
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced catalytic activity and thermal stability of lipase bound to oxide nanosheets
Akane Yamadaa,
Kai Kamada *a,
Taro Uedaa,
Takeo Hyodoa,
Yasuhiro Shimizua and
Nobuaki Sohb
*a,
Taro Uedaa,
Takeo Hyodoa,
Yasuhiro Shimizua and
Nobuaki Sohb
aDepartment of Chemistry and Materials Engineering, Graduate School of Engineering, Nagasaki University, Nagasaki 852-8521, Japan. E-mail: kkamada@nagasaki-u.ac.jp
bFaculty of Agriculture, Saga University, Saga 840-8502, Japan
First published on 4th June 2018
Abstract
The present study reports the effects of binding of lipase, which is an inexpensive digestive enzyme (candida antarctica lipase) that catalyzes the hydrolysis reaction and is frequently utilized for artificial synthesis of a variety of organic molecules, to titanate nanosheets (TNSs) on their biocatalytic activities and stabilities under several lipase concentrations. TNSs were prepared through a hydrolysis reaction of titanium tetraisopropoxide (TTIP) with tetrabutylammonium hydroxide (TBAOH), resulting in formation of a colorless and transparent colloidal solution including TNSs with nanometric dimensions (hydrodynamic diameter: ca. 5.6 nm). TNSs were bound to lipase molecules through electrostatic interaction in an aqueous phase at an appropriate pH, forming inorganic-bio nanohybrids (lipase–TNSs). The enzymatic reaction rate for hydrolysis of p-nitrophenyl acetate (pNPA) catalyzed by the lipase–TNSs, especially in diluted lipase concentrations, was significantly improved more than 8 times as compared with free lipase. On the other hand, it was confirmed that heat tolerance of lipase was also improved by binding to TNSs. These results suggest that the novel lipase–TNSs proposed here have combined enhancements of the catalytic activity and the anti-denaturation stability of lipase.
Introduction
As is well-known, enzymes are protein-based biocatalysts that have high substrate specificity and reaction selectivity. Additionally, they work even under mild conditions, such as at atmospheric pressure and moderate temperature, different from inorganic catalysts.1 Therefore, various catalytic syntheses in chemical industries employ many kinds of enzymes. Among numerous types of enzymes, lipases that promote hydrolytic decomposition of lipids in living bodies have been practically applied to the preparation of enantiopure compounds from racemic pairs, prochiral or meso compounds and diastereomeric mixtures via ester hydrolysis, ester formation and ester transformation in the state-of-the-art organic syntheses.2–6 In addition, many researchers believe that these production routes contribute to development of clean synthetic chemistry strongly desired in modern pharmaceutical and chemical industries.Even though enzymes consisting of proteins are environmentally friendly catalysts, extraction and purification of enzymes from organisms has high costs compared to the production of inorganic catalysts. Moreover, enzymes show relatively low stability especially at high temperatures or nonphysiological (acidic or basic) conditions. These drawbacks have significantly hampered their practical application. To overcome them, in the past few decades, enzymes have been frequently immobilized on solid surfaces of physicochemically stable oxides or polymers (immobilized enzymes) to use them in chemical syntheses under non-biological conditions,7,8 for example, asymmetric syntheses9 and enzyme-based electrochemical sensors.10–12 The immobilization improves not only enzymatic stability but also recyclability and reusability, because fixation of tiny enzyme molecules to relative large supports can facilitate recovery from a reaction system by means of centrifugation, filtering and magnetic separation. Xie and co-workers have reported that lipases immobilized on iron based solid particles can be recycled by magnetic separation.13–16 However, immobilized enzymes generally have lower catalytic activity compared with natives in exchange for enhanced stability. This is caused by restriction of movement of enzyme molecules in a solution, reducing the possibility of colliding with substrates.
Among various categories of solid supports, inorganic nanosheets with thickness less than 1 nm that are generally formed by exfoliation of layered compounds in an aqueous solution may be one of the potential candidates as a support because of their high chemical stability, dispersibility and huge specific surface area.17,18 Kumar et al. firstly reported that some oxidoreductases were bound to zirconium phosphate (α-ZrP) nanosheets, and the obtained enzyme–α-ZrP retained a certain catalytic activity even at elevated temperatures up to 90 °C.19 Recently, we have reported enhanced enzymatic activity of horseradish peroxidase (HRP) interacting with nanometric titanate nanosheets (TNSs) due to peptization of agglomerated HRP molecules.20 Although these studies proposed a novel approach of inorganic nanosheets as supports, influence of TNSs on enzymatic activity was investigated mainly for oxidoreductases including HPR. Oxidoreductases have only limited catalytic synthetic utility from the practical point of view. In the present study, hence, the kind of enzyme used in the novel enzyme-nanosheets is extended to an aforementioned lipase, which is cheap and useful for a number of catalytic reactions both in an aqueous or non-aqueous phase, meaning higher utility than oxidoreductases in the field of synthetic chemistry. As a result of binding to TNSs, we demonstrated highly improved catalytic activity and heat tolerance of lipase at diluted concentrations. This protocol would provide novel and desirable synthetic approach in the field of immobilized enzymes. Moreover, the fact that catalytic activity of enzymes increased at diluted lipase concentrations, indicates that expensive enzymes extracted from living bodies can be effectively utilized for practical application such as chemical and food industries.
Experimental
Synthesis and characterization of colloidal solutions of TNSs
A colloidal solution of TNSs was synthesized by a hydrolysis reaction of pure titanium tetraisopropoxide (TTIP) liquid (99.999%) with a tetrabutylammonium (TBA+) hydroxide solution,21 where a molar ratio of TBA+ to Ti was set to unity. After the hydrolysis at room temperature, the basic mixture including white precipitate was aged at 60 °C for 2 h with vigorous shaking, forming a colourless and transparent colloidal solution of TNSs. The solution was dialyzed using centrifugal filter units (molecular weight cut off (MWCO): 3 kDa) to remove by-products such as isopropanol and excess TBA+ ions. Deionized water was added to the concentrated colloidal solution and then the dialysis was carried out again. The procedure was repeated to obtain a weakly basic colloidal solution of TNSs (pH = 9). The particle size was evaluated by dynamic light scattering (DLS). The crystal structure of TNSs was analyzed by X-ray diffraction (XRD) with CuKα radiation (λ = 0.15418 nm) and Raman scattering after drying the colloidal solution on a grass plate. Fourier-transformed infrared (FTIR) spectra of free lipase and lipase–TNSs (prepared by a protocol described below) powders were also measured using a conventional KBr matrix technique.Binding study between lipase and TNSs
Prior to investigation of a possible binding amount of lipase to TNSs at various pHs, lipase was modified with fluorescein 5-isothiocyanate (FITC, C21H11NO5S). Since FITC has an isothiocyanate group (–NCS), amino terminal residues of lipase are bound to the NCS group at moderate temperature and a neutral pH value, resulting in formation of covalent bonds of (FITC)–NH–CS–NH–(lipase). The modification with FITC allows us to estimate concentration of lipase in a solution with fluorescence measurement. Lipase (Candida antarctica lipase, MW: 64 kDa) dissolved in a 100 mM tris–HCl buffer solution (pH = 8, mlipase = 1.0 mg mL−1) was mixed with an equal volume of 0.1 mM FITC/100 mM tris–HCl buffer solution (pH = 8). The mixed solution was retained for 2 h at 40 °C to fabricate lipase modified with FITC (lipase–FITC), then the reactant solution was dialyzed to remove excess FITC. Pure water was added to the concentrated lipase–FITC solution and then the dialysis was conducted again. This procedure was repeated several times to eliminate unreacted FITC completely. The resultant lipase–FITC solution was diluted with 20 mM buffer solutions having several pH values (4 (acetate), 5 (citrate), 7, 8 and 9 (tris–HCl)). Final concentration of lipase was adjusted to be mlipase = 1.0 mg mL−1 by controlling the additive volume of these buffer solutions.In order to bind lipase to TNSs, the lipase–FITC solution (0.3 mL) was mixed with a TNSs colloidal solution ([Ti] = 7.3 mM, 0.3 mL) followed by vigorously shaking for 30 min at room temperature. Then, the mixture was centrifuged to settle down the lipase–FITC bound to TNSs. To estimate unbound lipase–FITC concentration, the supernatant was diluted with a 100 mM tris–HCl buffer solution (pH = 8) exactly 3 times in volume and then fluorescence intensity originated from FITC was measured with a microplate reader (excitation: 460 nm, emission: 530 nm). Finally, a binding amount of lipase to TNSs was determined by subtracting that of unbound lipase–FITC from the total.
