
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient green synthesis and photodynamic therapeutic study of hypericin and its derivatives†
Ying Zhang,
Kun Shang,
Xiaowen Wu,
Siyu Song,
Zebo Li,
Zhichao Pei and
Yuxin Pei*
and
Yuxin Pei*
Shaanxi Key Laboratory of Natural Products & Chemical Biology, College of Chemistry & Pharmacy, Northwest A&F University, Yangling, Shaanxi 712100, PR China. E-mail: peiyx@nwafu.edu.cn
First published on 13th June 2018
Abstract
A highly efficient synthetic pathway for hypericin (7a) was achieved under mild conditions with an overall yield over two steps of 92% using emodinanthrone as a starting material, where protohypericin, a key precursor of hypericin, was synthesized in water with microwave assistance, which was then photocyclized to hypericin with a high yield via 1 h irradiation in a visible light reactor equipped with 575 nm monochromatic lamps. In addition, the method could be used to synthesize hypericin derivatives (7b–d) with similar overall yields. Furthermore, their effects of photodynamic therapy (PDT) were evaluated on A431, HepG-2, and MCF-7 cell lines. The PDT of 7b was better than that of 7a, whereas 7c and 7d were worse. Unlike other cell lines, MCF-7 was not sensitive to any of 7a–d at the same concentrations.
Introduction
Hypericin is a natural polycyclic aromatic dianthraquinone present in the St. John's Wort plant (Hypericum perforatum).1 It has been used as a traditional herbal medicine since ancient times for its anti-viral, anti-bacterial, and anti-inflammatory properties as well as to treat depression.2–7 In recent years, it has attracted increasing interest due to its potential as an effective photosensitizer used in photodynamic therapy (PDT) to fight various cancers.8–10The isolation of pure hypericin from Hypericum perforatum is a tedious procedure due to its very low content in the plant (0.03 to 0.09%)11 and poor solubility in solvents.1 To meet the needs of hypericin demand both from academic and industrial areas, great efforts have been made in the past to develop a way for it to be chemically synthesized. The first step towards the successful chemical synthesis of hypericin dates back to 1957, when Brockmann and his co-workers for the first time reported a total synthetic pathway of hypericin. Their pathway started from 3,5-dimethoxybenzoic acid methyl ester and involved 12 steps in total that resulted in a low overall yield of hypericin of 6% to 9%.12 Other total synthetic methods that were later developed started with ethyl formate, chlorinated ketene or 2-methyl anthraquinone, and shortened the synthetic pathway to 6 to 8 steps, although none of these methods reached an overall yield that was higher than 10%.6,13–15 In addition, these methods were generally characterized by the usage of extremely high temperature, pressure and as having a very long reaction time (up to 10 days). Besides the total synthesis, semi-synthesis of hypericin was proposed by Falk and coworkers using emodin extracted from Cortex frangulae as the starting material.1 Falk's method only required 3 steps, where emodin was first reduced to emodinanthrone by SnCl2 in concentrated HCl with a high yield of 90%, and then emodinanthrone took place oxidative dimerization in pyridine to form protohypericin, which was finally photocyclized to hypericin upon irradiation of visible light overnight in a moderate yield of 56% (overall yield was 51.6%). Although the overall yield was later increased to 74% by the same group by introducing microwave assistance in the synthesis of protohypericin, the reaction was carried out in a toxic organic solvent DMF and in the presence of a corrosively organic base potassium t-butoxide.16 Additionally, the photocyclization step of protohypericin to hypericin took place overnight with a 500 W halogen lamp.17 Therefore, it is necessary to find an efficient pathway for the synthesis of hypericin, where the required reactions can be performed under mild conditions, the solvents involved in each step are less or non-toxic, easy to handle after the reactions, and would preferably be water based to comply with the standpoints of green chemistry.
In this paper, as shown in Scheme 1, a greatly more efficient, facile and environmentally friendly method for the synthesis of hypericin (7a) was developed. This method started with the easily available emodinanthraquinone, from where its precursor protohypericin was obtained in water by using microwaves and the irradiation of protohypericin in acetone with 575 nm visible light. This method resulted in a high hypericin overall yield. Furthermore, we delightedly found that the strategy could be successfully adapted for the synthesis of the derivatives of hypericin (7b–d), which are especially significant in new photosensitizer discovery18,19 used in cancer photodynamic therapy.
