
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFeasibility of polyethylene composites reinforced by distillers dried fibers with solubles (DDFS) after different generations of ethanol fermentation†
Bo Chena,
Xiaotong Shena,
Zhangfeng Luoa,
Huidong Chenbc,
Di Cai *a,
Changjing Chena,
Changwei Zhanga,
Peiyong Qina,
Hui Cao*a and
Tianwei Tana
*a,
Changjing Chena,
Changwei Zhanga,
Peiyong Qina,
Hui Cao*a and
Tianwei Tana
aNational Energy R&D Center for Biorefinery, Beijing University of Chemical Technology, Beijing 100029, PR China. E-mail: caidibuct@163.com; caohui@mail.buct.edu.cn
bCollege of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, PR China
cCenter for Process Simulation & Optimization, Beijing University of Chemical Technology, Beijing 100029, PR China
First published on 18th July 2018
Abstract
In order to effectively evaluate the distillers dried fibers with solubles (DDFS) obtained from biorefinery processes, sorghum distiller (SD), cassava distiller (CD) and corn cob distiller (CCD) residuals from different generation bioethanol plants were used as the reinforcing phase for polyethylene composites. The mechanical performances and the physical properties of the polyethylene/DDFS composites were evaluated. The results showed that the CCD reinforced specimen offered the best mechanical performances, with a flexural strength of 21.8 ± 2.2 MPa and a tensile strength of 39.7 ± 3.2 MPa. After multigelation, the retention ratios of the rupture modulus and the elasticity modulus of the CCD reinforced specimen reached 88.7 ± 6.7% and 84.1 ± 2.7%, while after 2000 h of xenon lamp weathering they reached 96.2 ± 4.7% and 82 ± 1.8%, respectively. Hybridizing the biorefinery process with the process of composites production was feasible.
Introduction
Due to concerns about environmental change and an energy crisis, there is increasing attention on biofuel production.1 Bioethanol is considered to be a clean and renewable fuel that offers environmental benefits.2 The production of bioethanol from renewable biomass resources can generally be classified into two generations.3 The first generation (1 G) biofuels based on edible food based feedstock can be questioned due to the trade-off between food and fuel supplies.4 The second generation (2 G) biofuels that are derived from lignocellulosic biomass materials are considered more broadly social acceptable and are being widely studied.5 Moreover, there is another generation of biofuels, namely the one and a half generation (1.5 G) biofuels, that utilize non-food starchy or sugar based raw materials. The 1.5 G biofuels are easier to produce than the 2 G biofuels since the latter suffer from difficulties with delignification.6No matter which generation of fuels is produced, a common bottleneck which needs to be properly addressed is the identification of an appropriate method for dealing with the solid residuals, to extend the value chain. The distillers dried grains with solubles (DDGS) from the 1 G plants can be applied as animal feed, and heat and electricity can be generated from the lignocellulosic residuals of the 2 G plants, but a more profitable strategy is to produce wood-plastic composites (WPCs) using these solid residuals as the reinforcing phase.7–9
WPC is a ‘green’ biomaterial with low density and good mechanical and physical properties, which could partly replace woody materials and avoid the severe environmental consequences of accelerated deforestation.10,11 In previous studies, DDGS, the residual after 1 G biofuel production, was used as the reinforcing phase in poly (butylene adipate-co-terephthalate) (PBAT) and polyhydroxyalkanoate (PHA) matrices.12,13 For the 1.5 G biorefinery residuals, composites from polyhydroxybutyrate (PHB) and potato peel waste fermentation residuals were prepared.14 In our previous works, renewable polylactic acid (PLA)/sweet sorghum bagasse composites were obtained using residuals from the 1.5 G bioethanol and 2 G biobutanol plants.9,15 Besides, a PBAT/microalgae biomass residual based biocomposite was also obtained.16
Although numerous research studies have focused on WPC production from biorefinery residuals, no research has ever focused on a comparison of the mechanical and physical properties of the WPCs reinforced by the residuals from different generations of biofuel plants. In fact, the WPCs could effectively maximize the economic value of a biorefinery. Hence, there is a need to compare the properties of the WPCs produced from the diverse biofuel production routes. Here, a comparative study based on polyethylene composites reinforced by distillers dried fibers with solubles (DDFS) from different generations of biofuels was conducted. After simultaneous saccharification and fermentation (SSF) for ethanol production, DDFS were obtained as solid residuals. The sorghum distiller (SD), cassava distiller (CD) and corn cob distiller (CCD) residuals from the 1 G, the 1.5 G and the 2 G biofuel plants were utilized as the reinforcing phase (Fig. 1). The composition and the characterization of the different DDFS residuals were analyzed, and the mechanical and physical properties of the composites were then evaluated. The ageing resistances of the WPCs were also compared. The results suggest that the polyethylene/DDFS composites are promising for practical application. The process for the production of composites has the potential to be combined with the biorefinery processes.
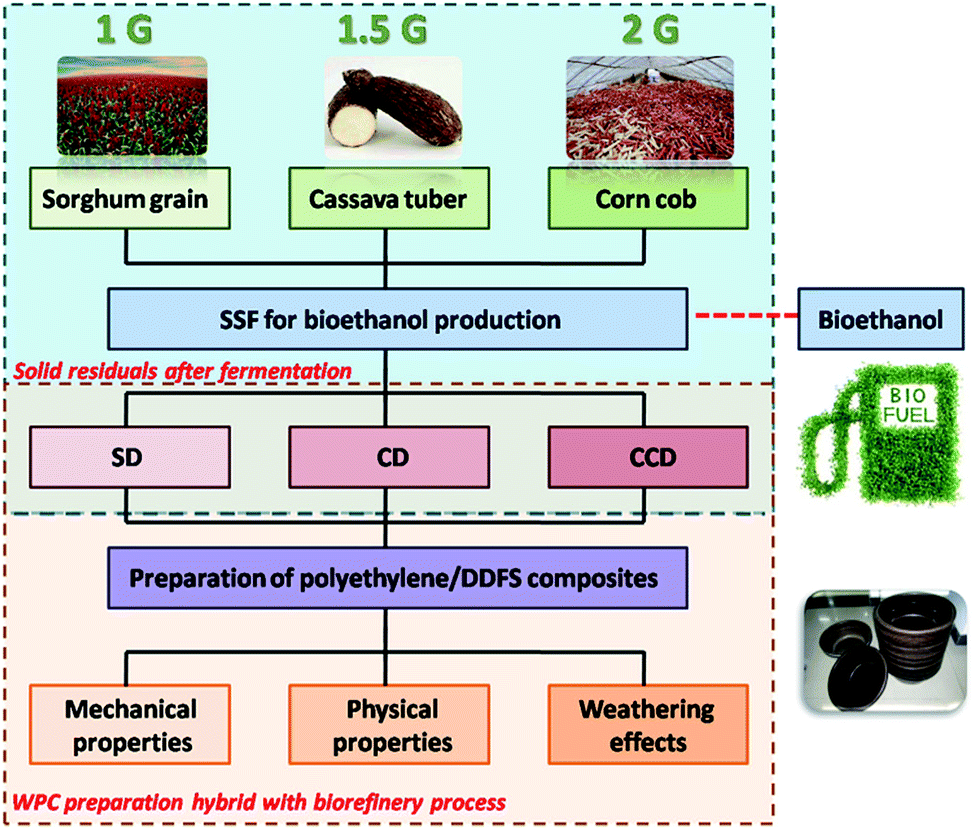 | ||
| Fig. 1 Flowchart of the process for the preparation of polyethylene/DDFS composites. Solid residuals from 1 G, 1.5 G and 2 G SSF bioethanol plants were used as the reinforcement. | ||
Experimental
DDFS preparation
The SD residual was obtained from the 1 G plant, and was kindly provided by local spirit distilleries in Shandong Province, China. It was derived from sorghum grain and was obtained after SSF. The CD residual was obtained from the 1.5 G plant, COFCO, China. The CCD residual derived from corn cob was kindly provided by the Longlive group located in Yucheng, Shandong Province, China. It was obtained at the end of SSF after a two-stage pretreatment. All the residuals were dried out overnight at 105 °C. Then, the solids were milled into a fine size (60–80 mesh) and were stored at −20 °C before use.Characteristics of DDFS residuals
The chemical compositions of the DDFS residuals were measured following the method of NREL.17 The functional groups of the DDFS residuals were analyzed by Fourier transform infrared spectroscopy (FT-IR) (Nicolet 6700, Thermo Fisher, USA). An X-ray diffractometer (D 8, Bruker, USA) was applied to analyze the crystalline cellulose in the DDFS residuals. A scan rate of 0.02° s−1 was used in the range of 3° to 60°. Results were calculated based on eqn (1):18
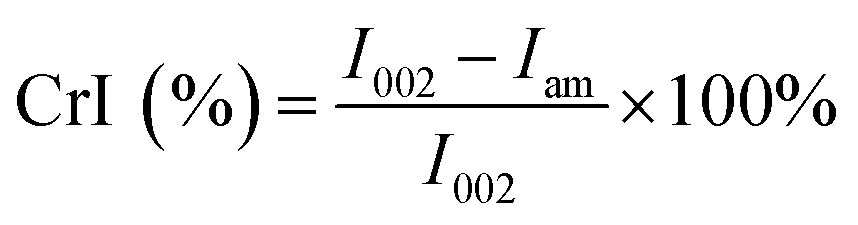 | (1) |
Preparation of polyethylene/DDFS composites
A co-rotating twin-screw extruder (Coperion ZSK, Werner & Pfleiderer, Germany) was used for the preparation of the polyethylene/DDFS composites. A mixture of 50 wt% of DDFS, 40 wt% of HDPE 600, 4 wt% of stearic acid, 3 wt% of MA-HDPE and 3 wt% of polyethylene wax was extruded under the zone temperatures of 140–175 °C as described in the study by Luo et al.19 Stearic acid was used as the outer lubricant, MA-HDPE was the interface modifying agent, and the polyethylene wax was the inner lubricant. The injection of a standard specimen was performed at 140–165 °C and 50 MPa using an injection moulding machine (HTF 120 X2, Haitian, China).Determination of the mechanical properties of the composites
The tensile and flexural strengths, the tensile and flexural moduli and the elongations at break of the composites were tested using a material testing machine (Instron 1185, USA) according to the test methods described in the government standard for commercial wood–plastic composites in China (GB/T 24508-2009).20 The speed used for the tensile tests was 20 mm min−1 and that for the flexural tests was 10 mm min−1. All the tests were performed in quadruplicate, and the average results are given in the results with error bars showing greatest absolute differences.Determination of the physical properties of the composites
The densities of the composites were determined from the weight/volume of the specimens.19 The water absorption rates of the composites were determined from the mass changes after submerging the specimens in distilled water (at 20 ± 5 °C).Thermal tests
The thermal stabilities of the composites were determined using a thermal gravimetric analyzer (TGA) (TGA/DSC/SF1100, Metter, Switzerland). Samples were placed in a platinum pan and heated from 20 °C to 600 °C at a heating rate of 10 °C min−1 in a nitrogen atmosphere. Differential scanning calorimetry (DSC) (TGA/DSC/SF1100, Metter, Switzerland) was used to evaluate the melting behavior of the polyethylene/DDFS composites. All the samples were heated from 30 °C to 170 °C, and then cooled down to 30 °C at a cooling rate of 10 °C min−1 in a nitrogen atmosphere. The thermograms of the second scans were used for analysis.The crystallinity (Xc%) of each composite was estimated using the following equation:
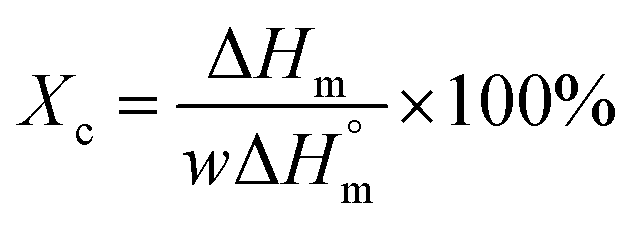 | (2) |
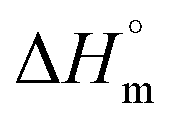 is the melting enthalpy for a 100% crystalline polymer (288.8 J g−1 for HDPE20), and w is the weight fraction of the polymer in the composite.
is the melting enthalpy for a 100% crystalline polymer (288.8 J g−1 for HDPE20), and w is the weight fraction of the polymer in the composite.
Weathering of the composites
Freeze–thaw cycles and xenon lamp weathering were performed according to GB/T 24508-2009 and GB/T 16422.2-1999.21 Specimens that had been submerged in water for one day were frozen at −30 °C for a second day. This procedure was repeated 3 times. The xenon lamp weathering was performed under 550 W m−2 of radiance (290–800 nm). The temperature and the moisture were maintained at 65 ± 5 °C and 50 ± 5%, respectively, during the xenon lamp weathering process.A differential scanning calorimeter (DSC 3+, Mettler Toledo, Switzerland) was used for determining the ΔHm and Tm (melting point) values of the composites before and after weathering. Scanning electron microscopy (SEM) (SU 1510, Hitachi High-Technologies Corporation, Japan) was used to take SEM images after weathering.
Results and discussions
Chemical compositions and characteristics of the DDFS residuals
The chemical compositions of the DDFS residuals were evaluated based on the method of NREL.17 As can be seen from Fig. 2, the DDFS residuals were mainly composed of soluble constituents, cellulose, hemicellulose, lignin and ash. Unlike the fresh lignocellulosic fibers, the DDFS residuals were obtained after SSF in bioethanol plants. Therefore, the soluble fractions in the DDFS residuals might be different from those in cellulosic agriculture residuals. Debris from yeast cells, proteins from saccharifying enzymes or cellulase, and salts from the fermentation media were included in the DDFS residuals. Compared with the CD and CCD, SD offered the highest soluble content of 42.9 ± 3.2%. This was caused by the higher level of soluble vegetable protein in the sorghum grain. Correspondingly, because of the low protein content in the raw corn cob material, the soluble component in the CCD was relatively low (11.6 ± 2.3%). There was a low level of hemicellulose contained in the SD (3.5 ± 0.6%). This was due to the low lignocellulose content in the raw sorghum grain. An even lower hemicellulose content (3.1 ± 0.6%) was obtained from the CCD, although hemicellulose is the largest constituent in corn cob.22 Due to the two-stage pretreatment process before SSF, cellulose was tested for cellulase adhesion by acid hydrolysis of the hemicellulose and delignification.23 In contrast, 17.4 ± 1.7% of hemicellulose was retained in the CD after SSF. Additionally, CCD provided the highest cellulose content (59.03 ± 3.3%). Therefore, this part of the cellulose was inaccessible to contact with cellulase. The lignin content in SD (33.4 ± 1.9%) was a little higher than that in CD (19.8 ± 2.3%) and CCD (18.5 ± 1.4%). Moreover, the CD had the highest ash content (21.8 ± 0.9%).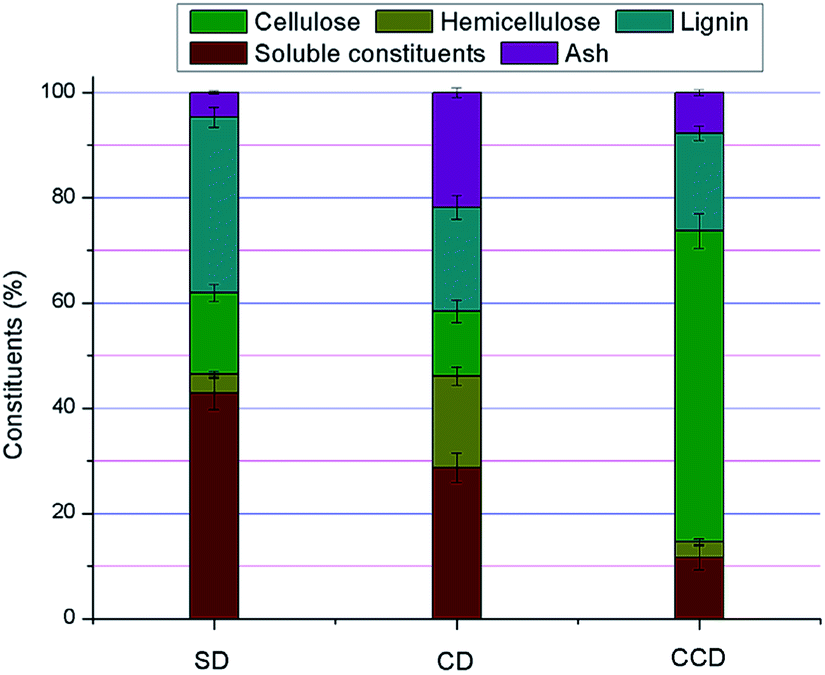 | ||
| Fig. 2 Chemical compositions of the DDFS residuals. | ||
In order to further analyze the structures and the functional groups on the surface of the DDFS residuals, FT-IR was performed. The results are shown in Fig. S1a.† The peaks between 3294 cm−1 and 3375 cm−1 were the –OH stretching vibrations.24 The peaks ranging from 2916 cm−1 to 2924 cm−1 were caused by the stretching vibrations of C–H from –CH and –CH2 groups. These peaks were also accordant with the cellulose and hemicellulose components in natural fiber.25 The vibrations at 1631–1655 cm−1 belonged to the conjugated carbonyl groups from lignin fractions of the DDFS residuals, and the peaks from 1513 to 1538 cm−1 and 1426 to 1458 cm−1 were caused by the benzene skeleton vibrations of lignin fractions. The peak located around 1240 cm−1 belonged to the C–O stretching vibrations, which are characteristic for hemicelluloses.26 The peaks between 1026 cm−1 and 1058 cm−1 were due to the C–O–C stretching vibrations of cellulose.
The crystallinity index (CrI) values of the fibers were used to analyze the changes of internal microstructure after chemical or physical treatment of the lignocellulosic materials. The crystallinity of cellulose is negatively related to the active –OH groups of the fibers while the hydrophilicity of the fibers is positively related to the active –OH.19 Fig. S1b† indicates the differences between SD, CD and CCD samples, when used as the reinforcing phases of WPCs. As can be seen from Table 1, the crystallinity of CCD was the highest compared with that of the other DDFS residuals. This was attributed to the higher cellulose content in CCD.
| DDFSs | SD | CD | CCD |
|---|---|---|---|
| I002 | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 047 047 |
7767 | 14457 |
| Iam | 10376 |
5845 | 6421 |
| CrI (%) | 26.1 | 24.7 | 55.6 |
As was indicated in the literature, the mechanical properties of the WPCs were hugely influenced by the constitution of the reinforcing fiber materials.19 Our previous study also demonstrated that the higher the cellulose and lignin contents in the fiber, the better the mechanical properties of the WPCs obtained.19 The hemicellulose content in fiber should be low due to its low stability and high hygroscopicity.27,28 Based on the above results of the DDFS chemical compositions and surface structures, we speculated that the SD and CCD, with their lower hemicellulose and higher cellulose contents, might result in better mechanical performances for their WPC products. This is proven in the following sections.
Mechanical properties of the polyethylene/DDFS composites
Fig. 3 shows the mechanical properties of composites reinforced by the different DDFS residuals. As illustrated in Fig. 3a, The SD and CCD reinforced composites had similar tensile strengths (22.3 ± 2.4 MPa for SD and 21.8 ± 2.2 MPa for CCD), which were a little higher than that of the CD composite (20.6 ± 2.6 MPa). Similar to the trend of tensile strength, a lower flexural strength was obtained in the CD reinforced composite (35.2 ± 1.9 MPa). These results demonstrated that the higher levels of cellulose contained in the reinforcement phase led to better mechanical properties, which was accordant with results in previous works.26 In addition, it proved that the hemicelluloses and lignin in the fibers had a significant effect on strength.19 The higher content of hemicellulose in CD might have contributed to the relatively poor strength properties of the related composite. In contrast, lignin with its hydrophobic performance and rigidity is compatible with the polyethylene.29,30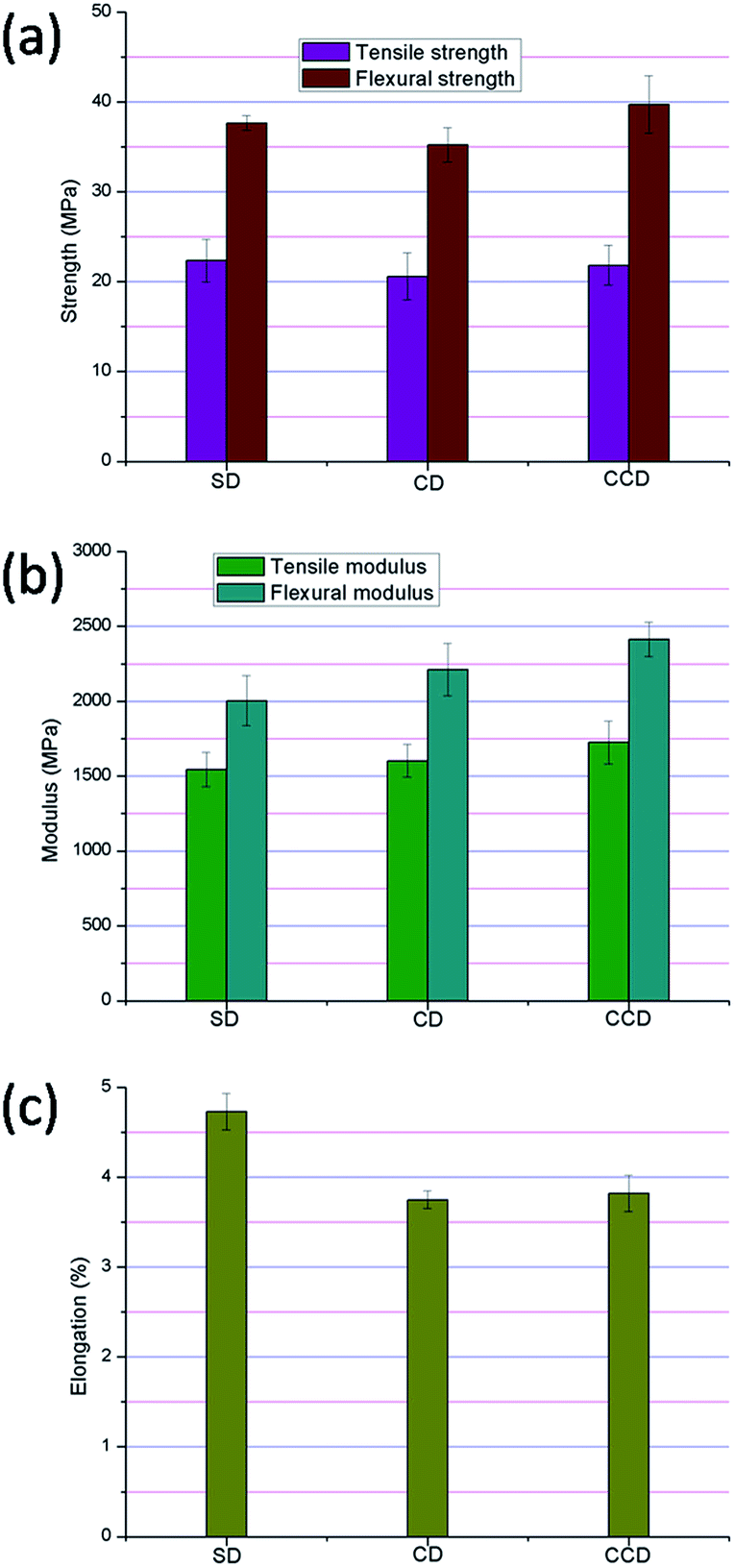 | ||
| Fig. 3 Mechanical properties of the polyethylene/DDFS composites: (a) flexural strength and tensile strength; (b) flexural modulus and tensile modulus; (c) elongation. | ||
For the modulus properties, an obviously negative correlation between the soluble fraction content and the modulus was obtained. The tensile modulus and the flexural modulus gradually increased in the order SD < CD < CCD. The maximum tensile modulus and flexural modulus were obtained for the CCD reinforced specimen (1725.4 ± 143.2 MPa and 2413.4 ± 115.2 MPa, respectively), which were 1.1 and 1.2 times higher than those of the SD composite (Fig. 3b). Therefore, the soluble fractions after SSF had negative effects on the mechanical properties of the composites, especially the moduli. However, the soluble fractions are a complex mixture of different types of compounds, and so the specific influence of the individual constituents in the soluble fractions on the composite properties calls for further investigation.
Fig. 3c shows the elongation at break of different types of DDFS reinforced composites. Similar elongations at break were obtained for the CD and CCD composites, and these were far lower than that of the SD group (4.7 ± 0.2%). The higher SD result might be caused by its higher lignin and soluble compound content. As demonstrated previously, a higher lignin content in fibers contributes to the flexural properties,31,32 and a higher content of soluble components in the reinforcing phase also positively affects the elongation properties of the composites.19
Physical properties of the polyethylene/DDFS composites
The physical properties of the polyethylene/DDFS composites were further compared. The water absorption rate is one of the most important parameters that influences the application of WPCs in practice.33 As can be seen from Fig. 4a, the water absorption rate of the specimens all met the national standard of China (below 3%), no matter which DDFS was used as the reinforcing phase. Specifically, 1.83 ± 0.2% and 1.32 ± 0.4% water was absorbed in the specimens reinforced by CCD and CD after immersion. By contrast, only 0.35 ± 0.2% water was absorbed in the specimen reinforced by SD. This phenomenon could be explained by the higher lignin content and lower hemicellulose content in the SD.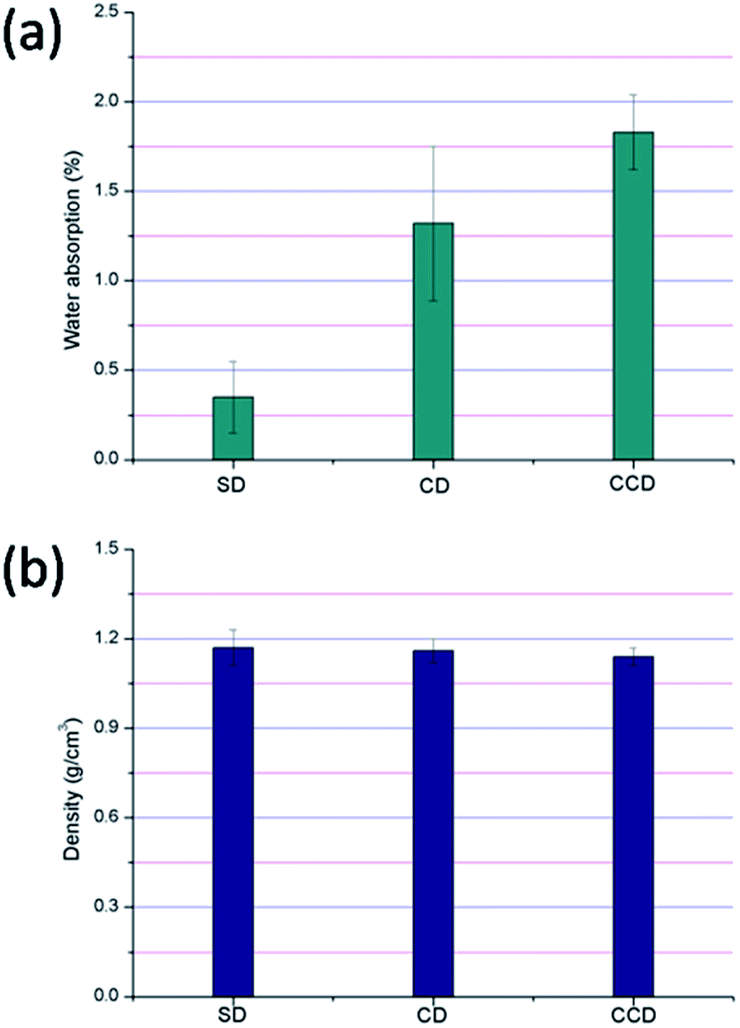 | ||
| Fig. 4 Physical properties of the polyethylene/DDFS composites: (a) water absorption rates; (b) densities. | ||
The higher water absorption rates in the CD and CCD reinforced specimens might be caused by their higher content of hemicellulose and cellulose. The –OH groups in hemicellulose and cellulose are likely to form hydrogen bonds. At the same time, the hydrophilic fractions in the DDFS and hydrophobic plastic matrix could allow the zones between the boundaries of the two phases to absorb water readily.14 The capillary mechanism of water diffusion also results in dimensional instability.34,35 On the contrary, lignin that has low numbers of –OH groups and poor hydrophilicity decreases water absorption.19,36 It is worth noting here that proteins in the DDFS samples would be denatured in the high temperature extruding process. Hence, the hydrophobic properties of the reinforcing phase might be increased due to the exposure of hydrophobic domains of the proteins after denaturing. It has been proven that the native soy protein can absorb >468% water.37 After the WPC manufacturing process, however, the hydrophobic denatured proteins might protect the specimens from water.
Fig. 4b shows the density of the polyethylene/DDFS composites. Generally, the density of the composites was maintained at 1.14–1.17 g cm−3, no matter which generation of DDFS residual was used. All the specimens met the national standard (GB/T 24508-2009) (>0.85 g cm−3). It has been demonstrated that a lower density WPC enhances the product performance and results in a reduction of production cost.19,38 Hence, the CCD reinforced composite that had the lowest density here (1.14 ± 0.03 g cm−3) and the best mechanical properties was the superior composite when compared with other specimens.
Thermal properties of the polyethylene/DDFS composites
TGA was carried out to evaluate the thermal properties of the polyethylene/DDFS composites. The thermograms obtained by TGA and differential thermal gravimetric (DTG) analysis are shown in Fig. S2.† The weight loss curves of the specimens were similar and two obvious weight loss stages were observed for all three composites. In the first weight loss stage of pyrolysis, the degradation of hemicelluloses appeared at 180–350 °C,39 the random cleavage of glycosidic linkages of cellulose occurred at 280–350 °C, and the degradation of lignin occurred at 250–500 °C.40 The second weight loss stage was mainly attributed to the degradation of HDPE.As shown in Fig. S2b,† the weight loss peak of the SD reinforced specimen was lower than that of the CD composite. The higher soluble content in the SD fiber might be the decisive factor in this performance. The soluble protein and fats in SD were likely degraded at relatively low pyrolytic temperatures. As a result, the first peak in the DTG curve of the SD composite was lower than that in the other samples. It is also demonstrated in Fig. S2b† that a shoulder peak arose in the curve of CCD (at about 250 °C). This is attributed to the lower pyrolytic temperature of hemicellulose (the predominant fraction in the CCD fiber) than those of lignin and hemicellulose. After reaching 600 °C, the pyrolysis residuals of the DDFS composites were obviously higher than the ash contents in the raw fibers. The low ash content of the DDFS samples in Fig. 2 was attributed to the oxygenolysis of the oxidizing fractions in the raw DDFS samples based on the NREL method for fiber composition analysis. Nevertheless, carbonization occurred during the TGA analysis, which is attributed to the oxygen-free nitrogen environment.
DSC results for the composites based on the different generation DDFS samples are shown in Fig. S3† and the key parameters are listed in Table 2. Generally, the Xc values of the specimens were higher than that of the pure HDPE; this indicated that the DDFS residuals acted as nucleating agents in providing initiation sites for crystallization.
| DDFS | SD | CD | CCD | HDPE |
|---|---|---|---|---|
| Tm (°C) | 128.92 | 128.63 | 129.02 | 130.59 |
| Tc (°C) | 117.81 | 117.37 | 118.39 | 116.24 |
| ΔHc (J g−1) | 73.62 | 65.45 | 75.28 | 146.84 |
| ΔHm (J g−1) | 71.83 | 63.09 | 70.43 | 148.95 |
| Xc (%) | 63.7 | 56.7 | 65.2 | 50.8 |
The Xc of polyethylene was 50.9%. The Xc of the composites reinforced by SD, CD and CCD were increased to 63.7%, 59.8% and 65.2%, respectively. Therefore, the Xc values of the composites were affected by the surface chemistry of the reinforcing phase. The CCD residuals acted as nucleating agents, provided initiation sites for crystallization, and further influenced the mechanical properties of the WPCs. In addition, the lower lignin content in the CD fiber might be the reason for the lowest Xc of the CD reinforced specimen. Here, the proportion of the reinforcing phase in the current work was 50%. Due to the relatively high fiber content, the wood fibers did not disperse well in the matrix, resulting in a reduction of heterogeneous nucleation and poor compatibility between the fibers and the matrix. Correspondingly, the melting temperatures (Tm) of the specimens were also obviously decreased. This might be the key reason for the slight differences of the crystallization temperatures (Tc) of the composites compared with that of pure HDPE.
Weathering effects on the polyethylene/DDFS composites
For practical applications, WPCs with good stability have advantages. Freeze–thaw and xenon lamp weathering were performed in this section according to GB/T 24508-2009 and GB/T 16422.2-1999,21 and the performances of the composites were compared.As can be seen from Fig. 5a, after 9 days of multigelation, the retention ratios of the modulus of rupture (MOR) were 91.1 ± 7.2%, 91.8 ± 6.1% and 88.7 ± 6.7% in the ageing specimens reinforced by SD, CD and CCD, respectively. Correspondingly, the retention ratios of the modulus of elasticity (MOE) were 80.3 ± 5.6%, 77.2 ± 6.2% and 96.2 ± 4.7%, respectively. The DSC results of the composites before and after multigelation were also evaluated. As can be seen from Table 3, the melting temperatures of the HDPE filled with the DDFS residuals ranged between 128.6 °C and 129.0 °C for the crude composites. After multigelation, the Tm of the composites was decreased except for that reinforced with CD. At the same time, the ΔHm of the SD composite was increased from 71.8 J g−1 for the crude specimen to 80 J g−1 for the weathered specimen, whilst the ΔHm of the CD and CCD groups were decreased from 63.1 J g−1 and 70.4 J g−1 in the crude specimens to 59.9 J g−1 and 61.7 J g−1 in the weathered specimens, respectively. Therefore, after freeze–thaw weathering, the crystallinity of the specimens was changed significantly. The decreases in the crystallinities of the CD and CCD specimens after multigelation might be caused by the rearrangement and recrystallization of the polymer matrix. The increase of crystallinity in the SD reinforced specimen after weathering might be assigned to the decomposition of amorphous regions in the composite, contributing to a higher percentage of crystalline content and a higher degree of crystallinity. In addition, the recrystallization of fractured fractions might contribute to the increase of crystallinity. Furthermore, the presence of lignin in the SD would help to reduce the degradation of polyethylene.
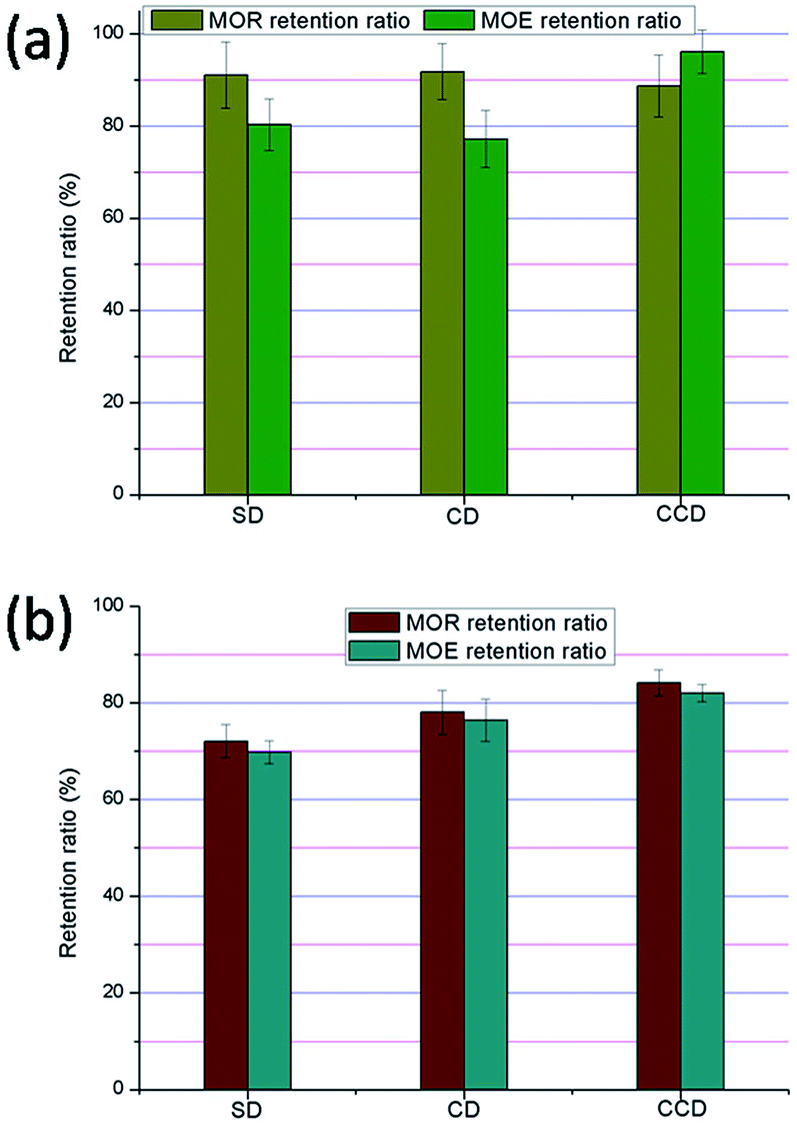 | ||
| Fig. 5 MOR and MOE retention ratios after (a) multigelation; (b) xenon lamp weathering. | ||
| DDFS | Before weathering | After multigelation | After xenon lamp weathering | |||
|---|---|---|---|---|---|---|
| Tm (°C) | ΔHm (J g−1) | Tm (°C) | ΔHm (J g−1) | Tm (°C) | ΔHm (J g−1) | |
| SD | 128.9 | 71.8 | 126.9 | 80.0 | 128.3 | 72.4 |
| CD | 128.6 | 63.1 | 129.1 | 59.9 | 129.4 | 49.5 |
| CCD | 129.0 | 70.4 | 128.1 | 61.7 | 129.3 | 60.1 |
The SEM images of the test specimens before and after multigelation are shown in Fig. S4 and S5.† As the images indicate, compared with the specimens before weathering, the surface morphologies of all composites were changed sharply by multigelation, although the polyethylene was still attached well to the reinforcing phase. Because of the intrusion of moisture, chain scissions of polyethylene occurred.41 In comparison with the small caves on the surface of the initial specimens, large cracks arose on the surface of the samples after weathering. In addition, parts of the specimen structures were swollen (see Fig. S2†). After multigelation, large gaps between the DDFS and the polyethylene matrix could be seen in the SEM images. Therefore, the decrease of the WPCs' mechanical properties was confirmed by testing the morphology of the composites. As can be seen from Fig. 5a, for both SD and CCD composites, the MOR retention ratios and the MOE retention ratios were above 80%. However, the sample reinforced with CD obtained from the 1.5 G biorefinery process was significantly worse than those reinforced with SD and CCD due to its lower MOE retention.
The performances of the WPCs after xenon lamp weathering were further evaluated. After 2000 h exposure in the radiance environment, 72.1 ± 3.4%, 78.1 ± 4.5% and 84.1 ± 2.7% of MOR and 69.8 ± 2.4%, 76.4 ± 4.4% and 82 ± 1.8% of MOE were retained in the specimens reinforced by SD, CD and CCD, respectively. Therefore, xenon lamp weathering showed a sharp influence on the mechanical performances of the specimens. Only the CCD sample exceeded the national standard after weathering.
The digital photographs of the specimens before and after weathering are also shown in Fig. 6. Before weathering, all of the samples were dark in color. After 100 h of weathering, the surface of the samples became rough. At the same time, the color of the SD and CD samples became lighter, which might be attributed to the photo-degradation of lignin.42,43 In good agreement with the stability of the CCD reinforced specimen displayed in Fig. 5b, there was no obvious color change after 100 h of weathering in the CCD sample. After 2000 h of weathering, the surface of the samples became rougher and large cracks were visible in the ageing specimens. The CCD sample also showed little cracks which corresponded to the results shown in Fig. 5b.
 | ||
| Fig. 6 Surface morphology and color changes of the polyethylene/DDFS composites before and after xenon lamp ageing. The left columns: before weathering; the middle columns: after 100 h of weathering; the right columns: after 2000 h of weathering. | ||
DSC results before and after xenon lamp weathering are listed in Table 3. ΔHm values for the CD and CCD reinforced specimens were decreased sharply by the weathering while the SD reinforced specimen still showed an increasing trend. Because of the relatively high content of lignin, the crystallinity of CD was low. Correspondingly, the ΔHm of the initial CD reinforced specimen was also low. After weathering, due to the photo-degradation of lignin, the ΔHm was hugely decreased. Similar results were also obtained in the CCD reinforced specimen. The reason for the decrease of ΔHm in the CCD reinforced specimen was similar to that for the multigelation results in Table 3; it might be caused by the rearrangement and recrystallization of the polyethylene in the composite.
Comparisons with other works, discussion and perspectives
Taking all results into consideration, CCD, the solid residual of the 2 G generation biofuel plant, was the most favorable choice for further WPC production. The high performance of the CCD reinforced specimen might be due to its high crystallinity and high content of cellulose and lignin.19 The novel method for WPC production shows promise for turning waste into wealth when coupled with the conventional biorefinery processes. Additional, environmental benefits would be generated at the same time.9Table 4 compares the currently available results on WPCs prepared using natural fibers as the reinforcing phase. It illustrates that the WPCs obtained in the current work are comparable with those obtained in other research using the crude fibers directly. Therefore, in comparison with other fibers, the DDFS after ethanol production shows good performances when used as the reinforcing phase in WPCs. The biggest advantage, however, is that a low-value solid residual can be used to produce a valuable biocomposite. This novel method could not only reduce the discharge of solid waste from biorefineries, but also extend the product chain, so improving the economic feasibility of conventional cellulosic ethanol production.
| Fiber | Fiber volume fraction (%) | Polymer | Flexural strength (MPa) | Flexural modulus (GPa) | Tensile strength (MPa) | Tensile modulus (GPa) | Elongation (%) | Reference |
|---|---|---|---|---|---|---|---|---|
| SD | 50 | HDPE | 37.7 ± 0.8 | 2.0 ± 0.2 | 22.4 ± 2.4 | 1.5 ± 0.1 | 4.7 ± 0.2 | This work |
| CD | 50 | HDPE | 35 ± 2 | 2.2 ± 0.2 | 20.6 ± 2.6 | 1.6 ± 0.1 | 3.8 ± 0.1 | This work |
| CCD | 50 | HDPE | 39.7 ± 3.2 | 2.4 ± 0.1 | 21.8 ± 2.2 | 1.7 ± 0.1 | 3.8 ± 0.2 | This work |
| Poplar | 50 | PP | 29.9 ± 1.4 | 1.7 ± 0.1 | 17.9 ± 2.2 | 1.1 ± 0.2 | — | 25 |
| Poplar | 40 | PE | — | — | 40.1 ± 0.3 | 4.2 ± 0.1 | 6.1 ± 0.4 | 31 |
| Scots pine | 50 | PP | 24.8 ± 1.2 | 3.8 ± 0.2 | 16.7 ± 1.3 | 3.2 ± 0.4 | — | 44 |
| Rubber wood | 55.8 | HDPE | 15.69 | 1.93 | 10.60 | 0.82 | — | 45 |
| Rubber wood | 45.8 | HDPE | 17.25 | 1.50 | 17.04 | 1.03 | — | 45 |
| Maple wood | 40 | HDPE | 39.0 ± 2.0 | 2.1 ± 0.2 | 65.8 ± 0.4 | 3.8 ± 0.1 | 5.4 ± 0.3 | 46 |
| Norway spruce | 58 | HDPE | 42.3 | 2.77 | 21.6 | 2.87 | — | 47 |
| Spruce, pine, fir | 47 | PP | 29.5 ± 1.9 | 2.9 ± 0.3 | 15.3 ± 0.9 | 2.9 ± 0.2 | — | 48 |
| Pinus pinaster | 55 | HDPE | 18.8 ± 0.1 | 3.2 ± 0.0 | 13.3 ± 0.4 | 3.2 ± 0.0 | — | 49 |
| Cotton stalk bark | 30 | PP | 48.4 ± 0.8 | 2.1 ± 0.0 | 32.9 ± 0.3 | 0.2 ± 0.0 | 7.3 ± 0.5 | 50 |
| Oak | 50 | HDPE | 45.7 ± 3.5 | 2.6 ± 0.3 | 33.0 ± 0.3 | 4.8 ± 0.3 | — | 51 |
| Paulownia | 50 | PP | 43.3 ± 3.4 | 4.8 ± 0.0 | 18.8 ± 2.2 | 5.3 ± 0.2 | — | 52 |
Future research should be focused on the flame retardant properties, and durability of the composites. The current WPCs were prepared from HDPE matrix and wood fiber, which are both flammable organic materials. Therefore, the flame retardant properties of the WPCs need to be enhanced to widen the applications for these composites.53 The weathering properties of the DDFS reinforced composites, especially the xenon lamp weathering, need to be improved. Several studies have indicated that the ageing properties can be influenced by photo-degradation, humidity or water from the environment, and thermal degradation, etc.54,55 Hence, the weathering properties should be improved by adding some additive agents or modifying the surface of the fibers.
Conclusions
DDFS residuals from 1 G, 1.5 G and 2 G biofuel plants could be used as the reinforcing phase in polyethylene composite production. Compared with SD and CD, CCD generated from the 2 G plant had higher crystallinity and a higher content of cellulose and lignin. Correspondingly, the CCD reinforced composite showed better mechanical and physical properties, and longer-term stability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (Grant No. 21676014, and 21706008), the China Postdoctoral Science Foundation (Grant No. 2017M610037; 2018T110036) and the Fundamental Research Funds for the Central Universities (Grant No. ZY 1832).References
- P. S. Nigam and A. Singh, Prog. Energy Combust. Sci., 2011, 37, 52–68 CrossRef.
- J. Baeyens, Q. Kang, L. Appels, R. Dewil, Y. Lv and T. Tan, Prog. Energy Combust. Sci., 2015, 47, 60–88 CrossRef.
- S. Ghosh, R. Chovdhury and P. Bhattacharya, Appl. Energy, 2017, 198, 284–298 CrossRef.
- M. C. Rulli, D. Bellomi, A. Cazzoli, G. De Carolis and P. D'Odorico, Sci. Rep., 2016, 6, 22521 CrossRef PubMed.
- Z. Losordo, J. McBride, J. Van Rooyen, K. Wenger, D. Willies, A. Froehlich, I. Macedo and L. Lynd, Biofuels, Bioprod. Biorefin., 2016, 10, 589–602 CrossRef.
- L. Zhao, S. Chang, H. Wang, X. Zhang, X. Ou, B. Wang and M. Wu, Energy, 2015, 83, 37–54 CrossRef.
- M. O. S. Dias, T. L. Junqueira, C. D. F. Jesus, C. E. V. Rossell, R. M. Filho and A. Bonomi, Energy, 2012, 43, 246–252 CrossRef.
- C. Böttger and K. H. Südekum, Anim. Feed Sci. Technol., 2017, 224, 66–77 CrossRef.
- J. Yu, T. Zhang, J. Zhong, X. Zhang and T. Tan, Biotechnol. Adv., 2012, 30, 811–816 CrossRef PubMed.
- M. Zahedi, T. Tabarsa, A. Ashori, M. Madhoushi and A. Shakeri, J. Appl. Polym. Sci., 2013, 3, 1491–1498 CrossRef.
- A. M. Youssef, A. El-Gendy and S. Kamel, Mater. Chem. Phys., 2015, 152, 26–33 CrossRef.
- S. Muniyasay, M. M. Reddy, M. Misra and A. Mohanty, Ind. Crops Prod., 2013, 43, 812–819 CrossRef.
- S. A. Madbouly, J. A. Schrader, G. Srinivasan, K. Liu, K. G. McCabe, D. Grewell, W. R. Graves and M. R. Kessler, Green Chem., 2014, 16, 1911–1920 RSC.
- L. Wei, S. Liang and A. G. McDonald, Ind. Crops Prod., 2015, 69, 91–103 CrossRef.
- J. Zhong, L. Zhang, J. Yu, T. Tan and X. Zhang, J. Appl. Polym. Sci., 2010, 117, 1385–1393 Search PubMed.
- S. Torres, R. Navia, R. C. Murdy, P. Cooke, M. Misra and A. K. Mohanty, ACS Sustainable Chem. Eng., 2015, 3, 614–624 CrossRef.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Laboratory Analytical Procedure, NREL, report no. TP-510-42618, 2008 Search PubMed.
- L. Segal, J. J. Creely, A. E. Martin and C. M. Conrad, Text. Res. J., 1959, 29, 786–794 CrossRef.
- Z. Luo, P. Li, D. Cai, Q. Chen, P. Qin, T. Tan and H. Cao, Ind. Crops Prod., 2017, 95, 521–527 CrossRef.
- S. Jose, A. S. Aprem, B. Francis, M. C. Chandy, P. Werner, V. Alstaedt and S. Thomas, Eur. Polym. J., 2004, 40, 2105–2115 CrossRef.
- H. Y. Wang, D. G. Li, Y. M. Wang and D. L. Lin, Appl. Mech. Mater., 2012, 170, 3175–3180 Search PubMed.
- P. Li, D. Cai, Z. Luo, P. Qin, C. Chen, Y. Wang, C. Zhang, Z. Wang and T. Tan, Bioresour. Technol., 2016, 206, 86–92 CrossRef PubMed.
- C. Lei, J. Zhang, L. Xiao and J. Bao, Bioresour. Technol., 2014, 167, 555–559 CrossRef PubMed.
- Y. Wu, F. Geng, P. R. Chang, J. Yu and X. Ma, Carbohydr. Polym., 2009, 79, 299–304 CrossRef.
- R. Nadlene, S. M. Sapuan, M. Jawaid, M. R. Ishak and L. Yusriah, Polym. Compos., 2018, 39, 274–287 CrossRef.
- R. Liu, Y. Peng, J. Cao and Y. Chen, Compos. Sci. Technol., 2014, 103, 1–7 CrossRef.
- O. Hosseinaei, S. Wang, A. A. Enayati and T. G. Rials, Composites, Part A, 2012, 43, 686–694 CrossRef.
- O. Hosseinaei, S. Wang, A. M. Taylor and J. W. Kim, Int. Biodeterior. Biodegrad., 2012, 71, 29–35 CrossRef.
- A. A. Morandim-Giannetti, J. A. M. Agnelli, B. Z. Lanças, R. Magnabosco, S. A. Casarin and S. H. P. Bettini, Carbohydr. Polym., 2012, 87, 2563–2568 CrossRef.
- H. D. Rozman, K. W. Tan, R. N. Kumar, A. Abubakar, Z. A. M. Ishak and H. Ismail, Eur. Polym. J., 2000, 36, 1483–1494 CrossRef.
- R. Ou, Y. Xie, M. P. Wolcott, F. Yuan and Q. Wang, Compos. Sci. Technol., 2014, 93, 68–75 CrossRef.
- R. Ou, Y. Xie, M. P. Wolcott, S. Sui and Q. Wang, Mater. Des., 2014, 58, 339–345 CrossRef.
- M. Boopalan, M. Niranjanaa and M. J. Umapathy, Composites, Part B, 2013, 51, 54–57 CrossRef.
- H. Dhakal, Z. Zhang and M. O. W. Richardson, Compos. Sci. Technol., 2007, 67, 1674–1683 CrossRef.
- J. S. Fabiyi, A. G. McDonald, J. J. Morrell and C. Freitag, Composites, Part A, 2011, 42, 501–510 CrossRef.
- K. B. Adhikary, S. Pang and M. P. Staiger, Chem. Eng. J., 2008, 142, 190–198 CrossRef.
- X. S. Sun, Plastics derived from starch and poly(lactic acids), Elsevier Academic Press, Burlington, MA, 2011, pp. 369–410 Search PubMed.
- Y. Li, K. Venkateshan and X. S. Sun, Polym. Int., 2010, 59, 1099–1109 CrossRef.
- H. S. Kim, S. Kim, H. J. Kim and H. S. Yang, Thermochim. Acta, 2006, 451, 181–188 CrossRef.
- D. G. Dikobe and A. S. Luyt, Thermochim. Acta, 2017, 654, 40–50 CrossRef.
- T. H. Yang, T. H. Yang, W. C. Chao and S. Y. Leu, Constr. Build. Mater., 2015, 88, 159–168 CrossRef.
- G. N. Salaita, F. Ma, T. C. Parker and G. B. Hoflund, Appl. Surf. Sci., 2008, 254, 3925–3934 CrossRef.
- D. Kocaefe, X. Huang, Y. Kocaefe and Y. Boluk, Surf. Interface Anal., 2013, 45, 639–649 CrossRef.
- N. Ayrilmis, A. Kaymakci and T. Güleç, Ind. Crops Prod., 2015, 74, 279–284 CrossRef.
- T. Ratanawilai and K. Taneerat, Constr. Build. Mater., 2018, 172, 349–357 CrossRef.
- B. Koohestani, I. Ganetri and E. Yilmaz, Composites, Part B, 2017, 11, 103–111 CrossRef.
- P. F. Sommerhuber, J. L. Wenker, S. Rüter and A. Krause, Resour., Conserv. Recycl., 2017, 117, 235–248 CrossRef.
- S. Y. Leu, T. H. Yang, S. F. Lo and T. H. Yang, Constr. Build. Mater., 2012, 29, 120–127 CrossRef.
- S. Chaudemanche, A. Perrot, S. Pimbert, T. Lecompte and F. Faure, Constr. Build. Mater., 2018, 162, 543–552 CrossRef.
- H. Wu, X. Liang, L. Huang, Y. Xie, S. Tan and X. Cai, Constr. Build. Mater., 2016, 118, 337–343 CrossRef.
- S. Bhattacharjee and D. S. Bajwa, Constr. Build. Mater., 2018, 172, 1–9 CrossRef.
- N. Ayrilmis and A. Kaymakci, Ind. Crops Prod., 2013, 43, 457–464 CrossRef.
- L. Kong, K. Tu, H. Guan and X. Wang, Appl. Surf. Sci., 2017, 407, 479–484 CrossRef.
- C. Croitoru, C. Spirchez, A. Lunguleasa, D. Cristea, I. C. Roata, M. A. Pop, T. Bedo, E. M. Stanciu and A. Pascu, Appl. Surf. Sci., 2018, 438, 114–126 CrossRef.
- J. Miklečić, H. Turkulin and V. Jirouš-Rajković, Appl. Surf. Sci., 2017, 408, 103–109 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03906b |
| This journal is © The Royal Society of Chemistry 2018 |