Binding effects of TNSs on enzymatic activity of lipase
We investigated catalytic activity of lipase at pH = 4 or 7 under the presence of lipase–TNSs. The TNSs colloidal solution ([Ti] = 7.3 mM, 0.75 mL, i.e., the total molar number of Ti: mTi = 5.5 μmol) was added to a 100 mM buffer solutions dissolving lipases with several concentrations (mlipase = 0.05 to 1.0 mg mL−1, 0.75 mL) in a transparent polystyrene cuvette. Then, the mixtures were vigorous shaken for 30 min at room temperature. Enzymatic activity of lipase was measured using a hydrolysis of p-nitrophenyl acetate (pNPA) as a substrate in an aqueous solution (Scheme 1). As a result of the enzymatic reaction, pale-yellowish p-nitrophenol with an absorbance maximum at λ = 400 nm (molar absorption coefficient: ε = 14.2 cm mM−1) is produced. Enzymatic reaction rate was calculated from an initial linear increasing rate of absorbance at λ = 400 nm and 310 K under vigorous stirring, immediately after the addition of 0.5 mL of a pNPA aqueous solution (5.5 mM) to the cuvette. Enzymatic activity of free lipase without TNSs, was also evaluated as a control experiment using the identical method except for using pure water in place of the TNSs colloidal solution.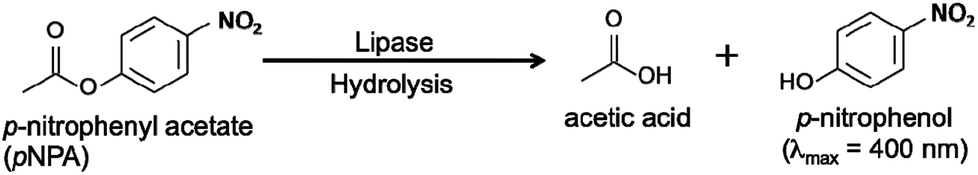 | ||
| Scheme 1 Hydrolysis reaction of pNPA in the presence of lipase in an aqueous phase. | ||
Evaluation of heat tolerance of lipase–TNS
In addition to the effect of the TNSs-binding on catalytic activity of lipase at moderate conditions, thermal stability of the lipase–TNSs was also studied to demonstrate usefulness of interaction with TNSs. The TNSs colloidal solution ([Ti] = 7.3 mM, 0.75 mL) was mixed with a lipase solution in 20 mM tris–HCl buffer solution (pH = 7, mlipase = 1.0 mg mL−1, 0.75 mL) in a microtube, followed by gentle shaking for 30 min at room temperature. The lipase–TNSs solution was heated at 90 °C for 20 min, and then cooled down to room temperature. A part of the heat-treated solution (1.4 mL) was injected into a transparent cuvette. Then, enzymatic activity of the lipase–TNS was evaluated for the hydrolysis of pNPA using the same procedure as described in the former paragraph. Needless to say, thermal stability of free lipase was also studied with the identical manner.Results and discussion
