
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCellulose triacetate synthesis via one-pot organocatalytic transesterification and delignification of pretreated bagasse†
Shiori Suzukia,
Yoshiki Shibataa,
Daisuke Hirosea,
Takatsugu Endob,
Kazuaki Ninomiyac,
Ryohei Kakuchi *d and
Kenji Takahashi*a
*d and
Kenji Takahashi*a
aFaculty of Natural System, Institute of Science and Engineering, Kanazawa University, Kakuma-machi, Kanazawa 920-1192, Ishikawa, Japan. E-mail: ktkenji@staff.kanazawa-u.ac.jp
bDepartment of Molecular Chemistry and Biochemistry, Faculty of Science and Engineering, Doshisha University, 1-3 Tatara Miyakodani, Kyoutanabe 610-0394, Kyoto, Japan
cInstitute for Frontier Science Initiative, Kanazawa University, Kakuma-machi, Kanazawa 920-1192, Ishikawa, Japan
dDivision of Molecular Science, Graduate School of Science and Technology, Gunma University, 1-5-1 Tenjin-cho, Kiryu 376-8515, Gunma, Japan. E-mail: kakuchi@gunma-u.ac.jp
First published on 13th June 2018
Abstract
Cellulose triacetate was synthesised by the transesterification reaction of mild acid-pretreated lignocellulosic biomass with a stable acetylating reagent (isopropenyl acetate, IPA) in an ionic liquid (1-ethyl-3-methylimidazolium acetate, EmimOAc) which enabled the dissolution of lignocellulose as well as the organocatalytic reaction. The homogeneous acetylation of pretreated sugar-cane bagasse was carried out under mild conditions (80 °C, 30 min), and the subsequent reprecipitation processes led to enriched cellulose triacetate with a high degree of substitution (DS; 2.98) and glucose purity (∼90%) along with production of lignin acetate.
1. Introduction
Cellulose-based materials (e.g. cellulose acetate) have been widely utilised for diverse applications1–3 such as membrane matrices,4 optical films,5 cigarette filters,6 etc.7–11 Their importance can also be acknowledged from the viewpoint of green and sustainable chemistry.12 Cellulose is a major component of lignocellulosic biomass, so-called lignocellulose, that is independent from the instinctive competition with human food supplies. Despite the inherent advantage of cellulose-based materials, their synthesis from the original lignocellulose typically involves multi-step transformations that require harsh conditions. This is primarily attributable to the insolubility of cellulose, which leads to a fairly high cost associated with the purification step as compared to the processing of petroleum-based chemicals.13 To be precise, although cellulose comprises water-soluble glucose as a repeating unit, it is nearly insoluble in both common organic and aqueous solvents because of the extraordinarily strong hydrogen bonding networks within its polymeric structures. It is also worth noting that cellulose generally needs to be isolated from lignocellulose containing hemicellulose and lignin14 prior to chemical modifications. The series of steps involved in this isolation process is known as pulping. The removal of lignin from lignocellulose, also called delignification,15 is one of the difficult tasks in the pulping system because of the low reactivity of lignin and the partial chemical linkages between the lignin and carbohydrates.16,17 Delignification is a multi-step process and requires a highly basic and oxidative medium at high temperatures.18,19 Furthermore, it suffers from poor selectivity and further purification by bleaching techniques can result in significant degradation of the constituent carbohydrates.20 Such multiple harsh delignification processes increase the cost and energy consumption in the entire cellulose purification process when compared to petroleum-based chemicals. Therefore, the production of highly pure cellulose-based materials from lignocellulose with simple and mild chemical treatments is in great demand to further supplement their inherent green chemistry attributes.In recent years, ionic liquids (ILs) have emerged as a novel platform with an excellent dissolution ability21,22 for otherwise insoluble materials including cellulose.23,24 Using ILs as solvents for cellulose bypasses the conventional binary solvent mixtures (e.g. N,N-dimethylacetamide/LiCl) and designer solvents can be developed by tuning the structure of ILs.25,26 Consequently, ILs have attracted significant attention for application in cellulose-related chemistry. In this context, we have previously reported that an ionic liquid, 1-ethyl-3-methylimidazolium acetate (EmimOAc), acts not only as a cellulose solvent, but also as an organocatalyst in the transesterification reaction of cellulose.27–29 This bifunctional employment of EmimOAc allowed cellulose to react efficiently under mild conditions without environmentally hazardous catalysts (e.g. Brønsted acids such as sulfuric acid and organometallics) and additional activating reagents such as acid anhydrides and carboxylic halides.
It is worth noting that EmimOAc can dissolve both pure cellulose30,31 and lignocellulose.32,33 This foreknowledge encouraged us to investigate the applicability of our cellulose modification protocol27 toward lignocellulose. Chen et al. have also utilised this attribute of EmimOAc to successfully obtain acylated bagasse.34 Although these authors demonstrated the direct conversion of lignocellulose, the isolation of cellulose derivatives and the loss of the lignin fraction was not addressed in detail. Therefore, it is still a challenging task to enrich the target cellulose derivatives directly from lignocellulose under simple and mild conditions.
In this study, the direct acetylation of bagasse was conducted using the bifunctional IL, EmimOAc, and the subsequent reprecipitation in methanol (MeOH) to separate both the polysaccharide and lignin acetyl derivatives was performed. In addition, we have proposed another synthetic process that allows enriching the cellulose derivatives from bagasse via a mild acid-pretreatment step, as depicted in Scheme 1. This involves (1) the selective acid hydrolysis of hemicellulose in bagasse, and (2) homogeneous acetylation of the pretreated bagasse and the subsequent reprecipitation process. This simple two-step process successfully afforded both enriched cellulose triacetate and lignin acetate.
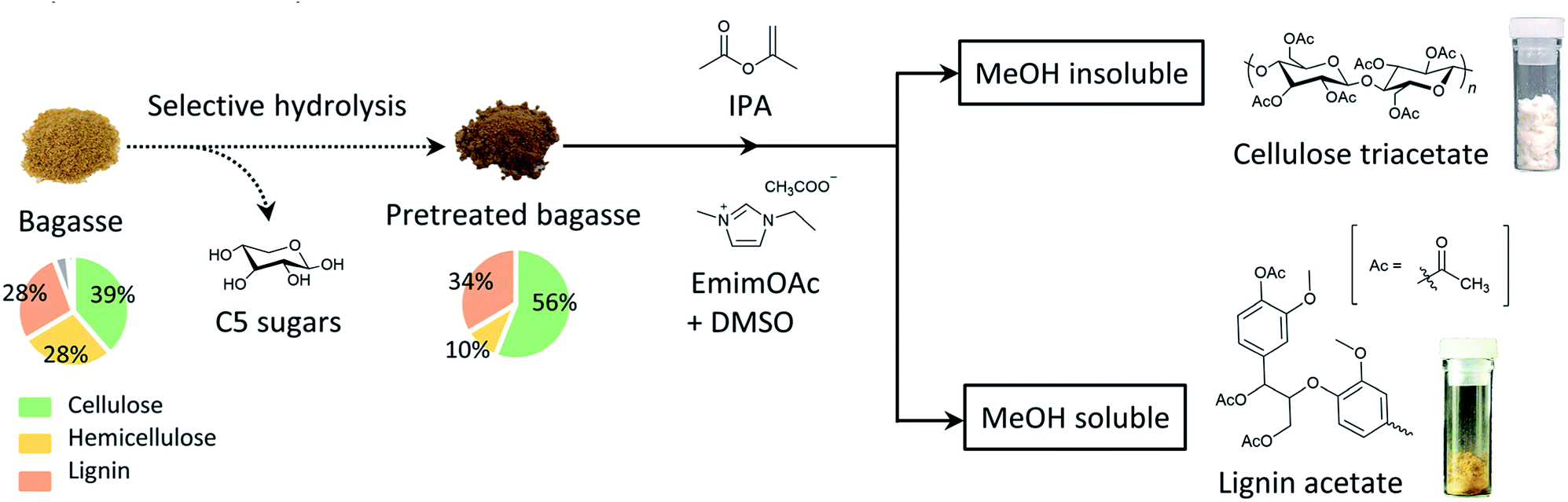 | ||
| Scheme 1 Schematic representation of the organocatalytic transesterification of mild acid-pretreated bagasse and successive fractionation into enriched cellulose triacetate and lignin acetate by solid–liquid separation. | ||
2. Experimental
2.1. General information
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 × g, 4 °C, 60 min) were conducted using a compact high-speed refrigerated centrifuge (KUBOTA 6930, Kubota Co., Ltd., Osaka, Japan). The molecular weights of the polymeric samples were determined by size exclusion chromatography (SEC, Prominence UFLC system, Shimadzu Co., Kyoto, Japan) based on polystyrene standards. All SEC measurements were carried out at 40 °C using TSK gel α-M (Tosoh Co., Tokyo, Japan), and 0.01 mol L−1 LiBr in dimethylformamide (DMF, HPLC grade, Kanto Chemicals Co., Inc., Tokyo, Japan) was used as an eluent at the flow rate of 1.0 mL min−1. The high-performance liquid chromatograph (HPLC) was equipped with a refractive index (RI) detector (Shimadzu Co., Kyoto, Japan) and a CARBOSep CHO-682 column (Tokyo Chemical Industry Co., Ltd., Tokyo, Japan). HPLC measurements were conducted at 85 °C with an ultra-pure water mobile phase and flow rate of 0.4 mL min−1. A DU® series 700 UV/VIS scanning spectrophotometer (Beckman Coulter, Inc, Tokyo, Japan) was used for UV measurements at 240 nm with a quartz cell (path length: 1 cm).
500 × g, 4 °C, 60 min) were conducted using a compact high-speed refrigerated centrifuge (KUBOTA 6930, Kubota Co., Ltd., Osaka, Japan). The molecular weights of the polymeric samples were determined by size exclusion chromatography (SEC, Prominence UFLC system, Shimadzu Co., Kyoto, Japan) based on polystyrene standards. All SEC measurements were carried out at 40 °C using TSK gel α-M (Tosoh Co., Tokyo, Japan), and 0.01 mol L−1 LiBr in dimethylformamide (DMF, HPLC grade, Kanto Chemicals Co., Inc., Tokyo, Japan) was used as an eluent at the flow rate of 1.0 mL min−1. The high-performance liquid chromatograph (HPLC) was equipped with a refractive index (RI) detector (Shimadzu Co., Kyoto, Japan) and a CARBOSep CHO-682 column (Tokyo Chemical Industry Co., Ltd., Tokyo, Japan). HPLC measurements were conducted at 85 °C with an ultra-pure water mobile phase and flow rate of 0.4 mL min−1. A DU® series 700 UV/VIS scanning spectrophotometer (Beckman Coulter, Inc, Tokyo, Japan) was used for UV measurements at 240 nm with a quartz cell (path length: 1 cm).2.2. Synthesis and isolation protocols
The MeOH filtrate obtained in the first precipitation step was concentrated using an evaporator at 40 °C, and a black viscous liquor containing EmimOAc, DMSO, and lignin derivatives was obtained. Acetone (100 mL) was added to the obtained solution to produce a clear homogeneous mixture. To remove Emim+, the homogeneous acetone solution was stirred at room temperature for 2 h along with a strong-acidic cation exchange resin (50 g, Amberlite® IRN-77, H+ form, Sigma-Aldrich Co., LLC., St. Louis, MO, USA). The used resin was removed by filtration and washed with acetone. The acetone filtrate was again evaporated at 40 °C, and the resultant mixture was added dropwise to distilled water (600 mL) to precipitate light brown coloured particles. After centrifugation, the residue was washed with distilled water and freeze dried for two days to obtain lignin acetate. The average yield (n = 3) was 144 mg.
The purified bagasse powder (3.0 g) was presoaked in a 3.0 mol L−1 sulfuric acid aqueous solution with a 3% (w/w) total solids loading at room temperature for 4 h. This slurry, which was prepared in a 250 mL polytetrafluoroethylene (PTFE) inner tube embedded within a pressure resistant stainless-steel vessel, was heated in a rotary type oven (RDV-TM2, SAN-AI Kagaku Co., Ltd., Aichi, Japan) at 160 °C for 20 min (160 °C temperature was reached in 50 min from room temperature). After the reaction container was cooled to room temperature in an ice bath, the acidic slurry was vacuum filtered. The red-brown coloured residue was washed with distilled water (100 mL) at least five times and dried in a vacuum oven at 70 °C for 24 h to obtain pretreated bagasse that mainly contained cellulose and lignin.
The homogeneous acetylation process using the EmimOAc/DMSO system and the subsequent separation of the synthesized cellulose triacetate in the MeOH-insoluble fraction and lignin acetate in the MeOH-soluble fraction were performed in the same manner for direct bagasse conversion as described in the experimental Section 2.2.1. The obtained cellulose triacetate was further purified by chloroform dissolution and filtration.§ The average yields (n = 3) were 423 mg (cellulose triacetate) and 197 mg (lignin acetate).
2.3. Composition analysis
Prior to composition analysis, deacetylation of the acetylated polysaccharides was conducted according to the literature procedure39 because this analytical method was intended for the chemically-unmodified lignocellulose.Polysaccharide acetate from bagasse, cellulose triacetate from pretreated bagasse, and cellulose acetate from Avicel were dried under vacuum at 70 °C for 24 h before use. The acetylated polysaccharide (200 mg) was weighed into a 30 mL vial. A mixed solvent (20 mL) of acetone/MeOH (1:1, v/v) was added and stirred at room temperature for 2 h. Deacetylation was conducted by stirring the mixture at room temperature for 24 h with a 1.0 mol L−1 sodium hydroxide aqueous solution (2.0 mL). The resultant solution was filtered after centrifugation. The pale yellow powdery residue was washed repeatedly with distilled water and collected after freeze drying for two days. The average yields (n = 3) of the deacetylated products were 105 mg (bagasse-derived polysaccharide acetate), 109 mg (pretreated bagasse-derived cellulose triacetate), and 116 mg (Avicel-derived cellulose acetate).
The composition analyses of the deacetylated polysaccharides and the starting materials (i.e., bagasse and pretreated bagasse) were conducted according to the standard biomass analytical method provided by the NREL.40 A simplified flow chart of the composition analysis protocol is depicted in Scheme 2. This procedure essentially involves the following two steps: (1) acid hydrolysis of the sample to fractionate into two quantifiable components depending on its solubility in an acidic solution, and (2) quantification of the hydrolysate obtained in the first step via HPLC and UV measurements.
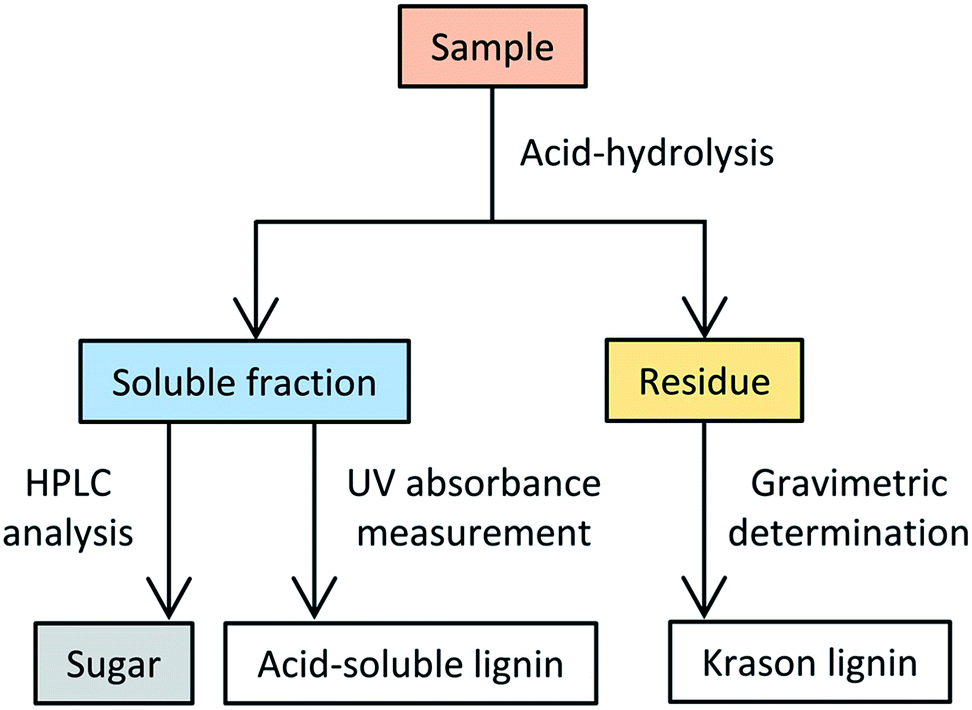 | ||
| Scheme 2 Flow chart of the composition analysis of lignocellulosic material according to the NREL analytical method.42 | ||
3. Results and discussion
3.1. Direct acetylation of bagasse and subsequent fractionation into polysaccharide acetate and lignin acetate
To synthesise cellulose derivatives with sufficient purity directly from lignocellulose, direct acetylation of bagasse was carried out. In this context, bagasse was subjected to a transesterification reaction in a mixed solvent of EmimOAc/DMSO, which enabled both lignocellulose dissolution and organocatalytic transesterification. Considering the common solubility behaviour of cellulose acetate,27,28 hemicellulose acetate,29 and lignin acetate,41 MeOH was selected as the anti-solvent for polysaccharide acetyl derivatives and as a good solvent for lignin acetate.¶ Additionally, chloroform was selected as good solvent for the obtained acetylated polysaccharide because it can solubilise cellulose acetate exceptionally well with a high degree of substitution (DS). Therefore, the obtained reaction mixture was precipitated into MeOH and then fractionated into MeOH-insoluble and soluble portions. The MeOH-insoluble portion containing the polysaccharide acetate was further purified by chloroform dissolution, and the isolated product yields are summarised in Table 1.| Fraction | Main component | Isolated yielda,b (%) |
|---|---|---|
| a Average yields (n = 3).b Theoretical yields were calculated from the composition of bagasse based on the hypothesis that hemicellulose was only composed of xylan.c Collected as a chloroform-soluble portion. | ||
| MeOH-insoluble | Polysaccharide acetate | 48 ± 3.8c |
| MeOH-soluble | Lignin acetate | 72 ± 2.1 |
The bagasse sample (600 mg) used in this study was revealed to contain 66 wt% polysaccharides corresponding to 398 mg (see Table S4 in ESI† for the composition of the starting materials). If all of the hydroxyl (OH) groups of the polysaccharides were acetylated, 683 mg of the polysaccharide acetate was expected to be generated, given that hemicellulose in the bagasse sample was essentially composed of xylan. Therefore, the collected weight of 326 mg corresponded to 48% isolated yield. Along with the carbohydrates, the lignin component was 28 wt% of the employed bagasse as determined by the composition analysis (Table S4 in ESI†). In addition, the alcoholic, phenolic, and carboxylic acid OH concentrations of lignin were separately estimated by quantitative 31P NMR analysis,42,43 which indicated that 4.53 mmol g−1 of total OH groups were present in bagasse-derived lignin (Tables S6–S7 in ESI† for the determination of the lignin OH concentration). Thus, 198 mg of lignin acetate was theoretically expected to be produced from bagasse via the acetylation process, thus providing a reasonable agreement with the experimental result of 144 mg (corresponding to 72% isolated yield). Therefore, both polysaccharide acetate and lignin acetate were obtained from bagasse in acceptable to moderate isolated yields.
To provide clear chemical information, both fractions were characterized by FT-IR and 1H NMR. In the FT-IR spectra (Fig. 1), a strong absorption at ∼1738 cm−1 corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching was observed for both fractions. Furthermore, the band at 1211 cm−1 (C–O–C ether stretching) was also detected for both fractions. These observations implied that the transesterification reaction of bagasse and subsequent reprecipitation processes afforded the acetylated polymeric fractions depending on their solubility in MeOH. A detailed insight into the chemical structures of these two fractions were provided by 1H NMR analysis, as shown in Fig. 2. In the spectrum of the MeOH-insoluble fraction (Fig. 2 (upper)), the chemical shifts corresponding to the acetyl protons were observed in the spectral range of 1.9–2.1 ppm. In addition, the signals in the range of 3.2–5.1 ppm corresponded to the sugar skeleton. As there were no clear peaks arising from the aromatic protons of lignin in the spectrum, it was concluded that the MeOH-insoluble fraction was mainly composed of polysaccharide acetate.
O stretching was observed for both fractions. Furthermore, the band at 1211 cm−1 (C–O–C ether stretching) was also detected for both fractions. These observations implied that the transesterification reaction of bagasse and subsequent reprecipitation processes afforded the acetylated polymeric fractions depending on their solubility in MeOH. A detailed insight into the chemical structures of these two fractions were provided by 1H NMR analysis, as shown in Fig. 2. In the spectrum of the MeOH-insoluble fraction (Fig. 2 (upper)), the chemical shifts corresponding to the acetyl protons were observed in the spectral range of 1.9–2.1 ppm. In addition, the signals in the range of 3.2–5.1 ppm corresponded to the sugar skeleton. As there were no clear peaks arising from the aromatic protons of lignin in the spectrum, it was concluded that the MeOH-insoluble fraction was mainly composed of polysaccharide acetate.
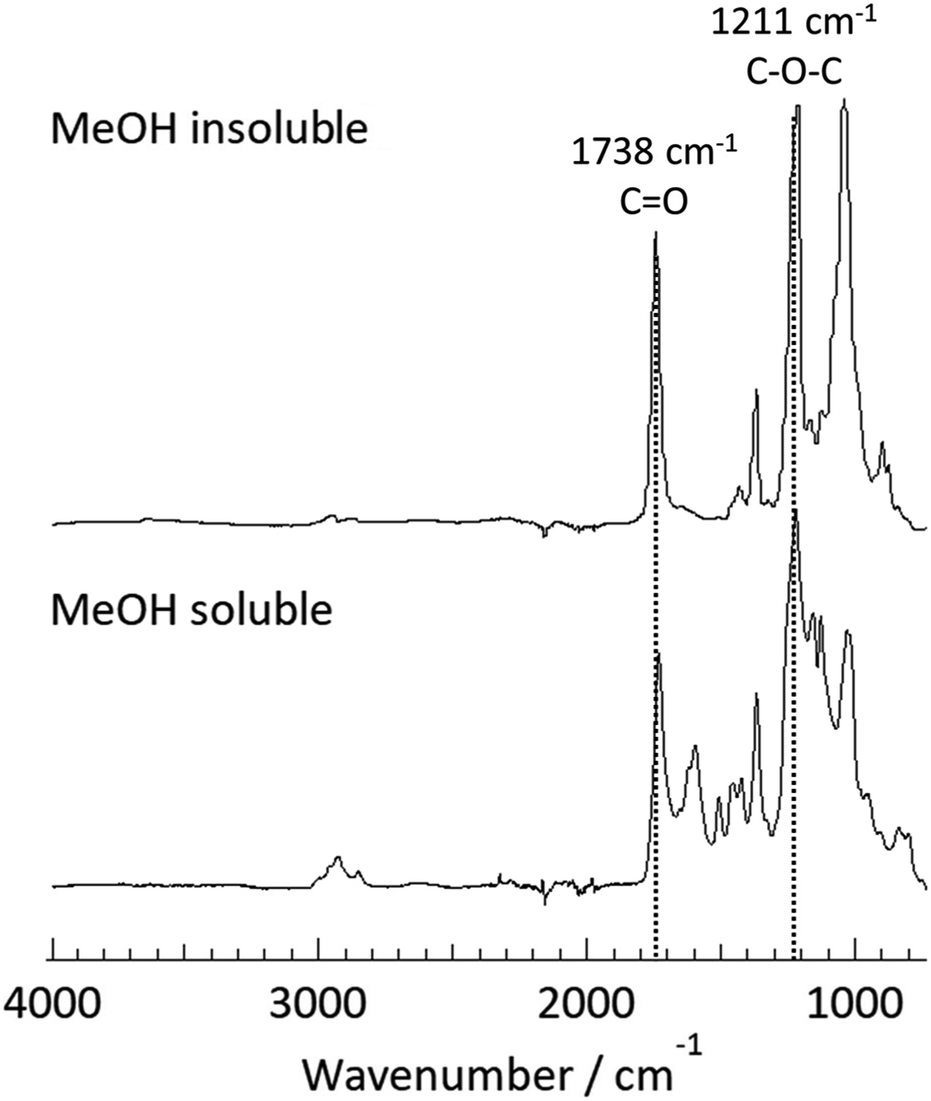 | ||
| Fig. 1 ATR-mode FT-IR spectra of the MeOH-insoluble fraction (upper) and the MeOH-soluble fraction (lower) isolated after the direct acetylation of bagasse at 80 °C for 30 min. | ||
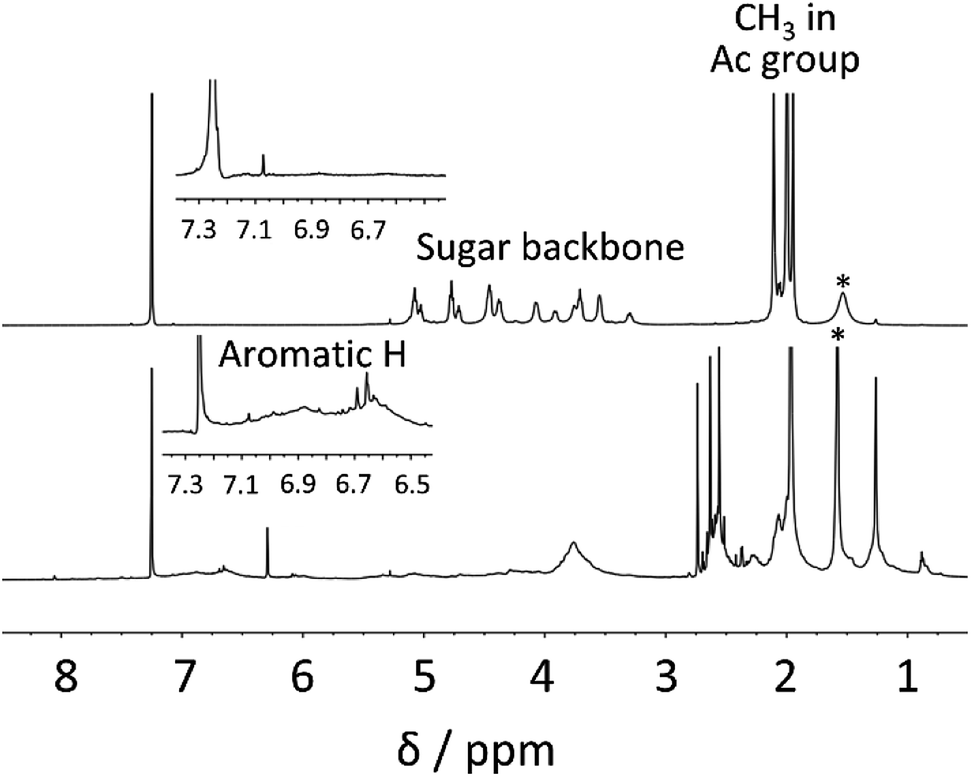 | ||
| Fig. 2 1H NMR spectra of the MeOH-insoluble fraction (upper) and the MeOH-soluble fraction (lower) isolated after the direct acetylation of bagasse at 80 °C for 30 min. Both 1H NMR spectra were measured in CDCl3 at 55 °C. | ||
In the same manner as that for the MeOH-insoluble fraction, peaks in the range of 1.9–2.4 ppm arising from the acetyl groups were detected in the spectrum of the MeOH-soluble fraction (Fig. 2 (lower)). The broad peaks in the spectral range of 6.5–7.2 ppm corresponded to the aromatic skeleton of lignin. It is noteworthy that there were no clear peaks attributable to polysaccharides. These experimental data indicated that the isolation of polysaccharide acetate from lignin acetate could be accomplished by a simple reprecipitation process with MeOH.
Next, a precise investigation of polysaccharide acetate was conducted to confirm the purity of the obtained polysaccharide acetate. Hence, the acetylated lignocellulosic components, namely, cellulose acetate, xylan acetate, and lignin acetate, were separately prepared as reference samples (see ESI† for the synthetic protocols). The 1H NMR spectrum of bagasse-derived polysaccharide acetate was compared with those of the reference samples, as depicted in Fig. 3. As expected from the chemical composition of bagasse, the above-mentioned 1H NMR spectrum was in a reasonable agreement with the superimposition of the spectra of cellulose acetate and that of xylan acetate without a distinct contribution from lignin acetate. In addition, the 13C NMR spectra also showed that the bagasse-derived polysaccharide acetate was composed of not only cellulose, but also other polymeric sugars (Fig. S1 in ESI†). These analyses demonstrated that the direct bagasse transesterification in EmimOAc/DMSO mixed system led to two fractions via a simple reprecipitation process, which corresponded to (1) the MeOH-insoluble polysaccharide-rich fraction mainly comprising cellulose acetate and xylan acetate, and (2) the MeOH-soluble fraction essentially containing lignin acetate.
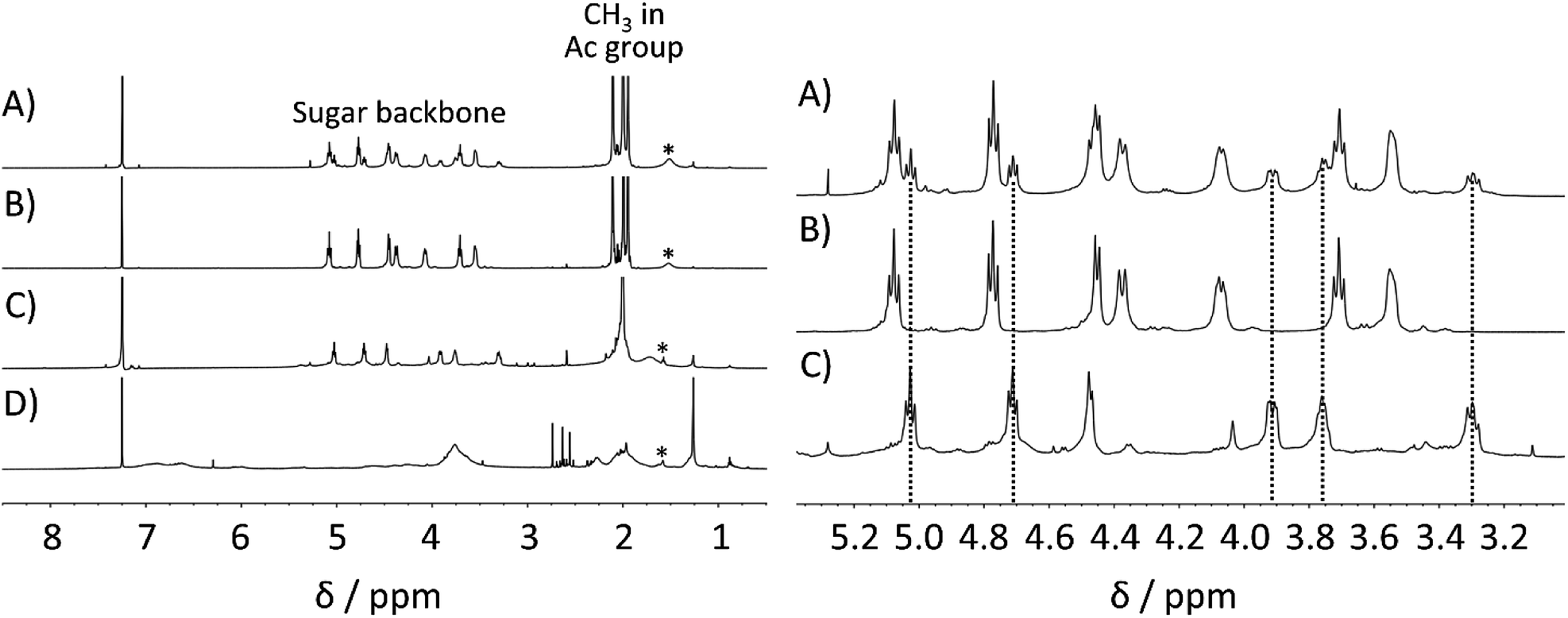 | ||
| Fig. 3 1H NMR spectra in CDCl3 at 55 °C (left) and the expansion (right) of (A) polysaccharide acetate in the MeOH-insoluble fraction after the direct acetylation of bagasse at 80 °C for 30 min, (B) cellulose acetate, (C) xylan acetate, and (D) lignin acetate as reference samples. | ||
3.2. Acetylation of mild acid-pretreated bagasse and enrichment of cellulose triacetate
As discussed in the previous section, the subsequent MeOH reprecipitation process after the direct modification of bagasse successfully afforded polysaccharide acetate without any harsh delignification treatments. Thus, the next challenge was to further separate cellulose acetate from hemicellulose acetate. The chemical structures of both cellulose and hemicellulose acetates are similar to each other and their solubility in common organic solvents is also similar in principle. It was expected that the similarity in properties would suppress the complete separation of cellulose acetate from hemicellulose acetate simply based on the solubility, as compared to the case of polysaccharide acetate and lignin acetate.We then hypothesised that a mild acid-pretreatment of the lignocellulose could lead to further separation of the lignocellulosic components, namely, cellulose and hemicellulose, because the mild acid hydrolysis could selectively destroy hemicellulose in the lignocellulose while cellulose and lignin remained intact.38 In order to chemically prove our concept, the mild acid-pretreated bagasse was subjected to transesterification in the EmimOAc/DMSO system in a manner similar to the bagasse transesterification mentioned above. After the reaction, the EmimOAc/DMSO reaction medium was also fractionated into the MeOH-insoluble and soluble portions. The isolated yields of the two fractions are summarised in Table 2 (the mass balance diagram is shown in Table S1 in ESI†), which were reasonable agreement with the corresponding theoretical yields estimated by the composition of pretreated bagasse (Table S4 in ESI†).
| Fraction | Main component | Isolated yielda,b (%) |
|---|---|---|
| a Average yields (n = 3).b Estimated on the basis of the results of the composition analyses of both pretreated bagasse and the acetylated products.c Collected as the chloroform-soluble portion. The amount of the chloroform-insoluble portion was negligible in the case of pretreated bagasse. | ||
| MeOH-insoluble | Cellulose triacetate | 61 ± 6.7c |
| MeOH-soluble | Lignin acetate | 82 ± 1.0 |
Furthermore, the 1H NMR spectrum of the MeOH-insoluble portion was in good agreement with that of cellulose acetate synthesized from Avicel (Fig. 4 (left)). Peaks corresponding to hemicellulose in the spectral range of 3.5–5.5 ppm and those attributable to lignin in the range of 6.2–7.2 ppm were not observed within the 1H NMR sensitivity range. The 13C NMR spectrum further confirmed that the MeOH-insoluble portion was correctly assigned as cellulose acetate (Fig. S2 in ESI†). In addition, the average DS (n = 3) of cellulose acetate obtained from pretreated bagasse was determined to be 2.98, corresponding to ∼99% OH group conversion (see ESI† for the DS determination). According to the US Federal Trade Commission, cellulose triacetate is defined as at least 92% of OH groups in the cellulose being acetylated.44 Thus, the cellulose acetate obtained from pretreated bagasse was assigned as cellulose triacetate.
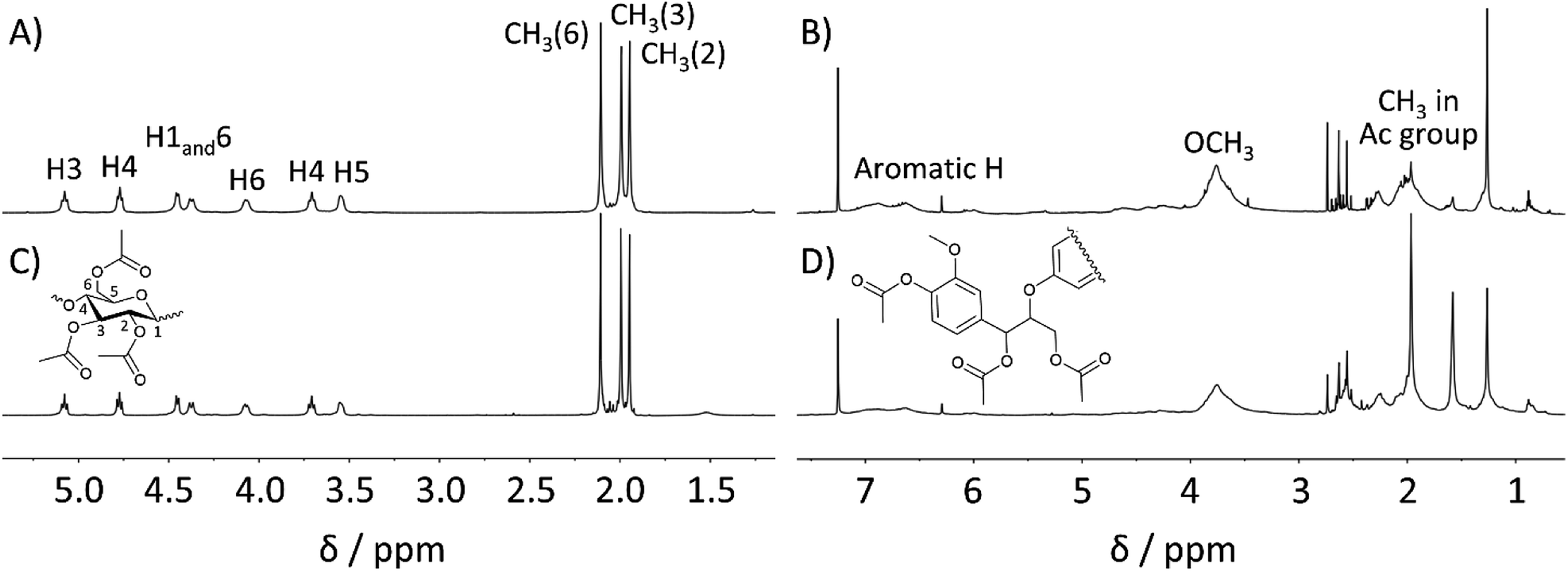 | ||
| Fig. 4 1H NMR spectra in CDCl3 at 55 °C of (A) cellulose triacetate in the MeOH-insoluble fraction, (B) lignin acetate in the MeOH-soluble fraction, which were isolated after direct acetylation of pretreated bagasse at 80 °C for 30 min (upper), (C) cellulose acetate synthesized from Avicel, and (D) lignin acetate sample synthesized from lignin isolated from bagasse as reference samples. | ||
On the other hand, the 1H NMR spectrum of the MeOH-soluble portion (depicted in Fig. 4 (right)) was also in good agreement with that of lignin acetate synthesised from lignin that was isolated from bagasse without any distinct peaks corresponding to the polysaccharides. The OH group conversion of lignin in pretreated bagasse was estimated to be 77% based on the quantitative 31P NMR measurements (Table S7 in ESI†). These results demonstrated the efficient acetylation of both cellulose and lignin in pretreated bagasse using the EmimOAc/DMSO system, and the subsequent reprecipitation processes enabled the synthesis of cellulose triacetate along with the production of lignin acetate. Considering the potential applications of lignin derivatives in diverse fields,19,45 this concurrent production of lignin acetate could be beneficial.
To further ensure the purity of the recovered cellulose triacetate, elemental analyses and SEC were measured. In addition, composition analyses by HPLC measurements were also performed for the monomeric sugars obtained after acid hydrolysis of the pretreated bagasse-derived cellulose triacetate. The elemental analysis of the cellulose triacetate was as follows: C, 49.19, H, 5.63, N, 0.20. This result was in good agreement with that of cellulose acetate obtained from Avicel (C, 49.18, H, 5.56, N, 0.16), demonstrating the easy purification method of cellulose triacetate from pretreated bagasse. Furthermore, SEC measurements were carried out for the cellulose triacetate and lignin acetate obtained from pretreated bagasse (Fig. 5, SEC charts and molecular weight (Mw) information of the other acetylated products are listed in ESI, Fig. S3–S4 and Table S2†). In general, cellulose acetate should be a linear polymer and form random coil conformations in the solution state. On the other hand, lignin acetate should have a branched structure and its hydrodynamic volume in solution is expected to be lower than that of cellulose acetate. As expected, the peak of cellulose triacetate obtained from pretreated bagasse did not overlap with that of lignin acetate (Fig. 5). The Mw of cellulose triacetate was 1.3 × 103 kg mol−1 and that of lignin acetate was determined to be 3.7 kg mol−1. These observations showed that cellulose triacetate was sufficiently separated from lignin acetate, and the obtained cellulose triacetate maintained its polymeric structure throughout the mild acid pretreatment.
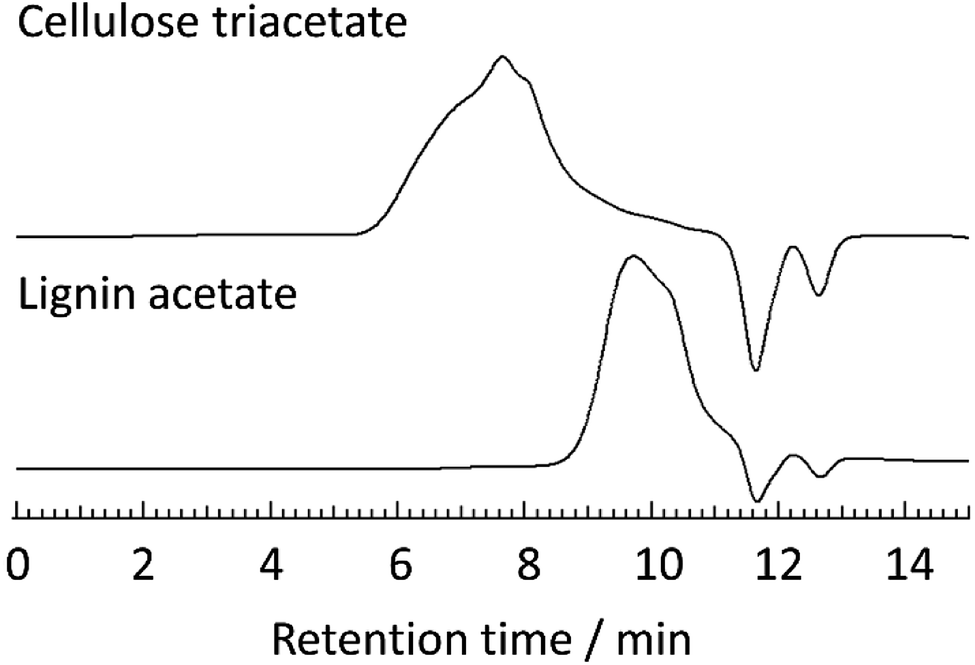 | ||
| Fig. 5 SEC charts of cellulose triacetate (upper) and lignin acetate (lower) from pretreated bagasse measured at 40 °C in DMF containing 0.01 mol L−1 LiBr. | ||
The monomeric structure of cellulose triacetate obtained from the pretreated bagasse was precisely determined by HPLC analysis. Specifically, the acid-hydrolysed polysaccharide samples were analysed by comparing with glucose, xylose, galactose, arabinose, and mannose as standard sugars (the details of composition analysis were summarized in ESI, please see Fig. S5–S6†). Fig. 6 shows the HPLC charts of the hydrolysates from cellulose triacetate that was obtained from both pretreated bagasse and Avicel and the determined compositions are summarised in Table 3. The contamination of hemicellulose (∼2.1 wt%) and lignin (∼5.2 wt%) was in the range observed for the Avicel-derived reference sample. Most importantly, the HPLC measurements proved a sufficiently high glucose purity (∼90%, Table S3 in ESI†) for cellulose triacetate obtained from pretreated bagasse. This glucose concentration reached the value for cellulose triacetate from Avicel. Overall, the simple synthesis of enriched cellulose triacetate was achieved by the combination of the mild acid-pretreatment of bagasse, direct transesterification in EmimOAc/DMSO, and subsequent reprecipitation in MeOH.
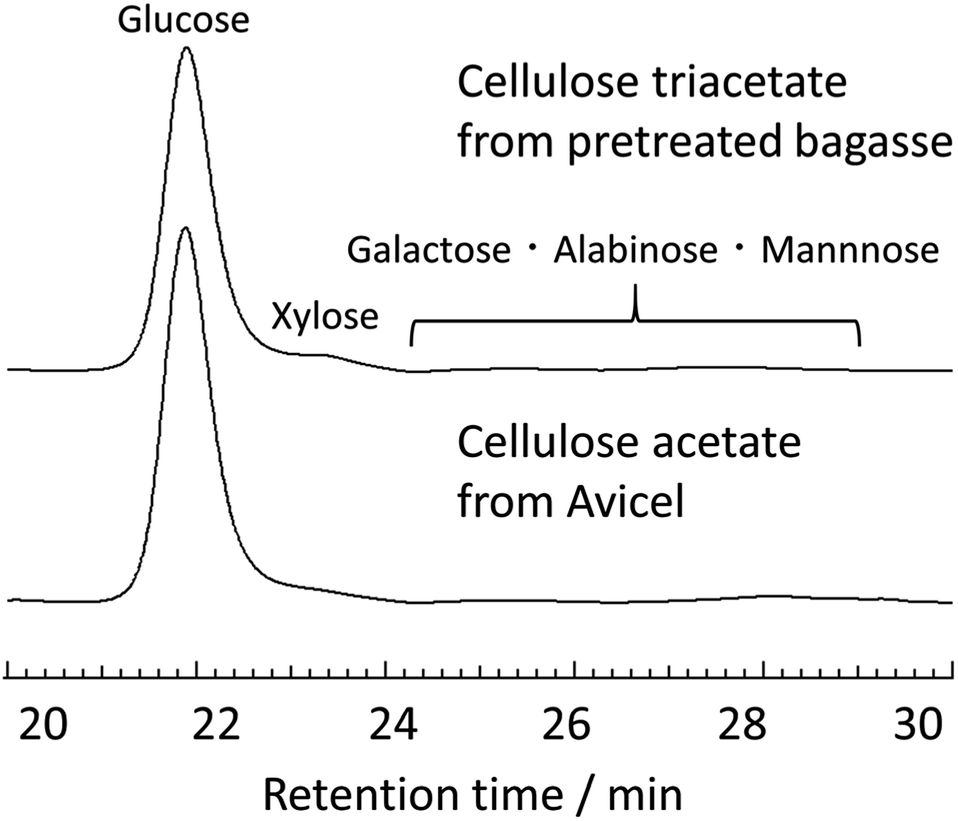 | ||
| Fig. 6 HPLC chromatograms of the acid-hydrolysed mixtures from cellulose triacetate from the pretreated bagasse (upper) and cellulose acetate from Avicel (lower) measured at 85 °C in ultra-pure water. | ||
| Material | Composition (wt%) | |||
|---|---|---|---|---|
| Cellulose | Hemicellulosea | Lignin | Acetyld | |
| a Composed of xylan as the main chain branched with galactose, arabinose, and mannose as minor monomeric sugars.b Average results (n = 3).c Although Avicel should not include lignin, the composition analysis of the Avicel-derived cellulose acetate included a slight amount (2.2%) of a component detected as lignin. This result can be rationalised by a small amount of remaining materials during the hydrolysis process, which was detected as “lignin”.d Calculated by the results of the DS values of the polysaccharides and lignin, determined via 1H NMR and quantitative 31P NMR analyses, respectively. | ||||
| Cellulose triacetate from pretreated bagasseb | 53.0 | 2.1 | 5.2 | 39.8 |
| Cellulose acetate from Avicelb,c | 55.9 | 1.4 | 2.2 | 40.3 |
4. Conclusions
The bifunctional use of EmimOAc enabled the homogeneous transesterification of lignocellulose without any corrosive reagents and additional catalysts. The reaction medium from the transesterification of bagasse was subjected to a simple reprecipitation in MeOH, essentially fractionating into polysaccharide acetate and lignin acetate. In addition, the mild acid-pretreated bagasse was employed as the starting biomass to realise the facile enrichment of cellulose triacetate with a high glucose purity (∼90%) without using a harsh delignification process. Therefore, we believe that the reported protocol can provide a direct path toward the production of important polymeric materials from biomass.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This study was promoted by the Center of Innovation Science and Technology based Radical Innovation and Entrepreneurship Program (COI stream) of the Science and Technology Agency of Japan (JST).Notes and references
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef PubMed.
- G. Chatel, K. De Oliveira Vigier and F. Jerome, ChemSusChem, 2014, 7, 2774–2787 CrossRef PubMed.
- F. Shafizadeh, Pure Appl. Chem., 1973, 35, 195–208 CrossRef.
- M. Ionita, L. E. Crica, S. I. Voicu, A. M. Pandele and H. Iovu, Polym. Adv. Technol., 2016, 27, 350–357 CrossRef.
- X. Fan, Z.-W. Liu, J. Lu and Z.-T. Liu, Ind. Eng. Chem. Res., 2009, 48, 6212–6215 CrossRef.
- D. Hoffmann, I. Hoffmann and K. El-Bayoumy, Chem. Res. Toxicol., 2001, 14, 767–790 CrossRef PubMed.
- T. C. Nirmale, I. Karbhal, R. S. Kalubarme, M. V. Shelke, A. J. Varma and B. B. Kale, ACS Appl. Mater. Interfaces, 2017, 9, 34773–34782 Search PubMed.
- H. Soeta, S. Fujisawa, T. Saito, L. Berglund and A. Isogai, ACS Appl. Mater. Interfaces, 2015, 7, 11041–11046 Search PubMed.
- K. Jedvert and T. Heinze, J. Polym. Eng., 2017, 37, 845–860 Search PubMed.
- S. Liu and K. J. Edgar, Carbohydr. Polym., 2017, 162, 1–9 CrossRef PubMed.
- S. Tanaka, T. Iwata and M. Iji, ACS Sustainable Chem. Eng., 2017, 5, 1485–1493 CrossRef.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- K. J. Edgar, C. M. Buchanan, J. S. Debenham, P. A. Rundquist, B. D. Seiler, M. C. Shelton and D. Tindall, Prog. Polym. Sci., 2001, 26, 1605–1688 CrossRef.
- W. O. S. Doherty, P. Mousavioun and C. M. Fellows, Ind. Crops Prod., 2011, 33, 259–276 CrossRef.
- R. Singh, A. Shukla, S. Tiwari and M. Srivastava, Renewable Sustainable Energy Rev., 2014, 32, 713–728 CrossRef.
- M. Alekhina, O. Ershova, A. Ebert, S. Heikkinen and H. Sixta, Ind. Crops Prod., 2015, 66, 220–228 CrossRef.
- T. Q. To, C. Kenny, S. Cheong and L. Aldous, Green Chem., 2017, 19, 2129–2134 RSC.
- J. S. Kim, Y. Y. Lee and T. H. Kim, Bioresour. Technol., 2016, 199, 42–48 CrossRef PubMed.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef.
- F. S. Chakar and A. J. Ragauskas, Ind. Crops Prod., 2004, 20, 131–141 CrossRef.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef PubMed.
- W. L. Hough and R. D. Rogers, Bull. Chem. Soc. Jpn., 2007, 80, 2262–2269 CrossRef.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef PubMed.
- C. Achtel, K. Jedvert, B. Kosan, O. A. El Seoud and T. Heinze, Macromol. Chem. Phys., 2017, 218, 1700208 CrossRef.
- T. Kakko, A. W. T. King and I. Kilpelainen, Cellulose, 2017, 24, 5341–5354 CrossRef.
- H. Hanabusa, E. I. Izgorodina, S. Suzuki, Y. Takeoka, M. Rikukawa and M. Yoshizawa-Fujita, Green Chem., 2018, 20, 1412–1422 RSC.
- R. Kakuchi, M. Yamaguchi, T. Endo, Y. Shibata, K. Ninomiya, T. Ikai, K. Maeda and K. Takahashi, RSC Adv., 2015, 5, 72071–72074 RSC.
- R. Kakuchi, R. Ito, S. Nomura, H. Abroshan, K. Ninomiya, T. Ikai, K. Maeda, H. J. Kim and K. Takahashi, RSC Adv., 2017, 7, 9423–9430 RSC.
- Q. V. Nguyen, S. Nomura, R. Hoshino, K. Ninomiya, K. Takada, R. Kakuchi and K. Takahashi, Polym. J., 2017, 49, 783–787 CrossRef.
- C. S. Lovell, A. Walker, R. A. Damion, A. Radhi, S. F. Tanner, T. Budtova and M. E. Ries, Biomacromolecules, 2010, 11, 2927–2935 CrossRef PubMed.
- S. Koehler, T. Liebert, M. Schoebitz, J. Schaller, F. Meister, W. Guenther and T. Heinze, Macromol. Rapid Commun., 2007, 28, 2311–2317 CrossRef.
- I. Kilpelaeinen, H. Xie, A. King, M. Granstrom, S. Heikkinen and D. S. Argyropoulos, J. Agric. Food Chem., 2007, 55, 9142–9148 CrossRef PubMed.
- N. Sun, M. Rahman, Y. Qin, M. L. Maxim, H. Rodriguez and R. D. Rogers, Green Chem., 2009, 11, 646–655 RSC.
- M.-J. Chen, R.-M. Li, X.-Q. Zhang, J. Feng, J. Feng, C.-F. Liu and Q.-S. Shi, ACS Sustainable Chem. Eng., 2017, 5, 360–366 CrossRef.
- B. Adney and J. Baker, Laboratory analytical procedure, 1996, 6, 1–8 Search PubMed.
- Y. Zhao, X. Liu, J. Wang and S. Zhang, J. Phys. Chem. B, 2013, 117, 9042–9049 CrossRef PubMed.
- J.-M. Andanson, E. Bordes, J. Devemy, F. Leroux, A. A. H. Padua and M. F. C. Gomes, Green Chem., 2014, 16, 2528–2538 RSC.
- C. Li, B. Knierim, C. Manisseri, R. Arora, H. V. Scheller, M. Auer, K. P. Vogel, B. A. Simmons and S. Singh, Bioresour. Technol., 2010, 101, 4900–4906 CrossRef PubMed.
- S. Hama, K. Nakano, K. Onodera, M. Nakamura, H. Noda and A. Kondo, Bioresour. Technol., 2014, 157, 1–5 CrossRef PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Laboratory analytical procedure, 2008, 1617, 1–16 Search PubMed.
- H. Sakai, K. Kuroda, S. Muroyama, T. Tsukegi, R. Kakuchi, K. Takada, A. Hata, R. Kojima, T. Ogoshi, M. Omichi, K. Ninomiya and K. Takahashi, Polym. J., 2018, 50, 281–284 CrossRef.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef.
- Y. Pu, S. Cao and A. J. Ragauskas, Energy Environ. Sci., 2011, 4, 3154–3166 Search PubMed.
- Fed. Regist., 2002, 67, 70835–70839 Search PubMed.
- S. Gillet, M. Aguedo, L. Petitjean, A. R. C. Morais, A. M. da Costa Lopes, R. M. Lukasik and P. T. Anastas, Green Chem., 2017, 19, 4200–4233 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation methods of lignin samples, synthetic protocol of cellulose acetate, xylan acetate, and lignin acetate, 13C NMR spectra, HPLC measurements for composition analysis, SEC measurement, determination of DS for the acetylated polysaccharides, quantitative 31P NMR for determination of OH content of lignin samples. See DOI: 10.1039/c8ra03859g |
| ‡ Since all the procedures were handled under either vacuum or Ar atmosphere, the water concentration of the reaction mixture was practically negligible. |
| § In the case of the acetylation of pretreated bagasse, only a small amount of the chloroform-insoluble material was obtained. |
| ¶ Among alcohol derivatives, MeOH was selected because of its low boiling point and low cost. In addition, other common organic solvents were not selected because acetone, for example, can partially dissolve polysaccharide acetate as well as lignin acetate. |
| This journal is © The Royal Society of Chemistry 2018 |