
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh throughput mass spectrometry-based characterisation of Arabidopsis thaliana group H glycosyltransferases†
Aishat Akere a,
Qian Liub,
Shibo Wua,
Bingkai Houb and
Min Yang*a
a,
Qian Liub,
Shibo Wua,
Bingkai Houb and
Min Yang*a
aThe School of Pharmacy, University College London, 29-39 Brunswick Square, London WC1N 1AX, UK. E-mail: min.yang@ucl.ac.uk
bKey Laboratory of Plant Cell Engineering and Germplasm Innovation, Ministry of Education of China, School of Life Sciences, Shandong University, Jinan, 250100, China
First published on 24th August 2018
Abstract
In this report, we cloned and characterised four members of group H glycosyltransferases (GTs) by studying their substrate specificities and kinetics. The formation of products and possible glycosylation position was confirmed using MS/MS. The results revealed that 76E1 and 76E5 have broader donor specificity, including UDP-glucose (UDPGlc), UDP-galactose (UDPGal) and UDP-N-acetylglucosamine (UDPGlcNAc) with various flavonoids as acceptor substrates. Pseudo-single substrate kinetics data showed a relatively low KM, indicating a high affinity for substrate UDPGlc and also supported that 76E5 is more of a galactosyl and N-acetylglucosamine transferase. Sequence alignment and site-directed mutagenesis studies indeed suggested that serine is a crucial residue in the UDPGlcNAc and UDPGal activity.
Introduction
Glycosylation is a widespread modification of plant secondary metabolites involved in various functions such as the regulation of hormone homeostasis, the detoxification of xenobiotics and the biosynthesis and storage of secondary compounds. In plants, these reactions are controlled by a specific subclass of the glycosyltransferase family. Family 1 plant UDP dependent glycosyltransferases (UGTs) are responsible for the transfer of sugar to many bioactive natural products1 such as alkaloids, terpenoids, flavonoids and phenylpropanoids. UDP activated sugars are mainly used as donor compounds by UGTs.2 Generally, transfer of sugar molecules can be either to macromolecules (such as proteins, lipids) or small molecules (such as plant secondary metabolites – natural products, oligosaccharides).3 However, the products of glycosylation catalysed by UGTs are glycosides of small molecules.4Numerous pharmacological activities have been reported for natural products containing sugar residues. This is because glycosylation improves their stability, solubility in water and bioavailability; hence it has become an important process in drug research and development.5 In addition, through this process, glycosides have become attractive compounds used as nutraceuticals and food additives.3 Leaves and seeds of Digitalis purpurea and Digitalis lanata are the most significant sources of cardiac steroids important in the treatment of various heart conditions. The aglycone steroid however, binds and dissociates too quickly from the receptor. Attachment of the sugar moiety to the aglycone steroid improves its binding stability to receptor, though the sugar itself has no cardiac activity. Hence, sugar addition enhanced the pharmacokinetic behaviour of the aglycone steroid, leaving us with low dissociation potent glycosides.6 New and effective methods to produce glycosylated natural products have been studied over the years.7 Since glycosyltransferases are important biocatalysts in sugar addition, it is imperative to study UGTs more in depth.