The hydrolysis of titanium tetraisopropoxide (TTIP) with a tetrabutylammonium (TBA+) hydroxide solution followed by aging at 60 °C for 2 h brought about formation of a colourless and transparent solution. This solution clearly scattered a beam of laser light, indicating existence of tiny solid particles. The colloidal solution could be stored without any precipitation over a half year. The particle size distribution curve of the colloidal solution measured by a dynamic light scattering method was given in Fig. 1(a). The curve consists of a single peak, indicating the solid particles have mono-dispersed size distribution. The mean hydrodynamic size was calculated to be ca. 5.6 nm, which is comparable to that of lipase (6 nm, estimated diameter from the MW). Considering that the produced solid particles could be anisotropic crystals (thin nanosheets) and continue to do Brownian motion in the solution, the calculated size would be a planar size of nanosheets.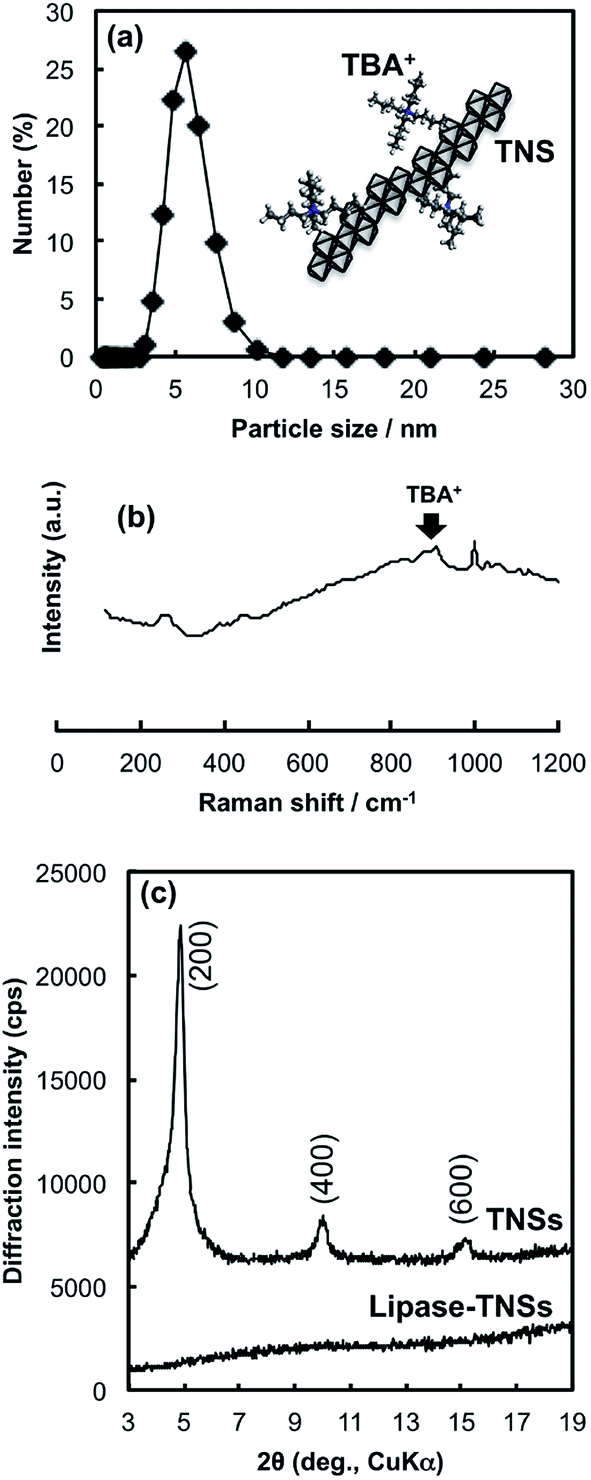 | ||
| Fig. 1 (a) A particle size distribution curve of a TNSs colloidal solution ([Ti] = 7.3 mM). The inset in (a) illustrates a schematic model of TNSs dispersed in a colloidal solution. (b) A Raman spectrum and (c) an XRD pattern of a TNSs colloidal solution after drying on a glass plate. An XRD pattern of dried lipase–TNSs prepared at pH = 4 is also displayed in (c). | ||
X-ray diffraction (XRD) and Raman scattering of the colloidal solution after drying on a glass plate were employed to reveal detailed crystal structure of the solid nanoparticles in the colloidal solution. According to Raman spectrum of the dried sample (Fig. 1(b)), the colloidal particles have crystal structure akin to layered tetratitanate (Ti4O92−), which was consistent with the literature reported by the other group.21 This fact demonstrates that the hydrolysis reaction of TTIP with TBAOH induced production of TNSs. Fig. 1(c) contains the XRD pattern of the dried TNSs. The pattern consists of sharp peaks indexed to (n00) planes of tetratitanate that correspond to basal planes of tetratitanate. During the drying process, TNSs possessing a negative charge seem to be stacked to form periodical layered structure with interstratifying TBA+ cations for charge compensation. Therefore, the d-spacing calculated from the peak position of the (200) plane allows us to approximate interlayer distance between TNSs (thickness of Ti4O92− monolayer: 0.75 nm). As a result, the distance was estimated to be 0.96 nm that is slightly larger than an ionic size of TBA+ (0.9 nm) due to accommodation of a certain amount of water molecules into the interlayer space. These results imply that TNSs in the colloidal solution exist as single layers attaching TBA+ as counter ions as illustrated in the inset of Fig. 1(a).