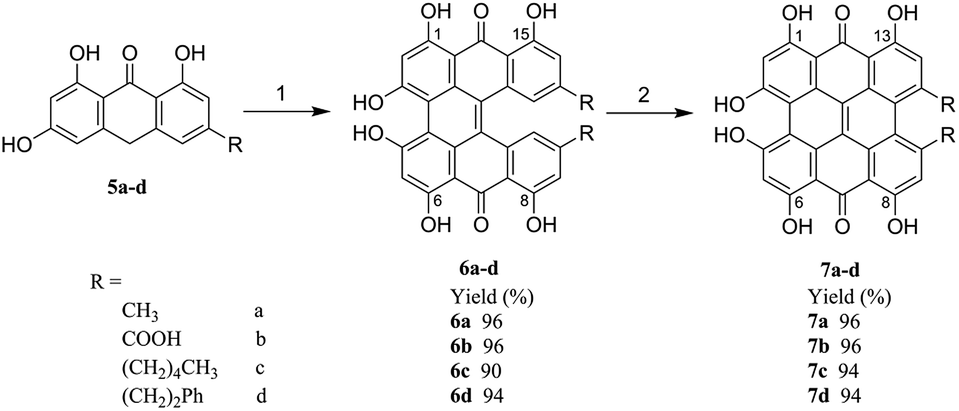 | ||
| Scheme 1 General synthetic pathway of hypericin (7a) and its derivatives (7b–d). Reagents and conditions: (1) 1.5% NaOH/H2O, pyridine-N-oxide, FeSO4·7H2O, 105 °C, 10 W, 70 min, N2 by microwave reactor; (2) 575 nm monochromatic light, acetone, N2, 60 min. | ||
Experimental
General remarks
All reagents and solvents were purchased from commercial suppliers and used without further purification unless specified. Dimethyl sulfate, N-bromosuccinimide (NBS), 18-crown-6 and benzoyl peroxide (BPO) were purchased from Energy Chemical Reagent Co. Triphenyl phosphine and tin(II) chloride dehydrate (SnCl2·2H2O) were purchased from Sinopharm Chemical Reagent Co. Butyraldehyde was purchased from TCI. Benzaldehyde was purchased from Aladdin. Ferrous sulfate heptahydrate (FeSO4·7H2O) was purchased from Tianjin Bodi Chemical Co., Ltd. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Kehao Biotech Co., Ltd. Flash chromatography was performed using silica gel with a grain size of 40–63 μm (Qingdao Haiyang Co., Ltd). RPMI 1640 medium, Dulbecco's Modified Essential Medium (DMEM) and trypsin–EDTA solution were obtained from Gibco BRL Co., Ltd. Penicillin/streptomycin was purchased from Nanjing KeyGen Biotech, fetal bovine serum was purchased from YuanhengJinma Co., Ltd.1H NMR and 13C NMR spectra were recorded on a Bruker 500 MHz Spectrometer with working frequencies of 500 MHz for 1H and 125 MHz for 13C, respectively, in DMSO-d6, MeOH-d4 or CDCl3. The residual signals from DMSO-d6 (1H: δ 2.50 ppm; 13C: δ 39.52 ppm), or MeOH-d4 (1H: δ 3.31 ppm; 13C: δ 49.00 ppm), CDCl3 (1H: δ 7.26 ppm; 13C: δ 77.00 ppm) were used as internal standards. HRMS (High Resolution Mass Spectrometer) analysis was performed on Agilent 1290-6540 UHPLC Q-TOF-HRMS. Microwave assisted syntheses were performed with Discover SP (USA). Photochemical reaction was performed with a Rayonet Chamber Reactor (Model RPR-100) equipped with 16 of 575 nm monochromatic lamps (25 W per lamp, 400 W in total, USA). The UV-Vis spectra were recorded with a Shimadzu 1750 UV-Visible spectrophotometer (Japan) at 298 K. The melting points were measured by a WRS-2 melting point apparatus (Shanghai, China) with a range of room temperature to 300 °C. The compounds 4, 12, 14 were prepared according to the published procedures20,21 (details can be found in ESI†).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) to give 5a as a pale yellow solid (1.22 g, 96%). 1H NMR (500 MHz, DMSO-d6) δ 12.37 (s, 1H), 12.21 (s, 1H), 10.82 (s, 1H), 6.77 (s, 1H), 6.67 (s, 1H), 6.42 (s, 1H), 6.22 (d, J = 4.3 Hz, 1H), 4.29 (s, 2H), 2.32 (s, 3H) ppm.23:1, v/v) to give 5b as a pale yellow solid (0.3 g, 92%). 1H NMR (500 MHz, DMSO-d6) δ 13.24 (br, 1H), 12.29 (s, 1H), 12.22 (s, 1H), 10.99 (s, 1H), 7.46 (s, 1H), 7.27 (s, 1H), 6.445–6.439 (m, 1H), 6.25 (d, J = 2.7 Hz, 1H), 4.42 (s, 2H) ppm.24:1, v/v) to give 5c as a pale yellow solid (33.6 mg, 71%). 1H NMR (500 MHz, DMSO-d6) δ 12.37 (s, 1H), 12.20 (s, 1H), 10.81 (s, 1H), 6.77 (s, 1H), 6.66 (s, 1H), 6.41 (d, J = 1.3 Hz, 1H), 6.23 (d, J = 2.2 Hz, 1H), 4.29 (s, 2H), 2.55 (t, J = 7.45 Hz, 2H), 1.60–1.54 (m, 2H), 1.34–1.23 (m, 4H), 0.86 (t, J = 7.0 Hz, 3H) ppm; 13C NMR (125 MHz, DMSO-d6) δ 191.0, 165.0, 164.5, 161.7, 151.7, 145.0, 141.9, 119.1, 114.4, 113.0, 108.4, 107.3, 100.9, 35.4, 32.3, 30.9, 29.8, 21.9, 13.9 ppm. HRMS: m/z calcd for C19H20O4 [M + H]+ 313.1440, found 313.1439. M.p.: 161.4–162.0 °C.:1, v/v) to give 5d as a pale yellow solid (265 mg, 90%). 1H NMR (500 MHz, DMSO-d6) δ 12.37 (s, 1H), 12.21 (s, 1H), 10.85 (s, 1H), 7.30–7.23 (m, 4H), 7.20–7.15 (m, 1H), 6.85 (s, 1H), 6.74 (s, 1H), 6.43 (d, J = 1.2 Hz, 1H), 6.22 (d, J = 2.1 Hz, 1H), 4.30 (s, 2H), 2.93–2.85 (m, 4H) ppm. 13C NMR (125 MHz, DMSO-d6) δ 191.1, 165.0, 164.6, 161.7, 150.7, 145.0, 142.0, 141.2, 128.4, 128.3, 126.0, 119.3, 114.6, 113.2, 108.4, 107.4, 101.0, 37.2, 36.0, 32.4 ppm. HRMS: m/z calcd for C22H18O4 [M + H]+ 347.1283, found 347.1281. M.p.: 196.7–197.1 °C.
:1, v/v) to give 5a as a pale yellow solid (1.22 g, 96%). 1H NMR (500 MHz, DMSO-d6) δ 12.37 (s, 1H), 12.21 (s, 1H), 10.82 (s, 1H), 6.77 (s, 1H), 6.67 (s, 1H), 6.42 (s, 1H), 6.22 (d, J = 4.3 Hz, 1H), 4.29 (s, 2H), 2.32 (s, 3H) ppm.23:1, v/v) to give 5b as a pale yellow solid (0.3 g, 92%). 1H NMR (500 MHz, DMSO-d6) δ 13.24 (br, 1H), 12.29 (s, 1H), 12.22 (s, 1H), 10.99 (s, 1H), 7.46 (s, 1H), 7.27 (s, 1H), 6.445–6.439 (m, 1H), 6.25 (d, J = 2.7 Hz, 1H), 4.42 (s, 2H) ppm.24:1, v/v) to give 5c as a pale yellow solid (33.6 mg, 71%). 1H NMR (500 MHz, DMSO-d6) δ 12.37 (s, 1H), 12.20 (s, 1H), 10.81 (s, 1H), 6.77 (s, 1H), 6.66 (s, 1H), 6.41 (d, J = 1.3 Hz, 1H), 6.23 (d, J = 2.2 Hz, 1H), 4.29 (s, 2H), 2.55 (t, J = 7.45 Hz, 2H), 1.60–1.54 (m, 2H), 1.34–1.23 (m, 4H), 0.86 (t, J = 7.0 Hz, 3H) ppm; 13C NMR (125 MHz, DMSO-d6) δ 191.0, 165.0, 164.5, 161.7, 151.7, 145.0, 141.9, 119.1, 114.4, 113.0, 108.4, 107.3, 100.9, 35.4, 32.3, 30.9, 29.8, 21.9, 13.9 ppm. HRMS: m/z calcd for C19H20O4 [M + H]+ 313.1440, found 313.1439. M.p.: 161.4–162.0 °C.:1, v/v) to give 5d as a pale yellow solid (265 mg, 90%). 1H NMR (500 MHz, DMSO-d6) δ 12.37 (s, 1H), 12.21 (s, 1H), 10.85 (s, 1H), 7.30–7.23 (m, 4H), 7.20–7.15 (m, 1H), 6.85 (s, 1H), 6.74 (s, 1H), 6.43 (d, J = 1.2 Hz, 1H), 6.22 (d, J = 2.1 Hz, 1H), 4.30 (s, 2H), 2.93–2.85 (m, 4H) ppm. 13C NMR (125 MHz, DMSO-d6) δ 191.1, 165.0, 164.6, 161.7, 150.7, 145.0, 142.0, 141.2, 128.4, 128.3, 126.0, 119.3, 114.6, 113.2, 108.4, 107.4, 101.0, 37.2, 36.0, 32.4 ppm. HRMS: m/z calcd for C22H18O4 [M + H]+ 347.1283, found 347.1281. M.p.: 196.7–197.1 °C.General procedure for the synthesis of protohypericin and its derivatives
To a microwave 10 mL tube, 5a, 5b, 5c, or 5d (0.5 mmol, 1 equiv.), pyridine-N-oxide (2.5 mmol, 5 equiv.), FeSO4·7H2O (10 mg, 36 μmol), and NaOH (40 mg, 1.0 mmol) were dissolved in 2 mL ultrapure water. The mixture was placed in a microwave reactor at 10 W, 105 °C under argon atmosphere for 70 min. The reaction mixture was then cooled to room temperature, and was acidified with 3% hydrochloric acid. The precipitate was filtered, washed with deionized water, and vacuum dried. The purification with column chromatography on silica gel by using petroleum ether/ethyl acetate/methanol (4:8:1, v/v/v) as eluent gave 6a–d, respectively.
General procedure for the synthesis of hypericin and its derivatives
To a 50 mL flask, 6a, 6b, 6c, or 6d (0.25 mmol, 1.0 equiv.) dissolved in 20 mL acetone was irradiated for 60 min by means of 575 nm monochromatic lamps. The color of the mixture changed from hyacinthine to prunosus. The solvent was removed under vacuum. The crude was purified by flash chromatography on silica gel by using petroleum ether/ethyl acetate/methanol (2:8:0.5, v/v/v) as eluent to give respective 7a–d.
Cell culture
The breast cancer cell line (MCF-7), human hepatoma cell line (HepG-2) and the human skin basal cell carcinoma (A431) were obtained from American Type Culture Collection. MCF-7 and A431 cells were cultured at 37 °C under a humidified 5% CO2 in DMEM (Dulbecco's Modified Eagle Medium) medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin; and HepG-2 cells were cultured in the same way except the culture medium used was RPMI 1640.Cell viability assay
The relative cell viability of compound 7a (7b, 7c, or 7d) was evaluated in vitro by MTT assay. HepG-2, MCF-7 or A431 cells were seeded respectively in 96-well plates at a density of 5 × 103 cells per well in 100 μL RPMI 1640 or DMEM medium, and grew for 24 h at 37 °C. Then the medium was replaced by 90 μL fresh medium and 10 μL 7a (7b, 7c, or 7d) in PBS (containing 0.2% DMSO-d6) at different concentrations (0.2, 0.4, 0.6, 0.8, 1.0 μM). After 4 h incubation, the cells were washed with PBS for 3 times, and then fresh medium was added. The cells were exposed to a LED array photosource (λ = 595–600 nm, 8.6 mW cm−2) for 30 min, and further cultured for 24 h at 37 °C. Then 10 μL fresh medium containing MTT (5 mg mL−1) was added to each well. After 4 h incubation, 100 μL DMSO was added to each well to dissolve formazam crystals. Finally, the plate was gently shaken for 10 min and the absorbance at 490 nm was recorded with a microplate reader.Results and discussion
Reaction optimization for synthesis of protohypericin
Protohypericin was the key precursor to the synthesis of hypericin, therefore the dimerization of emodinanthrone leading to protohypericin was recognized as the key step in this method.16 The synthesis of protohypericin has been extensively studied, which can be summarized in two methods based on starting materials, (1) emodin-type:20,26 by using potassium hydroxide as a base, protohypericin can be synthesized in the presence of hydroquinone in water. The drawbacks are extremely long reaction time (7–20 days) and low yield (25–72%); (2) emodinanthrone-type:16,17 by using potassium t-butoxide or piperidine as a base, protohypericin can be obtained in a moderate yield (70–78%) in a short time. While the drawbacks here are the requirement of notorious organic solvents (DMF or pyridine) and a still unsatisfying yield. Based on the discoveries previously reported in the literature, we chose emodinanthrone as a starting material for our method in order to pursue the development of an economic, green, and efficient synthetic method of protohypericin. In our method, the dimerization of emodinanthrone was performed in a microwave reactor by using cheap and easy to handle inorganic bases (such as NaOH, KOH, and LiOH) as a replacement for the organic bases, and water as solvent instead of the organic solvents. The reaction conditions were varied to study their effect on the yield, and the results were summarized in Table 1.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Reaction conditions | Yieldc (%) | ||||
| PNO (equiv.) | Catalyst (equiv.)b | Base | T (°C) | t (min) | ||
| a The optimized conditions: 5a, 120 mg, 1 equivalent; microwave, 10 W; water (2 mL); N2 atmosphere.b The catalyst was FeSO4·7H2O unless specified.c Isolated yield. N.D: no detection.d The catalyst was FeCl3.e The catalyst was CuCl2.f O2 atmosphere. | ||||||
| 1a | 5.0 | 0.08 | 1.5% NaOH | 105 | 70 | 96 |
| 2 | — | 0.08 | 1.5% NaOH | 105 | 70 | 6 |
| 3 | 5.0 | — | 1.5% NaOH | 105 | 70 | N.D |
| 4 | 3.2 | 0.08d | 6.0% NaOH | 105 | 70 | 94 |
| 5 | 5.2 | 0.08e | 6.0% NaOH | 105 | 70 | Very low |
| 6 | — | 3.0d,f | 6.0% NaOH | 105 | 70 | N.D |
| 7 | 3.5 | 0.08 | 1.5% NaOH | 105 | 70 | 93 |
| 8 | 4.5 | 0.08 | 1.5% NaOH | 105 | 70 | 91 |
| 9 | 5.0 | 0.08 | 0.5% NaOH | 105 | 70 | 77 |
| 10 | 5.0 | 0.08 | 1.0% NaOH | 105 | 70 | 94 |
| 11 | 5.0 | 0.08 | 2.0% NaOH | 105 | 70 | 96 |
| 12 | 5.0 | 0.08 | 5.0% NaOH | 105 | 70 | 76 |
| 13 | 5.0 | 0.08 | 1.5% LiOH | 105 | 70 | 71 |
| 14 | 5.0 | 0.08 | 3.6% LiOH | 105 | 70 | 95 |
| 15 | 5.0 | 0.08 | 1.5% KOH | 105 | 70 | 77 |
| 16 | 5.0 | 0.08 | 2.1% KOH | 105 | 70 | 93 |
| 17 | 5.0 | 0.08 | 1.5% NaOH | 90 | 70 | 72 |
| 18 | 5.0 | 0.08 | 1.5% NaOH | 100 | 70 | 90 |
| 19 | 5.0 | 0.08 | 1.5% NaOH | 120 | 70 | 88 |
| 20 | 5.0 | 0.08 | 1.5% NaOH | 105 | 50 | 86 |
| 21 | 5.0 | 0.08 | 1.5% NaOH | 105 | 60 | 90 |
| 22 | 5.0 | 0.08 | 1.5% NaOH | 105 | 80 | 89 |
| 23 | 5.0 | 0.08 | 1.5% NaOH | 105 | 90 | 90 |
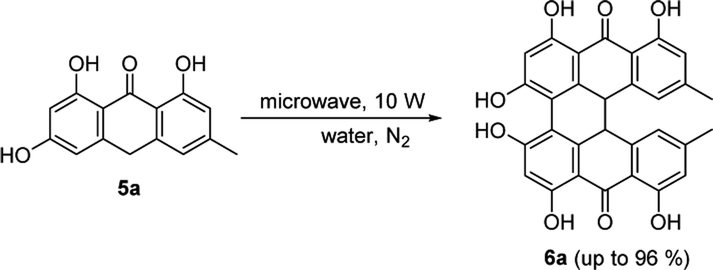
The various parameters, including the amount of pyridine N-oxide (PNO), catalyst, FeSO4·7H2O, base, reaction temperature and time, were thoroughly investigated. As shown in Table 1, the presence of pyridine N-oxide (PNO) and a suitable redox catalyst in the reaction were crucial for the reaction, with either absence leading to failure. For example, in the absence of PNO, 6a was obtained with only 6% yield, which demonstrated that PNO was an indispensible oxygen transfer reagent in this reaction system. While the absence of FeSO4·7H2O led to no reaction at all.3 In addition, ferric chloride could be used to substitute FeSO4·7H2O without compromise of the yield4 but copper chloride could not.5 The variation of the amount of PNO did not influence the yield much provided that the other reaction conditions were kept unchanged,1,7,8 where the best yield of 96% was obtained when PNO was 5 equivalents;1,11 The base, such as NaOH, helps to dissolve emodinanthrone in the reaction mixture and has obvious influence on the yield. A concentration of NaOH lower than 0.5% or higher than 5% decreased the yield from over 90% to about 76%. It's worth to mention that LiOH and KOH, which have similar chemophysical properties as NaOH, gave more or less equally good yields (95% for LiOH13 and 93% for KOH15), although the concentration of the two hydroxides used was slightly higher than that of NaOH. The study on the effect of reaction temperature disclosed that the yield increased with the temperature and reached to a peak value (96% at 105 °C, see Falk et al.1); further increase of the temperature did not benefited a higher yield, for instance, 88% was obtained at 120 °C.17 This might be ascribed to the possibility that high temperature led to side reactions, such as the oxidation of hydroxyl groups in emodinanthrone. The screening on reaction time proved that the yield increased with the reaction time18,19 and reached the best yield of 96% at 70 min.1 After this point, the yield decreased slightly with the reaction time with about 6%,20,26 which may due to the decomposition of protohypericin in the hot basic solution.
Notably, the reaction can be carried out in gram-scale with 91.6% yield under the optimized conditions (see ESI†).
Synthesis of hypericin and its derivatives
It was known that a major method to obtain hypericin was the photocyclization of protohypericin irradiated with visible light. The light sources reported previously include sunlight,27 halogen,17 glow,20 and high-/low-pressure mercury lamps.1 The reaction time varied from 10 min claimed by Falk et al.1 to 24 h by Kim et al.20 as well as the yield from 31% to 92%. We speculated the enormous variation of the reaction time and yield might result from the emission difference of the light sources.The UV-Vis spectra of 6a and 7a were shown in Fig. 1a. Considering20,27,28 protohypericin has a strong absorption band at 525–590 nm while no obvious absorption for 7a at 575 nm, we chose monochromatic lamps with a maximum emission peak at 575 nm as visible light source (Fig. 1b) to perform the photocyclization of 6a. The monitoring of the reaction by TLC displayed that the reaction was completed at 1 h, where an isolated yield of 96% was obtained by flash chromatography. The reaction has very good reproducibility and can be performed on large scale with little compromise to the yield (89.8%, see ESI†).
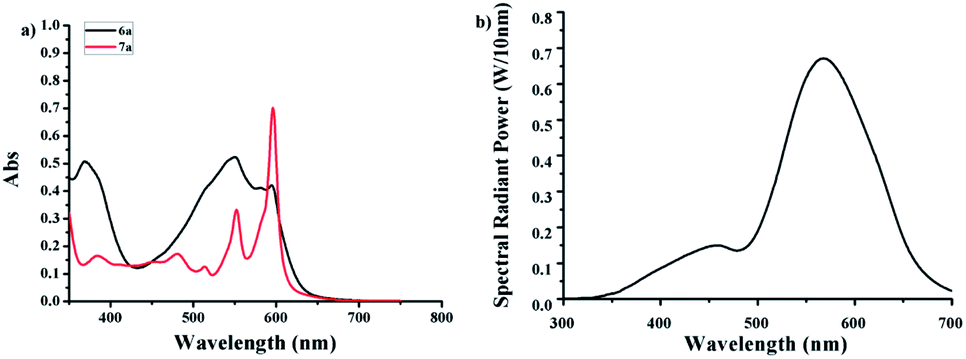 | ||
| Fig. 1 (a) UV-Vis absorption spectra of 6a and 7a (0.04 M in acetone); (b) emission spectrum of the monochromatic lamp with a maximum peak at 575 nm. | ||
To study the applicability of the synthetic pathway developed above, the derivatives of hypericin with different substituents were synthesized accordingly, where methyl groups on 10- and 11- positions in hypericin (7a) were replaced with carboxyl groups for 7b, pentyl groups for 7c, and phenethyl groups for 7d, respectively. As descripted in Scheme 1, starting with 5b–c, the analogues of emodinanthrone 5a, respective 6b–d were successfully synthesized with satisfying to excellent isolated yields (90–96%) under the optimized conditions screened for 5a, which were subjected to irradiation of visible light to give corresponding 7b–d with excellent yields (94–96%). This indicates that the synthetic pathway developed in the present work has good applicability for the derivatives of hypericin, which is significant in photosensitizer discovery based on natural bioactive molecules.
Photodynamic therapy study
Photodynamic therapy (PDT) is a noninvasive technique that is used to treat and detect small and superficial tumors.29–34 Hypericin has been regarded as an effective natural photosensitizer,35–37 and has gained increasing attention as a potential alternative treatment for various cancers due to its unique photochemical properties and photobiological activities.9,38–41 To explore the potential of the three derivatives 7b–d as photosensitizer for PDT, A431, HepG-2, and MCF-7 cell lines were used for cell cytotoxicity evaluation at a concentration range of 0.2–1.0 μM with MTT assay. For comparison, hypericin was used as a control. As displayed in Fig. 2, 7b showed better cytotoxicity to the test cell lines than 7a, while the cytotoxicity of 7c and 7d were much less than that of 7a at the same concentrations. This may be attributed to the better water solubility of 7b. The results imply that the properties of the substituents on position of 10- and 11- in hypericin molecules are strongly related to its photocytotoxicity: changing methyl groups to hydrophilic ones (such as –COOH for 7b) can enhance the hydrophilicity of a resulting compound and hence create a better photocytotoxicity; on contrary, the more hydrophobic and steric the substituents, the worse the photocytotoxicity (for instance, comparing methyl groups in 7a with pentyl in 7c or phenethyl in 7d).10,42–46 These founding are in line with those reported in the literature.21,40,47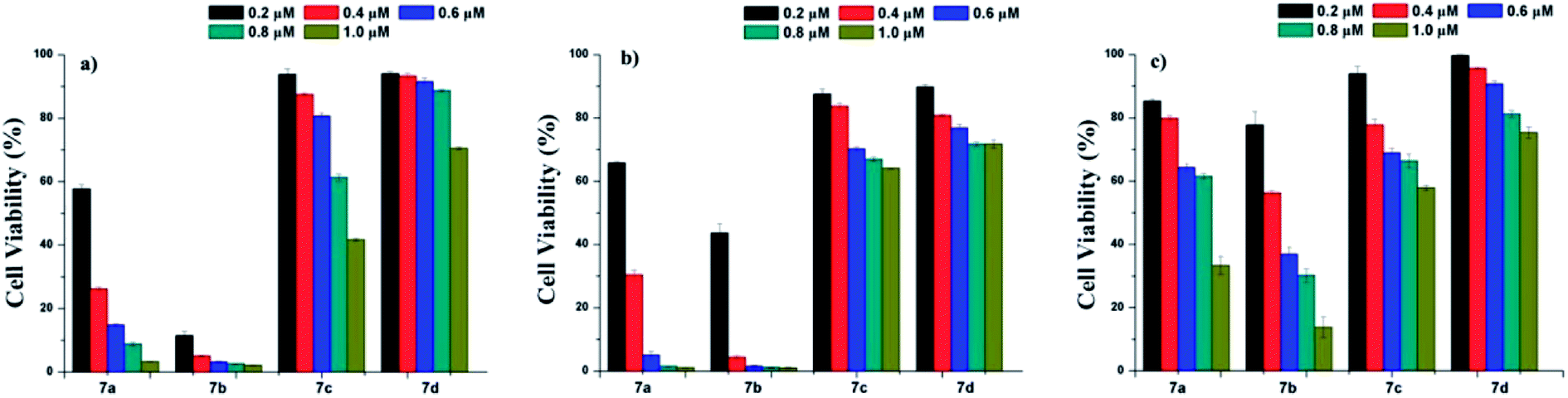 | ||
| Fig. 2 Cell viability of A431 cells (a); HepG2 cells (b); and MCF-7 cells (c) determined by MTT cell viability assay. The cells were incubated with compound 7a–d, respectively, and irradiated with 595-600 nm light for 30 min. | ||
Conclusions
In summary, we developed an economic, green and highly efficient synthetic pathway for preparing hypericin and its derivatives under mild reaction conditions. Water was used as the solvent in the key step of emodinanthrone dimerization. The reaction time of the photocyclization of protohypericin was greatly shortened to 1 h. Furthermore, the PDT effect of 7b is better than those of 7a, 7c and 7d. These results indicate the derivatives of hypericin synthesized in the present work have the potential as photosensitizers for fighting cancers.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research work was supported by the National Natural Science Foundation of China (21772157, 21572181 and 21174113) to the Project of Science and Technology of Social Development in Shaanxi Province (2016SF-029) and Yangling Demonstration Zone (2016SF-029).References
- H. Falk, From the photosensitizer hypericin to the photoreceptor stentorin-the chemistry of phenanthroperylene quinones, Angew. Chem., 1999, 38, 3117–3136 Search PubMed.
- J. Barnes, L. A. Anderson and J. D. Phillipson, St John's Wort (Hypericum perforatum L.): a review of its chemistry, pharmacology and clinical properties, J. Pharm. Pharmacol., 2001, 53, 583–600 CrossRef PubMed.
- K. Souvik, L. Marc, Z. Sebastian and S. Michael, An endophytic fungus from Hypericum perforatum that produces hypericin, J. Nat. Prod., 2008, 71, 159–162 CrossRef PubMed.
- P. S. Martin, C. Anthony, J. C. Marie, C. D. Jean and N. Michel, Hypericin activates l-type ca2+ channels in cardiac myocytes, J. Nat. Prod., 2007, 70, 510–514 CrossRef PubMed.
- K. Souvik, Z. Sebastian, K. Ján, C. Eva and S. Michael, Light-independent metabolomics of endophytic thielavia subthermophila provides insight into microbial hypericin biosynthesis, J. Nat. Prod., 2009, 72, 1825–1835 CrossRef PubMed.
- N. Adolf and B. Veronika, Lessons learned from herbal medicinal products: the example of St. John's Wort, J. Nat. Prod., 2010, 73, 1015–1021 CrossRef PubMed.
- K. Michalska, H. Fernandes, M. Sikorski and M. Jaskolski, Crystal structure of Hyp-1, a St. John's Wort protein implicated in the biosynthesis of hypericin, J. Struct. Biol., 2010, 169, 161–171 CrossRef PubMed.
- A. Kamuhabwa, P. Agostinis, B. Ahmed, W. Landuyt, B. Van Cleynenbreugel, H. Van Poppel and P. de Witte, Hypericin as a potential phototherapeutic agent in superficial transitional cell carcinoma of the bladder, Photochem. Photobiol. Sci., 2004, 3, 772–780 Search PubMed.
- K. Gyuraszova, J. Mikes, A. Halaburkova, R. Jendzelovsky and P. Fedorocko, YM155, a small molecule inhibitor of survivin expression, sensitizes cancer cells to hypericin-mediated photodynamic therapy, Photochem. Photobiol. Sci., 2016, 15, 812–821 Search PubMed.
- J. T. Wessels, A.-C. Busse, M. Rave-Fraenk, S. Zaenker, R. Hermann, E. Grabbe and G.-A. Mueller, Photosensitizing and radiosensitizing effects of hypericin on human renal carcinoma cells in vitro, Photochem. Photobiol., 2007, 84, 228–235 Search PubMed.
- P. Mauri and P. Pietta, High-performance liquid chromatography/electrospray mass spectrometry of Hypericum perforatum extracts, Rapid Commun. Mass Spectrom., 2000, 14, 95–99 CrossRef PubMed.
- H. Brockmann, F. Kluge and H. Muxfeldt, Total synthesis of hypericin, Chem. Ber., 1957, 90, 2302–2318 CrossRef.
- J. Banville and P. Brassard, Reactions of ketene acetals. 8. Simple syntheses of the methyl ester-ethers of the anthraquinones endocrocin, ptilometric acid, and clavorubin, J. Org. Chem., 1976, 41, 3018–3020 CrossRef.
- Y. Hirose, M. Kuroiwa, H. Yamashita, T. Tanaka and T. Megumi, Chemical studies on the natural anthraquinones. I. Syntheses of munjistin, emodin, and 3-hydroxy-2-methylanthraquinone, Chem. Pharm. Bull., 1973, 21, 2790–2793 CrossRef.
- J. Zhao, Z. Zhang, H. Chen and X. Chen, Studies on synthesis and anti-HIV RT activity of hypericin and ethylhypericin, Yaoxue Xuebao, 1998, 33, 67–71 Search PubMed.
- S. Aigner and H. Falk, A microwave-assisted synthesis of phenanthroperylene quinones as exemplified with hypericin, Monatsh. Chem., 2008, 139, 991–993 CrossRef.
- Y. Mazur, H. Bock and D. Lavie, Preparation of Hypericin, US Pat. 5120412A, 1992 Search PubMed.
- L. Yip, J. B. Hudson, E. Gruszecka-Kowalik, L. H. Zalkow and T. G. H. Neil, Antiviral activity of a derivative of the photosensitive compound hypericin, Phytomedicine, 1996, 3, 185–190 CrossRef PubMed.
- M. Waser, Y. Popova, C. W. Klampfl and H. Falk, 9,12-Dibenzothiazolylhypericin and 10,11-Dibenzothiazolyl-10,11-Didemethylhypericin: Photochemical Properties of Hypericin Derivatives Depending on the Substitution Site, Monatsh. Chem., 2005, 136, 1791–1797 CrossRef.
- S. W. Kim, J. H. Park, S. D. Yang, M. G. Hur, Y.-S. Kim, J.-S. Chai, Y. S. Kim and K. H. Yu, Facile synthesis and radioiodine labeling of hypericin, Bull. Korean Chem. Soc., 2004, 25, 1147–1150 CrossRef.
- D. Geisslmeir and H. Falk, ω,ω′-Appended nucleo-base derivatives of hypericin, Monatsh. Chem., 2008, 139, 1127–1136 CrossRef.
- H. Falk and T. N. H. Tran, Synthesis and properties of an ω,ω′-appended eighteen carbon chains hypericin derivative, Monatsh. Chem., 1996, 127, 717–723 CrossRef.
- H. Falk and G. Schoppel, A synthesis of emodin anthrone, Monatsh. Chem., 1991, 122, 739–744 CrossRef.
- H. J. Banks, D. W. Cameron and W. D. Raverty, Chemistry of Coccoidea. II. Condensed polycyclic pigments from two Australian pseudococcids (Hemiptera), Aust. J. Chem., 1976, 29, 1509–1521 CrossRef.
- H. Falk and G. Schoppel, On the Synthesis of Hypericin by Oxidative Trimethylemodin Anthrone and Emodin Anthrone Dimerization: Isohypericin, Monatsh. Chem., 1992, 123, 931–938 CrossRef.
- D. Spitzner, Synthesis of photohypericin from emodin, Angew. Chem., 1977, 89, 55–56 CrossRef.
- E. Gruszecka-Kowalik and L. H. Zalkow, An improved synthesis of hypericin and related compounds, Org. Prep. Proced. Int., 2000, 32, 57–61 CrossRef.
- J. Motoyoshiya, Y. Masue, Y. Nishi and H. Aoyama, Synthesis of hypericin via emodin anthrone derived from a two-fold Diels-Alder reaction of 1,4-benzoquinone, Nat. Prod. Commun., 2007, 2, 67–70 Search PubMed.
- J. Berlanda, T. Kiesslich, V. Engelhardt, B. Krammer and K. Plaetzer, Comparative in vitro study on the characteristics of different photosensitizers employed in PDT, J. Photochem. Photobiol., B, 2010, 100, 173–180 CrossRef PubMed.
- Y. D. Liu, X. L. Liu, Y. Xiao, F. M. Chen and F. N. Xiao, A multifunctional nanoplatform based on mesoporous silica nanoparticles for imaging-guided chemo/photodynamic synergetic therapy, RSC Adv., 2017, 7, 31133–31141 RSC.
- H. Fan, G. Yan, Z. Zhao, X. Hu, W. Zhang, H. Liu, X. Fu, T. Fu, X. Zhang and W. Tan, A smart photosensitizer-manganese dioxide nanosystem for enhanced photodynamic therapy by reducing glutathione levels in cancer cells, Angew. Chem., 2016, 55, 5477–5482 CrossRef PubMed.
- L. B. Meng, W. Zhang, D. Li, Y. Li, X. Y. Hu, L. Wang and G. Li, pH-Responsive supramolecular vesicles assembled by water-soluble pillar[5]arene and a BODIPY photosensitizer for chemo-photodynamic dual therapy, Chem. Commun., 2015, 51, 14381–14384 RSC.
- H. H. Fan, X. B. Zhang and Y. Lu, Recent advances in DNAzyme-based gene silencing, Sci. China: Chem., 2017, 60, 591–601 CrossRef.
- H.-M. Meng, H. Liu, H. Kuai, R. Peng, L. Mo and X.-B. Zhang, Aptamer-integrated DNA nanostructures for biosensing, bioimaging and cancer therapy, Chem. Soc. Rev., 2016, 45, 2583–2602 RSC.
- M. C. Montanha, L. L. Silva, F. B. B. Pangoni, G. B. Cesar, R. S. Goncalves, W. Caetano, N. Hioka, T. T. Tominaga, M. E. L. Consolaro, A. Diniz and E. Kimura, Response surface method optimization of a novel hypericin formulation in P123 micelles for colorectal cancer and antimicrobial photodynamic therapy, J. Photochem. Photobiol., B, 2017, 170, 247–255 CrossRef PubMed.
- B. Kucharova, J. Mikes, R. Jendzelovsky, J. Vargova, L. Mikesova, Z. Jendzelovska, J. Koval and P. Fedorocko, Potentiation of hypericin-mediated photodynamic therapy cytotoxicity by MK-886: focus on ABC transporters, GDF-15 and redox status, Photodiagn. Photodyn. Ther., 2015, 12, 490–503 CrossRef PubMed.
- J. Mikes, R. Jendzelovsky and P. Fedorocko, in Cellular aspects of photodynamic therapy with hypericin, Nova Science Publishers, Inc., 2013, pp. 111–147 Search PubMed.
- Y. Xu, D. Wang, Z. Zhuang, K. Jin, L. Zheng, Q. Yang and K. Guo, Hypericin-mediated photodynamic therapy induces apoptosis in K562 human leukemia cells through JNK pathway modulation, Mol. Med. Rep., 2015, 12, 6475–6482 CrossRef PubMed.
- M. Maslanakova, L. Balogova, P. Miskovsky, R. Tkacova and K. Stroffekova, Anti- and Pro-Apoptotic Bcl2 Proteins Distribution and Metabolic Profile in Human Coronary Aorta Endothelial Cells Before and After HypPDT, Cell Biochem. Biophys., 2016, 74, 435–447 CrossRef PubMed.
- A. M. Lima, C. D. Pizzol, F. B. F. Monteiro, T. B. Creczynski-Pasa, G. P. Andrade, A. O. Ribeiro and J. R. Perussi, Hypericin encapsulated in solid lipid nanoparticles: phototoxicity and photodynamic efficiency, J. Photochem. Photobiol., B, 2013, 125, 146–154 CrossRef PubMed.
- D. Feinweber, T. Verwanger, O. Brueggemann, I. Teasdale and B. Krammer, Applicability of new degradable hypericin-polymer-conjugates as photosensitizers: principal mode of action demonstrated by in vitro models, Photochem. Photobiol. Sci., 2014, 13, 1607–1620 Search PubMed.
- A. Kubin, F. Wierrani, U. Burner, G. Alth and W. Gruenberger, Hypericin – the facts about a controversial agent, Curr. Pharm. Des., 2005, 11, 233–253 CrossRef PubMed.
- P. Agostinis, A. Vantieghem, W. Merlevede and P. A. M. de Witte, Hypericin in cancer treatment: more light on the way, Int. J. Biochem. Cell Biol., 2002, 34, 221–241 CrossRef PubMed.
- L. M. Davids, B. Kleemann, S. Cooper and S. H. Kidson, Melanomas display increased cytoprotection to hypericin-mediated cytotoxicity through the induction of autophagy, Cell Biol. Int., 2009, 33, 1065–1072 CrossRef PubMed.
- G. Seitz, S. W. Warmann, S. Armeanu, H. Heitmann, P. Ruck, R. M. Hoffman, J. Fuchs and J. T. Wessels, In vitro photodynamic therapy of childhood rhabdomyosarcoma, Int. J. Oncol., 2007, 30, 615–620 Search PubMed.
- J. Zuschrader, W. Schoefberger and H. Falk, A carbohydrate-linked hypericinic photosensitizing agent, Monatsh. Chem., 2008, 139, 1387–1390 CrossRef.
- I. Teasdale, M. Waser, S. Wilfert, H. Falk and O. Brueggemann, Photoreactive, water-soluble conjugates of hypericin with polyphosphazenes, Monatsh. Chem., 2012, 143, 355–360 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03732a |
| This journal is © The Royal Society of Chemistry 2018 |