Presently, 122 UGT encoding genes are found in the model plant Arabidopsis thaliana.8 Phylogenetic analysis of conserved amino acids in Arabidopsis UGTs led to the classification into 14 groups9 (ESI Fig. 1†). Although these enzymes have been studied for many years, to date only a few have been characterized in planta. Of the 19 genes found in group H, only 2 are fully characterised.8 Varieties of glycosides found in plants in vivo signify that plant UGTs can glycosylate diverse compounds.10 Although UGTs were believed to be highly specific in substrate recognition in vivo, reports have disclosed broader range promiscuity against both substrates.11–17 This substrate recognition includes recognition of both UDP sugar donor as well as acceptor.1 Generally, UGTs are highly specific for sugar donor although UDP-glucose is most commonly used.18 The screening studies11–17 which looked into substrate specificity within Arabidopsis thaliana phylogenetic groups found that UGTs of the same group glycosylate acceptors belonging to very different compound classes. These early studies challenged efforts to solely base substrate specificity on phylogeny. Although biochemical characterisation of the substrate specificity is quite challenging as it entails testing diverse substrates to fully understand specificity of individual UGTs, it will advance prediction of substrate specificity based on phylogeny.1,19 Comprehensive facts on the in vivo and in vitro activity and specificity of UGTs are important in the design of enhanced UGTs with desired properties.20
In recent times, the importance of glycosyltransferases (GTs) has been in the spotlight. However, insufficient biochemical data on individual member enzyme has hindered further research into their functional understanding. Identifying and biochemically characterising some GT genes has helped in studying mechanism of glycosylation in planta.21 Biochemical data on substrate preferences has been relied on for prediction of substrate specificity based on phylogeny. Till date, most of Arabidopsis thaliana UGTs substrates remain unknown.22
Here, we report the substrate specificity and kinetic analysis of four Arabidopsis thaliana group H UGTs (76E1, 76E2, 76E5 and 76D1). Substrate screening showed that the UGTs added sugar to acceptors from different classes of secondary metabolites ranging from flavonoids, cinnamic acids, coumarins to alkaloids. They also recognised varying donor compounds, with all accepting UDPGlc. In addition, 76E1 and 76E5 recognise UDPGal and UDPGlcNAc. Using tandem mass spectrometry, likely glycosylation positions of the acceptors were proposed. Further kinetic study measured each enzyme binding affinity with donor substrates, indicating their donor preferences and catalytic efficiencies. Knowledge of the activity and specificity of UGTs would provide a basis for their potential use in the design of improved UGTs to produce bioactive molecules for pharmaceutical purpose.
Experimental
(a) Gene cloning procedure
Total RNA was extracted from the 14d seedlings of the Arabidopsis using the TRIzol reagent (Takara, Japan). First-strand cDNA was synthesized using PrimerScript RT reagent Kit with gDNA Eraser (Takara, Japan), pooled cDNA was used as PCR template. The cDNA sequences of UGT76E1, UGT76E2, UGT76E5 and UGT76D1 were amplified by TransStart Fast Pfu DNA Polymerase (TRANS, China). A BamHI and XhoI restriction site were included in the sequence of forward primer and the reverse primer used for PCR of UGT76E1 and UGT76E2. The PCR primers for UGT76E1 cloning are: forward GGATCCATGGAAGAACTAGGAGTGAAG and reverse CTCGAGCTACATGAAATTCATCATT; the PCR primers for UGT76E2 cloning are: forward GGATCCATGGAGGAAAACAAGTGAAG, reverse CTCGAGTCACATGGAATTAACAAAGTC. The BamHI and EcoRI were included in the sequence of forward primer and the reverse primer used for PCR of UGT76E5. The PCR primers for UGT76E5 cloning are: forward GGATCCATGGAGAAAAATGCAGAGAAG, reverse GAATTCTCAAGTATTTCTATACTCTGC. The BamHI and SacI were included in the sequence of forward primer and the reverse primer used for PCR of UGT76D1. The primers for UGT76D1 cloning are: forward CGGGATCCATGGCAGAGATTCGCCAG, reverse CGAGCTCTCATTGTTCGTCAATTTGCATC. The amplified products were cloned into the corresponding sites of the Blunt simple cloning vector, previously digested with appropriate restriction enzymes. After sequencing and alignment with reference sequence of the four genes shown in the Arabidopsis information resource, the right clones were chosen for further cloning. Lastly, the cDNA fragments of the target genes were inserted and ligated into the pGEX-3H plasmid previously digested with appropriate restriction enzymes, respectively. The resulting recombinant plasmids were transformed into competent E. coli BL cells.(b) Expression and purification of AtUGT76E1, AtUGT76E2 and AtUGT76E5