Binding stoichiometry between TNSs and lipase was studied in buffer solutions with several pHs. Identical volumes of the TNSs colloidal solution ([Ti] = 7.3 mM) and the lipase solution (mlipase = 1.0 mg mL−1) buffered at various pH values (pH = 4–9) were mixed together, then concentration of free lipase after eliminating the lipase–TNSs nanohybrids by centrifugation. Fig. 2 indicates that pH dependence of a binding amount of lipase on TNSs. The figure reveals the remarkable pH-dependent binding behavior between TNSs and lipase. That is, lipases were barely bound to TNSs more than pH = 5, while a relatively large amount was bound to TNSs at pH = 4.
 | ||
| Fig. 2 Dependence of pH on binding amounts of lipases to a constant amount of TNS (mTi = 5.5 μmol) at various pH values. | ||
Generally, the production of lipase–TNSs should take place through physical interaction.22 According to pH-controlled zeta potential measurements, isoelectric points were located at pH = ca. 2.0 and 4.3 for TNSs and lipase, respectively. Consequently, among the pH values investigated, both components have opposite surface charges only at pH = 4, in other words, positive lipases attract negative TNSs. Hence, the binding amount of lipase dramatically increased at pH = 4. Hence, the binding of lipases to TNSs could be induced through electrostatic interaction at pH = 4. In addition to the electrostatic binding stated above, hydrophobic interaction might somewhat contribute to the binding independent of pH, because lipase has a relatively hydrophobic surface and a TBA+ ion existing on TNSs has hydrophobic butyl groups. Actually, a small amount of lipase was bound to TNSs above pH = 4 as shown in Fig. 2. Furthermore, the lipase–TNSs prepared at pH = 4 did not release lipases even after neutralization thanks to the hydrophobic interaction, suggesting such hydrophobic interaction would inhibit dissociation of both components. An XRD pattern of the lipase–TNSs collected from the mixed solution at pH = 4 by centrifugation was displayed in Fig. 1(c). No discernible diffraction line was observed different from TNSs, indicating that tiny TNS crystals and lipase molecules were irregularly aggregated. Since the vibration bands of amide group of proteins are sensitive to microstructural environment,23 the FTIR spectra of native lipases and the lipase–TNSs were collected to follow a conformational change of lipase during the binding. There was no significant change both in the amide I (1656 cm−1) and the amide II (1541 cm−1) bands due to the binding, suggesting that the hybridization process took place without any deformation of the secondary structure of lipase.
