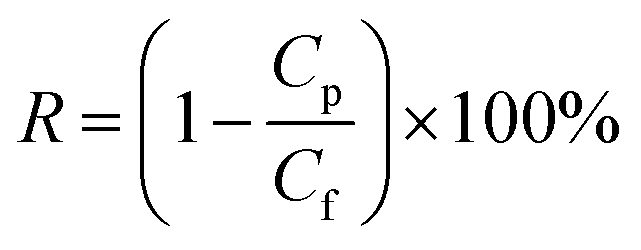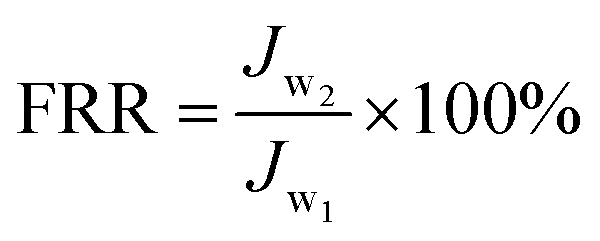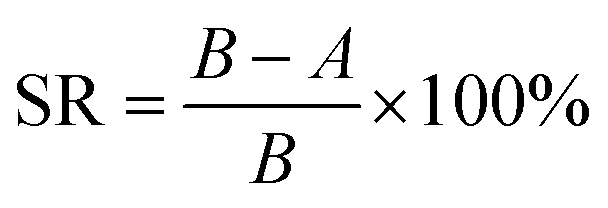DOI:
10.1039/C8RA03934H
(Paper)
RSC Adv., 2018,
8, 24690-24700
Hydrophilic and antimicrobial core–shell nanoparticles containing guanidine groups for ultrafiltration membrane modification
Received
8th May 2018
, Accepted 3rd July 2018
First published on 9th July 2018
Abstract
Physical blending is a common technique to improve the water flux and antifouling performance of ultrafiltration (UF) membranes. In the present work, a novel hydrophilic and antimicrobial core–shell nanoparticle was synthesized through the chemical grafting of poly(guanidine-hexamethylenediamine-PEI) (poly(GHPEI)) on the surface of silica nanoparticles (SNP). The synthesized core–shell nanoparticles, poly(GHPEI) functionalized silica nanoparticles (SNP@PG), were incorporated into polyethersulfone (PES) to fabricate hybrid UF membranes by a phase inversion process. The chemical composition, surface and cross section morphologies, hydrophilicity, water flux and protein rejection of the membranes were evaluated by a series of characterizations. Results show that the prepared PES/SNP@PG hybrid membrane exhibits not only improved water flux, which is around 2.6 times that of the pristine PES membrane, but also excellent resistance to organic fouling and biofouling.
1. Introduction
Water treatment is increasingly critical due to the global scarcity and severe pollution of water resources. As an efficient and environment-friendly technology, ultrafiltration (UF) is playing an important role in improving water quality.1 UF membranes, with surface pores of 1–100 nm, can be used to remove colloids, proteins, bacteria and other macromolecules from various aqueous media.2 Polymeric UF membranes are typically composed of hydrophobic polymers, including polyvinylidene fluoride (PVDF), polyethersulfone (PES), polysulfone (PSf), polyacrylonitrile (PAN), and so on.3 Although the hydrophobicity of the bulk membrane material helps to maintain the structural integrity in aqueous environments, the membranes often suffer severe surface fouling from bacteria and organic pollutants during the operation. Surface fouling can lead to temporary or permanent water flux decline, and thus increase energy consumption and shorten membrane lifespan.4
In recent years, various methods have been applied to construct fouling-resistant UF membrane surfaces, such as ultraviolet irradiation,5,6 surface coating,7–9 surface grafting,10,11 physical blending12,13 and surface segregation.14,15 To alleviate organic and bacterial fouling, the membrane surface should be well designed through using functional materials to avoid the attachment of foulants or inhibit the growth of microorganisms. Usually, the acceptable modification strategies should fulfill the enhanced antifouling ability without decreasing the water flux or solute selectivity of the membranes.
One of the most effective methods for membrane modification is to increase the surface hydrophilicity. Fang et al.7 introduced an iron–tannin-framework (ITF) complex to a PES casting solution as a hydrophilic additive to fabricate ITF/PES UF membrane with an increased hydrophilicity and porosity. When the ITF content was 0.3 wt%, the hybrid membrane exhibited a pure water flux of 319.4 L m−2 h−1 at 0.1 MPa and improved fouling resistance. In the study of Keskin et al.,9 the PS-b-P4VP diblock copolymer membrane was modified first with an ATRP initiator and then with a hydrophilic monomer 2-hydroxyethyl methacrylate to obtain a surface with antifouling property. Gao et al.12 blended novel sulfobetaine polyimides (PIs) into the PSf membrane and promoted the overall performance of UF membrane with enhanced hydrophilicity, porosity and antifouling properties. In the study of Li et al.,16 glycine-functionalized PVA was synthesized and grafted onto a polydopamine (PDA) coated UF membrane. The modified membrane surface showed a decreased roughness and an improved wettability, leading to a higher flux recovery in the fouling test. However, the pure water flux decreased by ∼27% after modified by PDA and PVA.
Besides, various antibacterial agents, including silver nanoparticles, graphene oxides (GO), and polymers with quaternary ammonium (N+) have been applied to UF membrane surface.12,17–20 It is common to prepare hybrid UF membranes via phase inversion process using antibacterial nanomaterials as the additives. Li et al.21 synthesized silver nanoparticles assembling on graphene oxide sheets (GO-Ag) and fabricated GO-Ag/PVDF membranes to enhance the antibacterial properties and inhibit the biofilm formation. The membrane permeability was also improved during the modification. In the study of Aani et al.,22 multi-walled carbon nanotubes (MWCNTs) was coated with silver nanoparticles using a microwave treatment, and then the nanocomposites were incorporated into PES UF membranes. The hybrid UF membranes achieved greatly improved antibacterial activity, with log kill of 4.24 against E. coli and 2.9 against S. aureus.
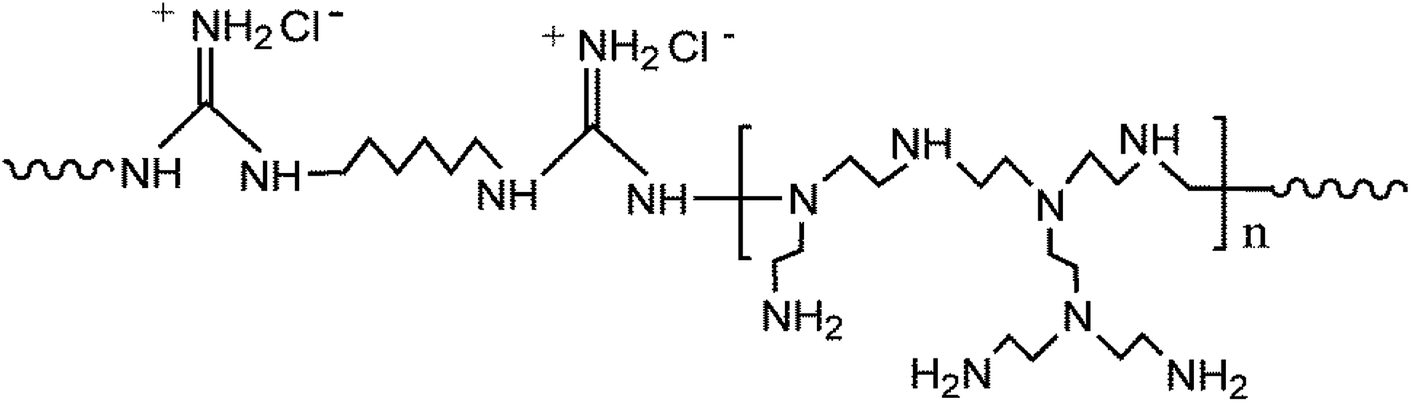
Silica nanoparticles (SNP) are widely used in fabricating hybrid membranes,23–25 owing to its proper chemical reactivity and high thermal resistance.26 Polymers with guanidine groups show outperforming antibacterial property with the broad-spectrum of antibacterial activity and low mammalian toxicity.27 Their antibacterial property against bacteria is generally ascribed to the electrostatic attractions between the cationic guanidine and the negative charged surface of bacterial cell, resulting in the leakage of intracellular contents and consequential bacterial death.27,28 In this study, a novel hydrophilic and antimicrobial core–shell nanoparticle was synthesized through the chemical grafting of poly(guanidine-hexamethylenediamine-PEI) (poly(GHPEI)) on the surface of SNP. As reported in our previous work,28 poly(GHPEI) is an efficient polymeric bactericide containing plenty of guanidine and amine groups. The chemical structure of poly(GHPEI) is shown in Fig. 1. The synthesized poly(GHPEI) grafted silica nanoparticles (SNP@PG) were incorporated into PES to fabricate hybrid UF membranes via phase inversion process. Chemical composition, surface and cross section morphologies, hydrophilicity and permeability of the nanocomposite membranes were detailedly evaluated by a series of characterizations.
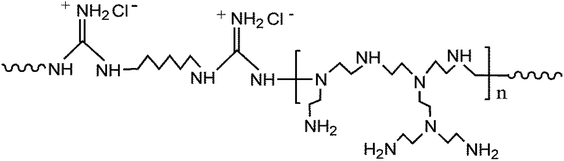 |
| | Fig. 1 The chemical structure of poly(GHPEI). | |
2. Experimental
2.1. Materials
PES (Ultrason E6020P, 58 kDa) was provided by BASF. 1-Methyl-2-pyrrolidone (NMP) was purchased from Real&lead Chemical Co., Ltd. (Tianjin, China). Guanidine hydrochloride (GH) (99.5%), 1,6-hexamethylenediamine (HMDA, 99.5%), tris(hydroxymethyl)aminomethane (Tris, 99%), 3-hydroxytyramine hydrochloride (dopamine, 98%) and polyethyleneimine (PEI, branched, 99%, Mw of ∼600 g mol−1) were supplied by Aladdin Reagent Co., Ltd. (Shanghai, China). Bovine serum albumin (BSA, 67 kDa) and egg albumin (EA, 43 kDa) were received from Beijing Solarbio Science & Technology Co., Ltd. Escherichia coli (E. coli) and Bacillus subtilis (B. subtilis) were purchased from Transgen Biotech (Beijing, China). All the reagents mentioned above were used without further treatment. Deionized water with a conductivity less than 15 μs cm−1 was produced by an ultrapure laboratory water purification system.
2.2. Synthesis and characterization of SNP@PG nanocomposites
SNP@PG nanocomposites were synthesized through the following steps.
Firstly, GH, HMDA and PEI were polymerized to prepare the guanidine-based polymer, namely poly(guanidine-hexamethylenediamine-PEI) (poly(GHPEI)). The synthesis process was conducted via a two-stage direct melt polycondensation according to our previous study.28 The poly(GHPEI) contained plenty of guanidine and amine groups and its minimum bactericidal concentration (MBC) was measured as 12.5 mg L−1. In addition, the weight-average molecular weight (Mw) and number-average molecular weight (Mn) of poly(GHPEI) are 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 144 and 11629, respectively.
144 and 11629, respectively.
Secondly, the SNPs were prepared according to the method reported in literature.29 Typically, 100 mL absolute ethanol, 3.4 g deionized water and 1.8 g ammonium hydroxide were mixed in a 250 mL three-necked flask which was put in a 30 °C thermostatic water-bath and stirred for 30 min. Then 0.52 g tetraethoxysilane (TEOS) was added to the flask, and the reaction was conducted for 3 h. After that, 54 g deionized water was added into the flask and vigorously mixed. Then 3.12 g TEOS was added and the reaction was kept under stirring for another 3 h. The silica nanoparticles were obtained by centrifuging the reacted liquid and washing with ethanol three times, and then drying in an oven.
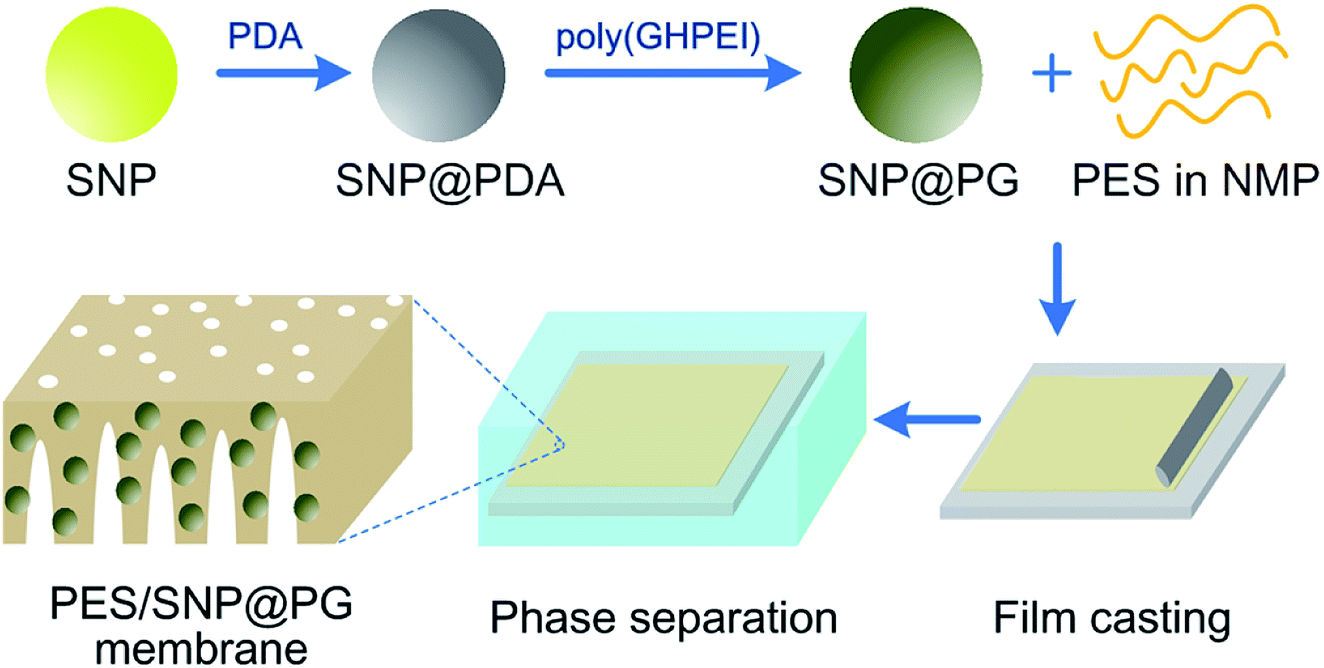
Next, a certain mass of SNP powder was dispersed into deionized water, followed by mixing with dopamine hydrochloride (2.0 g L−1) in tris(hydroxymethyl)aminomethane buffer (15 mM, pH 8.5). The reaction was kept for 12 h at room temperature with continuous stir. The remaining precipitate after the centrifugation was washed several times with deionized water and ethanol successively to obtain the gray powder, namely polydopamine modified silicon nanoparticles (SNP@PDA). Finally, a certain mass of SNP@PDA powder was added into 1 wt% poly(GHPEI) aqueous solution under vigorous stirring and kept for 12 h at room temperature. The black powder, namely poly(GHPEI) modified silica nanoparticles (SNP@PG), was then obtained after the centrifugation, washing and drying in sequence. Generally, the synthesis route of SNP@PG is shown in Fig. 2.
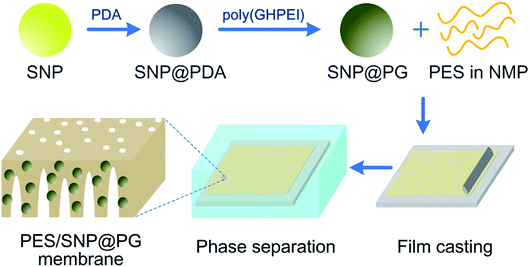 |
| | Fig. 2 Schematic diagram of SNP@PG synthesis route and PES/SNP@PG membrane fabrication process. | |
Chemical compositions of SNP and SNP@PG nanocomposites were characterized by Fourier transform infrared spectroscopy (FTIR, FTS-6000, Bio-Rad Inc., USA), X-ray photoelectron spectroscopy (XPS, PHI5000 VersaProbe, ULVAC-PHI Inc., Japan) and thermal gravimetric analysis (TGA, STA449f3, Netzsch Inc., Germany). Scanning electron microscope (SEM, NanoSEM430, FEI, USA) and transmission electron microscopy (TEM, JEM-100 CX, JEOL Ltd., Japan) was applied to observe the morphologies of SNP and SNP@PG nanocomposites.
2.3. Preparation of hybrid UF membranes
Membranes were prepared via non-solvent induced phase inversion process. For pristine PES membrane, the casting solution containing 15 wt% of PES in NMP was cast on a glass plate by using a casting knife with a gap of 200 μm and then exposed to the atmosphere for 30 s. It should be mentioned that the fabrication atmosphere was controlled at 25 °C and 20% relative humidity. Afterwards, the nascent film was carefully transferred into deionized water for precipitation. After precipitation, the membrane was rinsed and kept in deionized water for at least 24 h. For SNP or SNP@PG blended membranes, the casting solution contained 15 wt% and X wt% of SNP or SNP@PG in NMP. Hybrid membranes were prepared in the same way described above. The prepared hybrid membranes were denoted as PES/SNP-X or PES/SNP@PG-X membrane. Fig. 2 shows the PES/SNP@PG membrane fabrication process.
2.4. Characterization of membrane structure
The chemical composition of membranes was characterized by ATR-FTIR and XPS. Each membrane sample was dried at 60 °C for at least 12 h prior to the characterization.
The membrane surface and cross section morphologies were investigated by SEM. For cross section morphology observation, the membrane samples were pretreated by liquid nitrogen freezing and then fracturing. Before SEM analysis, the samples were dried and sputter-coated with a thin layer of gold. The membrane surface roughness was also tested by atomic force microscopy (AFM, Dimension Icon, Bruker) using a tapping mode in air.
The static water contact angle of membrane surface was measured by a contact angle goniometer (OCA15EC, Dataphysics, Germany) equipped with a video camera using sessile drop method. For each membrane sample, at least five water contact angle values were recorded.
2.5. Evaluation of membrane separation performance
2.5.1. Water flux and protein rejection measurement. The membrane separation performance was tested by using a cross-flow UF experimental apparatus. Each membrane coupon was cut into a circle with an effective area of 19.3 cm2 and supported by a piece of round non-woven fabric. Membrane compaction was firstly carried out under 0.30 MPa transmembrane pressure (TMP), 25 ± 2 °C and 0.22 m s−1 cross-flow velocity. After 30 min, a steady flux value was achieved and the TMP was adjusted to 0.20 MPa. The water flux under 0.20 MPa was calculated by eqn (1).| |
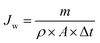 | (1) |
where Jw (L m−2 h−1) is the water flux, m (g) is the mass of the permeate, ρ is the water density at 25 °C (997 g L−1), A (m2) is the effective membrane area and Δt (h) is the permeation time.The rejection property of the membrane was evaluated from BSA (200 mg L−1) or EA (200 mg L−1) aqueous solution. The permeate and feed concentrations of BSA or EA were tested by using an UV-vis spectrophotometer. The protein rejection value was calculated by eqn (2).
| |
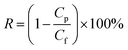 | (2) |
where
Cp and
Cf (mg L
−1) are the concentrations of protein in the permeate and feed solutions, respectively.
2.5.2. Antifouling property evaluation. Membrane antifouling property was tested in the filtration experiment of BSA (200 mg L−1) aqueous solution. After pre-compacted at 0.30 MPa TMP for 30 min, the initial water flux (Jw1) of was tested at 0.20 MPa TMP. Then 200 mg L−1 of BSA aqueous solution was fed into the feed tank and the flux was recorded at 0.20 MPa TMP, 25 ± 2 °C and 0.22 m s−1 cross-flow velocity. The antifouling behaviors of the membranes were tested through three fouling–cleaning cycles for over 400 min in total. During each cycle, the membrane coupon was fouled by BSA solution for 100 min and then cleaned with pure water for 30 min. The water flux of the membrane after three fouling–cleaning cycles was measured as Jw2. Flux recovery ratio (FRR) value was calculated by using eqn (3) to evaluate the antifouling property of the membrane.| |
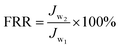 | (3) |
2.5.3. Antibacterial activity and anti-biofouling property evaluation. Membrane antibacterial activity was investigated by measuring the sterilization ratio. Firstly, the E. coli and B. subtilis strains were diluted to approximately 1.0 × 106 CFU mL−1. Then the diluted bacterial suspension of 100 μL was spread on the membrane surface with the help of a glass slide. After incubation at 37 °C for 2 h, the membrane together with the glass slide was washed repeatedly with 20 mL normal saline (0.9 wt% NaCl aqueous solution) to collect the bacterial cells. The obtained bacterial solution of 100 μL was uniformly dispersed on Luria–Bertani (LB) plate and kept at 37 °C for 16 h. Then the number of visible bacterial colonies was counted and the corresponding sterilization ratio was calculated by using eqn (4).| |
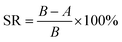 | (4) |
where SR is the sterilization ratio, A is the number of visible bacterial colonies after 2 h contact with membranes, and B is the number of visible bacterial colonies without contacting any membrane.Membrane anti-biofouling property was investigated in dynamic filtration operation. The bacterial feed solution was prepared by adding 10 mL of an overnight culture of E. coli broth into 2 L of saline. The flux was recorded under 0.20 MPa TMP, 25 ± 2 °C and 0.22 m s−1 cross-flow velocity for 90 min. The changes in water flux of the membrane samples were investigated to evaluate the anti-biofouling property.
3. Results and discussion
3.1. Characterization of SNP@PG nanocomposites
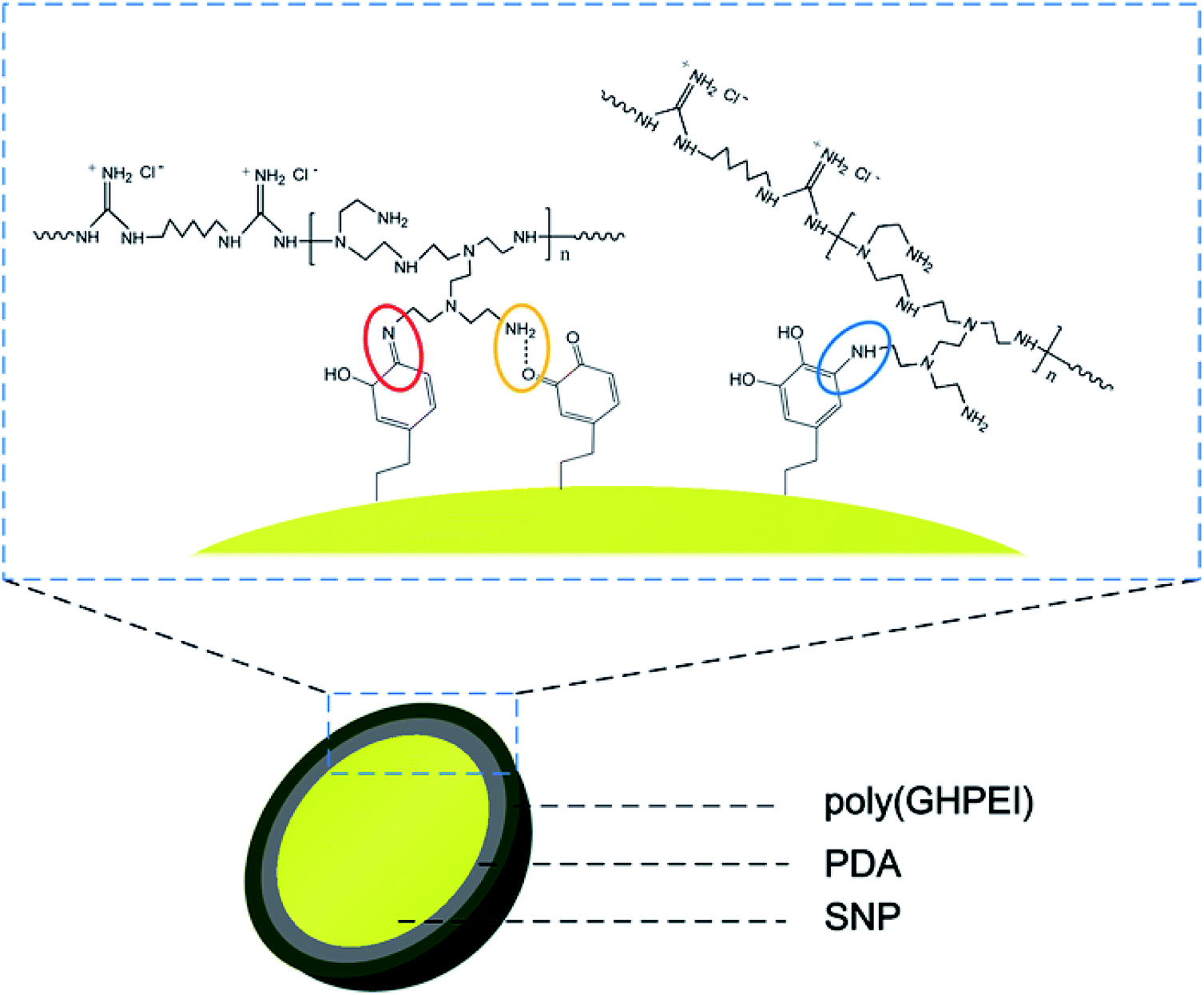
During the synthesis of SNP@PG, the adhesive and reductive PDA layer was used to assist the graft modification of poly(GHPEI) onto the surface of SNP. After the self-polymerization of dopamine, the primary amine group in poly(GHPEI) reacted with polydopamine through Michael-type addition or Schiff base reaction, leading to the formation of secondary amine group and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, respectively.28,30 In addition, the hydrogen bonding could be formed between the amine groups on poly(GHPEI) and catechol or quinones on PDA, according to literature.31,32 The chemical structure of SNP@PG shell layer is illustrated in Fig. 3.
N bond, respectively.28,30 In addition, the hydrogen bonding could be formed between the amine groups on poly(GHPEI) and catechol or quinones on PDA, according to literature.31,32 The chemical structure of SNP@PG shell layer is illustrated in Fig. 3.
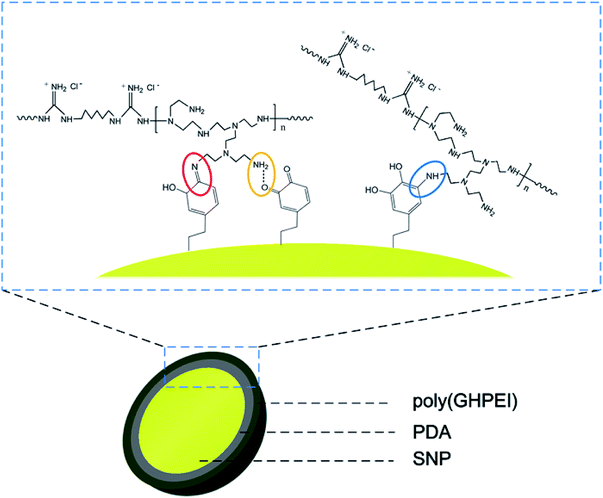 |
| | Fig. 3 Chemical structure of the shell layer on SNP@PG nanocomposites: Schiff base reaction (in red circle), Michael-type addition (in blue circle), and hydrogen bonding (in yellow circle). | |
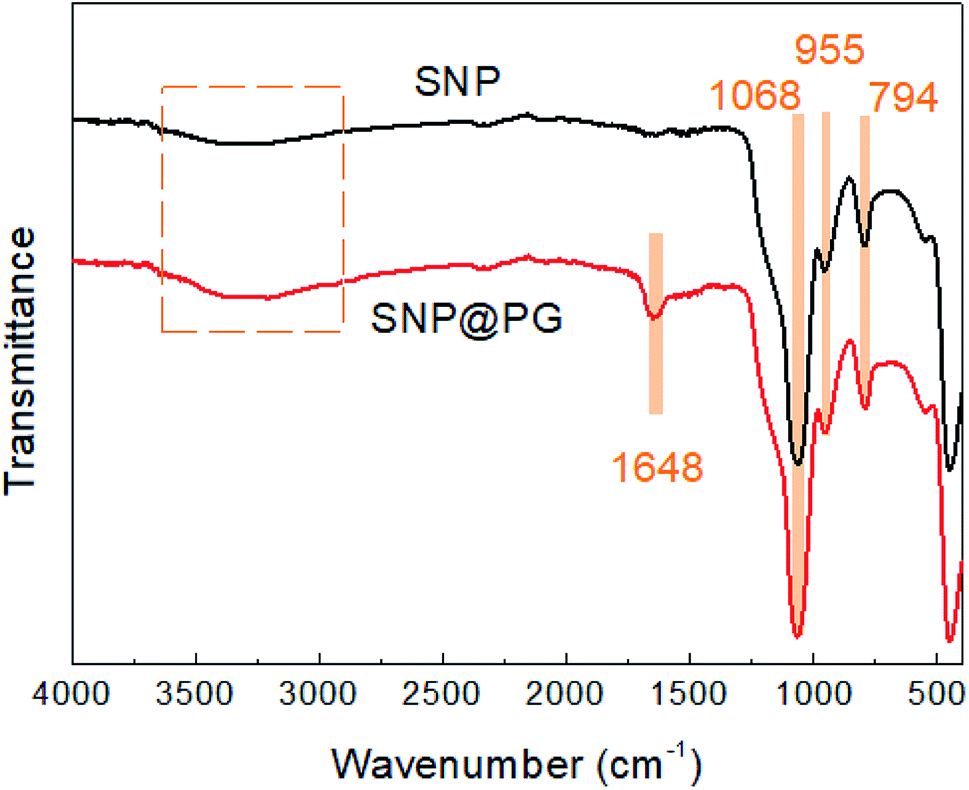
As shown in Fig. 4, the FTIR spectrum of SNP presents the characteristic adsorption peaks at 1068 cm−1 (Si–O–Si stretching vibration), 955 cm−1 (Si–OH bending vibration) and 794 cm−1 (Si–O stretching vibration), in agreement with the results reported in literature.29 After modification by PDA and poly(GHPEI), a new absorption peak appear at 1648 cm−1 for the spectrum of SNP@PG, which may be ascribed to the overlay of CN stretching vibration and –NHx bending vibration from poly(GHPEI).33,34
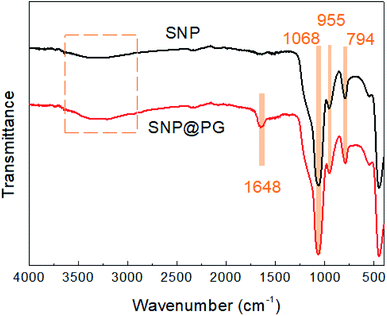 |
| | Fig. 4 FTIR spectra of SNP and SNP@PG nanocomposites. | |
Table 1 presents the element percentages of SNP and SNP@PG surfaces investigated from XPS. It can be seen that after the graft of poly(GHPEI), the C atomic percent increases largely from 5.29 at% to 59.08 at%, while the Si and O atomic percent decreases obviously. The N atom appears in the surface of SNP@PG. These results confirmed the successful graft of PDA and poly(GHPEI) onto SNP.
Table 1 The elemental percentages of SNP and SNP@PG surfaces obtained from XPS analysis
| Nanoparticles |
Atomic percent (at%) |
| C |
Si |
O |
N |
| SNP |
5.29 |
30.37 |
64.34 |
— |
| SNP@PG |
59.08 |
7.28 |
24.54 |
9.1 |
Fig. 5 shows the TG curves of SNP and SNP@PG tested from 50 to 900 °C. For SNP, the mass loss above 234 °C was ascribed to the fracture of Si–OH and the loss of hydroxyl group.35 For SNP@PDA or SNP@PG, the mass loss above 234 °C was caused by the fracture of Si–OH, as well as the decomposition of PDA or poly(GHPEI). When the samples were heated to 900 °C, the residual mass percentages for SNP, SNP@PDA and SNP@PG are 89.6%, 82.6% and 78.3%, respectively. These results indicated that the grafting contents of PDA and poly(GHPEI) were 7.7 wt% and 5.2 wt%, respectively.
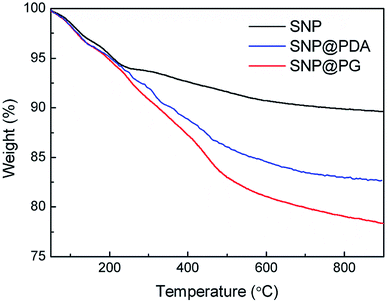 |
| | Fig. 5 TGA curves of SNP and SNP@PG nanocomposites. | |
As shown in Fig. 6, SNP shows regular spherical morphology and SNP@PG shows the core–shell structure. The diameter of SNP is 89 ± 9 nm, while that of SNP@PG slightly increases to 98 ± 12 nm with a rough shell layer. The hydrodynamic diameter and the dispersibility of nanoparticles were determined by DLS and shown in Table 2. It can be seen that the particle sizes of SNP and SNP@PG are around 124 ± 16 nm and 162 ± 25 nm, respectively. The particle dispersion indexes (PDI) of SNP and SNP@PG are both less than 0.1, indicating the well dispersion and narrow size distribution of SNP and SNP@PG. According to the zeta potential values, after the graft of poly(GHPEI), SNP@PG presents positive charge resulting from the introduction of amine and guanidine groups.
 |
| | Fig. 6 Morphological characterization of SNP (a and c) and SNP@PG (b and d) nanoparticles observed from SEM and TEM images. | |
Table 2 Hydrodynamic diameter, PDI and zeta potential of SNP and SNP@PG nanoparticles
| Nanoparticles |
Hydrodynamic diameter (nm) |
PDI |
Zeta potential (mV) |
| SNP |
124 ± 16 |
0.075 |
−42 ± 5 |
| SNP@PG |
162 ± 25 |
0.081 |
39 ± 4 |
3.2. Chemical characterization of the membranes
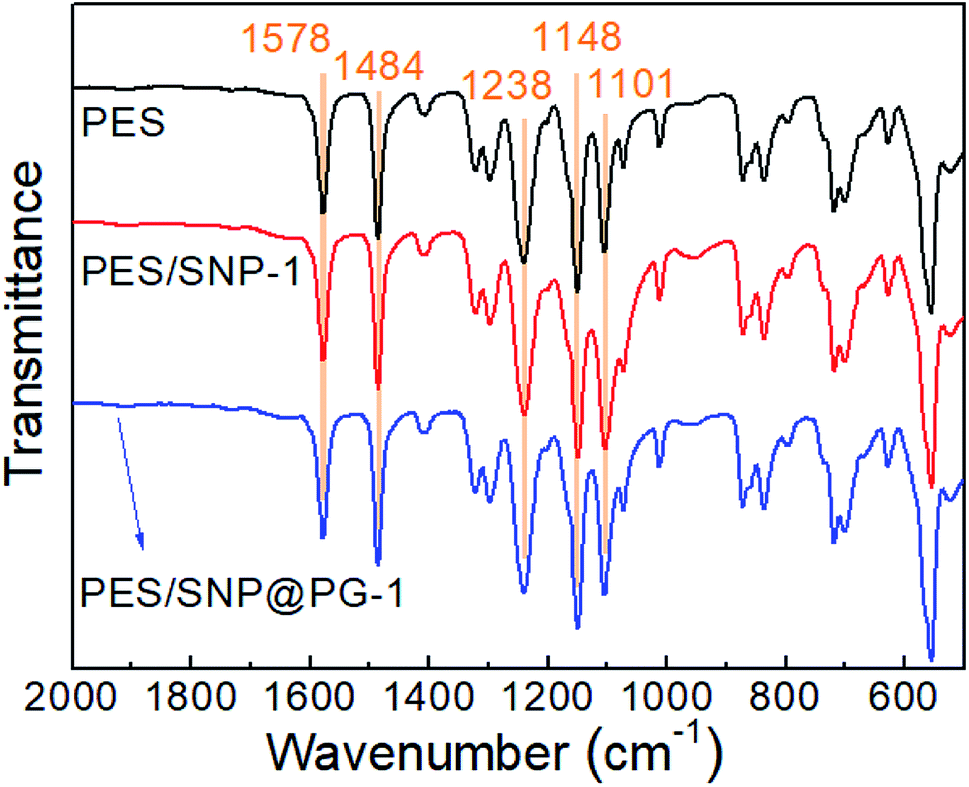
Fig. 7 shows ATR-FTIR spectra of the membranes. There is no obvious difference in the spectra of the membrane surfaces. Diameters of both SNP and SNP@PG are approaching only 100 nm and the blending contents are less than 2 wt%, thus the absorption peaks for SNP or SNP@PG are difficult to be detected. Characteristic peaks of PES can be seen at 1578 cm−1 (CC stretching vibration), 1484 cm−1 (C–C stretching vibration), 1238 cm−1 (C–O–C asymmetric vibration) and 1148 cm−1 (OSO stretching vibration), which are in accordance with literature.36,37
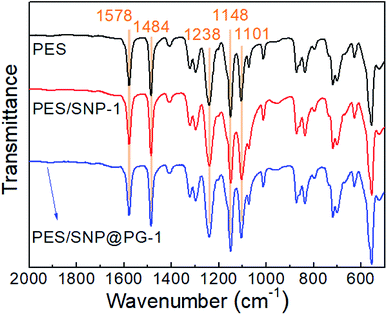 |
| | Fig. 7 ATR-FTIR spectra of PES, PES/SNP-1 and PES/SNP@PG-1 membrane surfaces. | |
Table 3 lists the elemental atomic percentages of the membranes. With the introduction of SNP or SNP@PG, Si element exists and O element increases obviously on membrane surface. Different from the pristine PES membrane and PES/SNP-1 membrane, the N element appears on the surface of PES/SNP@PG-1 membrane, confirming the successful graft of poly(GHPEI).
Table 3 The elemental percentages of PES, PES/SNP-1 and PES/SNP@PG-1 membrane surfaces obtained from XPS analysis
| Membrane |
Atomic percent (at%) |
| C |
Si |
O |
N |
S |
| PES |
72.48 |
— |
22.68 |
— |
4.84 |
| PES/SNP-1 |
71.38 |
0.92 |
23.12 |
— |
4.58 |
| PES/SNP@PG-1 |
71.10 |
0.61 |
23.23 |
0.65 |
4.41 |
3.3. Morphological characterization of the membranes
Fig. 8 shows the digital pictures of the membranes. Unlike the white PES and PES/SNP-1 membranes, PES/SNP@PG-1 membrane displays a brown color, which results from the graft of PDA and poly(GHPEI) layer on the nanoparticle surface. The membrane surface and cross section morphology at high magnification was investigated by SEM images and shown in Fig. 9. On one hand, the surface pores ranging from 6 to 15 nm are clearly observed from surface SEM images (a, c and e). The surface pore size and pore density were measured through ImageJ software and the results are listed in Table 4. In comparison with PES membrane, both PES/SNP-1 and PES/SNP@PG-1 membranes display surface pores with larger size and higher density, which was probably attributed to the enhancement in the thermodynamic demixing of the casting solution after incorporating hydrophilic SNP or SNP@PG into the casting solution.38 On the other hand, all the membrane cross sections exhibit similar asymmetric structure that includes a dense top layer, finger-like pores and macro pores. In addition, some nanoparticles could be observed in both the inner pores and the top layer of PES/SNP-1 and PES/SNP@PG-1 membranes, as shown in Fig. 9(d) and (f).
 |
| | Fig. 8 Digital pictures of PES (a), PES/SNP-1 (b), PES/SNP@PG-1 (c) membranes. | |
 |
| | Fig. 9 Surface and cross section SEM images of PES (a and b), PES/SNP-1 (c and d) and PES/SNP@PG-1 (e and f) membranes. Particles in the cross section are marked with yellow circles. | |
Table 4 Average surface pore sizes, surface pore densities, surface water contact angles and surface roughness values (Ra and Rq) of PES, PES/SNP-1 and PES/SNP@PG-1 membranes
| Membrane |
Surface pore size (nm) |
Surface pore density (number per μm2) |
Contact angle (°) |
Ra (nm) |
Rq (nm) |
| PES |
10.2 ± 1.8 |
18.6 ± 4.2 |
79.6 ± 4.5 |
9.8 |
11.2 |
| PES/SNP-1 |
12.4 ± 2.8 |
30.7 ± 3.5 |
71.4 ± 3.7 |
10.9 |
14.8 |
| PES/SNP@PG-1 |
12.8 ± 2.5 |
44.0 ± 4.1 |
57.3 ± 3.5 |
12.4 |
15.9 |
The hydrophilicity of membrane surface was investigated by water contact angle measurement. As shown in Table 4, the water contact angle of PES membrane is 79.6 ± 4.5°. The addition of SNP improved the membrane surface hydrophilicity with the contact angle of 71.4 ± 3.7°. SNP@PG further improved the membrane hydrophilicity, making the water contact angle decrease to 57.3 ± 3.5°. The improvement of membrane surface hydrophilicity was owing to better affinity to water with the help of hydrophilic nanoparticles in the top layer, which would also result in a higher water flux.
Membrane surface roughness was expressed as average value (Ra) and root mean square value (Rq), which were obtained through AFM analysis. As listed in Table 4, PES/SNP-1 and PES/SNP@PG-1 hybrid membranes have slightly greater roughness values in comparison with PES membrane. The addition of SNP or SNP@PG produced a rougher membrane surface probably because hydrophilic additives would improve the exchange rate between solvent and nonsolvent during the phase inversion.
3.4. Permeation flux and rejection
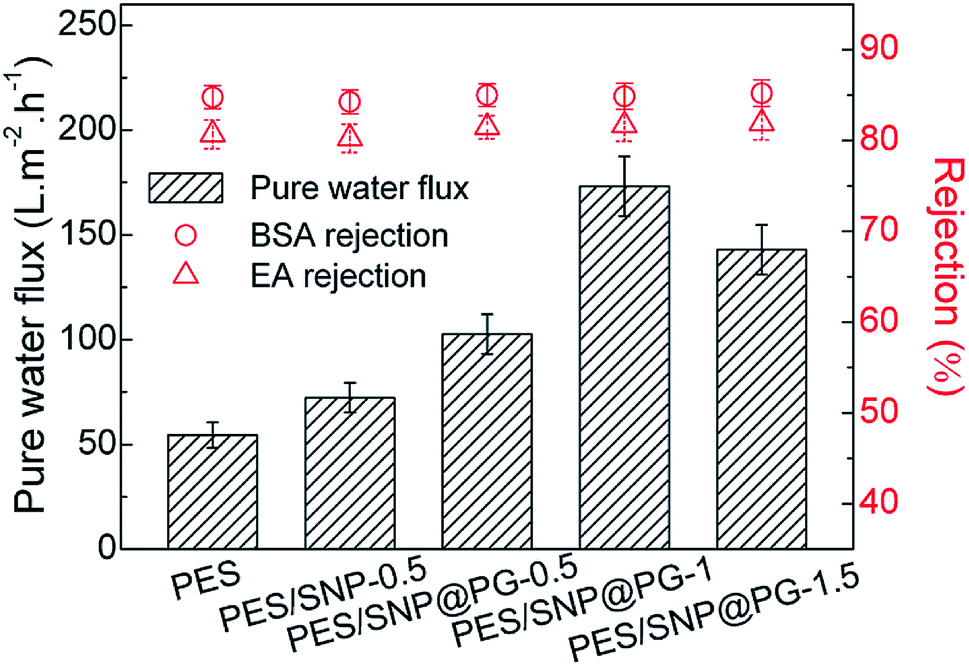
Fig. 10 shows the pure water fluxes and protein rejections of the membranes at 0.20 MPa TMP. In general, the incorporation of SNP or SNP@PG nanocomposites improves the water flux of the membranes obviously. Compared with PES membrane, the water fluxes of PES/SNP-0.5 and PES/SNP@PG-0.5 membranes show increase of 32.7% and 88.4% respectively. When increasing the loading contents of SNP@PG from 0.5 wt% to 1.5 wt%, the water flux of the PES/SNP@PG membranes increases quickly and then decreases. The optimal content for SNP@PG addition is 1 wt% only taking water flux into consideration, and the water flux of PES/SNP@PG-1 membrane is 173.2 ± 14.2 L m−2 h−1, 2.6 times that of the pristine PES membrane. All the membranes show similar rejections, with ∼85% of BSA rejection and ∼82% of EA rejection, probably due to their similar surface pore size ranging from 6 to 15 nm.
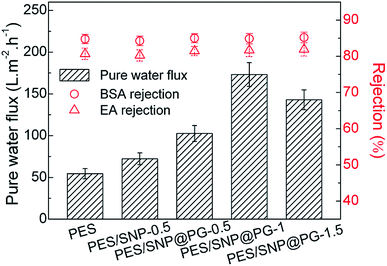 |
| | Fig. 10 The pure water fluxes, BSA rejections and EA rejections of PES, PES/SNP and PES/SNP@PG membranes at 0.20 MPa TMP. BSA or EA concentration in the feed solution: 200 ppm. | |
The change in water flux of the membranes can be explained as follows. Firstly, the addition of hydrophilic SNP@PG nanocomposites accelerated the phase separation, leading to the formation of surface pores with larger size and higher density, which was consistent with the results in Fig. 9 and Table 4. Secondly, as the loading content of SNP@PG further increases, the solvent and nonsolvent exchange was suppressed to some extent, leading to the formation of dense layer and thereby results in the decrease of water flux.39,40 Thirdly, there are more hydrophilic amine groups on the SNP@PG shell layer than the SNP surface. Thus, the addition of SNP@PG further improves the hydrophilicity of membrane surface and correspondingly facilitates water transport.
3.5. Antifouling property
Fig. 11 shows the permeate fluxes of the membranes during three fouling–cleaning cycles. It can be seen that in each fouling–cleaning cycle, permeation fluxes of the membranes drop dramatically during the initial 15 min filtration and then keep a slow decline. After the third fouling, the fluxes of PES, PES/SNP-1 and PES/SNP@PG-1 membranes have 49.7%, 39.0%, and 21.6% decrease, respectively, probably due to the adsorption or deposition of protein on membrane surface and pores. After the third cleaning, PES, PES/SNP-1 and PES/SNP@PG-1 membranes get 62.2%, 72.8%, and 84.4% of flux recovery, respectively. Among these three membranes, PES/SNP@PG-1 membrane shows the lowest flux decline ratio and the highest flux recovery ratio, thereby having the best antifouling property during BSA filtration. The improvement of antifouling property primarily resulted from the enhanced surface hydrophilicity after incorporating SNP@PG nanocomposites, which helps to create a hydrated layer that alleviates the adhesion of foulant on the membrane surfaces or pores.
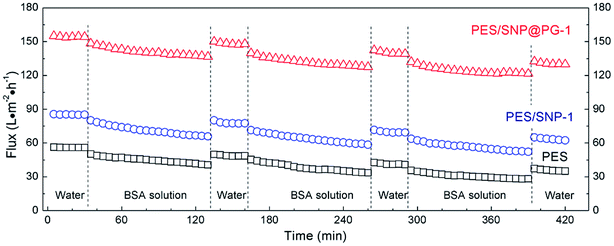 |
| | Fig. 11 The permeate fluxes of PES, PES/SNP-1 and PES/SNP@PG-1 membranes during BSA filtration at 0.20 MPa TMP. BSA concentration in the feed solution: 200 ppm. | |
3.6. Antibacterial activity of the membranes
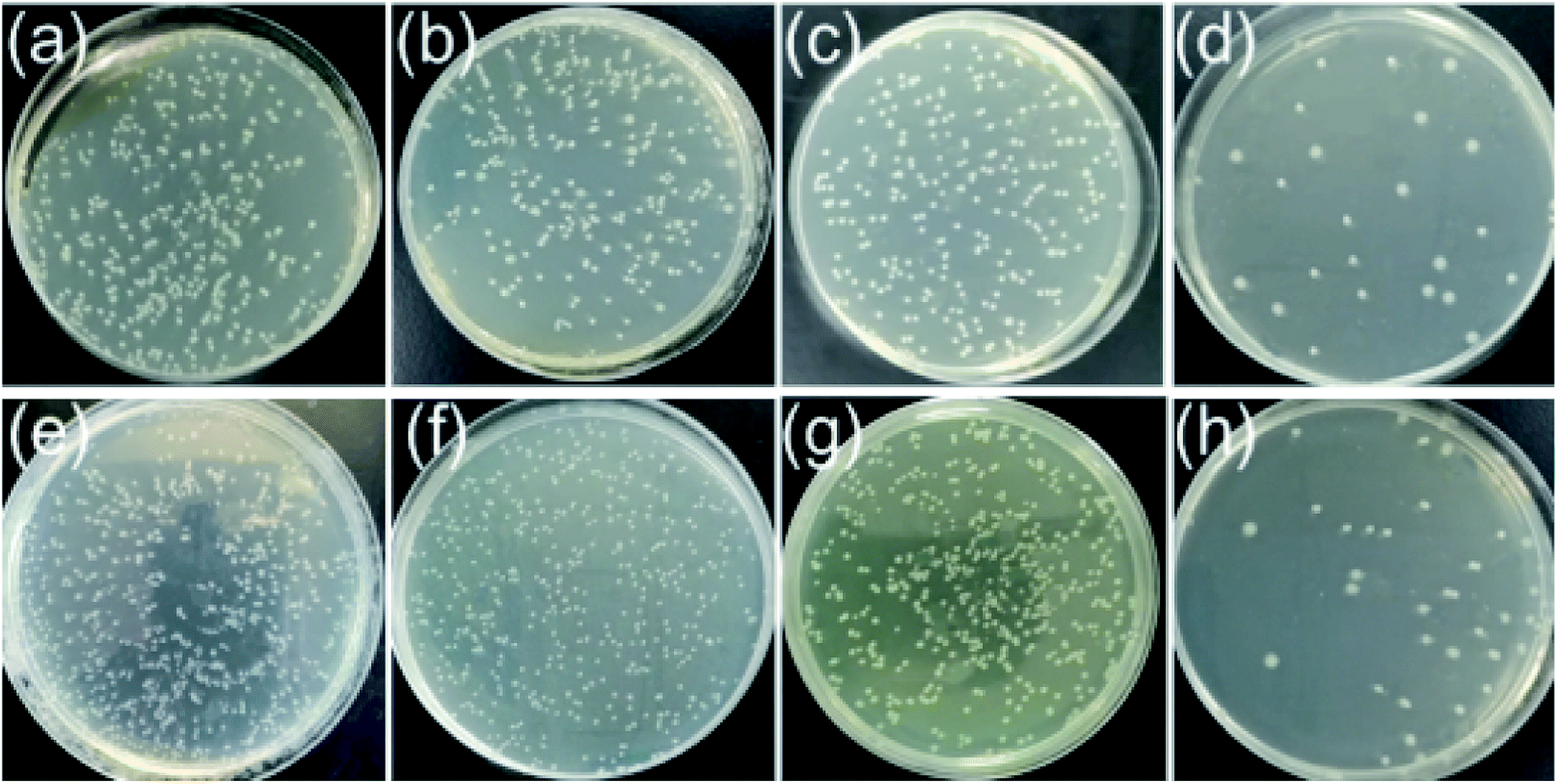
The antibacterial activities of PES, PES/SNP-1 and PES/SNP@PG-1 membranes were evaluated. Fig. 12 and Table 5 show the sterilization performance of the membranes against E. coli and B. subtilis. The sterilization ratios of PES membrane are 23.7 ± 3.3% and 21.3 ± 2.7% after 2 h contact with E. coli and B. subtilis suspensions, respectively. The values for PES/SNP-1 membrane show the similar level to PES membrane. While the sterilization ratios of PES/SNP@PG-1 membrane are 92.7 ± 2.1% and 92.0 ± 2.5% after 2 h contact with E. coli and B. subtilis suspensions, respectively. The decrease of bacterial colonies in PES or PES/SNP-1 membranes may be due to the natural death of cells in the condition that cell suspension solutions were prepared without adding nutrients. The improved antibacterial ability of PES/SNP@PG-1 membrane can be attributed to the introduction of guanidine enriched poly(GHPEI) through the blending of SNP@PG. During the phase inversion, SNP@PG nanocomposites spontaneously migrated to the membrane surface,22 and thereby PES/SNP@PG membrane could play the role of an efficient bactericide. In addition, after immersed the membrane into water for one month, the sterilization ratios of PES/SNP@PG-1 membrane could still keep around 91.7% and 91.2% against E. coli and B. subtilis, respectively, indicating the good stability of SNP@PG into the membrane surface.
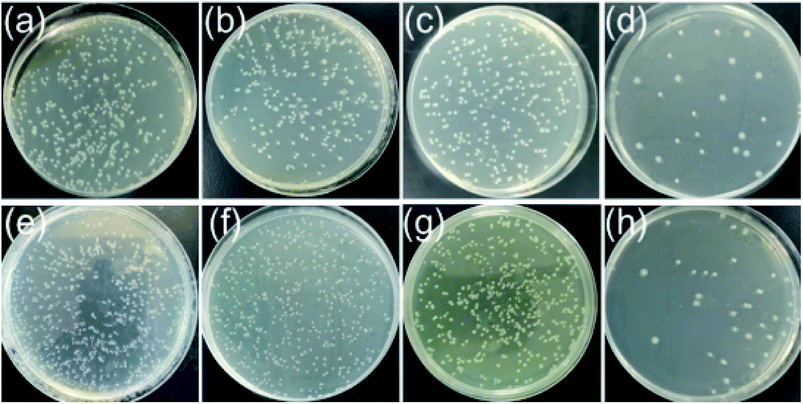 |
| | Fig. 12 The E. coli (top row) and B. subtilis (bottom row) colonies after 2 h contacting without any membrane (a and e), with PES membrane (b and f), with PES/SNP-1 membrane (c and g) and with PES/SNP@PG-1 membrane (d and h). LB plates were kept at 37 °C for 12 h. Inoculum levels were ca. 1.0 × 108 CFU m−2 for E. coli and 1.6 × 108 CFU m−2 for B. subtilis. | |
Table 5 The sterilization ratios of PES, PES/SNP-1 and PES/SNP@PG-1 membranes
| Membrane |
Sterilization ratio (%) |
| E. coli |
B. subtilis |
| PES |
23.7 ± 3.3 |
21.3 ± 2.7 |
| PES/SNP-1 |
23.5 ± 3.1 |
21.6 ± 2.8 |
| PES/SNP@PG-1 |
92.7 ± 2.1 |
92.0 ± 2.5 |
3.7. Anti-biofouling property
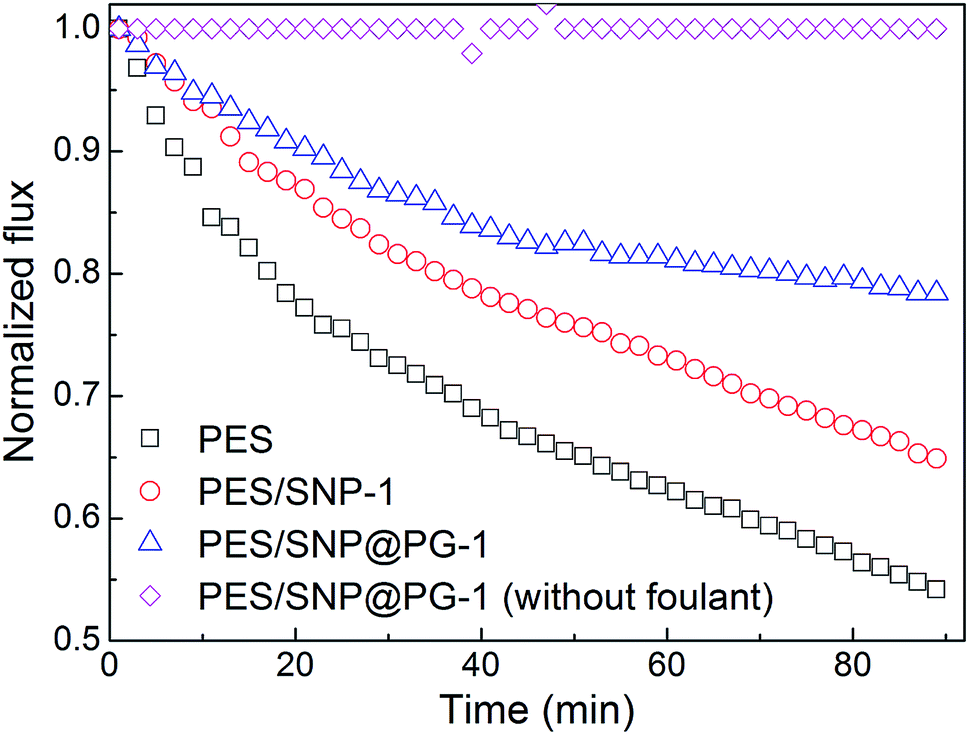
The biofouling experiment was conducted through the evaluation of membrane flux decline during bacterial suspension filtration. As shown in Fig. 13, the water fluxes of the membranes decrease quickly during 90 min biofouling operation. As the control group, water flux of PES/SNP@PG-1 membrane without foulant kept almost unchanged during 90 min test, indicating that the membrane exhibited stable water flux after compaction under 0.30 MPa. While under biofouling, the fluxes of PES membrane and PES/SNP-1 membrane decrease by 45.8% and 35.1%, respectively. In contrast, the PES/SNP@PG-1 membrane loses only 21.6% of the water flux after biofouling. The improved anti-biofouling property can be explained in the following aspects. Firstly, the addition of SNP@PG improved the membrane surface hydrophilicity, which could alleviate the adhesion of bacteria cells onto the membrane surface. Secondly, guanidine groups in SNP@PG surface could inhibit the growth of bacteria, based on the great antibacterial ability of poly(GHPEI).28 Thus, PES/SNP@PG-1 membrane could prevent the blockage of surface pores induced by bacterial attachment and exhibit the best anti-biofouling property compared with PES and PES/SNP membranes.
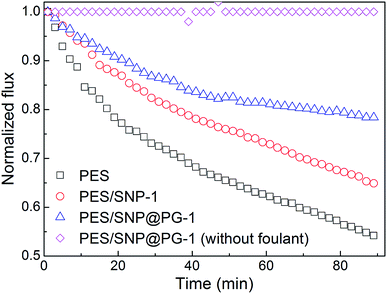 |
| | Fig. 13 The permeate flux decline of PES, PES/SNP-1 and PES/SNP@PG-1 membranes during bacterial fouling of E. coli. | |
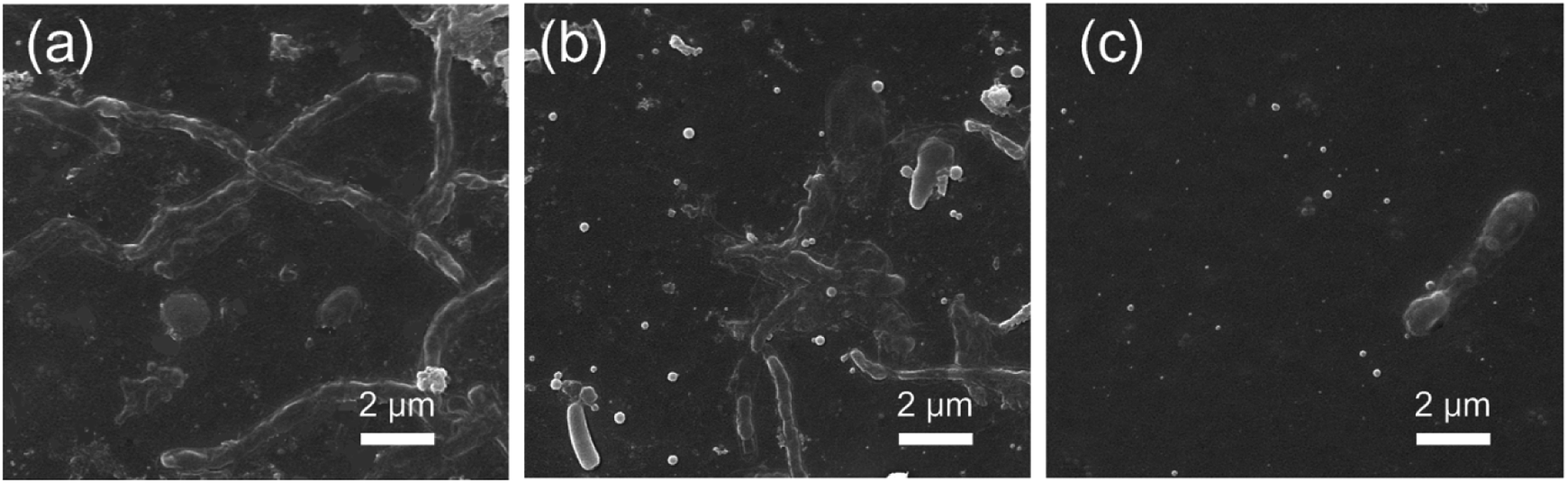
Fig. 14 shows the surface SEM images of PES, PES/SNP-1 and PES/SNP@PG-1 membranes after 90 min biofouling. It can be seen that lots of bacterial cells could be observed from the surface of PES and PES/SNP-1 membranes while PES/SNP@PG-1 surface is obviously cleaner.
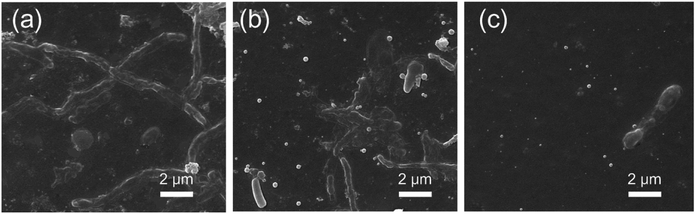 |
| | Fig. 14 The surface SEM images of PES (a), PES/SNP-1 (b), and PES/SNP@PG-1 (c) membrane after bacterial fouling tests. | |
4. Conclusions
In this study, novel hydrophilic and antimicrobial core–shell nanoparticles were prepared by grafting poly(GHPEI) onto the surface of silica via PDA immobilization. Then hybrid ultrafiltration membranes were fabricated by blending PES with poly(GHPEI) functionalized silica nanoparticles (SNP@PG). The obtained hybrid PES/SNP@PG membranes displayed not only improved water flux, but also excellent resistance to bacteria and organic fouling. The hydrophilicity of SNP@PG surface is beneficial to the formation of the membrane surface with better porous structure and enhanced hydrophilicity. According to the characterization results, both surface pore size and pore density improved obviously, and the water contact angle of membrane surface decreased from 79.6° to 57.3° after doping SNP@PG nanocomposites. Thus the PES/SNP@PG-1 membrane achieved an increased water flux, around 2.6 times that of the pristine PES membrane and maintained similar protein rejection. The antifouling property during BSA filtration was enhanced as well. Moreover, the guanidine and amine enriched surface of SNP@PG endowed the membrane surface with good antimicrobial activity and biofouling resistance. It can be concluded that the integrated hydrophilic and antimicrobial nanoparticles are applicable for surface modification of water treatment membranes and other coating materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by grants from National Natural Science Foundation of China (No. 21776205), Tianjin Research Program of Basic Research and Frontier Technology (No. 15JCQNJC43400), and the Program of Introducing Talents of Discipline to Universities (No. B06006). The authors gratefully acknowledge assistance in membrane characterization by Analysis Center of Tianjin University.
References
- H. Chang, H. Liang, F. Qu, B. Liu, H. Yu, X. Du, G. Li and S. A. Snyder, J. Membr. Sci., 2017, 540, 362–380 CrossRef.
- R. W. Baker, Membrane Technology and Applications,West Sussex, Wiley, 2004 Search PubMed.
- D. J. Miller, D. R. Dreyer, C. W. Bielawski, D. R. Paul and B. D. Freeman, Angew. Chem., Int. Ed. Engl., 2017, 56, 4662–4711 CrossRef PubMed.
- R. Zhang, Y. Liu, M. He, Y. Su, X. Zhao, M. Elimelech and Z. Jiang, Chem. Soc. Rev., 2016, 45, 5888–5924 RSC.
- J. Tian, C. Wu, H. Yu, S. Gao, G. Li, F. Cui and F. Qu, Water Res., 2018, 132, 190–199 CrossRef PubMed.
- R. Bernstein, C. E. Singer, S. P. Singh, C. Mao and C. J. Arnusch, J. Membr. Sci., 2018, 548, 73–80 CrossRef.
- X. Fang, J. Li, X. Li, S. Pan, X. Sun, J. Shen, W. Han, L. Wang and B. Van der Bruggen, J. Colloid Interface Sci., 2017, 505, 642–652 CrossRef PubMed.
- J. I. Clodt, V. Filiz, S. Rangou, K. Buhr, C. Abetz, D. Höche, J. Hahnet, J. Hahn, A. Jung and V. Abetz, Adv. Funct. Mater., 2013, 23, 731–738 CrossRef.
- D. Keskin, J. I. Clodt, J. Hahn, V. Abetz and V. Filiz, Langmuir, 2014, 30, 8907–8914 CrossRef PubMed.
- J. Zhang, T. Lin and W. Chen, Chem. Eng. J., 2017, 325, 160–168 CrossRef.
- P. Bengani-Lutz, R. D. Zaf and P. Z. Culfaz-Emecen, J. Membr. Sci., 2017, 543, 184–194 CrossRef.
- H. Gao, X. Sun and C. Gao, J. Membr. Sci., 2017, 542, 81–90 CrossRef.
- X. Wu, Z. Xie, H. Wang, C. Zhao, D. Ng and K. Zhang, RSC Adv., 2018, 8, 7774–7784 RSC.
- L. Zhou, K. Gao, Z. Jiao, M. Wu, M. He, Y. Su and Z. Jiang, Appl. Surf. Sci., 2018, 440, 113–124 CrossRef.
- N. Misdan, A. F. Ismail and N. Hilal, Desalination, 2016, 380, 105–111 CrossRef.
- F. Li, J. Ye, L. Yang, C. Deng, Q. Tian and B. Yang, Appl. Surf. Sci., 2015, 345, 301–309 CrossRef.
- C. Wu, Z. Wang, S. Liu, Z. Xie, H. Chen and X. Lu, J. Membr. Sci., 2018, 548, 50–58 CrossRef.
- J. Zhang, Y. N. Xu, S. Chen, J. Li, W. Han, X. Sun, D. Wu, Z. Hu and L. Wang, Appl. Surf. Sci., 2017, 434, 806–815 CrossRef.
- P. Biswas and R. Bandyopadhyaya, J. Colloid Interface Sci., 2017, 491, 13–26 CrossRef PubMed.
- A. K. Shukla, J. Alam, M. Alhoshan, L. A. Dass and M. R. Muthumareeswaran, Sci. Rep., 2017, 7, 41976 CrossRef PubMed.
- J. Li, X. Liu, J. Lu, Y. Wang, G. Li and F. Zhao, J. Colloid Interface Sci., 2016, 484, 107–115 CrossRef PubMed.
- S. Al Aani, V. Gomez, C. J. Wright and N. Hilal, Chem. Eng. J., 2017, 326, 721–736 CrossRef.
- S. Ghasemian, M. A. Sahari, M. Barzegar and H. Ahmadi Gavlighi, Food Chem., 2017, 230, 454–462 CrossRef PubMed.
- H. Yu, Y. Zhang, J. Zhang, H. Zhang and J. Liu, Desalin. Water Treat., 2013, 51, 3584–3590 CrossRef.
- C. Zhao, J. Lv, X. Xu, G. Zhang, Y. Yang and F. Yang, J. Colloid Interface Sci., 2017, 505, 341–351 CrossRef PubMed.
- K. Albert, X. C. Huang and H. Y. Hsu, Adv. Colloid Interface Sci., 2017, 249, 272–289 CrossRef PubMed.
- H. Du, Y. Wang, X. Yao, Q. Luo, W. Zhu, X. Li and Z. Shen, Polym. Chem., 2016, 7, 5620–5624 RSC.
- Y. Gao, S. Zhao, Z. Qiao, Y. Zhou, B. Song, Z. Wang and J. Wang, Desalination, 2018, 430, 74–85 CrossRef.
- S. Li, J. Wang, S. Zhao, W. Cai, Z. Wang and S. Wang, J. Mater. Sci. Technol., 2017, 33, 261–265 CrossRef.
- Y. Tian, Y. Cao, Y. Wang, W. Yang and J. Feng, Adv. Mater., 2013, 25, 2980–2983 CrossRef PubMed.
- P. Georgopanosa, E. Eichnerb, V. Filiza, U. A. Handgea, G. A. Schneiderc, S. Heinrichb and V. Abetz, Compos. Sci. Technol., 2017, 146, 73–82 CrossRef.
- J. Jiang, L. Zhu, L. Zhu, H. Zhang, B. Zhu and Y. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 12895–12904 CrossRef PubMed.
- X. Li, Y. Cao, H. Yu, G. Kang, X. Jie, Z. Liu and Q. Yuan, J. Membr. Sci., 2014, 466, 82–91 CrossRef.
- H.-C. Yang, K.-J. Liao, H. Huang, Q.-Y. Wu, L.-S. Wan and Z.-K. Xu, J. Mater. Chem. A, 2014, 2, 10225–10230 RSC.
- L. Yu, Y. Zhang, Y. Wang, H. Zhang and J. Liu, J. Hazard. Mater., 2018, 287, 373–383 CrossRef PubMed.
- S. Zhu, S. Zhao, Z. Wang, X. Tian, M. Shi, J. Wang and S. Wang, J. Membr. Sci., 2015, 493, 263–274 CrossRef.
- J. A. Prince, S. Bhuvana, V. Anbharasi, N. Ayyanar, K. V. Boodhoo and G. Singh, Water Res., 2016, 103, 311–318 CrossRef PubMed.
- S. Liang, G. Qi, K. Xiao, J. Sun, E. P. Giannelis, X. Huang and M. Elimelech, J. Membr. Sci., 2014, 463, 94–101 CrossRef.
- H. Sun, B. Tang and P. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 21473–21484 CrossRef PubMed.
- B. Díez, N. Roldán, A. Martín, A. Sotto, J. A. Perdigón-Melón, J. Arsuaga and R. Rosal, J. Membr. Sci., 2017, 526, 252–263 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Yunlong Qiab,
Wen Zhangab,
Xuefei Sunab,
Zhi Wang
*ab,
Yunlong Qiab,
Wen Zhangab,
Xuefei Sunab,
Zhi Wang