The recombinant plasmids were transformed into BL1 (DE3) cells for protein expression. Thereafter, they were verified by DNA sequencing via sanger sequencing (Source Bioscience Ltd). The bacterial cells were grown at 37 °C while being shaken at 200 rpm to an OD600 of 0.6–0.8. Isopropyl 1-thiogalactopyranoside (IPTG) was added to the cell culture to a final concentration of 0.1 mM to induce protein expression. The culture was grown overnight at 20 °C and collected by centrifugation at 4 °C. The GST-tagged recombinant protein was purified by affinity chromatography (columns) and quantified using the Bradford assay according to the standard procedure provided by the suppliers. Cell lysate before purification and purified protein were analysed on sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) gels to verify protein production (ESI Fig. 2a–d†).(c) In vitro UGT reaction assay
The UGT enzyme assay included the following components: 1 mM Tris 1 mM, MgCl2 (pH 8.0), 10 mM UDP-sugars, 10 mM acceptor compounds and purified target proteins. For the acceptor screening, the 42 compounds screened are shown in the acceptor library (ESI Table 1†). The 7 sugar donor compounds used in the donor screening are also indicated in Fig. 2. The reaction mixture was incubated at 37 °C for 3 and 15 hours at different times. The reaction was terminated with acetonitrile and centrifuged to remove proteins. The supernatant was then analysed with LCMS. Glycosylated products were identified by their molecular weights, and these target compounds were subsequently fragmented using MS/MS for confirmation.(d) UDP-Glo™ glycosyltransferase assay – for kinetics
Kinetic study was performed using the UDP-Glo™ glycosyltransferases Assay kit-following the procedure provided by the supplier. The UGT enzyme assay mixture for kinetics contained Tris buffer (50mM Tris, 5 mM MgCl2, pH 7.5), kaempferol (for 76E1 and 76E5) and quercetin (for 76E2 and 76D1). Kaempferol and quercetin were fixed at 20 μM. The donor substrates concentration; UDP Glc (five concentrations) was varied from 10–500 μM while UDP Gal and GlcNAc were varied from 0.1–10 μM. The UGT reaction was carried out at 25 °C and terminated using the UDP-Glo assay detection buffer. This kit detects UDP generated after UDP-sugar transfer by converting UDP to light (measured in relative luminescence units) in a luciferase type reaction. A linear standard curve using 0–25 μM UDP was performed (ESI Fig. 17†). Following the kit's protocol, the UGT assay was combined in a ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (10 μl:10 μl) with the UDP-Glo™ detection reagent in individual wells (white flat bottom 384-well plate [Corning]). Luminescence signal was measured after 1 hour incubation using a Pherastar microplate reader (BMG Labtech).
:1 (10 μl:10 μl) with the UDP-Glo™ detection reagent in individual wells (white flat bottom 384-well plate [Corning]). Luminescence signal was measured after 1 hour incubation using a Pherastar microplate reader (BMG Labtech).
(e) HPLC-MS/MS
Samples were analysed with Agilent 6400 triple quadruple mass spectrometer coupled with HPLC system using phenomenex-C18 column (50 × 4.6 mm, kinetex 5u, 100A). The solvents used were as follows: (A) HPLC grade water containing 0.1% formic acid and (B) acetonitrile containing 0.1% formic acid. For the MS scan, both positive and negative spectra were obtained and run at a flow rate of 0.4 ml min−1 in isocratic mode (10% A and 90% B). The injection volume was 10 μl, detection wavelength at 260 nm and column temperature of 20 °C. Other details include start and end mass of 100 and 1000 respectively, scan time of 500 and cell accelerator voltage of 7. The MS/MS (product ion scan) had a flow rate 0.5 ml min−1 at gradient mode (70% A: 30% B for 1 min, 55% A: 45% B for 1.50 min and lastly 70% A: 30% B for 2.50 min). The injection volume was maintained at 10 μl and only negative spectra were obtained here. The precursor ions were fragmented for confirmation within the range of m/z 100–100. Scan segment details are as follows: scan time 500 fragmentor 135, collision energy 15 and cell accelerator voltage 7. All analyses were done in duplicates.(f) Site directed mutagenesis
Primer design was done using NEBaseChanger v1.2.7 (http://nebasechanger.neb.com/) and the primers synthesized by Eurofin Genomics Ltd. Templates DNAs were extracted using QIAprep Spin miniprep kit protocol. Q5 Site-directed mutagenesis kit protocol (from NewEngland Biolabs) was observed to make mutants from the wild type template DNAs. Mutagenesis was done in three stages of exponential amplification (polymerase chain reaction), digestion and transformation. Mutant plasmid DNAs were subsequently extracted via miniprep. The mutant DNAs were sent for sanger sequencing (SourceBioScience) to confirm mutations. The primer sequences are: 76E2N320S: Forward primer AGAGGAATTCAGTAGGTTGGTTTC, reverse primer GGTAAGGACTCTGTCCATTC; 76E1S318N: forward primer GGAGGAATTCAATAGGTTGGTTT, reverse primer GGTAAAGACTCTGTCCATTC; 76E5S311N: forward primer AGTGGAAGTCAATAAGATTGTCTC, reverse primer GGCATTGACTCTGTACCG. All WT and mutant sequences were confirmed using BLAST (ESI Fig. 11–17†).(g) Sequence alignments
The amino acid sequences of the wild type UGTs were aligned using clustal omega by European Bioinformatics Institute (https://www.ebi.ac.uk/Tools/msa/clustalo/). The mutant and wild type DNA sequences were compared using NCBI nucleotide BLAST (National Centre for Biotechnology Institute).Results and discussion
(a) MS based HTS method
The genes encoding 76E family and 76D1 were expressed as recombinant proteins in Escherichia coli and purified to electrophoretic homogeneity. In vivo, the functions of these UGTs are not known except for 76D1 whose functions have been recently published.23 The Green-Amber-Red ‘GAR’ screen11,24 was developed to probe potential substrates, in vitro, using a large acceptor and nucleotide-sugar donor library (Fig. 2, ESI Table 1†). A virtual colour, reflecting DNA micro-array practice, of Green-Amber-Red (GAR, green indicates positive, amber means not clear and red shows negative) is assigned reflecting the presence of selected ions of mass. This procedure using liquid chromatography triple quadrupole Mass Spectrometry (LC-MS/MS) was composed of two parts; a general MS scan to identify desired product peaks such as [M − H+]− or [M + Cl−]− in negative mode and [M + H+]+ or [M + Na+]+ in positive mode first, and then a MS/MS scan to confirm the presence of acceptor precursor which indicates the loss of the sugar moiety to confirm the reaction product (Fig. 1a). This method, similar to the previous NLCIT,11 could be used to determine the glycosylation site. No difference was noticed in the reaction products from 3 h and 15 h assay incubation. In addition, pseudo single substrate enzyme kinetics was measured by varying one substrate concentrations while saturating the other substrate using a commercial UDP-Glo® assay. An appropriate enzyme concentration was obtained prior to the kinetic study with fixed substrate concentrations (UDPGlc at 100 μM and KMP at 20 μM) while enzyme concentration varied. The formation of UDP was monitored in the function of time and initial rates were obtained. Kinetics parameters such as Vmax/Kcat and KM were generated using either Lineweaver–Burk or Michaelis–Menten equation (ESI Fig. 13–16†). | ||
| Fig. 1 MS based method and GAR screening result. (a) The MS/MS method to determine the formation of product using an example of UDPGlcNAc with kaempferol catalyzed by UGT76E5 (inset: reaction scheme and the fragment of product, product structure for illustration purpose only). (b) GAR acceptor screening result of enzymes (c) structures of the compounds showed positive reaction in the acceptor screening. | ||
(b) GAR substrate specificity
In this study, we have reported the acceptor substrate profile of UGTs 76E1, 76E2, 76E5 and 76D1 (Fig. 1b). Of the forty-two acceptor substrates screened, 76E1 recognised nine flavonoids and one coumarin. 76E2 only accepted flavonoids as substrates while 76E5 on the other hand, recognised one benzoic acid, one coumarin and two flavonoids. Similarly, 76D1 accepted two flavonoids and one benzoic acid. Although there seem to exist a flavonoid trend, recognition of benzoic acid by 76E5 and 76D1 and non-recognition of other acceptor substrate class reinforces the established finding that substrate recognition cannot be predicted solely based on phylogenetic grouping.1
Our results showed that although acceptor compounds from varying classes were recognised, flavonoids appeared to be a common factor. According to Parajuli et al.,25,26 expanded substrate study of a glycosyltransferase AtUGT89C1 revealed that it recognised five different classes of flavonoids and varying NDP-sugars. Another Arabidopsis thaliana glycosyltransferases, AtUGT78D1 glycosylated several compounds of structural types; flavonoids, flavonoid glycosides and others.26 Furthermore, Jones et al.10 reported flavonoid glucoside/rhamnoside production from UGT73C6. All these studies corroborated that AtUGTs seemed to generally favour flavonoids O-glycosylation. Flavonoid glycosides possess promising beneficial health activities27 and regioselective glycosyltransferases offer to overcome limitations in their pharmacological potency.28 This implies that UGTs tested in group H may be useful in producing useful flavonoids glycosides.
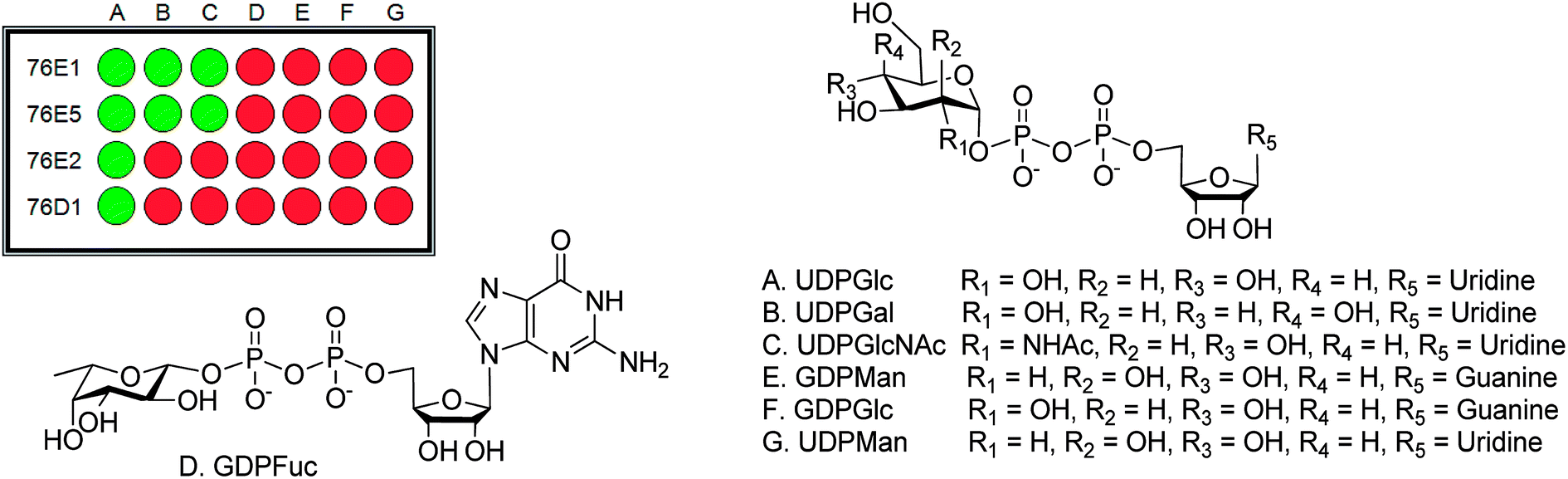 | ||
| Fig. 2 Donor screening result. (A) UDPGlc, (B) UDPGal, (C) UDPGlcNAc, (D) GDPFuc, (E) GDPMan, (F) GDPGlc and (G) UDPMan. | ||
Flavonoids have multiple glycosylation position which can change the product physical and biological activity. Mass spectra have been found to give information on structural characterization, especially glycosylation position. Most sugar molecules in flavonoid O-glycosides bind at 3-, 7- or 4′ positions (Fig. 1c), leaving others such as 5-OH which show very low activity to glycosylation.29 Differentiating between 3-, 7- and 4′ – positions can be quite problematic; however, MS has shown advantage to provide possible solutions. In the MS2 fragment of a glycosylated flavonoid, radical aglycone ions ([M–H+–H˙–Gly]˙−, 284/300) are found to be more abundant for flavonol 3-O glycosides than regular aglycone ions ([M–H+–Gly]−, 285/301) which leaves 284 (kaempferol) or 300 (quercetin) instead of 285 or 301 (Fig. 3a). The formation of a radical aglycone ion is due to the homolytic cleavage of the glycosidic bond between the O-linked sugar and the aglycone moiety.29
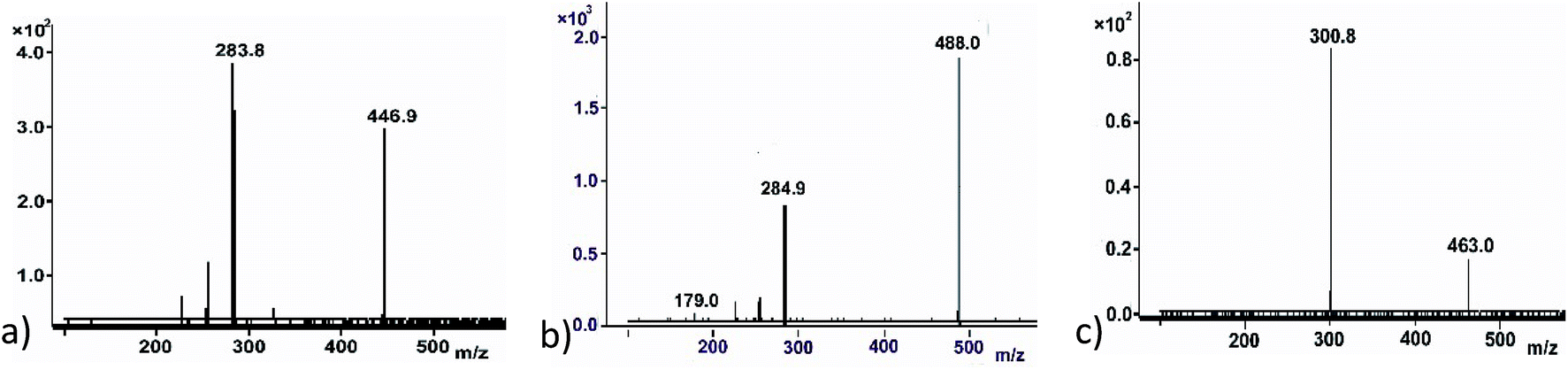 | ||
| Fig. 3 The MS/MS method to determine the formation of product and potentially glycosylation position. (a) An example to show the 3-O glycosylation – the formation of radical aglycone (283.8). (b) An example to show the glycosylation on 4′-O position – the formation of aglycone (284.9) and the presence of fragment of 179.0. (c) An example to show the 7-O glycosylation – the formation of aglycone (300.8) only. | ||
The presence of peak 179 (designated as the diagnostic ion, Fig. 3b) in retrocyclisation pathway involving loss of B-ring indicates specificity of 3′ or 4′ derivatives in negative ion ESI-MS/MS.30 Fig. 3 showed a few other examples of using the above rule to probe the glycosylation position. For UDPGlcNAc, peak 285 was more prominent, indicating glycosylation may not be at 3-O position. It will either be glycosylation at 7-O or 4′-O position. The presence of peak 179, a diagnostic ion for 4′-O glycosylation suggests that it might be kaempferol 4′-O glucoside (Fig. 3b). Another example showing a glycosylation position was seen in a quercetin glycoside. Here, the regular aglycone ion peak 301 is more prominent than radical aglycone ion 300, showing the glycosylation was not at the 3-O position. Since peak 179 is absent here, we infer that the product was a quercetin 7-O glucoside (Fig. 3c).
From the acceptor screening (Fig. 2) it indicated that 76E1 can be more flexible in accommodating acceptors (either flat or curved). The result indicates the glycosylation position can be on 7-OH but no further information can be assured without NMR data. Interestingly, 76E1 is also very flexible in donor binding pocket with three (3) UDP-sugars used as substrate, but all on the 7-O position. 76E2 performed similarly to 76E1, glucosylating quercetin at 7-OH position. Similar flexibility was demonstrated for 76E5 in its donor binding pocket as 76E1 although 76E1 can utilise little more acceptors than 76E5. When UDPGlc was used as donor, it glycosylated kaempferol at 3-OH but with UDPGlcNAc as donor, the glycosylation position is on 4′ or 7-OH. The acceptor screening prediction is challenging as it involves non-flavonoids (Table 1).
| Enzymes | Donor | Acceptor | Peaks used in identification | Glycosylation position |
|---|---|---|---|---|
| 76E1 | UDPGlc | Kaempferol | 285 | 7-OH |
| UDPGlc | Quercetin | 301 | 7-OH | |
| UDPGal | Kaempferol | 285 | 7-OH | |
| UDPGlcNAc | Quercetin | 301 | 7-OH | |
| 76E2 | UDPGlc | Quercetin | 301 | 7-OH |
| 76E5 | UDPGlc | Kaempferol | 284 | 3-OH |
| UDPGlc | Quercetin | 300 | 3-OH | |
| UDPGal | Kaempferol | 284 | 3-OH | |
| UDPGal | Quercetin | 300 | 3-OH | |
| UDPGlcNAc | Kaempferol | 285, 179 | 4′-OH | |
| UDPGlcNAc | Quercetin | 301 | 7-OH | |
| 76D1 | UDPGlc | Quercetin | 301 | 7-OH |
 | ||
| Fig. 4 Sequence alignment of UDPGlcNAc glycosyltransferases and mutant activities. (a) Sequence alignment showing serine thought to be involved in O-galactosylation and O-GlcNAcylation. (b) Sequence alignment showing serine in other Group H UGTs, thought to be involved in O-galactosylation and O-GlcNAcylation, (c) mutants' donor activity results: comparison between wild type enzymes 76E5, 76E1 and 76E2 and mutant enzymes 76E5 S311N, 76E1 S318N and 76E2 N320S activity with donor compounds (A) UDPGlc, (B) UDPGal, (C) UDPGlcNAc. 76E2 N320S mutant newly acquired UDPGal and UDPGlcNAc activities. | ||
Mass spectra from UGT assay catalysed by mutant enzymes 76E1 S318N, 76E5 S311N and 76E2 N320S are shown in ESI Fig. 18–24.† Total loss of glycosylation activity (UDPGlc, UDPGal and UDPGlcNAc) was observed in 76E1 S318N and 76E5 S311N as summarised in Fig. 4c. This indeed demonstrates the strong link between these activities and serine at this point. More interestingly, UDPGal and UDPGlcNAc activity was acquired by 76E2 N320S; the WT enzyme 76E2 only has UDPGlc activity (Fig. 4c). Although the exact mechanism by which serine acts is still unclear, this finding confirms our hypothesis that serine at this point is important in the UDPGlcNAc and UDPGal activity.