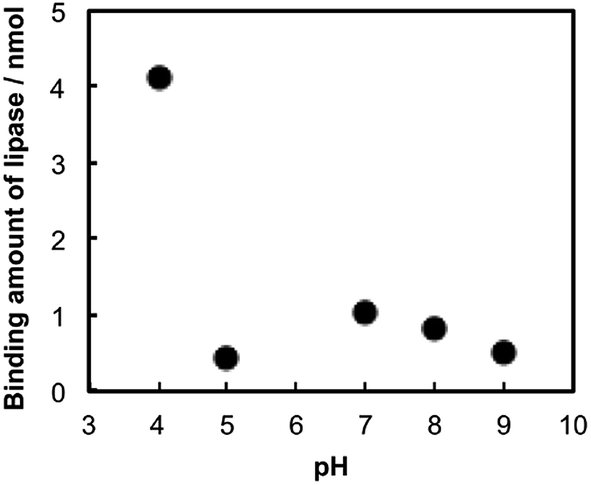
The influence of the binding to TNSs on an enzymatic reaction rate of lipase was investigated using a hydrolysis reaction of pNPA (Scheme 1), where the measurements were performed for several concentrations of lipase (mlipase = 0.02–0.38 mg mL−1) under a constant amount of TNSs at pH = 4 and 310 K. The obtained kinetic data (reaction velocity V (free lipase and lipase–TNSs) and relative V: calculated from Vlipase–TNSs divided by Vfree lipase) are summarized in Table 1. The relative V was plotted as a function of lipase concentration as shown in Fig. 3, where the dashed horizontal line indicates a position where the relative V becomes unity, meaning that enzymatic activity of free lipase and lipase–TNS are identical each other. At a high lipase concentration (mlipase = 0.38 mg mL−1), the coexistence (binding) of TNSs did not affect the enzymatic reaction velocity. At such a high concentration, the effect of TNSs might be ignored since most lipases did not interact with TNSs. On the contrary, it was found that diluting of the lipase concentration raised the enzymatic activity of lipase and the activities attained 8 times lager than those of free lipase at mlipase = 0.1 mg mL−1 or less. This fact implies that the binding to TNSs significantly increases the enzymatic activity of lipase especially at low concentrations. We have reported that several kinds of proteins including enzymes could be activated in the presence of TNSs at relative low concentrations, because peptization of aggregated proteins were caused by interacting with TNSs, in other words, an increase in apparent active centers of proteins.20,24 In the case of lipase, the analogous effect of TNSs appears to induce the activity improvement as depicted in Fig. 3. Since lipase is covered with many liposoluble amino acid residues, solubility is not so high in an aqueous phase. Combining with TBA+ existing on TNSs through the electrostatic and the hydrophobic interaction would enlarge the solubility of lipase because of hydrophilic character of TNSs, and hence the apparent enzymatic activity could be improved.
| mlipase/mg mL−1 | V/nM s−1 | Relative V | |
|---|---|---|---|
| Vfree lipase | Vlipase–TNSs | ||
| 0.38 | 3.68 | 4.68 | 1.27 |
| 0.19 | 2.46 | 8.67 | 3.52 |
| 0.09 | 2.03 | 16.50 | 8.13 |
| 0.04 | 1.77 | 13.70 | 7.74 |
| 0.02 | 1.76 | 14.10 | 8.01 |
 | ||
| Fig. 3 Relative enzymatic reaction velocity (V) of lipase–TNSs for pNPA hydrolysis as compared with unbound (free) lipase at pH = 4 and 310 K, where final concentrations of Ti and pNPA were fixed to 2.8 and 1.4 mM, respectively. | ||
Sustainable enzymatic activity under various conditions is a matter of no little interest for industrial uses of immobilized enzymes. Therefore, we evaluated thermal stability of lipase–TNSs to demonstrate their usefulness. Fig. 4 displays comparison of activity before and after heat treatment of free lipase and lipase–TNSs at 90 °C for 20 min. Unfortunately, regardless of non-binding and binding to TNSs, lipase was completely denaturated at pH = 4 after the heat treatment, so the evaluation was carried out at pH = 7. As shown in Fig. 2, a small amount of lipase could be attached to TNSs even at pH = 7. Before the heat treatment, the enzymatic reaction of lipase–TNSs proceeded more rapid than free lipase as similar to Fig. 3. As a result of the heat treatment, the reaction velocity of free lipase was significantly reduced to a level less than half (ca. 49%). Even though the treatment was achieved at fairly high temperature, the lipase–TNSs retained high activity more than three quarters of initial one (ca. 76%). These findings prove excellent thermal stability of fragile lipases bound to TNSs. According to the previous literatures,25,26 immobilized enzymes harvest enhanced thermal durability corresponded to an equilibrium binding constant between an enzyme and a stable support. Even in this study, lipases seem to be stabilized through contacting with TNSs as physicochemically stable supports. To our best knowledge, the present study firstly validates the improvement of catalytic activity and thermal stability of lipase simultaneously. The lipase–TNSs can be easily collected by centrifugation or filtration after a catalytic reaction different from free lipases, suggesting their reusability as similar to conventional immobilized enzymes.
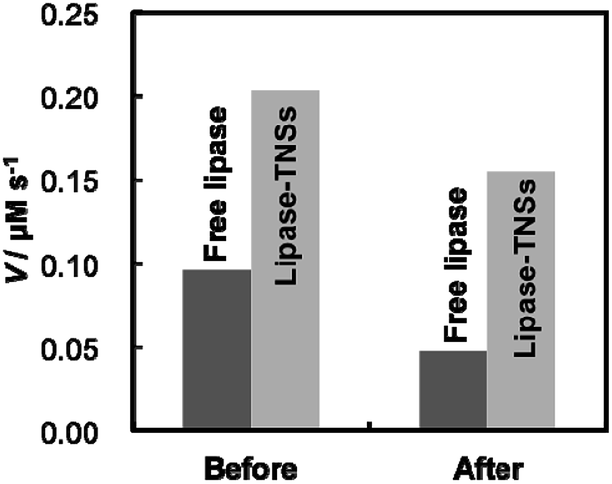 | ||
| Fig. 4 Changes in enzymatic reaction velocity (V) of free lipase and lipase–TNSs before and after heat treatment at 90 °C for 20 min. Both lipases were dispersed in a 20 mM tris–HCl buffer solution at pH = 7, then the heat treatment was carried out. Immediately after cooling down of the solutions to moderate temperature, enzymatic activity for pNPA hydrolysis was evaluated with the protocol in the same manner as Fig. 3. | ||
Lipases as digestive enzymes have high affinity to lipids like fatty acids dissolving in nonpolar solvents easily, therefore, the enzymes have been frequently applied to a number of catalytic syntheses such as ester hydrolysis and ester formation in organic solvents as stated already. Subsequently, well-dispersibility of lipases in organic solvents is another important feature from the practical point of view.27 We have already succeeded in production of inorganic nanosheets being dispersible in several solvents including hexane, formamide, etc., by using tetraalkylammonium (CnH2n+1)N+ hydroxides with longer alkyl chain lengths (n = 5 and 6) instead of TBA+ (n = 4). Hence, we will devote to evaluate catalytic activity of lipases bound to the nanosheets in these organic solvents in the near future.
Conclusions
The present study reported synthesis and characterization of novel lipase–immobilized titanate nanosheets (TNSs). The hydrolysis reaction of titanium tetraisopropoxide with tetrabutylammonium hydroxide resulted in formation of the nanosized TNSs colloidal solution having comparable size to lipase molecules. The simple mixing of TNSs and lipase caused binding of them via electrostatic and hydrophobic interaction. It was confirmed that the existence of TNSs provided more prominent catalytic activity of lipases than before the binding. Especially at diluted lipase solutions, the enzymatic activity (reaction velocity) of lipase–TNSs reached about 8 times faster than those of natives. The observed influence of TNSs was considered as enhanced dispersion stability and hydrophilicity of lipase with a hydrophobic surface. In addition, the thermal stability of lipase was also improved by the existence of TNSs. Those results indicate that the proposed lipase–TNSs may offer versatility for several catalytic syntheses under appease environmental constraints. We trust that the protocol reported here, that is, the binding of lipases to TNSs, can be extended to a variety of enzymes to be stabilized, and enables enzymatic reactions at various cruel conditions. Therefore, hybridization with TNSs will be applied to other enzymes and their activity evaluation will be attempted in our future study.Conflicts of interest
There are no conflicts to declare.Notes and references
- K. M. Koeller and C. H. Wong, Nature, 2001, 409, 232 CrossRef PubMed.
- E. Forró, R. Megyesi, T. Paál and F. Fülöp, Tetrahedron: Asymmetry, 2016, 27, 1213 CrossRef.
- X. Zhou, D. Zheng, B. Cui, W. Han and Y. Chen, Tetrahedron, 2015, 71, 4738 CrossRef.
- P. Hara, M. C. Turcu, R. Sundell, M. Tosa, C. Paizs, F. D. Irimie and L. T. Kanerva, Tetrahedron: Asymmetry, 2013, 24, 142 CrossRef.
- R. Lihammar, R. Millet and J. E. Bäckvall, J. Org. Chem., 2013, 78, 12114 CrossRef PubMed.
- J. Štambaský, A. V. Malkov and P. Kočovský, J. Org. Chem., 2008, 73, 9148 CrossRef PubMed.
- Y. Kuwahara, T. Yamanishi, T. Kamegawa, K. Mori and H. Yamashita, ChemCatChem, 2013, 5, 2527 CrossRef.
- Y. Kuwahara, T. Yamanishi, T. Kamegawa, K. Mori, M. Che and H. Yamashita, Chem. Commun., 2012, 48, 2882 RSC.
- P. Borowiecki, D. Paprocki, A. Dudzik and J. Plenkiewicz, J. Org. Chem., 2016, 81, 380 CrossRef PubMed.
- A. Pattammattel, I. K. Deshapriya, R. Chowdhury and C. V. Kumar, Langmuir, 2013, 29, 2971 CrossRef PubMed.
- C. V. Kumar and G. L. McLendon, Chem. Mater., 1997, 9, 863 CrossRef.
- R. Chowdhury, B. Stromer, B. Pokharel and C. V. Kumar, Langmuir, 2012, 28, 11881 CrossRef PubMed.
- W. Xie and X. Zang, Food Chem., 2018, 257, 15 CrossRef PubMed.
- W. Xie and X. Zang, Food Chem., 2017, 227, 397 CrossRef PubMed.
- W. Xie and M. Huang, Energy Convers. Manage., 2018, 159, 42 CrossRef.
- W. Xie and J. Wang, Energy Fuels, 2014, 28, 2624 CrossRef.
- L. Shi, Q. Gao, Y. Wu, Z. Chen and A. Liu, Biosens. Bioelectron., 2009, 25, 948 CrossRef PubMed.
- A. K. Sarkar, A. Saha, L. Midya, C. Banerjee, N. Mandre, A. B. Panda and S. Pal, ACS Sustainable Chem. Eng., 2017, 5, 1881 CrossRef.
- C. V. Kumar and A. Chaudhari, Chem. Commun., 1997, 2382 Search PubMed.
- K. Kamada, A. Yamada and N. Soh, RSC Adv., 2015, 5, 85511 RSC.
- T. Ohya, A. Nakayama, T. Ban, Y. Ohya and Y. Takahashi, Chem. Mater., 2002, 14, 3082 CrossRef.
- G. A. M. Hutton, B. Reuillard, C. M. Martindale, C. A. Caputo, C. W. Lockwood, J. N. Butt and E. Reisner, J. Am. Chem. Soc., 2016, 138, 16722 CrossRef PubMed.
- Q. Wang, Q. Gao and J. Shi, Langmuir, 2004, 20, 10231 CrossRef PubMed.
- K. Kamada, RSC Adv., 2014, 4, 43052 RSC.
- V. K. Mudjivarthi, A. Bhambhani and C. V. Kumar, Dalton Trans., 2007, 5483 RSC.
- P. M. Guto, C. V. Kumar and J. F. Rusling, J. Phys. Chem. B, 2007, 111, 9125 CrossRef PubMed.
- Q. Wang, Q. Gao and J. Shi, J. Am. Chem. Soc., 2004, 126, 14346 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |