
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotomechanical photochromism in a cetyltrimethylammonium isopolytungstate†
Aaron V. Colusso,
Andrew M. McDonagh,
Angus Gentle and
Michael B. Cortie *
*
Institute for Nanoscale Technology, University of Technology Sydney, PO Box 123, Broadway NSW 2007, Australia. E-mail: Michael.Cortie@uts.edu.au
First published on 23rd May 2018
Abstract
The photochromic properties of a hybrid compound comprised of the surfactant cation cetyltrimethylammonium [(C16H33)N(CH3)3]+ (CTA+) and the isopolytungstate anion [H2W12O40]6− is investigated. The compound, which has the nominal formula (CTA)7[H2W12O40]Cl·2H2O, changes from white to blue when exposed to UV radiation. The sample returns to the bleached state if stored in the dark-ambient. Application of XPS indicates that the coloring species are WV and WIV. The CTA+ component is found by XPS and FTIR to undergo progressive and irreversible oxidation during this cycle. Examination of FTIR suggests that the changes occur at multiple sites across the amphiphile. Surprisingly, the photochromic cycle is correlated with changes in the X-ray diffraction pattern, indicating partially reversible changes in the ordering of the Keggin ions and their spacing. In particular, application of the UV radiation causes the progressive accumulation of strain in the [001] direction. This is due to permanent oxidative changes in the CTA+ accumulating from cycle to cycle, resulting in an increase in interlamellar-distance due to less interdigitation of the chains. This provides a controllable photomechanical response.
Introduction
Photochromism presents a passive, energetically favorable process that can be used in many current and emerging smart technologies, including memory-based materials,1 gas-sensing devices,2–4 solar-energy storage5 and smart-windows.6 Diverse materials have been employed to serve in photochromic devices, for example, tungsten oxides such as WO3−δ (0 < δ < 0.3) are strongly photochromic, with UV irradiation causing a change from white (the normal or ‘oxidized’ state, δ ≈ 0) to blue (the ‘reduced’ state, δ > 0).7–11 The carrier concentration of the reduced form, WO3−δ, is much higher than the oxidized form because every oxygen vacancy is associated with two free electrons. Indeed, if sufficiently reduced, the oxide becomes metallic in nature.12 In general, exposure of the blue material to air will result in its slow reversion to the white bleached state. These photochromic properties are also found in complexes between WOx and various organic materials, including the surfactant-polyoxometalate (POM) frameworks.13–20 Here we address the photochromic properties of a surfactant-POM compound synthesized from the metatungstate anion, [H2W12O40]6−, and the quaternary-ammonium surfactant cetyltrimethyl-ammonium (CTA+) cation. For convenience, we have designated this compound as ‘CTA-W12’.21 Compounds of this type may be produced by an efficient and simple synthetic method:22–24 they self-assemble in aqueous solution via electrostatic and hydrophobic–hydrophilic interactions between the organic cationic surfactants and the inorganic anionic POMs. The outcome is a highly predictable hierarchical nanostructure.25–27 The POMs themselves are a class of inorganic macromolecules that are usually comprised of group V and VI transition metals and oxygen.24 The metal–oxygen octahedra in POMs form cyclic-geometries that optimize the transition metal's mixed-valance characteristics,28–30 resulting in favorable photo-redox properties31 and hence potential use in photochromic devices.The coloration mechanism of surfactant-polyoxometalate materials is often said to involve the creation of a charge-transfer complex via proton-transfer from hydroxyl,19 amine, carboxylic17 or other suitable proton-transfer groups within the organic species, to the activated bridging-oxygen (Ob) neighboring the photo-reduced WV site (which stabilizes the colored state).14,17,32,33 This deprotonation-mediated mechanism allows for a reversible back-reaction where the POM-attached proton simply transfers back to the organic during bleaching by atmospheric O2.34 However, possibly due to disorder within the hydrophobic–hydrophilic interfaces or oxidative side-reactions involving the organic matrix,20,33 these systems regularly suffer from decreased switchability upon each iteration of the photochromic cycle and hence cannot be classified as completely reversible.15,18,35 There is an incomplete understanding of the redox mechanisms associated with the organic-POM photochromic processes,36–38 especially for compounds based on quaternary ammonium surfactants. Complete and/or accelerated reversal of surfactant-POM and other POM-based photochromic materials is possible via heating,39,40 or prolonged exposure to increased O2 partial pressures, however such methods usually result in decreased change in optical properties upon future coloration (i.e. loss of photochromic performance).
Here we examine the photochromic properties of CTA-W12, in particular the mechanism of coloring, the rate and degree of bleaching (back-reaction of the photochromic process towards the colorless state), the change in structure that occurs during the photochromic cycle, and the cyclic photochromic performance of surfactant-POM compounds. The objective of this paper is (i) to determine the reasons for the incomplete reversibility, and hence decreased switchability, of surfactant-POM photochromic materials, using CTA-W12 as a prototype, and (ii) to explain the unexpected photomechanical response of this particular compound. Synchrotron powder X-ray diffraction (P-XRD) was used to track the structural changes instigated by UV-exposure and associated coloration in situ. Reflectance UV-vis spectroscopy was employed to measure the change in diffuse reflectance, ΔR, upon coloration and track this change across successive photochromic cycles. Chemical changes to CTA-W12 over coloration cycles were investigated with X-ray photo-electron spectroscopy (XPS), Fourier-transform infra-red spectroscopy (FTIR), and gas chromatography mass spectrometry (GC-MS).
Experimental section
Syntheses
CTA-W12 was prepared and characterized as described elsewhere.21Analytical techniques
Transmission electron microscope (TEM) images were collected on a JEM-2200FS Field Emission Microscope. A suspension of CTA-W12 in methanol was drop cast onto a lacey-carbon grid and allowed to dry in air for 24 h.X-ray diffraction (XRD) experiments were carried out at the Australian Synchrotron on the powder diffraction beam-line, with samples in a rotating 0.5 mm diameter silica capillary. A wavelength of 0.82481 Å was used, determined by refining against a mixed standard of LaB6 and Al2O3. FTIR spectra were collected using a Nicolet 6700 ATR Spectrometer. Raman spectroscopy was conducted on the Renishaw InVia Confocal Raman Microscope using a 633 nm laser.