| Enzyme | Substrate varied | KM (μM) | Kcat (s−1) | Kcat/KM (s−1 mM−1) |
|---|---|---|---|---|
| 76E1 | UDP glucose | 10.5 ± 1.38 | 0.00832 | 0.79 |
| 76E2 | UDP glucose | 8.75 ± 1.38 | 0.0107 | 1.23 |
| 76E5 | UDP glucose | 8.79 ± 1.64 | 0.0194 | 2.21 |
| UDP galactose | 0.57 ± 0.19 | 0.0130 | 22.8 | |
| UDPN-acetyl glucosamine | 0.78 ± 0.28 | 0.0166 | 21.3 | |
| 76D1 | UDP glucose | 25.77 ± 8.82 | 0.00476 | 0.185 |
Conclusion
Here, we have reported the substrate specificities and kinetics of four UGTs in group H of Arabidopsis thaliana (76E1, 76E2, 76E5 and 76D1). These UGTs added sugars to acceptors from different classes of secondary metabolites ranging from flavonoids, cinnamic acids, coumarins to alkaloids. They also recognised varying donors tested, with all recognising UDP glucose and 76E1 and 76E5 binding to UDP-galactose and UDPGlcNAc in addition. Using tandem mass spectrometry, the likely glycosylation positions on the acceptors were proposed. Further kinetic study measured each enzyme binding affinity with donor substrates, indicating their donor preferences and catalytic efficiencies. The distinctive amino acid sequences of both UGTs recognising UDP Gal and UDP GlcNAc was observed. Mutagenesis highlighted residue serine, which seem to be important for UDPGlcNAc and UDPGal activity. Knowledge of the activity and specificity of UGTs would provide a basis for their potential use to design improved UGTs to produce bioactive molecules for pharmaceutical purposes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Nigerian Government, under the Presidential Special Scholarship Scheme for Innovation and Development (PRESSID) supervised by the Federal Scholarship Board (FSB).References
- S. A. Osmani, S. Bak and B. L. Moller, Phytochemistry, 2009, 70, 325–347 CrossRef PubMed.
- E. K. Lim and D. J. Bowles, EMBO J., 2004, 23, 2915–2922 CrossRef PubMed.
- F. De Bruyn, J. Maertens, J. Beauprez, W. Soetaert and M. De Mey, Biotechnol. Adv., 2015, 33, 288–302 CrossRef PubMed.
- D. Bowles, E. K. Lim, B. Poppenberger and F. E. Vaistij, Annu. Rev. Plant Biol., 2006, 57, 567–597 CrossRef PubMed.
- G. Huang, M. Lv, J. Hu, K. Huang and H. Xu, Mini-Rev. Med. Chem., 2016, 12, 1013–1016 CrossRef.
- V. Kren and L. Martinkova, Curr. Med. Chem., 2001, 8, 1303–1328 CrossRef PubMed.
- G. Huang and X. Mei, Curr. Drug Targets, 2014, 15, 780–784 CrossRef PubMed.
- P. M. Coutinho and B. Henrissat, Carbohydrate-Active Enzymes server at URL, http://afmb.cnrs-mrs.fr/%7Ecazy/CAZY/index.html, http://afmb.cnrs-mrs.fr/CAZY/index.html.
- J. Ross, Y. Li, E. Lim and D. J. Bowles, Genome Biol., 2001, 2, REVIEWS3004 CrossRef PubMed.
- P. Jones, B. Messner, J. Nakajima, A. R. Schaffner and K. Saito, J. Biol. Chem., 2003, 278, 43910–43918 CrossRef PubMed.
- S. M. Patel, M. Fuente, S. Ke, A. Guimaraes, A. O. Oliyide, X. Ji, P. Stapleton, A. Osbourn, Y. Pan, D. J. Bowles, B. G. Davis, A. Schatzlein and M. Yang, Chem. Commun., 2011, 47, 10569–10571 RSC.
- M. Nielsen, M. M. D. Suits, M. Yang, C. S. Barry, C. Martinez-Fleites, L. E. Tailford, J. E. Flint, C. Dumon, B. G. Davis, H. J. Gilbert and G. J. Davies, J. Biol. Chem., 2011, 286, 15155–15164 CrossRef PubMed.
- M. Yang, G. J. Davies and B. G. Davis, Angew. Chem., Int. Ed., 2007, 46, 3885–3888 CrossRef PubMed.
- D. Bolam, S. Roberts, M. Proctor, J. Turkenburg, E. Dodson, C. Martinez-Fleites, M. Yang, B. G. Davis, G. Davies and H. Gilbert, Proc. Natl. Acad. Sci. U. S. A., 2007, 13, 5336–5341 CrossRef PubMed.
- W. Offen, C. Martinez-Fleites, M. Yang, E. Kiat-Lim, B. G. Davis, C. A. Tarling, C. M. Ford, D. J. Bowles and G. J. Davies, EMBO J., 2006, 25, 1396–1405 CrossRef PubMed.
- M. Yang, M. Brazier, R. Edwards and B. G. Davis, ChemBioChem, 2005, 6, 346–357 CrossRef PubMed.
- J. Flint, E. Taylor, M. Yang, D. N. Bolam, L. E. Tailford, C. Martinez-Fleites, E. J. Dodson, B. G. Davis, H. J. Gilbert and G. J. Davies, Nat. Struct. Mol. Biol., 2005, 12, 608–614 CrossRef PubMed.
- L. V. Modolo, J. W. Blount, L. Achnine, M. A. Naoumkina, X. Wang and R. A. Dixon, Plant Mol. Biol., 2007, 64, 499–518 CrossRef PubMed.
- T. Vogt and P. Jones, Trends Plant Sci., 2000, 5, 380–386 CrossRef PubMed.
- D. Bowles, J. Isayenkova, E. K. Lim and B. Poppenberger, Curr. Opin. Plant Biol., 2005, 8, 254–263 CrossRef PubMed.
- T. Mo, X. Liu, Y. Y. Liu, X. H. Wang, L. Zhang, J. Wang, Z. X. Zhang, S. P. Shi and P. F. Tu, RSC Adv., 2016, 6, 84616–84626 RSC.
- V. Veljanovski and C. P. Constabel, Phytochemistry, 2013, 91, 148–157 CrossRef PubMed.
- X. X. Huang, G. Q. Zhu, Q. Liu, L. Chen, Y. J. Li and B. K. Hou, Plant Physiol., 2018, 176, 3103–3119 CrossRef PubMed.
- M. Yang, M. Brazier, R. Edwards and B. G. Davis, ChemBioChem, 2005, 6, 346–357 CrossRef PubMed.
- P. Parajuli, R. P. Pandey, N. T. H. Trang, T. J. Oh and J. K. Sohng, Carbohydr. Res., 2015, 418, 13–19 CrossRef PubMed.
- P. Parajuli, R. P. Pandey, S. Darsandhari, Y. I. Park and J. K. Sohng, J. Carbohydr. Chem., 2016, 35, 367–377 CrossRef.
- H. Cao, X. Chen, A. R. Jassbi and J. Xiao, Biotechnol. Adv., 2015, 33, 214–223 CrossRef PubMed.
- K. Yonekura-Sakakibara, T. Tohge, R. Niida and K. Saito, J. Biol. Chem., 2007, 282, 14932–14941 CrossRef PubMed.
- F. Cuyckens and M. Claeys, J. Mass Spectrom., 2004, 39, 1–15 CrossRef PubMed.
- N. Fabre, I. Rustan, E. Hoffmann and J. Quetin-Leclercq, J. Am. Soc. Mass Spectrom., 2001, 12, 707–715 CrossRef PubMed.
- J. A. Capra and M. Singh, Bioinformatics, 2007, 23, 1875–1882 CrossRef PubMed.
- B. Hou, E. K. Lim, G. S. Higgins and D. J. Bowles, J. Biol. Chem., 2004, 279, 47822–47832 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03947j |
| This journal is © The Royal Society of Chemistry 2018 |