
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA practically simple, catalyst free and scalable synthesis of N-substituted ureas in water
Lata Tiwari,
Varun Kumar,
Bhuvesh Kumar and
Dinesh Mahajan *
*
Drug Discovery Research Center, Translational Health Science and Technology Institute, NCR Biotech Science Cluster, 3rd Milestone, Faridabad-Gurgaon Expressway, Faridabad-121001, India. E-mail: dinesh.mahajan@thsti.res.in; chemidinesh@gmail.com
First published on 13th June 2018
Abstract
A practically simple, mild and efficient method is developed for the synthesis of N-substituted ureas by nucleophilic addition of amines to potassium isocyanate in water without organic co-solvent. Using this methodology, a variety of N-substituted ureas (mono-, di- and cyclic-) were synthesized in good to excellent yields with high chemical purity by applying simple filtration or routine extraction procedures avoiding silica gel purification. The developed methodology was also found to be suitable for gram scale synthesis of molecules having commercial application in large volumes. The identified reaction conditions were found to promote a unique substrate selectivity from a mixture of two amines.
Introduction
Development of resource-efficient as well as environment-friendly synthetic processes for manufacturing of chemicals and intermediates being used in large quantities is a must for sustainable industrial development.1 Urea and its derivatives are an important class of molecules having diverse chemical and biological properties and hence being extensively employed in chemical,2 pharmaceutical3 and agrochemical4 industries. Because of their diverse applications, a large number of urea derivatives are high production volume chemicals (HPV).5 Being molecules of commercial interest, not only their use but also their manufacturing methods are a potential source of environment pollution. There are strict regulatory guidelines for use and application of HPV chemicals. Unfortunately, that is not the case for their manufacturing processes.6 In general, industrial manufacturing processes for high volume chemicals are influenced by economy and ease of execution providing little consideration for environmental impacts.7 Synthetic chemists have obligations and challenges to develop resource-efficient as well as environment-friendly production methods.1d N-Substituted ureas are an important subclass of urea derivatives, where one of the nitrogen atoms is substituted with an alkyl or aryl group. The N-substituted urea derivatives were not only found to have direct applications as agrochemicals4 or pharmaceutical3 agents but also serve as building blocks for various other important chemicals2 of high commercial interest.The most common method for the synthesis of N-substituted ureas involve the reaction of isocyanates or carbamoyl chlorides with ammonia.3f–i,8 The desired isocyanate and carbamoyl chloride can be generated by the reaction of corresponding amine with phosgene.8f,g This is neither environment friendly nor safe,9 but still one of the few methods which is assertively being used. Lately, phosgene was replaced with safer reagents such as triphosgene, 1,1-dicarbonyl imidazole and carbonates.10 Hoffmann, Curtius and Lossen rearrangement are complementary approaches which exploit in situ isocyanate generation.11 However compared to phosgene, these methods are less attractive for commercial production. Importantly, all these methods involving isocyanate need ammonia gas as one of the reactant, which led to safety issues and operational complexities. Direct alkylation or arylation of urea appears to be straightforward approach but need harsh conditions.12 Also, involvement of O-alkylation and/or dialkylation as competing reactions is a point of concern. Reaction of amines with 4-nitrophenyl-N-benzyl carbamate followed by hydrogenation,13 oxidation of thiourea derivatives,14 metal or base catalysed transamidation of urea,15 conversion of cyanamides via Tiemann rearrangement16 and transition metal catalysed carbonylation17 of amines are some recently developed less explored methods for urea synthesis. These methods are mostly employed for synthesis of substituted ureas and though added huge intellectual addition on academic understandings, but unfortunately found to have no or very limited applicability for industrial production of N-substituted ureas. Transamidation of urea is an attractive approach, but often requires catalyst or/and heating.15 Interestingly, nucleophilic addition of amine to metal isocyanate looks like a most facile and safe method for commercial production of N-substituted ureas.18 However, this is least explored method compared to all with very limited substrate scope.18d–f Moreover, most of the reports are limited to the use of either excess of acid or specialized acid as promoter and often requires high temperature or microwave irradiation.18a,b Requirement of harsh conditions and fancy promoters not only restrict the scope but also make the approach a resource inefficient and environmentally hazardous. Herein, we want to report a synthetic protocol for the synthesis of N-substituted ureas in water, which is practically simple, having broader substrate scope, found to be scalable and environment friendly as well. The newly developed method potentially has a possibility to be a method of choice for manufacturing of various N-substituted ureas, which are being used in industry as large volume commercial products and intermediates. Additionally, the developed methodology holds potential to provide a chemo-selectivity based on pKaH of amines.
Results and discussion
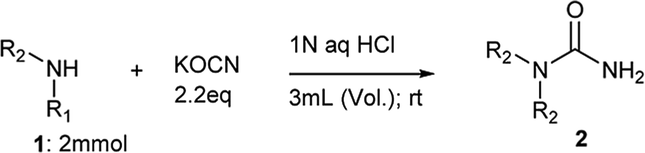
To facilitate internal drug discovery efforts, we were keen to develop a mild and efficient method for synthesis of N-substituted ureas exploiting nucleophilic addition of amines to potassium isocyanate. Accordingly, reaction of aniline (1a) and benzylamine (1b) was attempted individually in presence of hydrochloric acid using different solvent systems at 25 °C (Scheme 1). The hydrochloric acid was used primarily to activate potassium cyanate as an electrophile. The outcome of this study is tabulated in Table 1. We planned to use 4 M solution of HCl in dioxane for this reaction with and without any additional co-solvent such as tetrahydrofuran (THF) and acetonitrile (ACN) (entry 1, 2 and 3; Table 1). Reactions performed in either neat dioxane or using freshly dried THF as a co-solvent, gave no conversion for aniline and low conversion for benzylamine (entry 1 and 2) to corresponding ureas 2a and 2b respectively. Intriguingly, ACN as a co-solvent found to have a profound effect on reactivity of aniline and no effect on benzylamine unlike THF (entry 3 vs. entry 2). Using ACN as co-solvent not only gave better conversion for urea 2a but also demonstrated a reversal in reactivity order of two amines. Aniline, in spite of being less nucleophilic gave better conversion compared to benzylamine using ACN as solvent. We started looking for an explanation to describe this unusual effect of ACN. It was noticed that, the ACN used from shelf bottle has a small amount of residual water as mentioned by manufacturer label. This instigated us to do a new experiment with fresh lot of ACN that was dried over molecular sieves before employing in reaction (entry 4).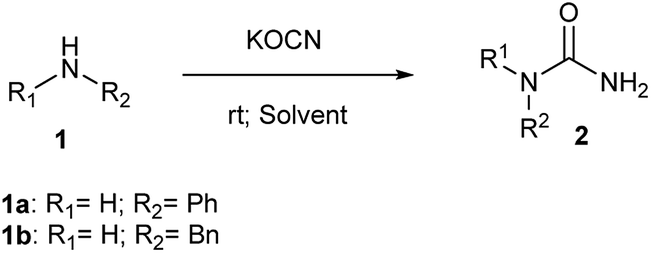 | ||
| Scheme 1 | ||
| Entry | Amine 1 | 4 M HCl (dioxane) | Co-solvent | Conversiona (%) 2 | Time (hrs) |
|---|---|---|---|---|---|
| a Based on 1H NMR.b 2.2 equivalent of KOCN used. | |||||
| 1 | 1a | 3 mL | No solvent | 0 | 12 |
| 1b | 17 | ||||
| 2 | 1a | 1 mL | THF (dry; 2 mL) | 0 | 12 |
| 1b | 29 | ||||
| 3 | 1a | 1 mL | ACN (2 mL) | 88 | 12 |
| 1b | 30 | ||||
| 4 | 1a | 1 mL | ACN (dry; 2 mL) | 18 | 12 |
| 1b | ND | ||||
| 5 | 1a | 1 mL | H2O (0.5 mL) | <5 | 12 |
| 1b | ND | ||||
| 6 | 1a | 1 mL | H2O (2 mL) | 81 | 12 |
| 1b | 98 | ||||
| 7 | 1a | NA | Aq. HCl (1 N, 3 mL) | >95 | 10 |
| 1b | >95 | ||||
| 8 | 1a | NA | Aq. HCl (1 N, 3 mL) | >95 | 6b |
| 1b | >95 | ||||
| 9 | 1a | NA | Aq. HCl (0.5 N,3 mL) | 66 | 12 |
| 10 | 1a | NA | Aq. HCl (4 N, 3 mL) | 31 | 12 |
| 11 | 1a | NA | Water 3 mL | 14 | 12 |
The use of dry ACN, was found to be detrimental for conversion of aniline to its corresponding urea (2a; 18%) but did not affect the overall conversion of benzylamine (2b; 30%) as mentioned in entry 4. This made us analyse and explore the role of water on reactivity of amine under given reaction conditions (entry 5, 6 and 7). For reactions of aniline and benzylamine, corresponding N-substituted ureas were observed in high yields when reaction was performed using water as co-solvent (entry 6) or using water only as the only solvent (entry 7). Actually, rate of reaction, product conversion and isolated yields were found to be better when reaction was performed exclusively in water without any co-solvent (entry 7 vs. entry 6). Water a universal solvent, found to be a good promoter for higher yield of N-substituted urea. It was also observed that 2 equivalents of KOCN afford better conversion and yields in shorter time compared to 1.1 equivalents (entry 7 vs. entry 8). The critical role of HCl (entry 11 vs. entry 6) and its optimum amount (1 N, 3 mL aqueous solution of HCl) was quickly established by varying the concentration of HCl (entry 9 and 10). We attempted to build on this and planned for a substrate scope evaluation using conditions of entry 8 of Table 1 using different amines. The outcome of this study is presented in Table 2.
