). The computational active spaces used in this work are described in Section II. All energies (in kcal mol−1) are given with respect to the reactant (Ir-S0-Rea). For the crucial points of the CASSCF optimized structures, see Fig. 7. For more information, see the text. | ||
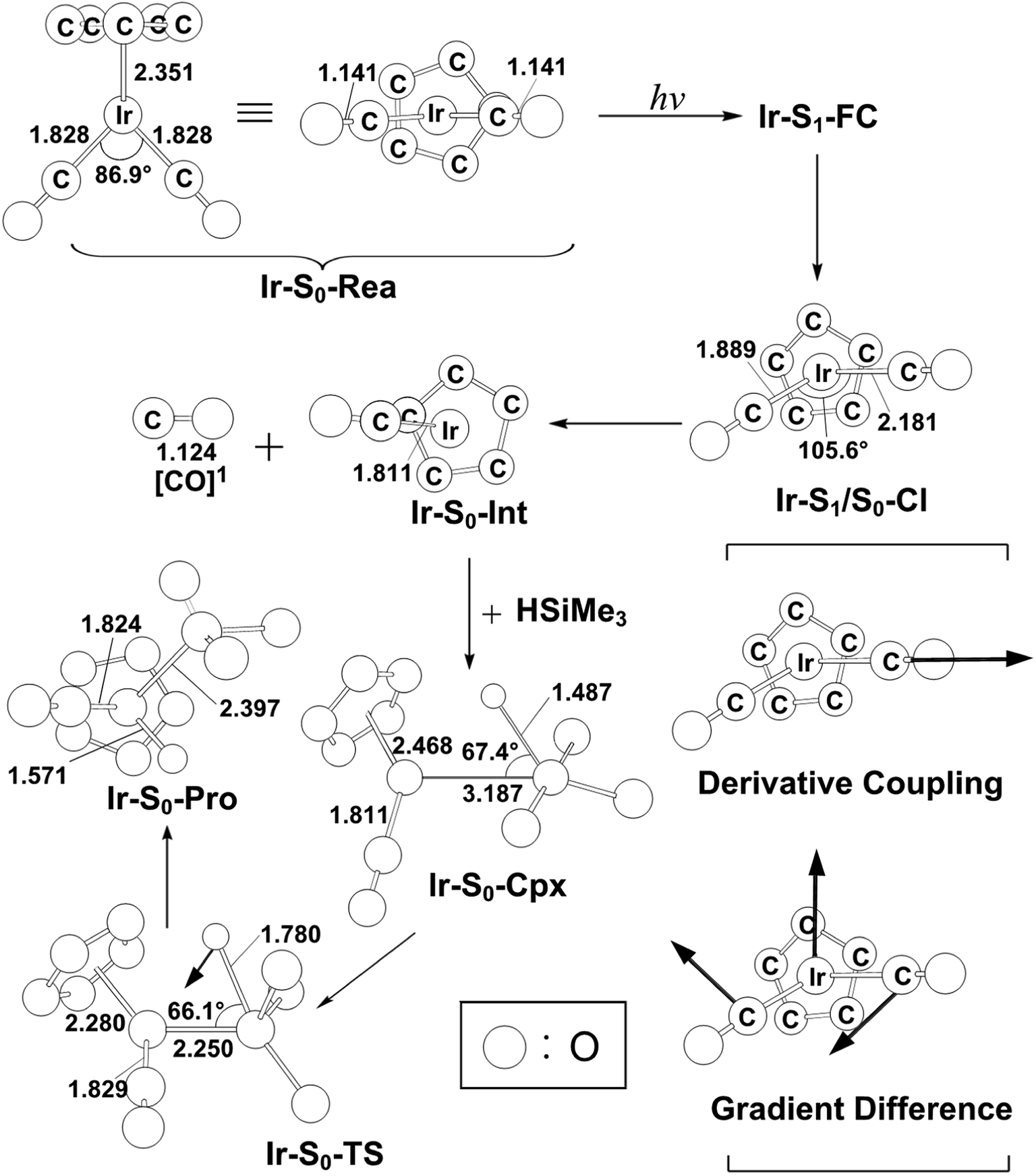 | ||
| Fig. 7 The CASSCF geometries (in Å and deg) for photo-oxidative-addition reactions of η5-CpIr(CO)2 (Ir-S0-Rea). The computational active spaces used in this work are described in Section II. Their relative energies are given in Fig. 6. Some hydrogen atoms are omitted for clarity. | ||
On the basis of the MP2-CAS computational data, Fig. 6 demonstrates that the lowest vertically excited energy state in the FC region is Ir-S1-FC (91.8 kcal mol−1), whose energy is notably lower than that of its irradiation of light (96.9 kcal mol−1). That is to say, the present theoretical observations indicate that the half-sandwich reactant complex (Ir-S0-Rea) is initially irradiated to its lowest lying singlet excited state (Ir-S1-FC). In the current work, we thus concentrate below on the singlet state channel for the photochemical activation reaction of Ir-S0-Rea with Si(CH3)3(H) from now on.
Subsequently, from the Ir-S1-FC point, this excited species relaxes to the conical intersection point (Ir-S1/S0-CI), whose energy is calculated to be about 52 kcal mol−1 relative to that of its corresponding reactant. Funneling through the Ir-S1/S0-CI point, several reaction routes on the ground-state energy surface may be foreseen by following its derivative coupling and the gradient difference vectors, which are already collected in Fig. 7. It is apparent from Fig. 7 that the main contribution of the gradient difference vector is the bending motions of two C–O bonds that result in a vibrationally hot Ir-S0-Rea molecule, whereas the derivative coupling vector corresponds to an Ir–CO bond cleavage. Therefore, following the derivative coupling vector from Ir-S1/S0-CI (Fig. 7) can give rise to the generation of one singlet CO molecule and one singlet half-sandwich monocarbonyl η5-CpIr(CO) intermediate (Ir-S0-Int). The MP2-CAS computations, which are given in Fig. 6, reveal that this Ir-S0-Int + CO + Si(CH3)3(H) point is 32.0 kcal mol−1 above the corresponding singlet reactants. Then the Ir-S0-Int species interacts with Si(CH3)3(H) to yield a precursor complex, Ir-S0-Cpx, whose energy is estimated to be about 18 kcal mol−1 with respect to the corresponding singlet state species. Finally, this activated complex undergoes its oxidative addition reaction via a transition state (Ir-S0-TS) to lead to the formation of a photoproduct, Ir-S0-Pro, as demonstrated in Fig. 6. Our MP2-CAS data indicate that the energy barrier from Ir-S0-Cpx to Ir-S0-TS and the reaction enthalpy of Ir-S0-Pro are estimated to be about 26 and −12 kcal mol−1, respectively. From the reaction profile given in Fig. 6, it is clear that the Ir-S0-Rea molecule has sufficient internal energy (38 kcal mol−1) to overcome the energy barrier between Ir-S0-Cpx and Ir-S0-TS (26 kcal mol−1) to reach the final insertion product (Ir-S0-Pro), after the photoexcited Ir-S0-Rea relaxes to the CI point (Ir-S1/S0-CI). Accordingly, the mechanism of the photoactivation reaction of η5-CpIr(CO)2 with trimethylsilane can be described as follows: [Si]1 stands for Si(CH3)3(H) at the singlet state.
| Ir system: Ir-S0-Rea + [Si]1 + hν → Ir-S1-FC + [Si]1 → Ir-S1/S0-CI + [Si]1 → Ir-S0-Int + [CO]1 + [Si]1 → Ir-S0-Cpx + [CO]1 → Ir-S0-TS + [CO]1 → Ir-S0-Pro + [CO]1 |
IV. Conclusion
The photochemical Si–H bond activation reactions by the group 9, d8 organometallic molecules, η5-CpM(CO)2 (M = Co, Rh, and Ir), have been studied by using the CAS as well as MP2-CAS computational methods. Taking all the model conclusions obtained in this work together yield the following summary:(1) Our model computations demonstrate that upon absorption of a photon of light, η5-CpCo(CO)2 (Co-S0-Rea) is promoted vertically to one higher excited state and then relaxes to the triplet FC point, Co-T1-FC. From this point, this cobalt complex returns to the triplet minimum species, Co-T1-Min. After its photochemical decarbonylation, three important reaction pathways (paths I, II, and III) have been considered to examine its oxidative addition of a Si–H bond with Si(H)Me3. The theoretical observations indicate that Co-S0-Rea initiates from the excited triplet state surface and finally changes to the singlet ground-state pathway. In other words, the intersystem crossing between the triplet (T1) and singlet (S0) surfaces must play a vital role in describing the mechanisms for the photochemical CO-extrusion and oxidative addition reactions of the Co-S0-Rea complex. Our theoretical findings strongly suggest that path II is the most energetically favorable reaction route for such alkylsilane activation reactions. In particular, the computational examinations reveal that the triplet monocarbonyl species ([η5-CpCo(CO)]3) plays a dominant role, but the corresponding singlet intermediate ([η5-CpCo(CO)]1) plays no role, in the photochemical CO-extrusion reaction and its subsequent activation reaction for a Si–H bond in alkylsilane. The above theoretical conclusions have previously been verified by some experimental findings.24–36
(2) On the other hand, after the CO-photoextrusions of η5-CpRh(CO)2 (Rh-S0-Rea) and η5-CpIr(CO)2 (Ir-S0-Rea), our theoretical findings demonstrate that their half-sandwich monocarbonyl intermediates (Rh-S0-Int and Ir-S0-Int) should still proceed on the singlet energy surfaces. In other words, the conical intersection plays a central role in determining the mechanisms of their photochemical oxidative addition reactions with alkylsilanes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the National Center for High-Performance Computing of Taiwan for generous amounts of computing time, and the Ministry of Science and Technology of Taiwan for the financial support. One of the author (Ming-Der Su) also wishes to thank Professor Michael A. Robb, Dr Michael J. Bearpark, Dr S. Wilsey, (University of London, UK) and Professor Massimo Olivucci (Universita degli Studi di Siena, Italy) for their encouragement and support during his stay in London. Special thanks are also due to reviewers 1 and 2 for very helpful suggestions and comments.References
- P. P. Deutsch and R. Eisenberg, Stereochemistry of Hydrogen Oxidative Addition and Dihydride-Transfer Reactions Involving Iridium(I) Complexes, Chem. Rev., 1988, 88, 1147–1161 CrossRef.
- L. M. Rendina and R. J. Puddephatt, Oxidative Addition Reactions of Organoplatinum(II) Complexes with Nitrogen-Donor Ligands, Chem. Rev., 1997, 97, 1735–1754 CrossRef PubMed.
- G. Zeni and R. C. Larock, Synthesis of Heterocycles via Palladium-Catalyzed Oxidative Addition, Chem. Rev., 2006, 106, 4644–4680 CrossRef PubMed.
- L. Souillart and N. Cramer, Catalytic C–C Bond Activations via Oxidative Addition to Transition Metals, Chem. Rev., 2015, 115, 9410–9464 CrossRef PubMed.
- R. H. Crabtree, Dihydrogen Complexation, Chem. Rev., 2016, 116, 8750–8769 CrossRef PubMed.
- J. Y. Corey, Reactions of Hydrosilanes with Transition Metal Complexes, Chem. Rev., 2016, 116, 11291–11435 CrossRef PubMed.
- H. G. Schuster-Woldan and F. Basolo, Kinetics and Mechanism of Substitution Reactions of π-Cyclopentadienyldicarbonylrhodium, J. Am. Chem. Soc., 1966, 88, 1657–1663 CrossRef.
- W. P. Weiner, M. A. White and R. G. Bergman, Direct Observation of Alkyl/Nitrosyl Migratory Insertion in an Organotransition Metal Complex, J. Am. Chem. Soc., 1981, 103, 3612–3614 CrossRef.
- A. H. Janowicz, J. H. Bryndza and R. G. Bergman, Phosphine Substitution in (η5-cyclopentadienyl)bis(triphenylphosphine)cobalt(I): Evidence for a Dissociative Mechanism, J. Am. Chem. Soc., 1981, 103, 1516–1518 CrossRef.
- A. H. Janowicz and R. G. Bergman, Carbon-Hydrogen Activation in Completely Saturated Hydrocarbons: Direct Observation of M + R-H → M(R)(H), J. Am. Chem. Soc., 1982, 104, 352–354 CrossRef.
- J. K. Hoyano and W. A. G. Graham, Oxidative Addition of the Carbon-Hydrogen Bonds of Neopentane and Cyclohexane to a Photochemically Generated Iridium(I) Complex, J. Am. Chem. Soc., 1982, 104, 3723–3725 CrossRef.
- W. D. Jones and F. J. Feher, Mechanism of arene carbon-hydrogen bond activation by (C5Me5)Rh(PMe3)(H)Ph. Evidence for arene precoordination, J. Am. Chem. Soc., 1982, 104, 4240–4242 CrossRef.
- E. P. Wasserman, R. G. Bergman and C. B. Moore, IR Flash Kinetic Spectroscopy of Transients Generated by Irradiation of (η5-C5H5)Co(CO)2 in the Gas Phase and in Solution, J. Am. Chem. Soc., 1988, 110, 6076–6084 CrossRef PubMed.
- A. H. Janowicz and R. G. Bergman, Activation of Carbon-Hydrogen Bonds in Saturated Hydrocarbons on Photolysis of (η5-C5Me5)(PMe3)IrH2. Relative Rates of Reaction of the Intermediate with Different Types of Carbon-Hydrogen Bonds and Functionalization of the Metal-Bound Alkyl Groups, J. Am. Chem. Soc., 1983, 105, 3929–3939 CrossRef.
- J. K. Hoyano, A. D. McMaster and W. A. G. Graham, Activation of Methane by Iridium Complexes, J. Am. Chem. Soc., 1983, 105, 7190–7191 CrossRef.
- B. H. Weiller, E. P. Wasserman, R. G. Bergman, C. B. Moore and G. C. Pimentelt, Time-resolved IR Spectroscopy in Liquid Rare Gases: Direct Rate Measurement of an Intermolecular Alkane Carbon-Hydrogen Oxidative Addition Reaction, J. Am. Chem. Soc., 1989, 111, 8288–8290 CrossRef.
- T. P. Dougherty and E. J. Heilweil, Transient Infrared Spectroscopy of (η5−C5 H5)Co(CO)2 Photoproduct Reactions in Hydrocarbon Solutions, J. Chem. Phys., 1994, 100, 4006–4009 CrossRef.
- A. A. Bengali, R. G. Bergman and C. B. Moore, Evidence for the Formation of Free 16-Electron Species Rather than Solvate Complexes in the Ultraviolet Irradiation of CpCo(CO)2 in Liquefied Noble Gas Solvents, J. Am. Chem. Soc., 1995, 117, 3879–3880 CrossRef.
- A. J. Rest, I. Whitwell, W. A. G. Graham, J. K. Hoyano and A. D. McMaster, Photoactivation of Methane by η5-Cyclopentadienyl and Substituted η5-Cyclopentadienyl Group 8 Metal Dicarbonyl Complexes, [M(η5-C5R5)(CO)2] (M = Rh or Ir, R = H or Me), and Dicarbonyl(η5-indenyl)iridium: A Matrix Isolation Study, J. Chem. Soc., Dalton Trans., 1987, 16, 1181–1190 RSC.
- E. P. Wasserman, C. B. Moore and R. G. Bergman, Gas-Phase Rates of Alkane C–H Oxidative Addition to a Transient CpRh(CO) Complex, Science, 1992, 255, 315–318 CrossRef PubMed.
- W.-S. Lee and H. Brintzinger, Reactive Intermediates in the Photolysis of cyclopentadienylcobalt(I) Dicarbonyl and in the Reduction of Cyclopentadienylcobalt (III) Carbonyl Diiodide, J. Organomet. Chem., 1977, 127, 87–92 CrossRef.
- W. Jetz and W. A. G. Graham, Silicon-transition Metal Chemistry. I. Photochemical Preparation of Silyl(transition metal) Hydrides, Inorg. Chem., 1971, 10, 4–9 CrossRef.
- S. T. Belt, F. W. Grevels, W. E. Klotzbuecher, A. McCamley and R. N. Perutz, Intermediates in the Time-resolved and Matrix Photochemistry of (η5-Cyclopentadienyl)rhodium Complexes. Roles of Alkane Activation and Rhodium-Rhodium Bond Formation, J. Am. Chem. Soc., 1989, 111, 8373–8382 CrossRef.
- K. T. Kotz, H. Yang, P. T. Snee, C. K. Payne and C. B. Harris, Organometallic Complexes for Nonlinear Optics: Part 19. Syntheses and Molecular Quadratic Hyperpolarizabilities of Indoanilino–alkynyl–ruthenium Complexes, J. Organomet. Chem., 2000, 596, 183–192 CrossRef.
- P. T. Snee, C. K. Payne, K. T. Kotz, H. Yang and C. B. Harris, Triplet Organometallic Reactivity under Ambient Conditions: An Ultrafast UV Pump/IR Probe Study, J. Am. Chem. Soc., 2001, 123, 2255–2264 CrossRef PubMed.
- H. Yang, K. T. Kotz, M. C. Asplund, M. J. Wilkens and C. B. Harris, Ultrafast Infrared Studies of Bond Activation in Organometallic Complexes, Acc. Chem. Res., 1999, 32, 551–560 CrossRef.
- J. P. Lomont, S. C. Nguyen, J. P. Schlegel, M. C. Zoerb, A. D. Hill and C. B. Harris, Ultrafast Observation of a Solvent Dependent Spin State Equilibrium in CpCo(CO), J. Am. Chem. Soc., 2012, 134, 3120–3126 CrossRef PubMed.
- B. Sztáray and T. Baer, Dissociation Dynamics and Thermochemistry of Energy-Selected CpCo(CO)2+ Ions, J. Am. Chem. Soc., 2000, 122, 9219–9226 CrossRef.
- D. P. Drolet and A. J. Lees, Photochemistry of (η5-C5H5)Rh(CO)2 in Phosphine Solutions: Evidence for an Associative Photosubstitution Mechanism, J. Am. Chem. Soc., 1990, 112, 5878–5879 CrossRef.
- Z. Hu, R. J. Boyd and H. Nakatsuji, Molecular structures and Excited States of CpM(CO)2 (Cp = η5-C5H5); M = Rh, Ir) and [Cl2Rh(CO)2]-. Theoretical Evidence for a Competitive Charge Transfer Mechanism, J. Am. Chem. Soc., 2002, 124, 2664–2671 CrossRef PubMed.
- X. Li, G. M. Bancroft, R. J. Puddephatt, Y.-F. Hu and K. H. Tan, Variable-Energy Photoelectron Spectroscopy of CpM(CO)2 (M = Co, Rh, Ir):
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Molecular Orbital Assignments and an Evaluation of the Difference in Ground-State Orbital Characters, Organometallics, 1996, 15, 2890–2904 CrossRef.
Molecular Orbital Assignments and an Evaluation of the Difference in Ground-State Orbital Characters, Organometallics, 1996, 15, 2890–2904 CrossRef. - J. A. Banister, A. I. Cooper, S. M. Howdle, M. Jobling and M. Poliakoff, Solvent-Free” Photochemical Activation of CH4, C2H4, and C2H6 by (C5Me5)Ir(CO)2 in Supercritical Fluid Solution, Organometallics, 1996, 15, 1804–1812 CrossRef.
- K. H. G. Mak, P. K. Chan, W. Y. Fan, R. Ganguly and W. K. Leong, Photochemical Reaction of Cp*Ir(CO)2 with C6F5X (X = CN, F): Formation of Diiridium(II) Complexes, Organometallics, 2013, 32, 1053–1059 CrossRef.
- P. K. Chan and W. K. Leong, Reaction of Cp*Ir(CO)2 with Activated Perfluoroaromatic Compounds: Formation of Metallocarboxylic Acids via Aromatic Nucleophilic Substitution, Organometallics, 2007, 27, 1247–1253 CrossRef.
- T. Ziegler, V. Tschinke, L. Fan and A. D. Becke, Theoretical Study on the Electronic and Molecular Structures of (C5H5)M(L) (M = Rhodium, Iridium; L = Carbonyl, Phosphine) and M(CO)4 (M = Ruthenium, Osmium) and Their Ability to Activate the Carbon-Hydrogen Bond in Methane, J. Am. Chem. Soc., 1989, 111, 9177–9185 CrossRef.
- T. R. Ward, O. Schafer, C. Daul and P. Hofmann, Geometry of Coordinatively Unsaturated Two-Legged Piano Stool Complexes with 16 Valence Electrons: A Theoretical Study, Organometallics, 1997, 16, 3207–3215 CrossRef.
- P. Hofmann, Unsaturated Organometallic Intermediates: Electronic Structure and Structural Dynamics of (η5-C5H5)Mn(CO)2, Angew. Chem., Int. Ed. Engl., 1977, 16, 536–537 CrossRef.
- M.-D. Su and S.-Y. Chu, Theoretical Model for Insertion of the 16-Electron Species (η5-C5H5)M(L) into Saturated Hydrocarbons. A (η5-C5H5)M(CO) + CH4 (M = Ru-, Os-, Rh, Ir, Pd+, Pt+) Case Study, Organometallics, 1997, 16, 1621–1627 CrossRef.
- M.-D. Su and S.-Y. Chu, An Energetically Feasible Mechanism for the Activation of the C–H Bond by the 16-Electron CpM(PH3)(CH3)+ (M = Rh, Ir) Complex. A Theoretical Study, J. Am. Chem. Soc., 1997, 119, 5373–5383 CrossRef.
- V. Gandon, N. Agenet, K. P. C. Vollhardt, M. Malacria and C. Aubert, Cobalt-Mediated Cyclic and Linear 2:1 Cooligomerization of Alkynes with Alkenes: A DFT Study, J. Am. Chem. Soc., 2006, 128, 8509–8520 CrossRef PubMed.
- N. Agenet, V. Gandon, K. P. C. Vollhardt, M. Malacria and C. Aubert, Cobalt-Catalyzed Cyclotrimerization of Alkynes: The Answer to the Puzzle of Parallel Reaction Pathways, J. Am. Chem. Soc., 2007, 129, 8860–8871 CrossRef PubMed.
- V. Gandon, N. Agenet, K. P. C. Vollhardt, M. Malacria and C. Aubert, Silicon–Hydrogen Bond Activation and Hydrosilylation of Alkenes Mediated by CpCo Complexes: A Theoretical Study, J. Am. Chem. Soc., 2009, 131, 3007–3015 CrossRef PubMed.
- P. Hofmann and M. Padmanabha, Electronic and Geometric Features of (η5-C5H5)ML 16-Electron Fragments. A Molecular Orbital Study of Ligand Effects, Organometallics, 1983, 2, 1273–1284 CrossRef.
- J. Song and M. B. Hall, Theoretical Studies of Inorganic and Organometallic Reaction Mechanisms. 6. Methane Activation on Transient Cyclopentadienylcarbonylrhodium, Organometallics, 1993, 12, 3118–3126 CrossRef.
- D. G. Musaev and K. Morokuma, Ab Initio Molecular Orbital Study of the Mechanism of H-H, C-H, N-H, O-H and Si-H Bond Activation on Transient Cyclopentadienylcarbonylrhodium, J. Am. Chem. Soc., 1995, 117, 799–805 CrossRef.
- M. Couty, C. A. Bayse, R. Jiménez-Cataño and M. B. Hall, Controversial Exothermicity of the Oxidative Addition of Methane to (Cyclopentadienyl)rhodium Carbonyl, J. Phys. Chem., 1996, 100, 13976–13978 CrossRef.
- P. E. M. Siegbahn, Comparison of the C−H Activation of Methane by M(C5H5)(CO) for M = Cobalt, Rhodium, and Iridium, J. Am. Chem. Soc., 1996, 118, 1487–1496 CrossRef.
- M.-D. Su and S.-Y. Chu, A Theoretical Model for the Orientation of 16-Electron CpML Insertion into the C-H Bond of Propane and Cyclopropane and Its Regio- and Stereoselectivity, Chem.–Eur. J., 1999, 5, 198–206 CrossRef.
- M.-D. Su and S.-Y. Chu, A New Aspect for the Insertion of the 16-Electron Species (η5-C5H5)ML into Saturated Hydrocarbons. A (η5-C5H5)ML + CH4 (M = Rh, Ir; L = CO, SH2, PH3) Case Study, J. Phys. Chem. A, 1997, 101, 6798–6806 CrossRef.
- M. J. Frisch; G. W. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani; V. Barone; B. Mennucci and G. A. Peterssonet al., Gaussian, Inc., Wallingford CT, 2013.
- F. Weigend and R. Ahlrichs, Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- P. Hofmann, Unsaturated Organometallic Intermediates: Electronic Structure and Structural Dynamics of (η5-C5H5)Mn(CO)2, Angew. Chem., Int. Ed. Engl., 1977, 16, 536–537 CrossRef.
- P. Hofmann and M. Padmanabhan, Electronic and Geometric Features of (η5-C5H5)ML 16-Electron Fragments. A Molecular Orbital Study of Ligand Effects, Organometallics, 1983, 2, 1273–1284 CrossRef.
- T. R. Ward, O. Schafer, C. Daul and P. Hofmann, Geometry of Coordinatively Unsaturated Two-Legged Piano Stool Complexes with 16 Valence Electrons: A Theoretical Study, Organometallics, 1997, 16, 3207–3215 CrossRef.
- M. J. Bearpark, M. A. Robb and H. B. Schlegel, A Direct Method for The Location of The Lowest Energy Point on a Potential Surface Crossing, Chem. Phys. Lett., 1994, 223, 269–274 CrossRef.
- M.-D. Su, Model Study of The Photochemical Rearrangement Pathways of 1,2,4-Oxadiazole, ChemPhysChem, 2014, 15, 2712–2722 CrossRef PubMed.
- M.-D. Su, A Model Study on the Photochemical Isomerizations of Isothiazoles and Thiazoles, Phys. Chem. Chem. Phys., 2014, 20, 17030–17042 RSC.
- M.-D. Su, A Theoretical Investigation of Photochemical Reactions of an Isolable Silylene with Benzene, Chem.–Eur. J., 2014, 16, 9419–9423 CrossRef PubMed.
- M.-D. Su, A Model Study on The Photochemical Isomerizations of Cyclic Silenes, Phys. Chem. Chem. Phys., 2015, 17, 5039–5042 RSC.
- M.-D. Su, Excited-State Photolytic Mechanism of Cyclopentene Containing A Group 14 Element: A MP2-CAS//CASSCF Study, J. Phys. Chem. A, 2015, 119, 8611–8618 CrossRef PubMed.
- M.-D. Su, A Mechanistic Study of the Addition of Alcohol to a Five-Membered Ring Silene via a Photochemical Reaction, Phys. Chem. Chem. Phys., 2016, 18, 8228–8234 RSC.
- S.-H. Su and M.-D. Su, Mechanistic Investigations of CO-Photoextrusion and Oxidative Addition Reactions of Early Transition-Metal Carbonyls: (η5-C5H5)M(CO)4 (M = V, Nb, Ta), Phys. Chem. Chem. Phys., 2016, 18, 16396–16403 RSC.
- Z.-F. Zhang, H.-C. Hua, S.-H. Su and M.-D. Su, Mechanistic Investigations on the Photorearrangement Reactions of M(CO)4(CS) (M = Group 6 Metal), Inorg. Chem., 2016, 55, 9017–9025 CrossRef PubMed.
- M.-D. Su, Mechanistic Investigations of the Photochemical Isomerizations of [(CO)5MC(Me)(OMe)] (M = Cr, Mo, and W) Complexes, ACS Omega, 2017, 2, 5395–5406 CrossRef.
- Z.-F. Zhang and M.-D. Su, A Mechanistic Study for The Photochemical Reactions of d6 M(CO)5(CS) (M = Cr, Mo, and W) Complexes, ACS Omega, 2017, 2, 2813–2826 CrossRef.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhas, Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef.
- B. Metz, H. Stoll and M. Dolg, Small-Core Multiconfiguration-Dirac–Hartree–Fock-Adjusted Pseudopotentials for Post-d Main Group Elements: Application to PbH and PbO, J. Chem. Phys., 2000, 113, 2563–2569 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-energy Formula into a Functional of the Electron Density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
- F. E. Jorge, A. C. Neto, G. G. Camiletti and S. F. Machado, Contracted Gaussian Basis Sets for Douglas–Kroll–Hess Calculations: Estimating Scalar Relativistic Effects of Some Atomic and Molecular Properties, J. Chem. Phys., 2009, 130, 064108–064113 CrossRef PubMed.
- N. X. Wang and A. K. Wilson, Effects of Basis Set Choice upon the Atomization Energy of the Second-Row Compounds SO2, CCl, and ClO2 for B3LYP and B3PW91, J. Phys. Chem. A, 2003, 107, 6720–6724 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03866j |
| This journal is © The Royal Society of Chemistry 2018 |