X-ray photoelectron spectroscopy (XPS) measurements were performed using a Thermo Scientific ESCALAB250Xi utilizing monochromated Al Kα (1486.68 keV) radiation at 120 W. The spectrometer was calibrated using Au 4f7, Ag 3d5 and Cu 2p3 lines at 83.96, 368.21 and 932.62 eV respectively, and a C 1s binding-energy reference (284.8 eV for adventitious hydrocarbon).
Gas chromatography-mass spectrometry (GC-MS) was conducted using a Thermo Scientific GCMS, employing a TSQ-8120504 stationary phase and scanning for masses between 25 and 300 amu with a 0.2014 s scan time. The column was heated to 50 °C for 5 minutes, followed by a heating rate of 5 °C min−1 to 250 °C, followed by a heating rate of 10 °C min−1 to 280 °C and finally held at 280 °C for 10 minutes. The MS transfer line temperature was held at 280 °C and the ion source at 290 °C.
Photo-coloration and bleaching
UV exposure experiments utilized a DYNAMAX BlueWave 50 light source with a peak intensity at 315 nm of 3.5 W cm−2. The effect of photochromic cycling was determined in a custom-apparatus in which an immobile powder sample was periodically irradiated while also being measured using an optical spectrometer. The UV radiation in the cycling experiment was applied for 2 hours, and then bleaching was allowed to occur for 48 to 72 hours before the next cycle was initiated.Results and discussion
The structure of CTA-W12, which consists of surfactant-POM complexes that are packed into layers, has been discussed elsewhere.21 A schematic depiction is shown here in Fig. 1 for convenience. TEM images (Fig. S1†) showed a lamellar structure with an interplanar distance of 24.5 Å. An interplanar distance of 24.5 Å was also measured using low-angle powder XRD.21 The length of one CTA+ surfactant ion is reported to be 24 Å,41 and the metatungstate anion has a diameter of 10 Å,42 indicating that a significant degree of interdigitation of the CTA+ tailgroups occurs within the structure. In a related POM structure,38 (CTA)5(CH3CN)4[HxSiMo12O40], the CTA+ tail groups were interdigitated by an average of 13.3 carbons per chain with some CTA+ cations completely interdigitated (16 carbons) and others only by 9 carbons. In the present work, the CTA-W12 exceeds the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 POM:surfactant ratio required for completely ordered lamellar surfactant-polyoxometalate materials37,38 and therefore a different degree of interdigitation is expected. The colored and bleached forms of CTA-W12 (after one cycle) are shown Fig. 2a, and the corresponding UV-vis-NIR diffuse reflectance spectra are provided in Fig. 2b. In the bleached form, the material exhibited significantly more reflectance between 450 and 1650 nm, as expected from its lighter color. The absorption edge, located at 380 nm, is a consequence of oxygen-to-metal (O → M) ligand-to-metal-charge transfer (LMCT) from the highest occupied molecular orbital (HOMO), which is mainly comprised of O 2p orbitals, to the lowest unoccupied molecular orbital (LUMO), mainly comprised of W 5d orbitals.14,34 Upon exposure to UV light, a large decrease in scattered light was observed in the range 500 to 1650 nm. A blue color developed, due to the formation of two absorbance bands at 780 and 1400 nm which are assigned to reduction of the tungsten center (WVI ↔ WV) by the O → M LMCT and population of the conducting W 5d orbital. The band at 783 nm, with a shoulder at 633 nm, is assigned to intervalence charge transfer (IVCT) mechanisms between reduced and oxidized tungsten sites (WV–Ob–WVI ↔ WVI–Ob–WV), across WO6 octahedra. The broad band at 1400 nm is assignable to d–d transitions within the partially populated W 5d orbital. Raman spectroscopy, Fig. S2,† revealed the presence of luminescent defects in the bleached material 72 hours after UV irradiation. Considering the lack of luminescent groups in CTA+ (or its likely photo-redox products), this emission is very likely due to a combination of structural defects and oxygen vacancies, as reported in the literature for WO3−δ.43 Fig. 3a shows the changes in absorbance at 783 nm as the result of the application of eighteen photochromic cycles. The difference between bleached and colored states initially decreases with the number of irradiation/recovery cycles indicating that irreversible processes are occurring, and then, after 4 cycles, seems to become constant. The bleached state, which is initially colorless, becomes progressively browner over a number of cycles. The changes with time during coloring or bleaching were modelled using an empirical relationship of the form:
:4 POM:surfactant ratio required for completely ordered lamellar surfactant-polyoxometalate materials37,38 and therefore a different degree of interdigitation is expected. The colored and bleached forms of CTA-W12 (after one cycle) are shown Fig. 2a, and the corresponding UV-vis-NIR diffuse reflectance spectra are provided in Fig. 2b. In the bleached form, the material exhibited significantly more reflectance between 450 and 1650 nm, as expected from its lighter color. The absorption edge, located at 380 nm, is a consequence of oxygen-to-metal (O → M) ligand-to-metal-charge transfer (LMCT) from the highest occupied molecular orbital (HOMO), which is mainly comprised of O 2p orbitals, to the lowest unoccupied molecular orbital (LUMO), mainly comprised of W 5d orbitals.14,34 Upon exposure to UV light, a large decrease in scattered light was observed in the range 500 to 1650 nm. A blue color developed, due to the formation of two absorbance bands at 780 and 1400 nm which are assigned to reduction of the tungsten center (WVI ↔ WV) by the O → M LMCT and population of the conducting W 5d orbital. The band at 783 nm, with a shoulder at 633 nm, is assigned to intervalence charge transfer (IVCT) mechanisms between reduced and oxidized tungsten sites (WV–Ob–WVI ↔ WVI–Ob–WV), across WO6 octahedra. The broad band at 1400 nm is assignable to d–d transitions within the partially populated W 5d orbital. Raman spectroscopy, Fig. S2,† revealed the presence of luminescent defects in the bleached material 72 hours after UV irradiation. Considering the lack of luminescent groups in CTA+ (or its likely photo-redox products), this emission is very likely due to a combination of structural defects and oxygen vacancies, as reported in the literature for WO3−δ.43 Fig. 3a shows the changes in absorbance at 783 nm as the result of the application of eighteen photochromic cycles. The difference between bleached and colored states initially decreases with the number of irradiation/recovery cycles indicating that irreversible processes are occurring, and then, after 4 cycles, seems to become constant. The bleached state, which is initially colorless, becomes progressively browner over a number of cycles. The changes with time during coloring or bleaching were modelled using an empirical relationship of the form:| y = a + be−cx | (1) |
 | ||
| Fig. 1 A schematic depiction of the structure of CTA-W12 showing planes of hexagonally-packed Keggin ions with intercalated and interdigitated cetyltrimethylammonium chains (see Colusso et al.21 for further discussion.). | ||
 | ||
| Fig. 2 Photochromic properties of CTA-W12. (a) Blue color developed when 315 nm UV source was directed onto the side of a sample of dry powder. (b) UV-vis diffuse reflectance spectra of CTA-W12 as-prepared and after 1 hour of UV exposure. The diffuse reflectance scale has been calibrated with a white TiO2 sample and converted to approximate RGB colors via the CIE L-a*-b* color scale. | ||
 | ||
| Fig. 3 Photochromic behavior of CTA-W12. (a) Plot of diffuse reflectance at 783 nm vs. photochromic cycles. (b) Diffuse reflectance at 783 nm vs. time for the 1st coloring cycle of a bulk powder sample showing fit of eqn (1). (c) Transmittance at 783 nm vs. time for 1st bleaching cycle of a thin coating of CTA W12 on a glass slide, showing fit of eqn (1) (the vertical scales are in arbitrary units due to the non-standard set-up of the multi-cycle experiments). | ||
Here the parameter c gives the rate of change in s−1 while a and b account for the scaling of the vertical axis. A characteristic time constant, the halfway complete time for the process can be obtained from
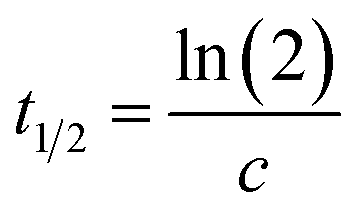 | (2) |
The value of c for coloring was of the order of 5 ± 3 × 10−4 s−1, which corresponds to a t1/2 of about 1400 seconds, Fig. 3b. A typical bleaching experiment is shown in Fig. 3c and the value for c in this case is about 6 ± 1 × 10−4 s−1, corresponding to a t1/2 of about 1500 seconds. The similarity of the two values is a coincidence since the former is, inter alia, determined by the flux and wavelength of the UV lamp used and hence a function of experimental setup whilst the latter value depends upon the oxygen content of the ambient environment. The long t1/2 has been attributed to the slow diffusion of O2 across the hydrophobic and hydrophilic layers.19,44,45 Long diffusion-mediated bleaching times have been observed in other organic-POM photochromic devices.34,36
XPS spectra showing binding energies for the W 4f energy level are shown in Fig. 4. Prior to exposure, the material exhibits strong signals assigned to WVI 4f7/2 and WVI 4f5/2 (37.3 and 39.5 eV, respectively) with much smaller signals assigned to WV 4f7/2, WV 4f5/2 and the WV p3/2 satellite at 35.5, 37.7 and 42.6 eV, respectively.
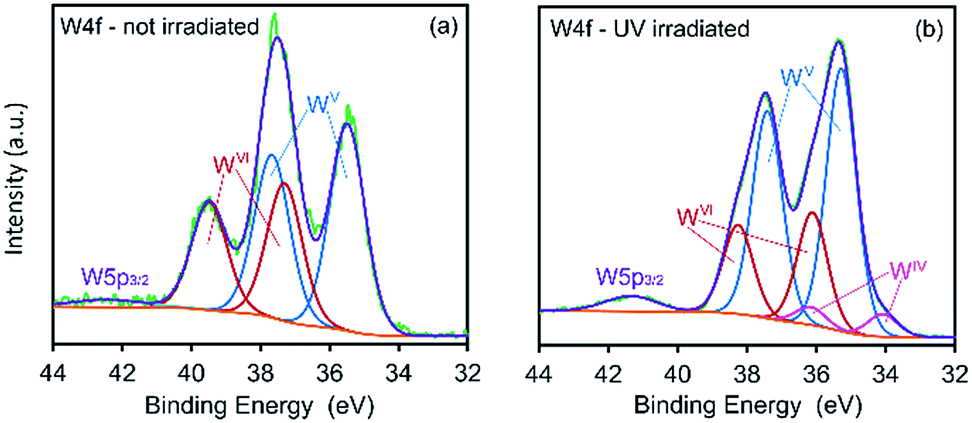 | ||
| Fig. 4 XPS spectra of CTA-W12 showing binding energies for W 4f energy levels in (a) un-irradiated and (b) UV-irradiated (3 h) material. | ||
The appearance of WV species in the un-irradiated specimen is attributed to photo-reduction of tungsten centers by the incident X-ray radiation, a process previously observed.26,27,29,32 After exposing the material to UV light for 3 hours, a significant decrease in the intensity of the WVI 4f7/2 and WVI 4f5/2 is observed together with an increase in the intensity of the WV f7/2 and WV 4f5/2 signals. Peaks at 34.1 and 36.2 eV, assigned to WIV 4f7/2 and WIV 4f5/2 states, respectively, are also apparent in the irradiated sample.34,46–49 Thus, in the material examined here, the coloration upon irradiation with UV light is due to photo-reduced WV and WIV centers as expected.16,30,49
Two carbon environments are apparent within the C 1s spectrum of pristine CTA-W12, Fig. 5a. These correspond to carbon atoms within the methyl-groups that are attached to the cationic-nitrogen head-groups of CTA+ (C–N), and to carbon atoms within the alkyl-chains (C–C) at 286.1 and 284.8 eV, respectively.50,51 Upon irradiation, two new bands appear in the C 1s spectrum, Fig. 5b, centered at 288.98 and 287.58 eV. We assign the new peaks to carbon atoms in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O groups,52 respectively. Thus, upon irradiation, some degree of oxidation of the carbon atoms in the CTA+ group occurs. Two bands in the O 1s spectrum of pristine CTA-W12, Fig. 5c, are assigned to oxygen atoms located within the Keggin cage (O–W, 531.9 eV) and adsorbed moisture (O–H, 530.5 eV). A new band emerged in the spectrum of irradiated material, Fig. 5d, at 533.4 eV. The additional peak is assigned to oxygen bonded to carbon.53 This is consistent with the C 1s spectrum that shows formation of CO and C–O groups. The intensity of the band assigned to O–H groups increased upon irradiation, which is consistent with the proton-transfer mechanism of photochromism.14 A band with binding-energy of 402.2 eV is observed in the N 1s spectrum of pristine CTA-W12 along with a side band at 403.0 eV, Fig. 5e. We assign the N 1s peaks to quaternary alkyl-ammonium (NR4+) groups of the CTA+ cation bound to the POM and to the small chloride content of the compound, respectively. An additional band is apparent in the spectrum of the irradiated material at 400.1 eV, Fig. 5f, which is assigned to nitrogen bonded to hydrogen.
O and C–O groups,52 respectively. Thus, upon irradiation, some degree of oxidation of the carbon atoms in the CTA+ group occurs. Two bands in the O 1s spectrum of pristine CTA-W12, Fig. 5c, are assigned to oxygen atoms located within the Keggin cage (O–W, 531.9 eV) and adsorbed moisture (O–H, 530.5 eV). A new band emerged in the spectrum of irradiated material, Fig. 5d, at 533.4 eV. The additional peak is assigned to oxygen bonded to carbon.53 This is consistent with the C 1s spectrum that shows formation of CO and C–O groups. The intensity of the band assigned to O–H groups increased upon irradiation, which is consistent with the proton-transfer mechanism of photochromism.14 A band with binding-energy of 402.2 eV is observed in the N 1s spectrum of pristine CTA-W12 along with a side band at 403.0 eV, Fig. 5e. We assign the N 1s peaks to quaternary alkyl-ammonium (NR4+) groups of the CTA+ cation bound to the POM and to the small chloride content of the compound, respectively. An additional band is apparent in the spectrum of the irradiated material at 400.1 eV, Fig. 5f, which is assigned to nitrogen bonded to hydrogen.
 | ||
| Fig. 5 XPS spectra of CTA-W12 showing binding energies for C 1s energy levels in (a) un-irradiated and (b) UV-irradiated (3 h) material, O 1s energy levels in (c) un-irradiated and (d) UV irradiated, and N 1s energy levels in (e) un-irradiated and (f) UV-irradiated. | ||
In situ low-angle synchrotron powder XRD patterns were collected during UV irradiation of CTA-W12, Fig. 6. As mentioned earlier, the peak in the diffraction pattern at d = 24.51 Å is assigned to interlamellar spacing of the sheets of pseudo-hexagonally-packed Keggin ions. The peak at d = 9.80 Å is assigned to diffraction between Keggin ions within each plane.21
 | ||
| Fig. 6 In situ low-angle synchrotron P-XRD patterns of CTA-W12 continually irradiated by UV light. The 2θ values are scaled for comparison with Cu Kα data. The lamellar (001) and pseudo-hexagonal (110) POM peaks appear at d = 24.51 Å and d = 9.80 Å, respectively. Inset. Interlamellar (001) and pseudo-hexagonal (110) peak height as a function of exposure time. | ||
Previous work has shown that two Keggin ions in a unit cell with a = 12.40 ± 0.10 Å, b = 16.45 ± 0.10 Å, c = 24.90 ± 0.20 Å, α = 90°, β = 101.5 ± 0.5° and γ = 90° will produce a diffraction pattern similar to that observed.21 The peaks at 24.5 Å and 9.80 Å correspond to (001) and (110) respectively if indexed on the above unit cell. The intensity of both of these peaks decreased rapidly upon exposure to UV light (Fig. 6, inset). After 220 minutes of UV exposure, the lamellar and pseudo-hexagonal peaks had decreased in intensity by 76% and 43%, respectively (the effect of irradiation conditions on the area under of the (001) peak is shown in the ESI, Fig. S3.†). These measurements indicate increasing disorder within the material upon exposure to UV light. Although the intensity of this XRD peak is initially restored after the UV lamp is turned on, we expect that, in the dark state, this increase would eventually be overwhelmed by damage induced by the X-rays alone. Fig. 7a shows the intensity of the (001) peak when the UV source is turned on and then off at 25 minute intervals. For comparison, the data for continuous X-rays plus UV, and X-rays only, are also shown. It is evident that there was also a slow decrease of peak intensities during the continuous exposure to X-rays alone. The individual segments of the UV-on/UV-off sequences can be superimposed on each other by (i) correcting for the gradual baseline decrease in intensity due to X-rays alone and (ii) translating each pair of treatments (along the time and intensity scales) so that they start at a common time and share a common intensity at the changeover from UV-on to UV-off, Fig. 7b. This maneuver permits the overall rate to be examined even if the times and intensities of individual segments are different. The aggregated data for decline of intensity of the (001) peak while the UV lamp was on can also be fitted with eqn (1). In this case, a 95% confidence interval (CI) for t0.5 of about 600 to 1500 seconds is indicated. This is of the same order as the t0.5 value of ∼1400 seconds obtained earlier from the change in optical properties. Next we note that, when the lamp is turned off, there is a process of dynamic recovery in the intensity. Evidently, the Keggin ions (which are responsible for this diffraction peak) are pushed out of their ordered positions when the lamp is switched on but return over the time scale of several minutes when the lamp is off. In this case a 95% confidence interval (CI) for t0.5 of 600 to 4000 seconds is indicated by fitting eqn (1). This brackets the t0.5 value reported earlier for bleaching. Based on the available evidence, we suggest that the processes that decrease and then restore the XRD peak intensity are respectively directly proportional to the degrees of coloring and bleaching. The further implication is that the coloring process is associated with displacement and disordering of the Keggin ions while some degree of re-ordering of the Keggin ions occurs during bleaching.
 | ||
| Fig. 7 Changes on application of UV-irradiation during continuous X-ray diffraction measurement. (a) Change in intensity over entire range of experiment. (b) Intensity change for each pair of UV-on/UV-off measurements. Data for each pair of measurements have been baselined against the X-ray only trend line, then translated to a common start time and mid-sequence intensity to provide the overlay. The curve fits based on eqn (1) are superimposed. | ||
An examination of the interlamellar spacing during cycling, Fig. 8, is also informative. The disordering associated with the coloring process is also associated with an expansion in the interlamellar gallery, while the re-ordering that occurs during bleaching is associated with a contraction. The effect of the small temperature changes that occur during the UV-on/UV-off sequence can also be seen in this data: the coefficient of thermal expansion in the [001] direction of CTA-W12 between RT and 70 °C is high (about 3.56 × 10−4 K−1, with a 95% confidence interval of 2.81 × 10−4 to 4.39 × 10−4 K−1) (ESI, Fig. S4†). Analysis of the reversible lattice expansion and contraction that occurred when the UV lamp was turned on and off respectively indicated that application of the lamp caused a ΔT of about +6 °C. The temperature evidently stabilized in the silica capillaries within two or three minutes, and further changes in lattice parameter must have occurred isothermally.
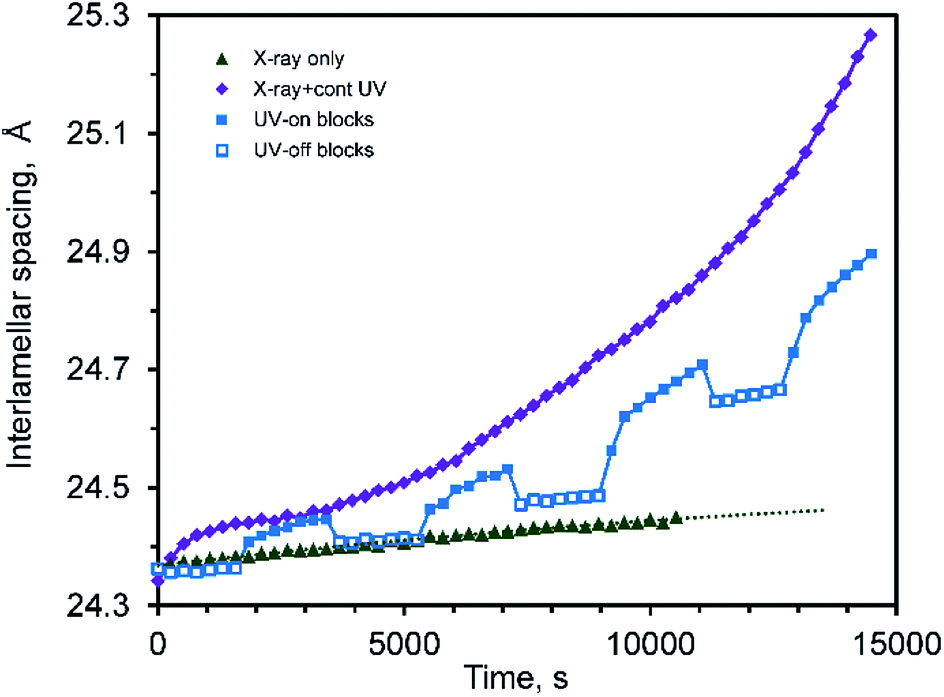 | ||
| Fig. 8 Interlamellar spacing of CTA-W12 as a function of time irradiated with X-rays only, X-rays plus UV, and intermittent UV. | ||
Overall, the effect of the UV irradiation is to cause a rapid transient increase in interlamellar spacing when the UV lamp was first turned on due to heating, followed by a slower but monotonic increase in interlamellar spacing and concurrent decrease in crystalline order of the sheets of Keggin ions. When the UV lamp is turned off there is an immediate decrease in interlamellar spacing due to thermal contraction, followed by a slower decrease due to the re-ordering of the Keggin ions.
FTIR and Raman spectroscopy was used to investigate the chemical changes accompanying photochromic cycling. FTIR spectra are presented in Fig. 9a. Upon irradiation with UV light, bands assigned to –CH2– and –CH3 groups diminished while new bands assigned to oxygen-containing species (CO, C–OH) appeared (Fig. S5†). After one irradiation cycle, a broad band emerged at 1685–1734 cm−1 assigned to CO species.54 This band increased in intensity after each irradiation cycle, Fig. 9b. The exposure-induced chemical changes are permanent and increased in proportion with exposure time. Fig. 9b displays the ratio of the area of the ν(CO) band to that of the ν(O–Wb–O) band (889 cm−1), the latter band being unaffected by UV (ESI, Fig. S5 and S6†). The FTIR data support the findings of the XPS experiments and indicate that redox reactions occur between the POM and the interdigitated CTA+ alkyl-chain species at multiple sites. Similar behavior has been reported in other POM-organic photochromic materials55,56 where carbonyl and hydroxyl formation was observed.
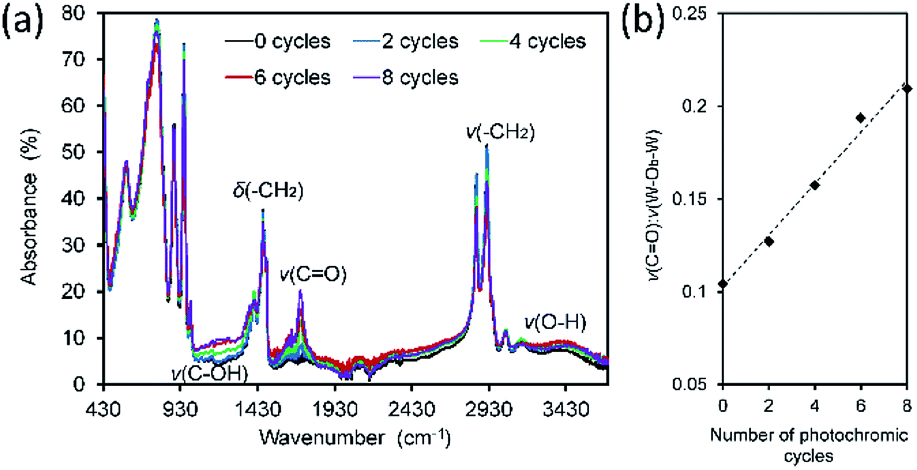 | ||
| Fig. 9 (a) FTIR spectra of CTA-W12 and after 0, 2, 4, 6 and 8 photochromic cycles (1 cycle = 1 h UV exposure followed by 23 h in dark ambient). (b) Plot of ν(CO):ν(W–Ob–W) peak area ratio versus number of photochromic cycles. | ||
Pure CTA-Br was also irradiated for one hour followed by 23 hours in the dark for a total of eight cycles. After eight exposure cycles, a small peak at 1736 cm−1 attributable to ν(CO) was observed, which is slightly blue-shifted relative to CTA-W12 due to the absence of the POM, Fig. S7a.† The growth of the ν(CO) band was an order of magnitude slower in the experiment using CTA-Br, showing that an interaction between the POM and CTA greatly accelerates the oxidation of the organic phase.
A sample of CTA-W12 was irradiated for 48 hours and then washed with chloroform and the extracted material was analyzed by GC-MS, Fig. S8.† Hexadecylamine and 1-hexdecanol were detected, with hexadecylamine the major product and 1-hexadecanol present in trace amounts. The presence of these compounds indicates that photo-redox processes between CTA+ and [H2W12O40]6− occur in the vicinity of the head-group of CTA+ and the C–N bonds therein. Cleavage of C–N bonds between the methyl-groups of the head-group and nitrogen atoms results in the formation of hexadecylamine. The detection of photo-redox products containing N–H bonds is consistent with the photochromic mechanism of quaternary-ammonium–POM hybrid compounds as proposed by T. Yamase57 and T. Zhang.15 Methylamine was not detected, however, this is unsurprising considering its volatility (bp ≈ 5 °C). Cleavage of the C–N bond between the nitrogen and C16 alkyl-chain followed by reaction with oxygen or water may form 1-hexadecanol. No soluble products were detected by GC-MS in the un-irradiated CTA-W12, Fig. S8(d).† The mass of the material decreased by about 9% upon UV irradiation, which is consistent with the formation of volatile products.
The absence of oxygenated products suggests that the photo-redox processes that form these groups do not mutually form a product that is soluble in CHCl3. This is likely due to the carbonyl-containing CTA+ photo-redox products remaining cationic. Indeed, oxygenating photo-redox reactions between POMs and organic components, such the Mars-van Krevelen mechanism58,59 or formation of organic-radicals and their subsequent reaction with O2 or H2O,60 would not lead to reacted CTA+ becoming non-cationic. It is evident therefore, that a number of photo-redox interactions occur between the POM and CTA+ in CTA-W12 during photochromism.
The abstraction of alkyl groups from the head group of CTA+, and/or the oxidation of –CH2 groups to CO or C–OH is consistent with the amorphization and increasing lattice parameter of CTA-W12 with increasing irradiation time. The formation of CO and C–OH groups or removal of methyl groups from the head group or scission of the head group would disrupt the CTA+ interdigitation within the interlamellar space, thereby causing disruptions to the ordered packing of the Keggin ions and the observed changes in the XRD patterns.
Considering the chemical changes to CTA+, the photochromic process in CTA-W12 may be described as proceeding via an irreversible photo-redox reaction between [H2W12O40]6− and CTA+, rather than by the reversible proton-transfer mediated mechanism described for many other photochromic alkyl-ammonium POM materials.14,61,62 This irreversible behavior has been reported for analogous systems: [N(C4H9)]4[W10O32] in acetonitrile produced 1-butane, butyraldehyde and tributylamine,57 while [(CH3)2N(C18H37)2]3[PW12O40] in air produced 1-octadecylene.63 Thus, the coloration of CTA-W12 should continue until no oxidizable organic species remain. There is, however, the possibility that the production of secondary and tertiary amines (facilitating a reversible protonation–deprotonation mechanism, known from other systems) would extend the lifetime of CTA-W12 as a photochromic material.
If the observed carbonyl species are due to a Mars-van Krevelen-type mechanism whereby cluster oxygen of the POM is consumed,58,59 and the O2 diffusion rate is slow, then WV sites deep within the material will remain for extended periods of time. In our work, the concentration of stable WV species is constant after a small number of photochromic cycles, Fig. 3.
In general, many surfactant-POM and similar photochromic materials suffer from decreases in ΔR upon successive coloration cycles.15,18,35,64 We have shown that one cause of this is unavoidable and irreversible oxidative side reactions to organic species whilst bleaching can be very slow due to slow O2 diffusion dynamics.
Conclusions
The photochromic properties of a surfactant-POM compound constructed from the isopolyanion [H2W12O40]6− and the surfactant cetyltrimethylammonium cation were investigated. CTA-W12 turned blue and darkened upon exposure to UV radiation, and returned to the bleached state after an hour of exposure to normal ambient conditions. XPS analyses indicated that coloring was due the formation of WV and WIV species, and that the CTA+ within CTA-W12 underwent oxidation when the compound was colored. This oxidation was permanent, occurred at multiple CTA+ sites, and progressively increased with each successive coloration cycle, as confirmed by FTIR. Persisting WV sites in the material after prolonged bleaching times were identified with UV-vis and Raman spectroscopy and were ascribed to the formation of a more stable-charge transfer complex.In situ synchrotron XRD showed that prolonged exposure of CTA-W12 to UV caused the interlamellar distance to increase and the compound to be amorphized due to alteration of the structure-supporting CTA+ backbone by progressive oxidation, creating disorder in the interlamellar space and in the pseudo-hexagonal order of the POMs. Partial recovery of the inter-lamellar distance occurred when the UV illumination was removed. The associated photomechanical strain was strongest in the [001] direction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Annette Dowd (University of Technology Sydney) for TEM, Dr ‘Bill’ Gong (University of New South Wales) for XPS, Dr J. Kimpton and Dr Qinfen Gu (Australian Synchrotron) for assistance with the in situ XRD experiments, and Dr Verena Taudte and Mr Alex Angleloski for assistance with analytical chemical techniques. A portion of this research was undertaken on the Powder Diffraction beamline at the Australian Synchrotron, part of ANSTO.References
- G. Poirier, M. Nalin, L. Cescato, Y. Messaddeq and S. Ribeiro, J. Chem. Phys., 2006, 125, 161101–161103 CrossRef PubMed.
- F. Nourmohammadian, T. Wu and N. Branda, Chem. Commun., 2011, 47, 10954–10956 RSC.
- V. Bren, A. Dubonosov, V. Minkin, A. Tsukanov, T. Gribanova, E. Shepelenko, Y. Revinsky and V. Rybalkin, J. Phys. Org. Chem., 2007, 20, 917–928 CrossRef.
- J. Grate, L. Moore, D. Janzen, D. Veltkamp, S. Kaganove, S. Drew and K. Mann, Chem. Mater., 2002, 14, 1058–1066 CrossRef.
- L. Palilis, M. Vasilopoulou, A. Douvas, D. Georgiadou, S. Kennou, N. Stathopoulos, V. Constantoudis and P. Argitis, Sol. Energy Mater. Sol. Cells, 2013, 114, 205–213 CrossRef.
- R. Baetens, B. Jelle and A. Gustavsen, Sol. Energy Mater. Sol. Cells, 2010, 94, 87–105 CrossRef.
- S. K. Deb, Philos. Mag., 1973, 801–822 CrossRef.
- T. H. Fleisch and G. J. Mains, J. Chem. Phys., 1982, 76, 780–786 CrossRef.
- M. U. Sun, N. Xu, Y. W. Cao, J. N. Yao and E. G. Wang, J. Mater. Sci. Lett., 2000, 19, 1407–1409 CrossRef.
- T. He and J. Yao, J. Mater. Chem., 2007, 17, 4547–4557 RSC.
- V. B. Kumar and D. Mohanta, Bull. Mater. Sci., 2011, 34, 435–442 CrossRef.
- K. Manthiram and A. P. Alivisatos, J. Am. Chem. Soc., 2012, 134, 3995–3998 CrossRef PubMed.
- T. Yamase, N. Takabayashi and M. Kaji, J. Chem. Soc., Dalton Trans., 1984, 1984, 793–799 RSC.
- T. Yamase, Chem. Rev., 1998, 98, 307–325 CrossRef PubMed.
- T. Zhang, W. Feng, R. Lu, X. Zhang, M. Jina, T. Lia, Y. Zhao and J. Yao, Thin Solid Films, 2002, 402, 237–241 CrossRef.
- W. Feng, Y. Ding, Y. Liu and R. Lu, Mater. Chem. Phys., 2006, 98, 347–352 CrossRef.
- T. He and J. Yao, Prog. Mater. Sci., 2006, 51, 810–879 CrossRef.
- L. Aia, W. Fenga, J. Chena, Y. Liua and W. Cai, Mater. Chem. Phys., 2008, 109, 131–136 CrossRef.
- A. S. Poulos, D. Constantin, P. Davidson, M. Impéror, B. Pansu, P. Panine, L. Nicole and C. Sanchez, Langmuir, 2008, 24, 6285–6291 CrossRef PubMed.
- X. Luo and C. Yang, Phys. Chem. Chem. Phys., 2011, 13, 7892–7902 RSC.
- A. C. Colusso, M. B. Cortie, A. Dowd and A. McDonagh, Dalton Trans., 2017, 46, 11053–11062 RSC.
- A. Bouly, G. Cooper and L. Cronin, Polyoxometalate Chemistry, World Scientific Publishing Co Pte Ltd, Glasgow, 2013 Search PubMed.
- T. Zhang, J. Brown, R. Oakley and C. Faul, Curr. Opin. Colloid Interface Sci., 2009, 14, 62–70 CrossRef.
- D. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105–121 RSC.
- P. Yin, D. Li and T. Liu, Chem. Soc. Rev., 2012, 41, 7368–7383 RSC.
- A. Nisar and X. Wang, Dalton Trans., 2012, 41, 9832–9845 RSC.
- Y. Yan and L. Wu, Isr. J. Chem., 2011, 51, 181–190 CrossRef.
- J. Li, J. Cluster Sci., 2001, 13, 137–163 CrossRef.
- M. Pope, Inorg. Chem., 1972, 11, 1973–1974 CrossRef.
- J. Altenau, M. Pope, R. Prados and H. So, Inorg. Chem., 1975, 14, 417–421 CrossRef.
- P. Gomez-Romero and N. Casan-Pastor, J. Phys. Chem., 1996, 100, 12448–12454 CrossRef.
- M. Jiang, E. Wang, G. Wei, L. Xu and Z. Li, J. Colloid Interface Sci., 2004, 275, 596–600 CrossRef PubMed.
- Z. Ku, S. Jin and H. Zhou, Koord. Khim., 2009, 35, 195–199 CrossRef.
- T. He and J. Yao, Prog. Mater. Sci., 2006, 51, 810–879 CrossRef.
- D. Shen, S. Li, H. Liu, W. Jiang, Q. Zhang and G. Gao, J. Mater. Chem. C, 2015, 3, 12090–12097 RSC.
- A. Poulos, D. Constantin, P. Davidson, M. Imperor, B. Pansu, P. Panine, L. Nicole and C. Sanchez, Langmuir, 2008, 24, 6285–6291 CrossRef PubMed.
- M. Nyman, D. Ingersoll, S. Singh, F. Bonhomme, T. Alam, C. Brinker and M. Rodriguez, Chem. Mater., 2005, 17, 2885–2895 CrossRef.
- M. Nyman, M. Rodriguez, T. Anderson and D. Ingersoll, Cryst. Growth Des., 2009, 9, 3590–3597 Search PubMed.
- M. Wang, G. Xu, Z. Zhang and G. Guo, Chem. Commun., 2010, 46, 361–376 RSC.
- M. J. de Oliveira, A. L. de Souza, J. Schneider and U. Rodrigues-Filho, Chem. Mater., 2011, 23, 953–963 CrossRef.
- A. Stein, M. Fendorf, T. Jarvie, K. Mueller, A. Benesi and T. Mallouk, Chem. Mater., 1995, 7, 304–313 CrossRef.
- L. Baker and D. Glick, Chem. Rev., 1998, 98, 3–49 CrossRef PubMed.
- A. Karuppasamy and A. Subrahmanyam, J. Appl. Phys., 2007, 101, 113522 CrossRef.
- J. Chen, Y. Liu, D. Xiong, W. Feng and W. Cai, Thin Solid Films, 2008, 516, 2864–2868 CrossRef.
- C. Bechinger, G. Oefinger, S. Herminghaus and P. Leiderer, J. Appl. Phys., 1993, 74, 4527–4533 CrossRef.
- S. Santucci, C. Cantalini, M. Crivellari, L. Lozzi, L. Ottaviano and M. Passacantando, J. Vac. Sci. Technol., A, 2000, 18, 1077–1082 Search PubMed.
- F. Xie, L. Gong, X. Liu, Y. Tao, W. Zhang, S. Chen, H. Meng and J. Chen, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 112–118 CrossRef.
- R. Dixon, J. Williams, D. Morris, J. Rebane, F. Jones, R. Egdell and S. Downes, Surf. Sci., 1998, 399, 199–211 CrossRef.
- G. Leftheriotis, S. Papaefthimiou, P. Yianoulis and A. Siokou, Thin Solid Films, 2001, 384, 298–306 CrossRef.
- X. Yu, Microporous Mesoporous Mater., 2007, 98, 70–79 CrossRef.
- R. Blundell and P. Licence, Phys. Chem. Chem. Phys., 2014, 16, 15278–15288 RSC.
- G. Beamson and D. Briggs, High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database, Wiley, New York, 1st edn, 1992 Search PubMed.
- X. Luo and C. Yang, Phys. Chem. Phys. Chem., 2011, 13, 7892–7902 RSC.
- J. B. Lambert, Introduction to Organic Spectroscopy, Macmillan, New York, 1987 Search PubMed.
- X. Luo and C. Yang, Phys. Chem. Chem. Phys., 2011, 13, 7892–7902 RSC.
- H. Guan, Y. Tong and X. Cheng, Polym. Bull., 2007, 59, 577–585 CrossRef.
- T. Yamase, N. Takabayashi and M. Kaji, J. Chem. Soc., Dalton Trans., 1984, 1984, 793–799 RSC.
- A. Khenkin and R. Neumann, Angew. Chem., Int. Ed., 2000, 39, 4088–4090 CrossRef PubMed.
- A. Khenkin, L. Weiner, Y. Wang and R. Neumann, J. Am. Chem. Soc., 2001, 123, 8531–8542 CrossRef PubMed.
- T. Yamase, Catal. Surv. Asia, 2003, 7, 203–217 CrossRef.
- Y. Huang, Q. Pan, X. Dong and Z. Cheng, Mater. Chem. Phys., 2006, 97, 431–436 CrossRef.
- W. Feng, T. Zhang, Y. Liu, R. Lu, T. Li, Y. Zhao and J. Yao, J. Mater. Res., 2002, 17, 133–136 CrossRef.
- T. Zhang, W. Feng, Y. Fu, R. Lu, C. Bao, X. Zhang, B. Zhao, C. Sun, T. Li, Y. Zhaoa and J. Yao, J. Mater. Chem., 2002, 12, 1453–1458 RSC.
- P. Mialane, G. Zhang, I. Mbomekalle, P. Yu, J. Compain, A. Dolbecq, J. Marrot, F. Secheresse, B. Keita and L. Nadjo, Chem.–Eur. J., 2010, 16, 5572–5576 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TEM image of the studied compound, Raman, FTIR and GC-MS spectra of the compound as prepared and photo-coloured, comparison between the effects of irradiation types on lamellar spacing, determination of the compound's coefficient of thermal expansion, trend depicting growth of photo-oxidized groups against exposure time and FTIR of pristine and UV-irradiated CTA-Br. See DOI: 10.1039/c8ra03731k |
| This journal is © The Royal Society of Chemistry 2018 |