|
|||
|---|---|---|---|
| Entry | Product | Time (h) | Isolated yield (%) |
| a Un-reacted amine recovered.b Amine was used in excess; NR = no reaction.c Yields based on 1H NMR as product could not be isolated from starting amine. | |||
| 1 | 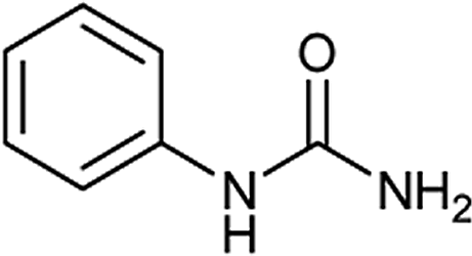 |
6 | 94 |
| 2 |  |
8 | 90 |
| 3 | 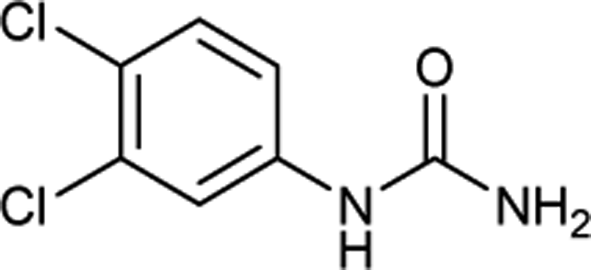 |
12 | 71a |
| 4 | 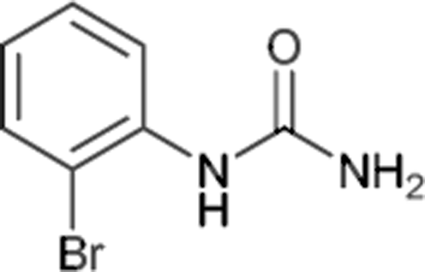 |
12 | 93 |
| 5 | 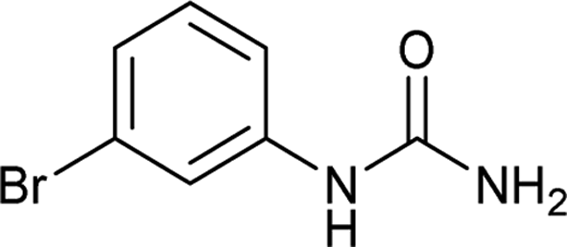 |
12 | 82 |
| 6 | 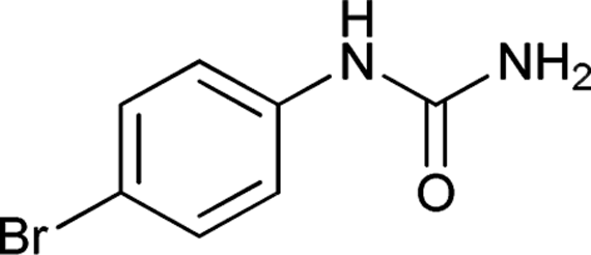 |
12 | 80 |
| 7 | 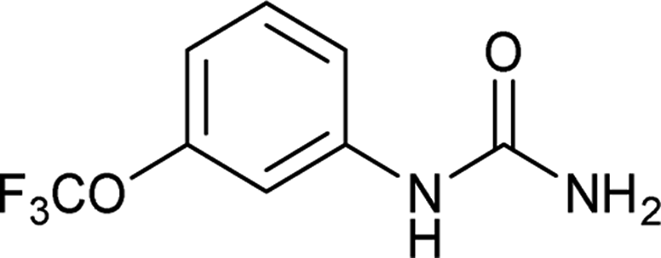 |
12 | 80a |
| 8 | 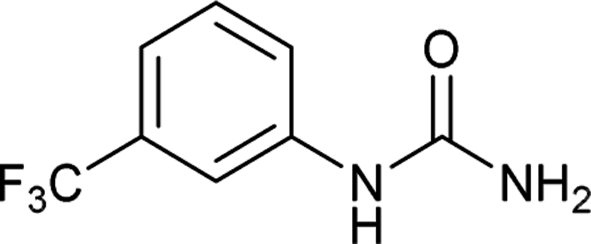 |
12 | 63a |
| 9 | 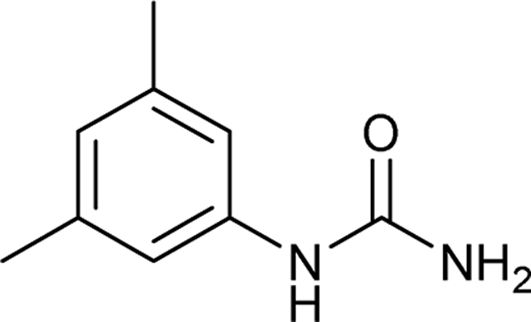 |
4 | 97 |
| 10 | 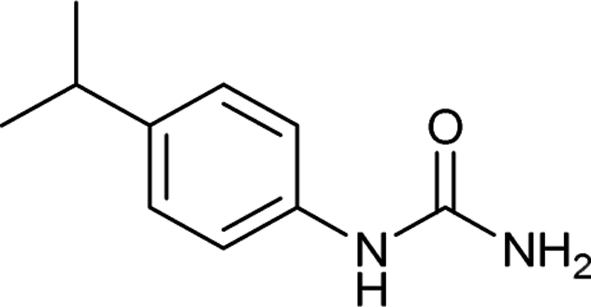 |
4 | 88 |
| 11 | 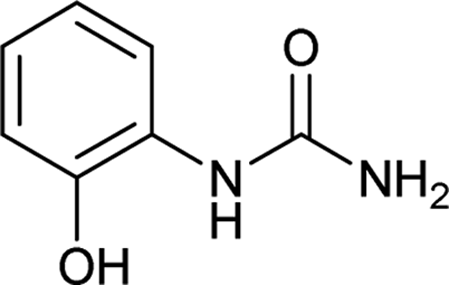 |
12 | 90 |
| 12 | 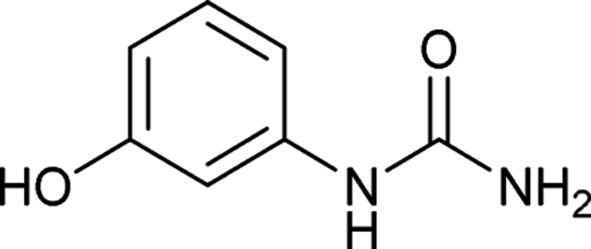 |
12 | 67a |
| 13 | 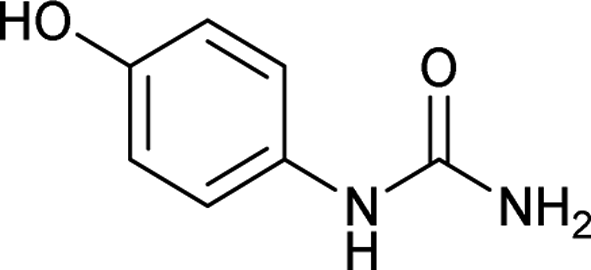 |
12 | 89 |
| 14 | 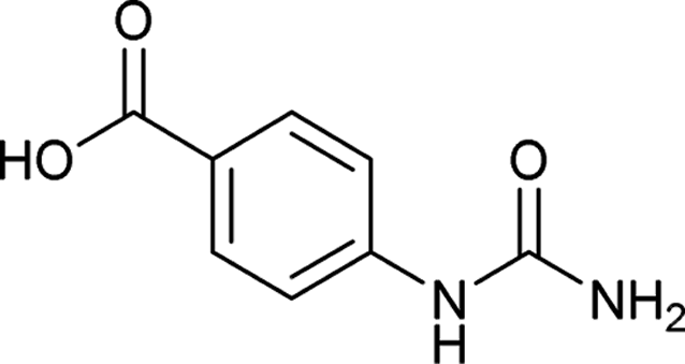 |
12 | 83 |
| 15 | 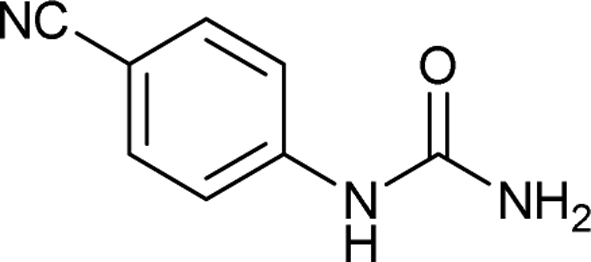 |
12 | 47a |
| 16 | 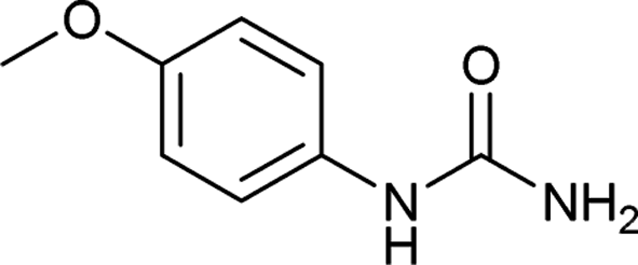 |
12 | 70a |
| 17 | 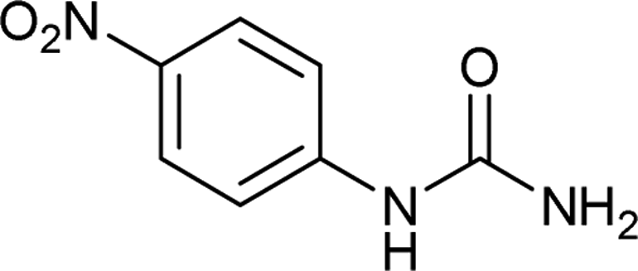 |
24 | NRa |
| 18 | 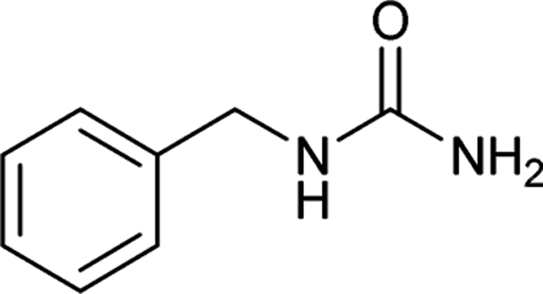 |
6 | 89 |
| 19 | 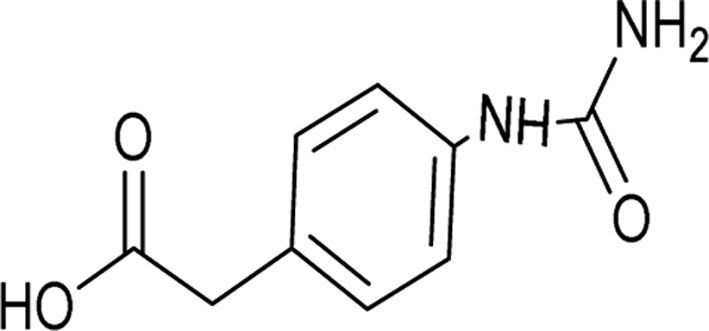 |
12 | 81a |
| 20 | 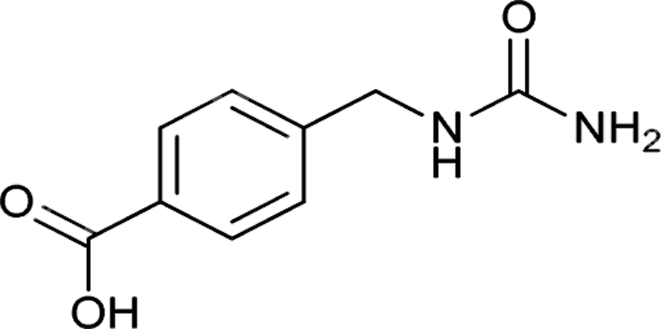 |
12 | 73a |
| 21 | 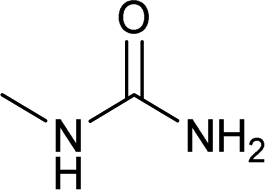 |
5 | 74 |
| 22 | 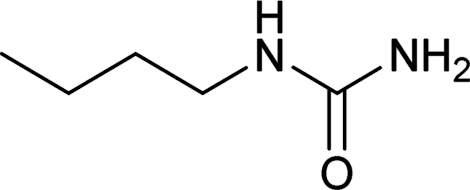 |
12 | 52a |
| 23 | 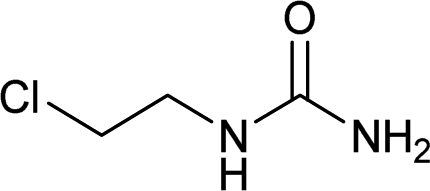 |
12 | 72c |
| 24 | 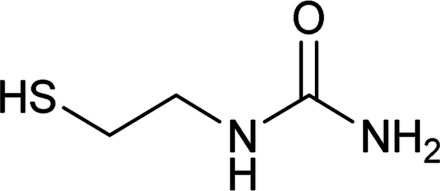 |
12 | 68c |
| 25 | 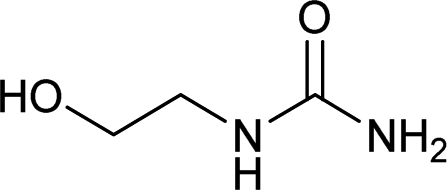 |
12 | 68a |
| 26 | 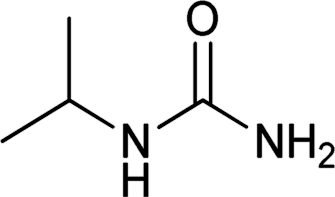 |
12 | 44a |
| 27 | 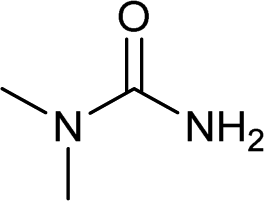 |
12 | 71a |
| 28 | 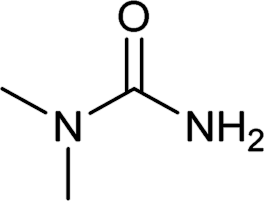 |
12 | 91b |
| 29 | 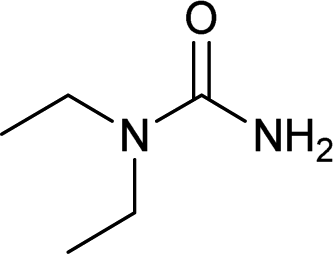 |
12 | 73a |
| 30 | 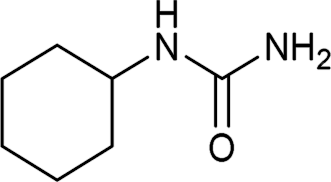 |
12 | 70a |
| 31 | 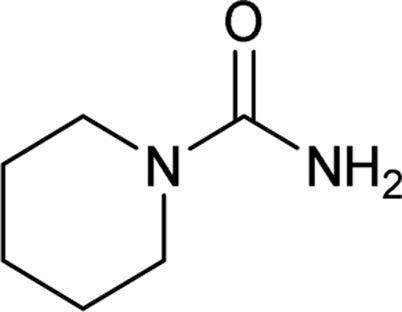 |
12 | 60a |
| 32 | 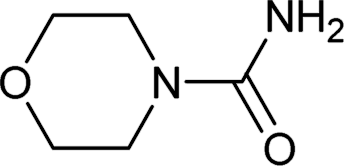 |
12 | 75a |
| 33 | 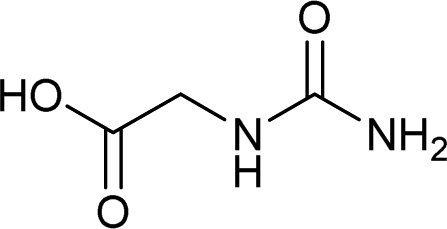 |
12 | 68a |
| 34 | 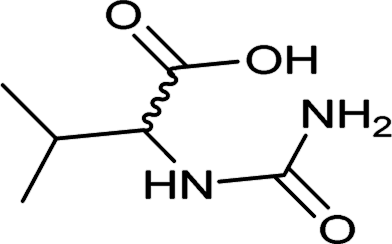 |
12 | 53a |
| 35 | 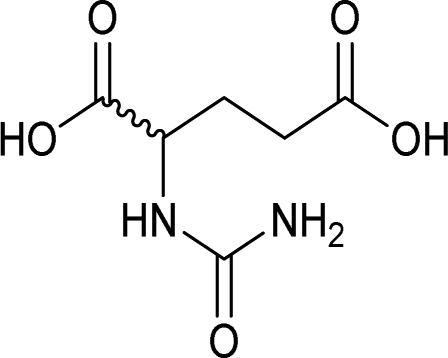 |
12 | 18a |
| 36 | 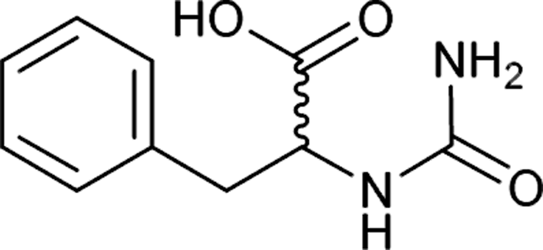 |
12 | 63a |
| 37 | 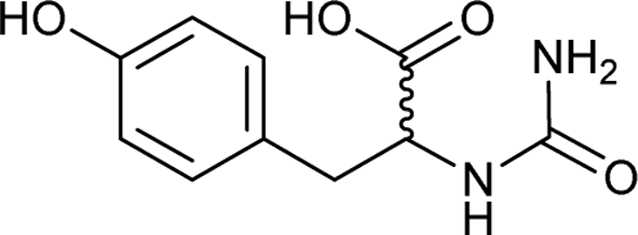 |
12 | 80 |
| 38 | 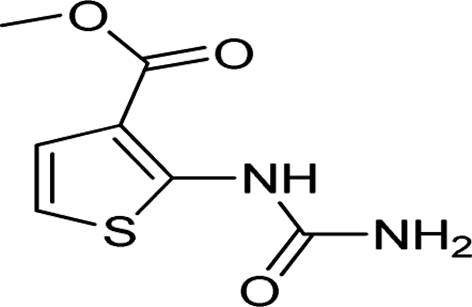 |
12 | 30a |
| 39 | 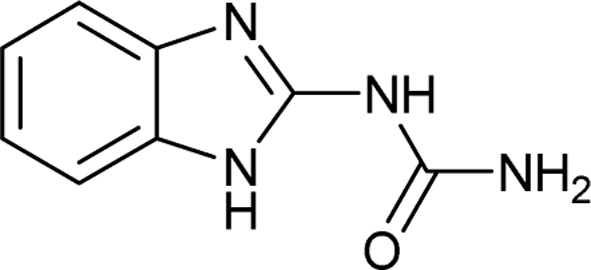 |
12 | 70a |
| 40 |  |
12 | 70a |
We tried to generalize reaction with different aromatic, aliphatic and hetero aromatic amines using 1 N aqueous HCl as solvent. All the reactions of Table 2 were done using 2 mmol of amine, 2.2 equivalent of KOCN and 3 mL of 1 N aqueous HCl. Most of the tested amines provided good to excellent isolated yields of the corresponding N-substituted ureas. The reaction profile was very clean for all the evaluated substrates irrespective of the amine type used. It was either desired N-substituted urea as a single product or a mixture of desired product with unreacted starting amine at the end of the reaction time. Reactions involving aniline derivatives were very facile and it was practically very easy to isolate the final products. The corresponding urea product of all tested anilines tends to precipitate out with progress of the reaction, which can be filtered out easily (entry 1 to 17). Compared to this, aliphatic ureas did not precipitate out as efficiently from the reaction mixture requiring an extraction with ethyl acetate in all reported cases. The reactions involving most of the aromatic amines proceeded smoothly and afforded good to excellent yields of the product within short reaction time (6–12 hours) except entries 3, 8, 15, 16, and 17. The higher reaction rate and better isolated yields were obtained for aniline substituted with electron donating groups e.g. entry 9 and 10. The lower conversion for entries 3, 8, 15 to 17 in Table 2 was attributed to the presence of electron withdrawing groups (i.e. Cl, CF3, CN, OMe and NO2) which seems to hamper the reactivity of the aniline. In contrast to aromatic amines, most of the aliphatic amines evaluated in Table 2 demonstrated slower reaction rates, which was a surprise, considering the higher nucleophilicity of aliphatic amines (entry 21, 22 and 28 to 32) compared to aniline. It is worth to mention that all of the reported reactions were very clean and one can recover unreacted aliphatic amines along with desired N-substituted ureas when there was an incomplete conversion. No chemical interference from other reactive groups such as –SH (entry 24), aliphatic –OH (entry 25) and phenolic –OH (entries 11 to 13) was found to be another pleasant outcome of the developed methodology. In addition, the developed methodology found to be a suitable approach to prepare N-carbamoylation of amino acids to generate corresponding ureas.18c It is important to mention that, we only tested racemic counterpart of few selected amino acids for this study (entries 33 to 37). This methodology was found to work well for heterocyclic amines affording good yield of corresponding N-substituted ureas in high purity (entries 38 to 40), however yields were found to be low due to in-complete conversion of the starting amines.
The plausible mechanism for this reaction involves formation of a reactive isocyanic acid (I) from KOCN in presence of aqueous acidic conditions (eqn (1); Fig. 1). This is followed by a nucleophilic attack of amine resulting in the formation of corresponding N-substituted urea (eqn (2)). We tried to reason out the slow rate of reaction for aliphatic amines (entry 21, 22 and 28–32) compared to anilines (entry 1–11). We argued that, it is the higher basicity of the aliphatic amines or higher pKa of their conjugate acids which is playing critical role in determining the rate of reaction. The average pKaH (pKa of conjugate acids) of aniline and its derivatives is low (range 2 to 5) compared to aliphatic amines (range of 8–12).19 Aliphatic amines such as diethyl amine (entry 29; Table 2) cyclohexyl amine (entry 30; Table 2), piperidine (entry 31; Table 2) and morpholine (entry 32; Table 2) tend to exist more as ammonium salt (eqn (3)) in aqueous HCl reaction conditions (pH between 2 to 4) because of their higher pKaH values. Hence, higher population of the basic aliphatic amine exists as ammonium salt (1 HCl) and not as free nucleophilic amine (1) and this led to slow rate of reaction and poor conversion of desired product. Plausible reaction mechanism
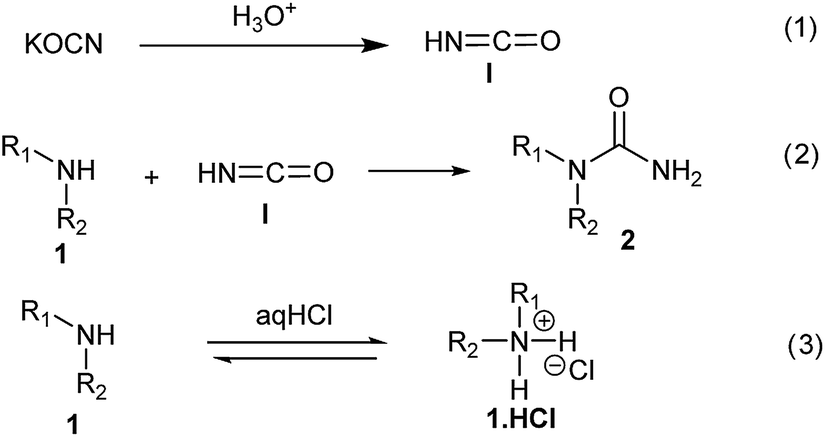 | ||
| Fig. 1 Plausible reaction mechanism. | ||
We planned to test this argument. A reaction of KOCN (1.1 equiv.) was performed with an equimolar mixture of aniline (1a) and morpholine (1c) as mentioned in Scheme 2. After a reaction time of four hours, we observed exclusive formation of N-substituted urea (2a) of aniline in reaction mixture. There was no reaction of morpholine and hence no trace of corresponding urea 2c after reaction time of 4 hours. The un-reacted morpholine was recovered in aqueous layer after filtration of solid precipitates of 2a. Although we did observe traces of 2c i.e. urea of morpholine after a long reaction time of 6 hours as studied by 1H NMR, but this was not significant. Exclusive reaction of aniline with KOCN even in presence of more nucleophilic morpholine supported our proposed hypothesis and role of pKaH. We attempted two additional reactions of aniline, one each in presence of benzylamine (1b) and piperidine (1d). The results were same in these two cases as well. There was exclusive formation of aniline urea (2b) and we could recover unreacted benzylamine and piperidine respectively in aqueous layer after filtration of pure 2b. This demonstrated that, the developed reaction conditions offered unique chemo-selectivity influenced by pKaH of the amines.
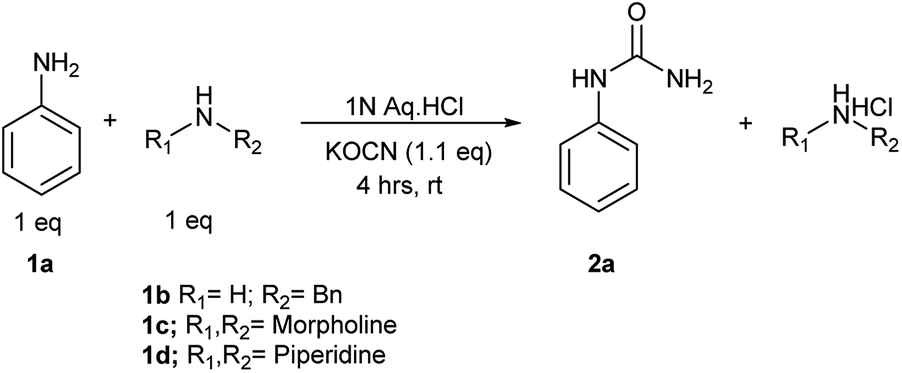 | ||
| Scheme 2 | ||
After successful demonstration of given protocol for synthesis of wide range of N-substituted ureas, we wanted to test the synthetic utility of the new protocol for industrial application. Accordingly, synthesis of few selected ureas was performed at incremental scale (1, 10 and 20 mmol) and yields of higher scale were compared to low mmol scales as mentioned in Table 3. For this study, amines were selected based on the commercial utility of their corresponding N-substituted ureas. For example, the N-substituted urea 2e of 4-p-aminocumene (1e, entries 1–3; Table 3) is a key intermediate for Isoproturon whereas, N-substituted urea of 3,4-dichloro aniline (entry 9 & 10) is a key structural motif for linuron and diuron.4a Isoproturon, linuron and diuron are three herbicides which are extensively being used for crop protection. The N-substituted urea of methylamine (entry 7 & 8) and aniline (entries 4–6) are extensively being used as key raw material for various target molecules.20 In all the scale up reactions of four different amines (Table 3), we found no detrimental effect on rate of reaction or overall isolated yields. In fact, in case of 4-isopropyl aniline, reaction gave considerably better isolated yield at 20 mmol scale compared to 1 or 10 mmol batch size. It is important to mention that a high level of chemical purity of the final product was obtained in 20 mmol reaction just by simple filtration without any additional workup or column chromatographic purification.
| Entry | Product | Scale (mmol) | Time (h) | Isolated yield (%) |
|---|---|---|---|---|
| 1 | 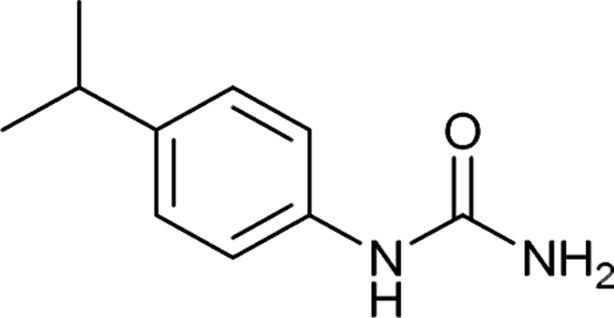 |
1 | 4 | 88 |
| 2 | 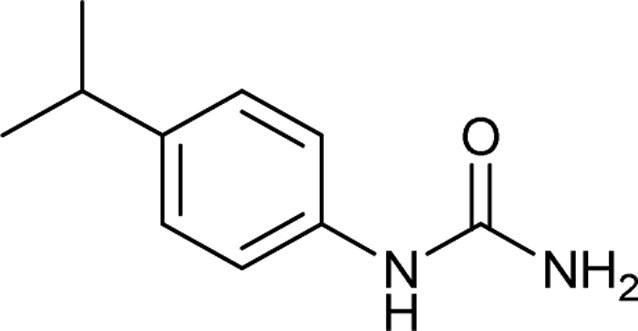 |
10 | 3 | 95 |
| 3 | 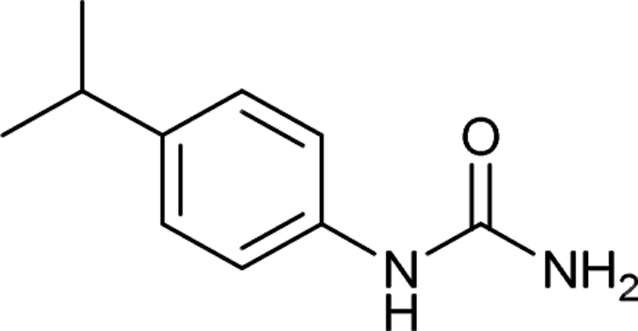 |
20 | 2 | 99 |
| 4 |  |
1 | 6 | 90 |
| 5 | 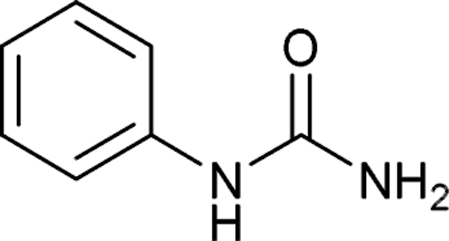 |
10 | 5 | 89 |
| 6 | 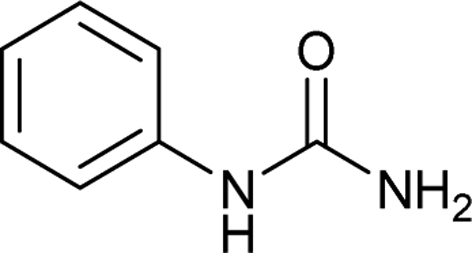 |
20 | 5 | 90 |
| 7 | 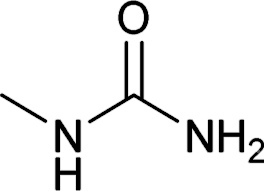 |
1 | 5 | 74 |
| 8 | 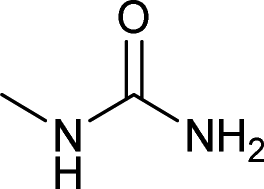 |
20 | 5 | 78 |
| 9 | 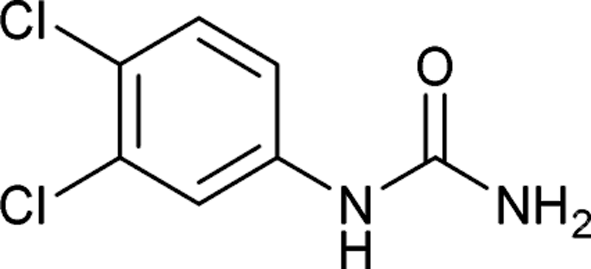 |
1 | 12 | 71 |
| 10 | 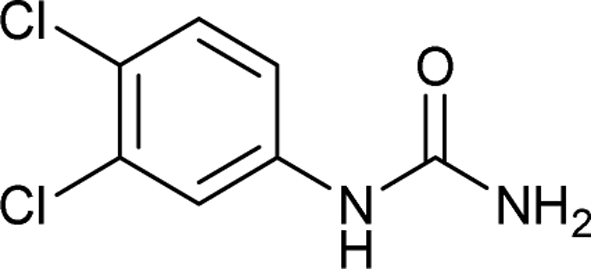 |
20 | 12 | 72 |
The industrial applicability of the developed protocol was further established by executing a one-pot synthesis of Isoproturon in water as mentioned in Scheme 3. Isoproturon is an agrochemical which is produced and used in large quantities. The N-substituted urea 2e produced by developed methodology was used as a key intermediate and reacted in situ with dimethyl amine (2 molar THF solution) in same pot to afford isoproturon as major product. This is the first reported one-pot synthesis of Isoproturon using water as a solvent avoiding catalyst or isocyanate chemistry.21
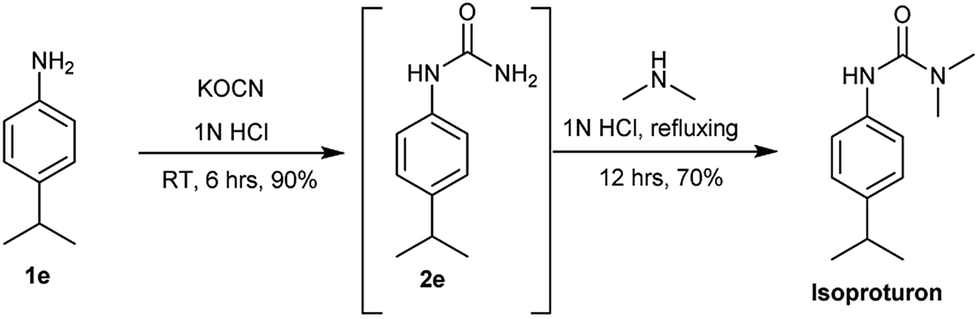 | ||
| Scheme 3 | ||
Experimental
General information
Reagents and starting materials were obtained from commercial sources and used as received. All reactions were performed under an inert atmosphere. Flash chromatography was carried out using silica gel (100–200 mesh). Thin layer chromatography was performed on silica gel and was visualized by staining with KMnO4 or iodine vapours. The 1H and 13C NMR spectra were recorded using a Bruker Avance 300 (300 MHz for 1H and 75 MHz for 13C) spectrometer in CDCl3 or DMSO-d6 or acetone-d6 using TMS as an internal standard. The chemical shifts (δ) for 1H and 13C are given in ppm relative to residual signals of the solvent. Coupling constants are given in Hz. The following abbreviations are used to indicate the multiplicity: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; brs, broad signal. The reactions were monitored by TLC (Merck). Evaporation of solvents was performed under reduced pressure using a rotary evaporator. Mass spectra were recorded on Advion Mass Express CMS instrument. Most of the compounds reported in this manuscript are known, and accordingly referred for their analytical data for characterization. For these known compounds, only 1H NMR data is provided along with physical characteristics. However, for new molecules, 1H, 13C NMR and mass spectrometry data is provided along with their physical characterization.General procedures for Scheme 1 and optimization studies of Table 1
To a stirring solution of 1 mmol amine (1a or 1b) separately in 3 mL solvent (4 M dioxane HCl and co-solvent if any as mentioned in Table 1), 2.2 equivalent of potassium cyanate was added. The reaction was allowed to stir for specific time at room temperature. The progress of reaction was monitored by TLC. The reaction aliquot was extracted and dried under vacuum. The 1H-NMR analysis of this vacuum dried aliquot was done to determine the quantification of the product.Procedures for synthesis of 1-phenylurea (2a) from aniline (1a)
To a stirring solution of aniline (1a, 0.18 mL; 2 mmol), and 1 N aqueous HCl (3 mL), potassium cyanate (4.4 mmol; 357 mg; 2.2 equiv.) was added. The reaction mixture was allowed to stir at room temperature for 6 hours. The progress of the reaction was monitored by TLC. The precipitates thus formed at the end of reaction time was filtered off and washed with aqueous 1 N HCl (5 mL). The solid residue was air dried and collected as white solid of 1-phenylurea18a (250 mg, 94%); 1H NMR (300 MHz, DMSO-d6): δ 5.81 (s, 2H, NH), 6.88 (t, J = 15 Hz, 1H), 7.21 (t, J = 15 Hz, 2H), 7.38 (d, J = 6 Hz, 2H), 8.49 (s, 2H).The compounds from Table 2 (entries 2–16, 19, 20, 36, 39, 40) were synthesized based on this method. The analytical data of these compounds is mentioned below:
Procedures for synthesis of N-benzyl urea (2b) from benzylamine (2a)
To a stirring solution of benzylamine (1b, 2 mmol), and 1 N aqueous HCl (3 mL), potassium cyanate (4.4 mmol; 2.2 equiv.) was added. The reaction mixture was allowed to stir at room temperature for 6 hours. The progress of the reaction was monitored by TLC. The reaction mixture was diluted with 1 N aqueous HCl (3 mL) and extracted with dichloromethane (20 mL). Organic layer was washed with water (5 mL), dried over anhydrous sodium sulphate and concentrated under reduced pressure to afford pure as white solid (270 mg, 89%). 1H NMR (300 MHz, DMSO-d6): δ 7.34–7.29 (m, 2H), 7.29–7.28 (m, 3H), 6.40 (t, J = 12 Hz, 1H), 5.51 (s, 2H, NH), 4.17 (d, J = 6 Hz, 2H).22Compound from Table 2 (entries 17, 21–25, 27–29, 31–35, 37, 38) were prepared by this method.
The above compound was not purified by column chromatography, here we have reported conversion through NMR.
Specific procedure for Scheme 2
To a stirring solution of aniline (1a; 0.09 mL; 1 mmol) and morpholine (1c; 0.086 mL, 1 mmol), was added 1 N aqueous HCl (3 mL). To this stirring reaction mixture, 1.1 mmol of potassium cyanate (89 mg) was added. The reaction mixture was stirred at room temperature for 6 hours. An aliquot were extracted after 4 hours and analysed for conversion of amines to corresponding N-substituted ureas using HPLC analysis and 1H NMR spectroscopy. The reaction mixture was filtered off after 6 hours to collect off white precipitates of phenyl urea (2b). The collected white solid was washed with 1 N HCl (5 mL). The collected solid as well as filtrate was analysed using HPLC and 1H NMR spectroscopy for quantification of N-substituted urea and left over amine. Other reactions of Scheme 2 were done using similar protocol.Specific procedures for Table 3 for 20 mmol scale of phenyl urea (1a, entry 6, Table 3)
To a stirring solution of aniline (1a, 20 mmol, 1.83 mL), and 1 N aqueous HCl (30 mL), potassium cyanate (44.0 mmol; 2.2 eq., 3.56 g) was added. The reaction mixture was allowed to stir at room temperature for 6 hours. The progress of the reaction was monitored by TLC. The precipitates thus formed at the end of reaction time was filtered off and washed with aqueous 1 N HCl (50 mL). The solid residue was air dried and collected as white solid of 1-phenylurea (yield: 2.4 g, 90%).One pot synthesis of isoproturon21 (Scheme 3)
4-Isopropylaniline (p-aminocumene; 1e; 1 mmol, 178 mg) was mixed in 1 N aqueous HCl (3 mL) and potassium cyanate (2.2 mmol, 178 mg) was added. The reaction mixture was stirred at room temperature and reaction was monitored using TLC for maximum consumption of aniline (6 hours). Further, dimethylamine (3 mmol) was added to reaction mixture and the reaction was refluxed for additional 12 hours. The progress of the reaction was monitored by TLC. Reaction mixture was cooled to room temperature, diluted with water (5 mL) and extracting with dichloromethane (25 mL). The organic layer was collected, dried over anhydrous sodium sulphate and concentrated under vacuum to afford crude product. The crude was purified by column chromatography using ethyl acetate and hexane as eluent to afford pure product as light yellow solid (175 mg; 70%). 1H NMR21 (300 MHz, CDCl3), δ 7.23 (d, J = 9 Hz, 2H), 7.09 (d, J = 9 Hz, 2H), 6.24 (s, 2H, NH), 2.98 (s, 6H), 2.75–2.86 (m, 1H), 1.18 (d, J = 6 Hz, 6H).Conclusion
We demonstrated a green method for the synthesis of N-substituted ureas, which are molecules of commercial interests and being produced and used in large quantities. The developed approach is mild, does not require any heating or pressure and reactions can be performed using water as a solvent without any organic co-solvent. In fact, water as a solvent found to provide a unique reactivity and selectivity under identified conditions. The developed methodology demonstrated a large substrate scope and practical ease for product isolation. The reactions can be done at gram scale using this methodology without any effect on yields. All these characters make this approach as an industrial amenable approach for manufacturing of N-substituted ureas.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors would like to acknowledge THSTI for research facility.Notes and references
- (a) K. Sanderson, Nature, 2011, 469, 18 CrossRef PubMed; (b) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC; (c) C. J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197 CrossRef PubMed; (d) J. H. Clark, Green Chem., 1999, 1, 1 RSC; (e) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998, p. 30 Search PubMed.
- (a) Q. Haijun, L. Xuejian, M. Fan and L. Xufeng, Beilstein J. Org. Chem., 2013, 9, 2846 CrossRef PubMed; (b) S. J. Connon, Chem. Commun., 2008, 2499 RSC; (c) M. H. Rodriguez, C. G. Avila-Ortiz, J. M. del Campo, D. H. Romero, M. J. R. Hoz and E. Juaristi, Aust. J. Chem., 2008, 61, 364 CrossRef; (d) S. J. Connon, Chem.–Eur. J., 2006, 12, 5418 CrossRef PubMed.
- (a) A. D Jagtap, N. B. Kondekar, A. A. Sadani and J. Wang Chern, Curr. Med. Chem., 2017, 24(6), 622 CrossRef PubMed; (b) A. K. Belfrage, J. Gising, F. Svensson, E. Akerblom, C. Skold and A. Sandstrom, Eur. J. Org. Chem., 2015, 978 CrossRef; (c) H. Gong, M. Yang, Z. Xiao, A. M. Doweyko, M. Cunningham, J. Wang, S. Habte, D. Holloway, C. Burke, D. Shuster, L. Gao, J. Carman, J. E. Somerville, S. G. Nadler, L. S. Cid, J. C. Barrish and D. S. Weinstein, Bioorg. Med. Chem. Lett., 2014, 24, 3268 CrossRef PubMed; (d) H. K. Lee, E. B. Choi and C. S. Pak, Curr. Top. Med. Chem., 2009, 9, 482 CrossRef PubMed; (e) J. Das, C. Liu, R. V. Moquin, J. Lin, J. A. Furch, S. H. Spergel, K. W. McIntyre, D. J. Shuster, K. D. ODay, B. Penhallow, C. Y. Hung, S. B. Kanner, T. A. Lin, J. H. Dodd, J. C. Barrish and J. Wityak, Bioorg. Med. Chem. Lett., 2006, 16, 2411 CrossRef PubMed; (f) M. Kurihara, A. Shara Shamsur Rouf, H. Kansui, H. Kagechika, H. Okudaa and N. Miyatac, Bioorg. Med. Chem. Lett., 2004, 14, 4131 CrossRef PubMed; (g) Darvesh, Sultan, et al., Bioorg. Med. Chem., 2010, 18(6), 2232 CrossRef PubMed; (h) C. L. Cioffi, N. Dobri, E. E. Freeman, M. P. Conlon, P. Chen, D. G. Stafford, D. C. Schwarz, K. C Golden, L. Zhu, D. B. Kitchen, K. D. Barnes, B. Racz, Q. Qin, E. Michelotti, C. L. Cywin, W. H. Martin, P. G. Pearson, G. Johnson and K. Petrukhin, J. Med. Chem., 2014, 57(18), 7731 CrossRef PubMed; (i) J. W. Mickelson, E. J. Jacobsen, D. B. Carter and J. D. petke, J. Med. Chem., 1996, 39(1), 158 CrossRef PubMed.
- (a) F. Muller and A. P. Applebyki, Weed Control, 2. Individual Herbicides, in Ullmann's Encyclopedia of Industrial Chemistry, 2012, DOI:10.1002/14356007.o28_o01; (b) C. D. S. Tomlin, The Pesticide Manual, 11th ed., British Crop Protection Council, Farnham, UK, 1997 Search PubMed; (c) H. C. Bucha and C. W. Todd, Science, 1951, 114, 493 Search PubMed.
- For OCED list of High Production Chemicals (HPV) see; OECD Environment, Health and Safety Publications Series on Testing and Assessment No. 112, env/jm/mono (2009) 40, 23 Oct, 2009.
- (a) L. Trasande, JAMA, 2016, 15, 1565 CrossRef PubMed; (b) B. Harvey, Environ. Health Perspect., 2005, 12, A828 Search PubMed.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437 RSC.
- (a) C. M. Sanabria, M. T. do Casal, R. B. A. de Souza, L. C. S. de Aguiar and M. C. S. de Mattos, Synthesis, 2017, 49(7), 1648 Search PubMed; (b) D. L. Browne, M. O. Brien, P. Koos, P. B. Cranwell, A. Polyzos and S. V. Ley, Synlett, 2012, 23(9), 1402 CrossRef; (c) S. E. Reisman, J. M. Ready, M. M. Weiss, A. Hasuoka, M. Hirata, K. Tamaki, T. V. Ovaska, C. J. Smith and J. L. Wood, J. Am. Chem. Soc., 2008, 130, 2087 CrossRef PubMed; (d) W. Lutz and W. Sundermeyer, Chem. Ber., 1979, 112(6), 2158 CrossRef; (e) E. S. Sherman, S. R. Chemler, E. Schaumann, D. Geffken, M. A. Kçllner, A. Studer, W. Maison, T. Vogler, I. O. Neil, U. Wille and P. Rademacher, Sci. Synth., 2009, 40b, 1285 Search PubMed; (f) C. Six and F. Richter, Isocyanates, organic, in Ullmann's encyclopedia of industrial chemistry, 2012, DOI:10.1002/14356007.a14_611; (g) P. Jager, C. N. Rentzea and H. Kieczka, Carbamates and carbamoyl chlorides, in Ullmann's encyclopedia of industrial chemistry, 2012, DOI:10.1002/14356007.a05_051.
- A. K. Vaish, S. Consul, A. Agrawal, S. C. Chaudhary, M. Gutch, N. Jain and M. M. Singh, J. Emergencies Trauma Shock, 2013, 6(4), 271 CrossRef PubMed.
- F. Bigi, R. Maggi and G. Sartori, Green Chem., 2000, 2, 140 RSC.
- (a) D. Landsberg and M. Kalesse, Synlett, 2010, 7, 1104 Search PubMed; (b) P. Dube, N. F. Nathel, M. Vetelino, M. Couturier, C. L. Aboussafy, S. Pichette, M. L. Jorgensen and M. Hardink, Org. Lett., 2009, 11(24), 5622 CrossRef PubMed; (c) N. Narendra, G. Chennakrishnareddy and V. V. Sureshbabu, Org. Biomol. Chem., 2009, 7, 3520 RSC; (d) P. Gogoi and D. Konwar, Tetrahedron Lett., 2007, 48, 531 CrossRef; (e) H. Lebel and O. Leogane, Org. Lett., 2005, 7(19), 4107 CrossRef PubMed.
- (a) R. Sharma, P. Samadhiya, P. D. Srivastava and S. k. Srivastava, Helv. Chim. Acta, 2012, 95, 514 CrossRef; (b) P. Samadhiya, R. Sharma, S. K. Srivastava and S. D. Savitri, Chin. J. Chem., 2011, 29, 1745 CrossRef; (c) S. Breitler, N. J. Oldenhuis, B. P. Fors and S. L. Buchwald, Org. Lett., 2011, 13, 3262 CrossRef PubMed; (d) M. V. Nandakumar, Tetrahedron Lett., 2004, 45, 1989 CrossRef; (e) A. G. Sergeev, G. A. Artamkina and I. P. Beletskaya, Tetrahedron Lett., 2003, 44, 4719 CrossRef; (f) G. A. Artamkina, A. G. Sergeev and I. P. Beletskaya, Russ. J. Org. Chem., 2002, 38, 538 CrossRef.
- Q. Liu, N. W. Luedtke and Y. Tor, Tetrahedron Lett., 2001, 42, 1445 CrossRef.
- S. Sahua, P. R. Sahooa, S. Patelb and B. K. Mishraa, J. Sulfur Chem., 2011, 32(2), 171 CrossRef.
- (a) T. C. Wang and J. X. Qiao, Tetrahedron Lett., 2016, 57, 1941 CrossRef; (b) R. Zhang, L. Guo, J. Chen, H. Gan, B. Song, W. Zhu, L. Hua and Z. Hou, ACS Sustainable Chem. Eng., 2014, 2, 1147 CrossRef; (c) J. W. Wu, Y. D. Wu, J. J. Dai and H. J. Xu, Adv. Synth. Catal., 2014, 2429 CrossRef.
- C. H. Wang, T. H. Hsieh, C. C. Lin, W. H. Yeh, C. A. Lin and T. C. Chien, Synlett, 2015, 26, 1823 CrossRef.
- (a) S. R. Jagtap, Y. P. Patil, A. G. Panda and B. M. Bhanage, Synth. Commun., 2009, 39, 2093 CrossRef; (b) D. L. Diaz, A. K. Darko and L. W. McElwee, Eur. J. Org. Chem., 2007, 4453 CrossRef.
- (a) A. R. Sardarian and I. M. Inaloo, RSC Adv., 2015, 5, 76626 RSC; (b) L. D. Luca, A. Porcheddu, G. Giacomelli and I. Murgia, Synlett, 2010, 2439 CrossRef; (c) J. Taillades, L. Boiteau, I. Beuzelin, O. Lagrille, J. Biron, W. Vayaboury, O. V. Trambouze, O. Giani and A. Commeyras, J. Chem. Soc., Perkin Trans. 2, 2001, 2, 1247 RSC; (d) X. Min, J. Liu, T. Dong and M. Hussain, Synthesis, 2018, 50, 341 CrossRef; (e) S. W. King, R. Natarajan, R. Bembi and T. H. Fife, J. Am. Chem. Soc., 1992, 10715 CrossRef; (f) R. Laudien and R. Mitzner, J. Chem. Soc., Perkin Trans. 2, 2001, 2, 2226 RSC.
- http://evans.rc.fas.harvard.edu/pdf/evans_pKa_table.pdf.
- (a) H. Lehmann, Green Chem., 2017, 19(6), 1449 RSC; (b) T. J. Kohn, X. Du, S. Lai, Y. Xiong, R. Komorowski, M. Veniant, Z. Fu, X. Jiao, V. Pattaropong, D. Chow, M. Cardozo, L. Jin, M. Conn, W. E. DeWolf, C. F. Kraser, R. J Hinklin, M. L. Boys, J. C. Medina, J. B. Houze, P. J. Dransfield and P. Coward, ACS Med. Chem. Lett., 2016, 7(7), 714 CrossRef PubMed; (c) X. K. Wang, S. R. Yan, Z. H. Li, K. N. Fan, M. Q. Kang and S. Y. Peng, Korean J. Chem. Eng., 2004, 21(2), 378 CrossRef; (d) R. Juarez, A. Corma and H. Garcia, Top. Catal., 2009, 52(12), 1688 CrossRef.
- K. Adeppa, D. C. Rupainwar and K. Misra, Synth. Commun., 2012, 42, 714 CrossRef.
- B. P. Bew, R. A. Brimage, G. H. Gipson, S. V. Sharma and S. Thurston, Org. Lett., 2009, 11(12), 2483 CrossRef PubMed.
- M. Miyahara, M. Nakadate and S. Kamaiya, Chem. Pharm. Bull., 1985, 33(2), 497 CrossRef.
- S. A. Konovalova, A. P. Avdeenko, M. V. Polishchuk, E. N. Lysenko, V. N. Baumer, I. V. Omel chenko and S. A. Goncharova, Russ. J. Org. Chem., 2015, 51(12), 1739 CrossRef.
- R. L. Wang, W. M. Li, M. Y. Liu and W. R. Xu, J. Chin. Chem. Soc., 2009, 56, 34 CrossRef.
- S. Breitler, N. J. Oldenhuis, B. P. Fors and S. L. Buchwald, Org. Lett., 2011, 13(12), 3262 CrossRef PubMed.
- E. Artuso, I. Degani, R. Fochi and C. Magistris, Synthesis, 2007, 22, 3497 Search PubMed.
- R. Zhang, L. Guo, J. Chen, H. Gan, B. Song, W. Zhu, L. Hua and Z. hou, ACS Sustainable Chem. Eng., 2014, 2, 1147 CrossRef.
- L. B. Figueroa, A. O. Porras and D. G. Sanche, J. Org. Chem., 2014, 79(10), 4544 CrossRef PubMed.
- J. C. Lorenz, C. A. Busacca, X. W. Feng, N. Grinberg, N. Haddad, J. Johnson, S. Kapadia, H. Lee, A. Saha, M. Sarvestani, E. M. Spinelli, R. Varsolona, X. Wei, X. G. Zeng and C. H. Senanayake, J. Org. Chem., 2010, 75(4), 1155 CrossRef PubMed.
- K. Yu. Chegaev, A. N. Kravchenko, O. V. Lebedev and Y. A. Strelenko, Mendeleev Commun., 2001, 11(1), 32 CrossRef.
- G. Verardo, P. Geatti and P. Strazzolini, Synth. Commun., 2007, 37(11), 1833 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |