DOI:
10.1039/C8RA03379J
(Paper)
RSC Adv., 2018,
8, 24740-24749
Pre-clinical pharmacokinetic-pharmacodynamic modelling and biodistribution studies of donepezil hydrochloride by a validated HPLC method†
Received
19th April 2018
, Accepted 24th June 2018
First published on 10th July 2018
Abstract
A simple, sensitive and robust HPLC–PDA assay was developed and validated for rapid determination of donepezil hydrochloride (DNP), a potent acetylcholinesterase inhibitor in rat plasma and tissues. All biological samples were prepared by the solid-phase extraction method using loratadine as an internal standard. Separation of the analytes was achieved on a Waters Nova-Pak C18 column (3.9 × 150 mm, 4 μm) using an isocratic mobile phase of acetonitrile and ammonium formate (pH 6.4; 0.01 M) (62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :38% v/v) at a flow rate of 1 mL min−1. All validation parameter results were within the acceptable range described in the guidelines for bioanalytical method validation. The method showed linearity in the concentration range of 50–5000 ng mL−1 with LOD of 20 ng mL−1 and LLOQ of 50 ng mL−1. Moreover, the advantage of this method over previously published methods is the short analysis run time of 6 min in HPLC itself, alongside its application not only for plasma samples but also in tissues, with low LLOQ. The method was successfully applied for studying the compartmental pharmacokinetics, tissue distribution and pharmacodynamics. A two-compartmental micro model was statistically fitted for the assessment of pharmacokinetic parameters. The tissue distribution studies suggest that the kidneys, lungs and liver are the primarily responsible organs for metabolism and elimination of DNP. Pharmacodynamic studies were performed by measuring acetylcholinesterase inhibitory activity of DNP, which indicated that the pharmacokinetic and pharmacodynamic data are in correlation with each other.
:38% v/v) at a flow rate of 1 mL min−1. All validation parameter results were within the acceptable range described in the guidelines for bioanalytical method validation. The method showed linearity in the concentration range of 50–5000 ng mL−1 with LOD of 20 ng mL−1 and LLOQ of 50 ng mL−1. Moreover, the advantage of this method over previously published methods is the short analysis run time of 6 min in HPLC itself, alongside its application not only for plasma samples but also in tissues, with low LLOQ. The method was successfully applied for studying the compartmental pharmacokinetics, tissue distribution and pharmacodynamics. A two-compartmental micro model was statistically fitted for the assessment of pharmacokinetic parameters. The tissue distribution studies suggest that the kidneys, lungs and liver are the primarily responsible organs for metabolism and elimination of DNP. Pharmacodynamic studies were performed by measuring acetylcholinesterase inhibitory activity of DNP, which indicated that the pharmacokinetic and pharmacodynamic data are in correlation with each other.
1. Introduction
Globally, ageing is accompanied by an increasing substantial social and financial impact owing to ageing-related diseases. Dementia is one among them, a syndrome which leads to deterioration in cognition, thinking, behavior and ability to carry out daily routine work.1 As per an estimate, there were 46.8 million people worldwide living with dementia in 2015 and this number may reach 131.5 million by 2050.2 Alzheimer's Disease (AD) is the most typical cause of dementia and it contributes to 60–70% of the reported cases. As per a recent investigation by the Alzheimer's association, every 65 s one person in the United States (US) develops AD. It is a chronic and progressive neurodegenerative disease. There are drugs approved by the US Food and Drug Administration (US FDA) to condense its symptoms, which fall under acetylcholinesterase inhibitors (AChEI) and N-methyl-D aspartate (NMDA) receptor antagonists. Tacrine was the first AChEI approved by the FDA and later it was withdrawn due to its side effects.3 Second generation AChEIs are DNP, galantamine and rivastigmine. Memantine which belongs to NMDA receptor antagonist was approved by FDA for the treatment of moderate and severe AD.4,5 All these drugs just delay the progression of the disease, dementia cannot be cured.6,7 At present the aim of the treatment is to maintain the quality of life as long as possible. DNP is effective in the treatment of cognitive impairment and memory loss in patients with AD. Moreover, due to the immense therapeutic potential and pharmacological activities of DNP, it is essential to develop a quick and accurate quantification method of DNP for conducting biostudies.
To the best of our knowledge, none of the methods have been applied either for estimation of DNP after intravenous (i.v.) administration or for compartmental pharmacokinetics, tissue distribution and pharmacodynamic studies. However, some methods have been reported in literature (Table 1), which are having more run time and require large sample volume with either a complex sample preparation procedure or use of sophisticated instruments like LC-MS/MS that hamper routine analysis for various applications. Abonassif, et al. developed a HPLC-RF method for the estimation of DNP in human plasma using a sample volume of 1000 μL with a total run time of 15 min, using protein precipitation technique (PP) by acetonitrile (ACN) as extracting solvent.8 Furukori, et al. have reported a HPLC-UV method using liquid–liquid extraction (LLE) process by n-hexane and IPA (97:3), with a run time of 15 min and sample volume 1000 μL of human plasma.9 A simultaneous method has also been developed for the estimation of DNP and memantine in plasma.10 A UPLC-MS/MS method has been reported for the estimation of DNP in human serum, using PP by ACN.11 Another study was performed using LC-ESI-MS/MS with a sample volume of 500 μL of human plasma, using solid-phase extraction (SPE) at a total run time of 10 min.12 LC-MS/MS method is highly recommended procedure, significantly enhances the sensitivity of the method; but maintenance cost of the instrument is more, requires trained personnel and also not available in every research laboratory. However, DNP has been reported to be administered at a dose ranging from 5–10 mg kg−1 in humans.13 Hence, the quantification upto 50 ng mL−1 is highly sufficient, HPLC based methods are equally helpful in estimation of DNP in pharmacokinetic samples. Moreover, the advantage of this method over previously published methods is the short analysis run time of 6 min in HPLC itself, its application not only for plasma samples but also in tissues and pharmacodynamic study.
Table 1 Examples of extraction procedures and analytical techniques for estimation of DNP in biological samplesa
| S. No |
Biological Sample |
Extraction method |
Linearity (ng mL−1) |
System |
Run time (min) |
% Recovery |
Reference |
| *NR, not reported. |
| 1 |
1000 μL human serum |
PP by acetonitrile |
5–2000 |
HPLC-RF |
15 |
NR* |
8 |
| 2 |
200 μL of rat plasma |
PP by acetonitrile |
50–2000 |
HPLC-UV |
20 |
NR* |
14 |
| 3 |
250 μL of human plasma |
PP by acetonitrile |
1–300 |
UPLC-MS/MS |
5 |
79 |
10 |
| 4 |
1000 μL human plasma |
LLE by n-hexane and IPA (97:3) |
3–90 |
HPLC-UV |
15 |
NR* |
9 |
| 5 |
100 μL of rat plasma |
LLE by n-hexane and IPA (95:5) |
5–500 |
HPLC-RF |
20 |
NR* |
15 |
| 6 |
500 μL of human plasma |
SPE by acetic acid and methanol (2:98) |
0.339–51.87 |
LC-ESI-MS/MS |
10 |
68.9 |
12 |
| 7 |
1000 mL of human serum |
SPE by 0.025 M ammonium formate (pH:2.5) and methanol (50:50) |
5–160 |
HPLC with UV detector |
15 |
55.7 |
16 |
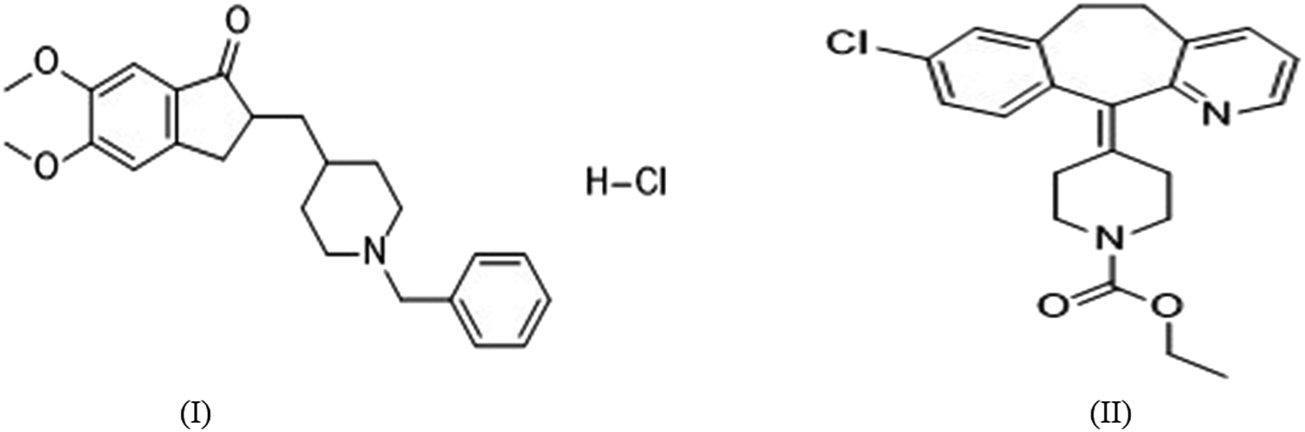
The present work describes method development and validation details for quantification of DNP in rat plasma and tissues within the range of 50–5000 ng mL−1 and elucidating the compartmental pharmacokinetics, tissue distribution and pharmacodynamic profiles. The proposed method has been developed using solid-phase extraction (SPE) method using methanol (2 mL) as an eluting solvent, and with a total run time of 6 min. Loratadine (LRD) was selected as internal standard (I.S) (Fig. 1). Complete validation was carried out including selectivity, lower limit of quantification (LLOQ), limit of detection (LOD), precision, accuracy, carry over effect, dilution integrity and stability as per guidelines on bioanalytical method validation. Stability studies were performed to determine the stability of stock solutions and in plasma samples that were exposed to different storage conditions which includes freeze-thaw, autosampler, bench top, short-term and long-term storage. Moreover, using this method we have generated a complete pharmacokinetic profile for DNP in rats. Hereby we report a new HPLC–PDA method for the estimation of DNP in rat plasma and in other biological tissues also. In addition to this, we also explored pharmacodynamic studies of DNP by estimation of acetylcholinesterase (AChE) levels.
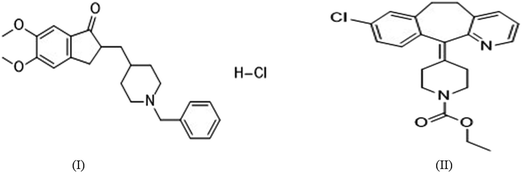 |
| | Fig. 1 Chemical structures of (I) DNP and (II) LRD. | |
2. Materials and methods
2.1 Chemicals, materials and reagents
DNP and LRD standards were kind gift samples from Vasudha Pharma Chem Limited (Hyderabad, India). ACN, methanol (HPLC grade) and sodium phosphate were procured from Merck Limited (Mumbai, India). Ammonium formate, acetylthiocholine iodide and 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) were procured from Sigma Aldrich (St. Louis, MO, USA). Solid-phase cartridges, Clearnert PEP-3 SPE was procured from Agela Technologies (Wilmington, USA). Bovine serum albumin (BSA) was purchased from Sisco Research Laboratories Pvt. Ltd. (Mumbai, India). Water was produced by millipore Milli-Q Plus water treatment system (Millipore Bedford Corp., Bedford, MA, USA).
2.2 Experimental animals
All experimental animals (Wistar rats; male; 8–10 weeks; weighing around 200–220 g), were housed in polyacrylic cages, and all procedures involving sample collection and their care were conducted in conformity with the institutional guidelines that are in compliance with Institutional Animal Ethics Committee (IAEC) of BITS-Pilani university with approved protocol (IAEC/RES/22/05). All animals were housed in a temperature (20–22 °C) and humidity (65%) controlled animal facility and maintained on a 12/12 h light/dark cycle. The temperature and relative humidity were recorded daily. Both feed and water were provided ad libitum throughout the acclimatization and study period.
2.3 Liquid chromatographic conditions
HPLC analysis were performed on Shimadzu liquid chromatograph system (Kyoto, Japan) equipped with a binary pump (LC-10AT), Photo Diode Array (PDA) detector (SPD-M20A), column oven (CTO-10AS) and auto sampler (SIL-HT, Shimadzu, Japan) was used to inject the samples. The HPLC system was equilibrated for approximately 30 min before starting of analysis and chromatography was carried out at 40 ± 0.5 °C. Eluents were monitored at a wavelength of 268 nm using a PDA detector. DNP and LRD were separated on Waters Nova-Pak C18 column (3.9 × 150 mm, 4 μm) with a mobile phase consisting of acetonitrile and ammonium formate (pH 6.4; 0.01 M) (62:38% v/v) in an isocratic mode at a flow rate of 1 mL min−1. The injection volume was 50 μL and retention time for DNP and LRD were 3.31 and 3.99 min respectively. Control of hardware, data acquisition and elaboration were performed using LC solution software version 1.24 SP1.
2.4 Preparation of stock solutions, calibration curve standards (CS) and quality control (QC) samples
Standard stock solution of DNP was prepared by dissolving accurately weighed amount in methanol to obtain a final concentration of 5 mg mL−1. Further, same volumes of DNP stock solution (5 mg mL−1) was diluted with Milli-Q water to make 400 μg mL−1 working stock solution. From 400 μg mL−1 working stock solution, working standard solutions of 1, 2, 6, 20, 40, 70 and 100 μg mL−1 were prepared by serial dilutions in Milli-Q water. The concentrations of working QC standard solutions were prepared as 1.6, 3 and 80 μg mL−1. CS and QC samples were prepared by spiking with 10 μL of corresponding working standard solutions in 190 μL of blank rat plasma. The final CS were prepared as 50, 100, 300, 1000, 2000, 3500 and 5000 ng mL−1 in plasma matrix. The QC were prepared by using QC standard solutions at four concentration levels, such as limit of quantification (LOQQC, 50 ng mL−1); low QC (LQC, 80 ng mL−1), medium QC (MQC, 1500 ng mL−1) and high QC (HQC, 4000 ng mL−1). All the stock and working solutions were stored in the refrigerator at −20 °C till analysis.
2.5 Sample preparation
Several extraction techniques were tested, such as protein precipitation with organic solvents (methanol, acetonitrile) and by liquid–liquid extraction using dichloromethane, n-hexane:IPA (97:3) and n-hexane:IPA (95:5). These procedures allow good recovery for IS, but plasma interference was observed with low recovery of analyte.
SPE method was also tested in order to reduce solvent consumption, maximize the analytes recovery, and to optimize the sample clean-up process. A 200 μL aliquot of plasma sample containing DNP was taken in 2 mL capacity of polypropylene disposable tube, followed by the addition of 20 μL of 100.0 μg mL−1 of LRD (I.S) solution (I.S solution was prepared in ACN) and was vortexed for 1 min. Then 200 μL of ammonium formate buffer was added and vortexed for 2 min on a vortex mixer. The samples were then loaded into the SPE cartridges using a semi-automated SPE system (ESI; Table S1†). The SPE eluents were collected in glass tubes and evaporated to dryness at 40 ± 0.5 °C under a gentle stream of nitrogen gas. The residue was reconstituted with 200 μL of mobile phase and vortexed for 30 s. Finally, 50 μL of sample was injected into HPLC through autoinjector for quantification.
2.6 Method validation procedures
The proposed method was validated as per guidelines US FDA and European Medical Agency guidelines of bioanalytical method validation.17,18
2.6.1 System suitability. System suitability is typically a system performance test, which was carried out by injecting the same concentration sample by six times for checking the column efficiency, reproductivity and resolution. It was performed to verify the holistic function of the chromatography system on a day-to-day basis.
2.6.2 Selectivity. The selectivity of the method was carried out to evaluate potential chromatographic interference from rat plasma matrix at retention time (RT) of the drug and IS. The rat blank plasma samples were collected randomly from selected six different Wistar rats and analyzed as per described chromatographic conditions.
2.6.3 Linearity and calibration curve. The calibration curve of an analyte (DNP) was prepared using six calibration standards in a range of 50 to 5000 ng mL−1. Six calibration curves were prepared for plasma and tissue samples by plotting peak area ratios (drug/I.S) on Y axis versus nominal plasma concentrations on X-axis. Highest calibration standard sample (ULOQ, 5000 ng mL−1) was 100 times less than the lowest limit of calibration point (LLOQ, 50 ng mL−1), so it was necessary to perform weighted linear regression on calibration data. All the different weighting factors were applied to the data obtained from least-squares regression analysis and the best weighting factors were chosen according the percentage relative error (% RE).
2.6.4 Limit of detection (LOD) and lower limit of quantitation (LLOQ). The limit of detection (LOD) is defined as the lowest concentration of analytes that can be detected and differentiated from the noise level.The lower limit of quantitation (LLOQ) is defined as the lowest concentration of analytes that can be quantified with accepted accuracy and precision recommendations (not exceeding ±20%).
LOD and LLOQ were determined by using signal to noise ratio method. First the signal to noise ratio was checked for standard samples at system suitability step. LOD and LLOQ were calculated based on eqn (1). Desired S/N values for LOD was 3 and for LLOQ was 10.
| |
 | (1) |
Generally, LLOQ is the first point in calibration curve and analyzed plasma samples by spiking with LLOQ concentration in six replicates (n = 6).
2.6.5 Precision and accuracy. The intra-day and inter-day assay precision and accuracy were determined by analyzing six replicates at three different QC levels (LQC, MQC, HQC) as described in Section 2.4 and LLOQ. The intra-day assay precision and accuracy were determined in six replicates by analyzing samples on same day, while inter-day assay precision and accuracy were determined in six replicates by analyzing each QC and LLOQ samples on five different days. The acceptance criteria for intra and inter-day precision (RSD, %) was limited to ≤15% and accuracy was required to be limited within ±15% except for LLOQ, where it should not exceed ±20% for accuracy and precision.
2.6.6 Recovery. The percentage recovery of DNP through SPE were determined by comparing the peak area of analyte spiked plasma QC samples with those of unextracted neat standards that is standard solution with equivalent concentrations.
2.6.7 Carry over effect and dilution integrity. Carry over effect was determined by injecting highest calibration standard sample (ULOQ, 5000 ng mL−1) followed by blank sample. As per guidelines, the sample dilution must be performed to ensure the integrity of plasma samples. In dilution integrity, blank plasma was spiked with 2 times higher than the ULOQ concentration (10000 ng mL−1) and was diluted 5 times and 10 times with blank plasma (n = 6) to obtain a concentration of 2000 ng mL−1 and 1000 ng mL−1. Dilutes samples were processed and analyzed with freshly spiked calibration curve in plasma. The accuracy and precision values should be within ±15%.
2.6.8 Stability studies. Stability of analytes in plasma matrix were evaluated before and after subjecting to different conditions and temperatures that could encounter during regular analysis. Stability in plasma was evaluated in terms of freeze-thaw stability, bench top stability, short-term stability and autosampler stability. All stability studies were conducted in three replicates at each concentration of the three different QC levels (LQC, MQC, and HQC). Freeze thaw stability was performed after freezing (−80 ± 10 °C for 48 h) and thawing QC samples for three consecutive cycles within 2 days. Bench top stability was analyzed at 25 ± 2 °C for 24 h, short-term stability was evaluated at −80 ± 10 °C for 15 days and long-term stability was examined at −80 ± 10 °C for 3 months were performed in rat plasma with all QC levels. Replicate injections of extracted plasma samples were analyzed after 48 h to estimate auto sampler stability.All QC samples were extracted and quantified against fresh calibration curves and fresh QC samples. The acceptance criteria of accuracy and precision for all stability samples should be within ±15%.
2.7 Pharmacokinetic study
To investigate the developed method, pharmacokinetic studies of DNP was performed on young Wistar rats weighing around 200–220 g. An i.v. administration, at a dose of 5 mg kg−1 of DNP were given to animals. After dosing, blood samples were collected in 1.5 mL capacity of polypropylene disposable tubes for each time point at 0, 0.083, 0.166, 0.5, 0.75, 1, 3, 6, 9, 12, 15, 18, 24, 30 and 36 h. Blood samples were collected from the retro-orbital plexus into heparinized micro centrifuge tubes at pre-determined time interval. Plasma samples were harvested by centrifuging the blood at 6000 rpm for 15 min and stored at −80 ± 10 °C until analysis. DNP plasma concentration-time profiles were plotted.
2.8 Pharmacokinetic modeling
Pharmacokinetic parameters for DNP were performed by using compartmental analysis. Model selection was guided by visual inspection of the observed plasma profiles and Akaike's information(AIC) criterion.19 The lowest AIC value suggests best fit model. Various pharmacokinetic parameters such as concentration maximum (Cmax), area under curve (AUC), volume of distribution in central compartment (V1), volume of distribution in peripheral compartment (V2), plasmatic clearance (Cl), intercompartmental clearance (CLD2), alpha half-life (t1/2 α), beta half-life (t1/2 β), and elimination rate constants of two compartmental model (K10, K12, K21) were estimated Phoenix WinNonlin Certara™ (Pharsight, U.S.A; version:8.0).
2.9 Tissue distribution study in rats
Wistar rats were randomly divided into 2 groups, one group as control and for the other group DNP was given i.v. at a dose of 5 mg kg−1. Tissues (brain, heart, lungs, kidney, spleen and liver) were promptly harvested at 0.166, 0.75, 2, 6 h (three rats/time point) after the administration of dose and thoroughly rinsed in ice-cold saline to remove blood and other content and blotted dry with filter paper. An accurately weighed amount of each tissue samples were homogenized in 1:2 w/v of phosphate buffer saline solution by using tissue homogenizer. After homogenization, centrifugation was done at 10000 rpm for 15 min, clear supernatant was collected and stored at −80 °C until analysis.
2.10 Pharmacodynamic studies
The levels of AChE and butyl cholinesterase are the biological markers for AD.20 Cholinesterase inhibitors (ChEIs) enhances the breakdown of AChE in synaptic cleft, thus enhancing ACh levels. Pharmacodynamic effects of DNP on AChE activity in rats was explored. The estimation of AChE activity in plasma samples were performed as per method described by Ellman et al.21 In brief, the assay sample contained 0.05 mL of plasma, 3 mL of 0.01 M sodium phosphate buffer (pH 8), 0.10 mL of acetylthiocholine iodide and 0.10 mL of DTNB (Ellman reagent). The change in absorbance was measured immediately at 412 nm spectrophotometrically. Protein content in plasma samples was measured by the method of Lowry et al.22 using BSA (1 mg mL−1) as a standard. Results were calculated using molar extinction coefficient of chromophore (1.36 × 104 M−1 cm−1) and expressed as nanomoles per milligram protein.23 Maximal AChE inhibition time of DNP was estimated.24,25
3. Results
3.1 Method development
3.1.1 Optimization of liquid chromatographic conditions. A methodological approach was followed for the method development so as to obtain analyte in less time using simplest possible chromatographic conditions for obtaining an appropriate recovery, symmetric peaks and high resolution of the drugs. For that, liquid chromatographic conditions such as choice of mobile phase, composition of mobile phase, pH of buffer, column selection, flow rate and injection volume were optimized (ESI; Table S2†). After successive optimization, acetonitrile–ammonium formate (pH: 6.4; 0.01 M) was selected as mobile phase which gave best peak shape and good peak intensities. Different columns were used to optimize better resolution, selectivity and retention time. Waters Nova-Pak C18 column (3.9 × 150 mm, 4 μm) with acetonitrile–ammonium formate (pH: 6.4; 0.01 M) mobile phase in ratio of 62:38% v/v in isocratic mode at 1 mL min−1 flow rate was found to be most suitable for DNP analysis with a total run time of 6 min. The injection volume was 50 μL and optimized retention time for DNP and LRD (I.S) were 3.31 and 3.99 min respectively at a wavelength 268 nm.
3.1.2 Optimization of plasma sample preparation. Once the chromatographic conditions were optimized, extraction recovery of analyte was optimized by selecting an appropriate extraction method. The plasma sample clean-up procedure was also an important step in the method as it is directly proportional to sensitivity and selectivity of method. Higher recovery of analyte from plasma matrix can be obtained by minimum sample preparation processing, as well as proper choice of extraction method. Various problems have been encountered during the method optimization. Different methods for extracting drug from plasma matrix have been tried such as protein precipitation (with ACN and methanol), liquid–liquid extraction (with hexane, methylene chloride, IPA) and solid-phase extraction (SPE) (ESI; Table S3†). Out of all, SPE was found to produce higher recoveries, reproductivity and cleaner chromatogram.
3.2 Method validation
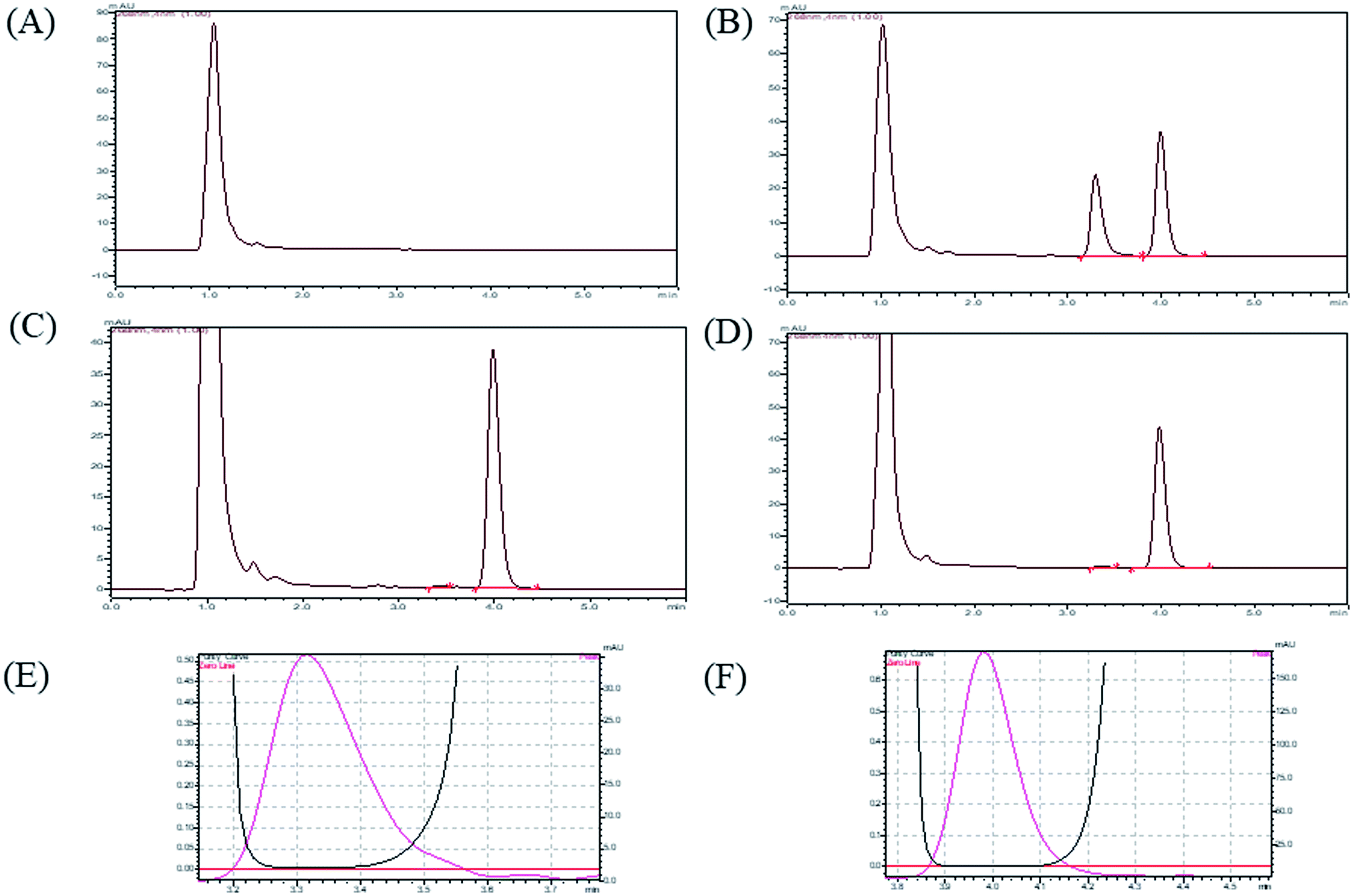
3.2.1 System suitability, selectivity and chromatographic separation. By performing the system suitability test, we conformed that system was working fine and it showed reproducibility with proper resolution. In described chromatographic condition, analytes were successfully separated with optimum retention time less than 6 min. (DNP and I.S retention times were 3.31 and 3.99). The sample clean-up procedure was so selective as there was no significant plasma matrix interference at retention time of analyte. A representative chromatogram of blank plasma, (plasma sample without analyte and IS), plasma sample with analyte and IS, LOD plasma sample (20 ng mL−1) and LLOQ plasma sample (50 ng mL−1) are shown in Fig. 2.
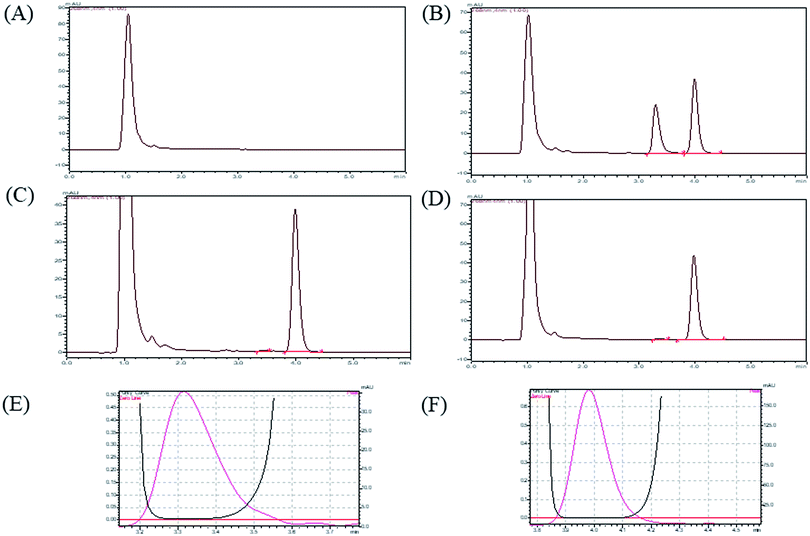 |
| | Fig. 2 HPLC chromatograms for (A) blank plasma, (B) calibration standard, (C) LOD (20 ng mL−1), (D) LLOQ (50 ng mL−1), (E) peak purity index of DNP, (F) peak purity index of I.S. | |
3.2.2 Linearity, calibration curve, LOD, LLOQ. All the calibration standard curves for plasma and tissue samples showed linearity and reproducibility over the selected concentration range of 50–5000 ng mL−1 with r2 values more than 0.9999. The r2 values, slopes and intercepts were calculated from five intra and inter day calibration curves using weighted linear regression analysis (1/var, 1/x, 1/x2, 1/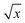 , 1/y, 1/y2, 1/
, 1/y, 1/y2, 1/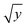 ) wherein, best weighting factors were chosen as 1/x2 as these showed minimum percentage relative error (% RE) (ESI; Table S4†). The observed mean back calculated concentrations with accuracy (% bias) and precision (% CV) are presented in Table 2. At LLOQ (n = 6) accuracy (% bias) was in the range of 3.74% with a % CV of 6.19%. LOD and LLOQ were established as 20 ng mL−1 and 50 ng mL−1 respectively.
) wherein, best weighting factors were chosen as 1/x2 as these showed minimum percentage relative error (% RE) (ESI; Table S4†). The observed mean back calculated concentrations with accuracy (% bias) and precision (% CV) are presented in Table 2. At LLOQ (n = 6) accuracy (% bias) was in the range of 3.74% with a % CV of 6.19%. LOD and LLOQ were established as 20 ng mL−1 and 50 ng mL−1 respectively.
Table 2 Precision and accuracy data of back calculated concentrations of calibration standard samples of DNP in rat plasma (n = 6)a
| Component |
Linear equation and coefficient of regression |
Nominal concentration (ng mL−1) |
Measured concentration (Mean ± SD, ng mL−1) |
Precision (% CV) |
Accuracy (% bias) |
| SD, standard deviation; CV, coefficient of variation. |
| Plasma |
y = 0.000384x − 0.0088, r2 = 0.9999 |
50 |
51.87 ± 3.21 |
6.19 |
3.74 |
| 100 |
95.30 ± 4.57 |
4.80 |
−4.70 |
| 300 |
304.26 ± 16.99 |
5.58 |
1.42 |
| 1000 |
999.58 ± 2.96 |
0.30 |
−0.04 |
| 2000 |
1983.08 ± 12.60 |
0.64 |
−0.85 |
| 3500 |
3464.12 ± 5.27 |
0.15 |
−1.03 |
| 5000 |
5042.00 ± 8.58 |
0.17 |
0.84 |
| Brain |
y = 0.0003x − 0.0058, r2 = 0.9998 |
50 |
55.26 ± 1.15 |
2.08 |
10.52 |
| 300 |
289.54 ± 15.48 |
5.35 |
−3.49 |
| 1000 |
939.40 ± 87.62 |
9.33 |
−6.06 |
| 2000 |
1950.68 ± 13.60 |
0.70 |
−2.47 |
| 3500 |
3335.07 ± 30.75 |
9.18 |
−4.71 |
| 5000 |
4872.22 ± 19.75 |
0.41 |
−2.56 |
| Heart |
y = 0.0003x − 0.0075, r2 = 0.9991 |
50 |
56.24 ± 6.54 |
11.63 |
12.48 |
| 300 |
314.36 ± 1.10 |
0.35 |
4.79 |
| 1000 |
1011.78 ± 18.88 |
1.87 |
1.18 |
| 2000 |
2147.01 ± 51.93 |
2.42 |
7.35 |
| 3500 |
3547.65 ± 4.37 |
0.12 |
1.36 |
| 5000 |
5262.01 ± 43.58 |
0.83 |
5.24 |
| Lungs |
y = 0.0003x − 0.0099, r2 = 0.9999 |
50 |
56.78 ± 4.23 |
7.45 |
13.56 |
| 300 |
316.62 ± 28.83 |
9.11 |
5.54 |
| 1000 |
1041.39 ± 93.03 |
8.93 |
4.14 |
| 2000 |
2168.05 ± 106.81 |
4.93 |
8.40 |
| 3500 |
3749.94 ± 206.25 |
5.50 |
7.14 |
| 5000 |
5372.00 ± 291.26 |
5.42 |
7.44 |
| Spleen |
y = 0.0003x + 0.0089, r2 = 0.9996 |
50 |
55.31 ± 3.32 |
6.00 |
10.62 |
| 300 |
289.15 ± 8.66 |
2.99 |
−3.62 |
| 1000 |
1049.53 ± 80.29 |
7.65 |
4.95 |
| 2000 |
2148.97 ± 146.01 |
6.79 |
7.44 |
| 3500 |
3600.13 ± 269.69 |
7.49 |
2.86 |
| 5000 |
5247.77 ± 32.88 |
0.63 |
4.96 |
| Kidney |
y = 0.0003x − 0.0008, r2 = 0.9995 |
50 |
46.42 ± 4.12 |
8.88 |
−7.16 |
| 300 |
309.21 ± 3.51 |
1.14 |
3.07 |
| 1000 |
1050.37 ± 85.23 |
8.11 |
5.04 |
| 2000 |
2243.01 ± 223.27 |
9.95 |
12.15 |
| 3500 |
3804.79 ± 163.19 |
4.29 |
8.71 |
| 5000 |
5355.65 ± 51.11 |
0.95 |
7.11 |
| Liver |
y = 0.0003x − 0.0126, r2 = 0.9999 |
50 |
52.94 ± 1.68 |
3.17 |
5.88 |
| 300 |
281.24 ± 25.88 |
9.20 |
−6.25 |
| 1000 |
935.92 ± 29.60 |
3.16 |
−6.41 |
| 2000 |
1880.22 ± 135.81 |
7.22 |
−5.99 |
| 3500 |
3240.65 ± 18.66 |
0.58 |
−7.41 |
| 5000 |
4703.54 ± 5.48 |
0.12 |
−5.93 |
3.2.3 Precision and accuracy. The intra and inter day accuracy, precision data obtained from QC samples at four different concentration levels (LLOQC, LQC, MQC, HQC) are shown in Table 3. The inter and intra-day precision values did not exceed 6.92%. The inter and intra-day % bias ranged from −3.11 to 7.08%. All the values were within the recommendation of international guidelines.
Table 3 Precision (% CV) and accuracy (% bias) of DNP in rat plasma samples at QC levels (n = 6)
| Level |
Nominal Conc. (ng mL−1) |
Inter-day |
Intra-day |
| Measured Conc. (Mean ± SD, ng mL−1) |
Precision (% CV) |
Accuracy (% bias) |
Measured Conc. (Mean ± SD, ng mL−1) |
Precision (% CV) |
Accuracy (% bias) |
| LLOQ |
50 |
53.54 ± 2.43 |
4.54 |
7.08 |
50.21 ± 3.19 |
6.35 |
0.42 |
| LQC |
80 |
79.74 ± 4.45 |
5.58 |
−0.33 |
77.51 ± 5.36 |
6.92 |
−3.11 |
| MQC |
1500 |
1543.40 ± 14.82 |
0.96 |
2.89 |
1513.29 ± 22.85 |
1.51 |
0.89 |
| HQC |
4000 |
4025.35 ± 25.97 |
0.65 |
0.63 |
3926.75 ± 65.42 |
1.67 |
−1.83 |
3.2.4 Recovery, carry over, dilution integrity. The mean recovery values from rat plasma samples are listed in Table 4. The mean absolute recovery for DNP was found to be 79.86 ± 1.55% (n = 12). No carry over effect was observed, there was absence of peaks of analytes in blank sample was injected after ULOQ. Dilution integrity was carried out by diluting 5 times and 10 times with the blank plasma (n = 6). The accuracy value after 5 times and 10 times dilution were found to be 98.60 ± 3.47 and 101.75 ± 2.81 respectively.
Table 4 Absolute recoveries (%) of DNP in rat plasma samples from QC levelsa
| Level |
Nominal concentration (ng mL−1) |
n |
Recovery (%) |
| Mean ± SD (n = 3) |
% CV |
| n, number of samples; SD, standard deviation; CV, coefficient of variation. |
| LLOQ |
50 |
3 |
80.22 ± 1.44 |
1.80 |
| LQC |
80 |
3 |
80.28 ± 1.18 |
1.47 |
| MQC |
1500 |
3 |
78.68 ± 1.62 |
2.06 |
| HQC |
4000 |
3 |
80.28 ± 1.95 |
2.43 |
| |
Mean |
12 |
79.86 ± 1.55 |
1.94 |
3.3 Pharmacokinetic study in rats
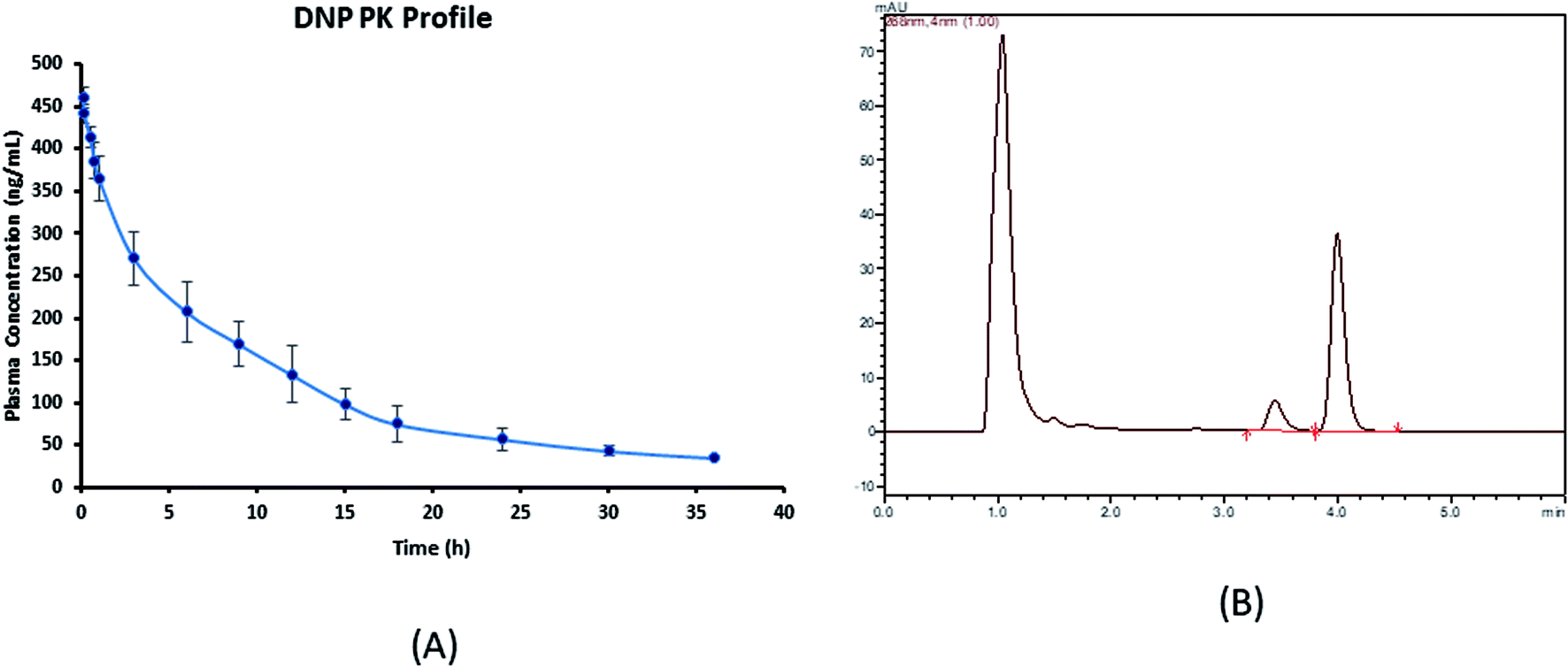
The developed assay was productively implemented for the pharmacokinetic study of DNP after the i.v. administration of DNP at a dose of 5 mg kg−1 in Wistar rats. The calibration range (50–5000 ng mL−1) of this assay method fitted to estimate all pharmacokinetic samples. The various pharmacokinetic parameters of DNP including observed maximum concentration (C0), half-life (t1/2) and area under the curve (AUC0−t) were calculated using compartmental pharmacokinetic analysis and represented in the Table 6. Mean ± SD plasma concentration versus time profile of DNP is shown in Fig. 3.
Table 6 Pharmacokinetic parameters for DNP after i.v. bolus (5 mg kg−1) administration in rats (n = 4)
| Pharmacokinetic parameters |
DNP 5 mg kg−1; i.v. (Mean ± SEM) |
| AIC of one compartment |
129.86 ± 2.61 |
| AIC of two compartment |
114.06 ± 5.36 |
| C0 (ng mL−1) |
468.02 ± 11.84 |
| AUC 0−∞ (ng h mL−1) |
4655.05 ± 352.53 |
| V1 (mL kg−1) |
10697.06 ± 274.35 |
| V2 (mL kg−1) |
4424.51 ± 816.59 |
| Cl (mL h−1 kg) |
1087.58 ± 89.00 |
| CLD2 (mL h−1 kg) |
2070.95 ± 599.09 |
| T1/2 α (h) |
1.21 ± 0.46 |
| T1/2 β (h) |
10.43 ± 0.53 |
| MRT (h) |
13.94 ± 0.95 |
| K10 (h−1) |
0.101 ± 0.0058 |
| K12 (h−1) |
0.196 ± 0.06 |
| K21 (h−1) |
0.554 ± 0.24 |
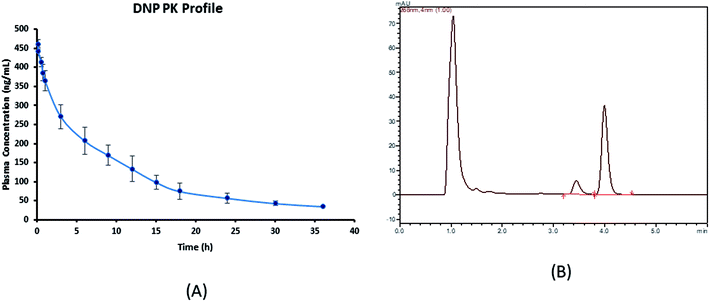 |
| | Fig. 3 (A) Plasma concentration–time profile of DNP (mean ± SD, n = 4) and (B) representative chromatogram of plasma sample obtained after 30 min of post drug administration.* *In these studies, DNP was administered by i.v. bolus at a single dose of 5 mg kg−1 each in rat (n = 4). | |
3.4 Pharmacokinetic modeling
Experimental plasma concentration-versus-time profiles were fitted to a two-compartmental micro model with i.v. bolus input and linear first order elimination from the central compartment using iterative weighted non-linear least squares regression using Phoenix WinNonlin Certara™ (Pharsight, U.S.A; version:8.0). The observed versus predicted concentrations did not differ from the line of identity, and no bias was observed (Fig. 4). DNP have shorter distribution half-life (T1/2 α) of 1.21 h, which indicates rapid distribution from systemic circulation into the extravascular tissue spaces. Moreover, smaller elimination rate constant (K10) of 0.1 h from central compartment, longer elimination half-life (T1/2 β) of 10.43 indicates behaved slow distribution and elimination characteristics.
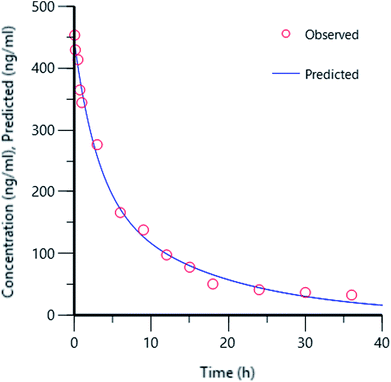 |
| | Fig. 4 Concentration plot of observed versus predicted. | |
3.5 Pharmacodynamic study
To examine the pharmacodynamic changes of DNP, the AChE inhibitory activity was determined (Fig. 5). Results have shown the correlation between pharmacokinetic profile, as the inhibitory activity was observed till 9 h thereafter, decrease in activity was noticed. The maximal effect of AChE inhibition was at 9.5 h, it clearly indicates the action of the DNP was matching with the pharmacokinetic data. Therefore, the established data in this study was reliable and highly suitable for pharmacokinetic-pharmacodynamic modeling.
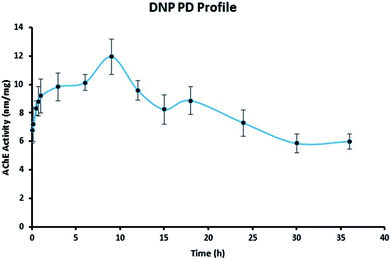 |
| | Fig. 5 Pharmacodynamic profile of DNP. | |
3.6 Tissue distribution study
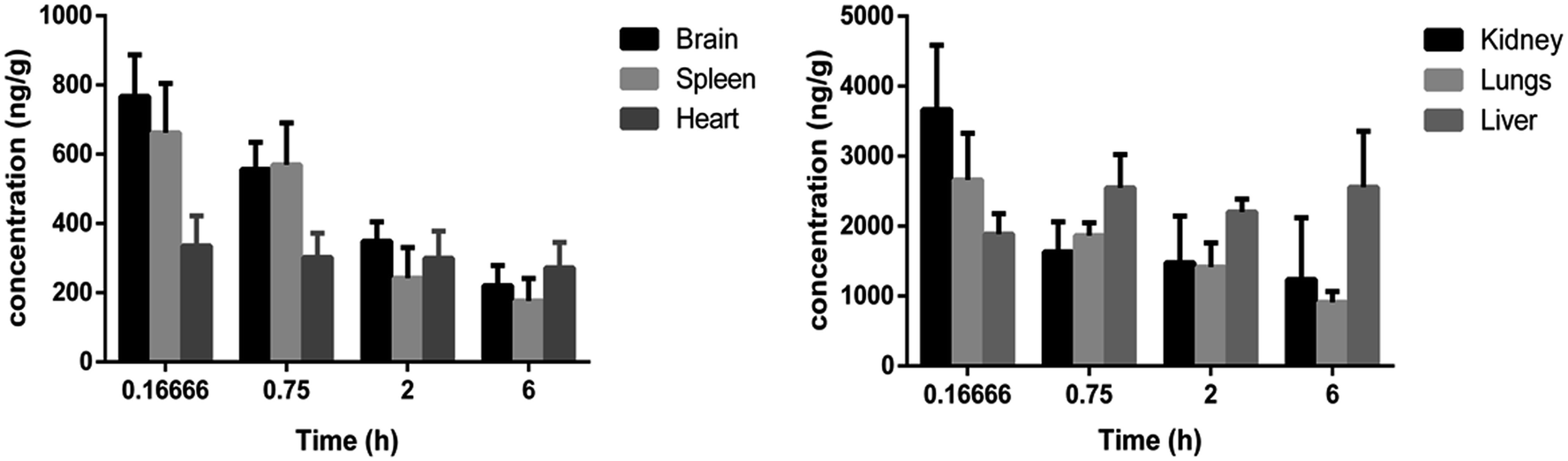
Tissue distribution study was carried out to identify the major distribution sites of DNP after i.v. administration at a dosage of 5 mg kg−1. The distribution of DNP in the various rat tissues are represented in Fig. 6 and the representative of chromatographs are shown in Fig. S1.† The results demonstrated that DNP has exhibited wide range of distribution to tissues within the time course examined. As per the data obtained, the DNP concentration in various organs showed the following order: kidney > lungs > liver > brain > spleen > heart. By comparing organ and plasma samples, the distribution level in the kidney was 7.95 folds higher than that in plasma, 5.77 folds higher in the lungs than that in plasma and 4.09 folds higher in the liver than that in plasma. This clearly suggests that kidney, lungs and liver were the organs primarily involved in metabolism of DNP, leading to deposition at these sites. Moreover, the lowest concentration was found in heart, which indicates DNP has less perfusion rate in heart. In summary, the distribution of DNP was highest in kidney and lowest in heart.
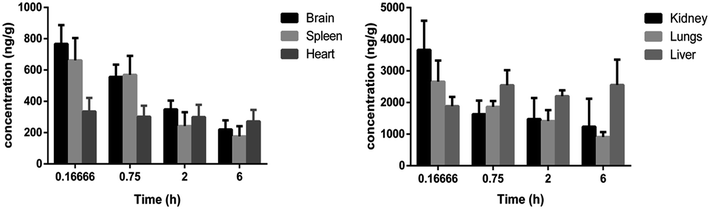 |
| | Fig. 6 Concentration (mean ± SD) levels of DNP in rat tissues at 0.1666, 0.75, 2 and 6 h.** **In these studies, DNP was administered by i.v. bolus at a single dose of 5 mg kg−1 each in rat (three/time point). | |
4. Conclusion
The aim of the present study was to develop an assay method for DNP in plasma matrix with simple extraction preparation techniques. Chromatographic separation was achieved on Waters Nova-Pak C18 column (3.9 × 150 mm, 4 μm) using isocratic mobile phase of acetonitrile and ammonium formate (pH 6.4; 0.01 M) (62:38% v/v) at flow rate of 1 mL min−1. Solid-phase extraction was selected as an optimized method for higher recovery and proper resolution. The assay recovery was ≥79 ± 1.55% with insignificant matrix effect. The calibration curves in plasma and other tissues were linear in a concentration range of 50–5000 ng mL−1 with LOD and LOQ was 20 ng mL−1 and 50 ng mL−1 for plasma samples, respectively. The advantages of the method include simple and rapid sample preparation process, freedom from matrix interference and requirement of small plasma sample volume (200 μL), can be applied within a wide concentration range (5000 ng mL−1) and a short total run time (06 min). Experimental plasma concentration-versus-time profiles were fitted to a two-compartmental micro model with i.v. bolus. The tissue distribution studies suggest, that kidney, lungs and liver are the primarily responsible organs for metabolism and elimination of DNP. Moreover, pharmacodynamic studies were conducted by estimating the inhibitory action of AChE was in correlation with pharmacokinetic profile. Our constructive studies provide valuable information to evaluate pharmacokinetic and pharmacodynamic studies for DNP.
Conflicts of interest
The authors have no conflict of interest.
Acknowledgements
Mr K. V. Krishna would like to acknowledge University Grant Commission (UGC), India for providing UGC-National fellowship. Vasudha Pharma Chem Limited (Hyderabad, India) is acknowledged for providing the gift samples of DNP and LRD.
References
- K. E. Stuart, A. E. King, C. M. Fernandez-Martos, M. J. Summers and J. C. Vickers, Environmental novelty exacerbates stress hormones and Aβ pathology in an Alzheimer's model, Sci. Rep., 2017, 7, 2764 CrossRef PubMed.
- M. Prince, A. Comas-Herrera, M. Knapp, M. Guerchet and M. Karagiannidou, World Alzheimer Report 2016 Improving healthcare for people living with dementia. Coverage, Quality and costs now and in the future, 2016, pp. 1–140, https://www.alz.co.uk/research/world-report-2016 Search PubMed.
- S. A. Eagger, R. Levy and B. J. Sahakian, Tacrine in Alzheimer's disease, Lancet, 1991, 337, 989–992 CrossRef.
- S. A. Lipton, Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond, Nat. Rev. Drug Discovery, 2006, 5, 160 CrossRef PubMed.
- B. Reisberg, R. Doody, A. Stöffler, F. Schmitt, S. Ferris and H. J. Möbius, Memantine in moderate-to-severe Alzheimer's disease, N. Engl. N. Engl. J. Med., 2003, 348, 1333–1341 CrossRef PubMed.
- J. C. de la Torre, Alzheimer's disease is incurable but preventable, J. Alzheimer's Dis., 2010, 20, 861–870 Search PubMed.
- J. Bond, C. Stave, A. Sganga, O. Vincenzino, B. O’connell and R. L. Stanley, Inequalities in dementia care across Europe: key findings of the Facing Dementia Survey, Int. J. Clin. Pract., 2005, 59, 8–14 CrossRef.
- M. Abonassif, M. Hefnawy, M. Kassem and G. Mostafa, Determination of donepezil hydrochloride in human plasma and pharmaceutical formulations by HPLC with fluorescence detection, Acta Pharm., 2011, 61, 403–413 Search PubMed.
- N. Yasui-Furukori, R. Furuya, T. Takahata and T. Tateishi, Determination of donepezil, an acetylcholinesterase inhibitor, in human plasma by high-performance liquid chromatography with ultraviolet absorbance detection, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2002, 768, 261–265 CrossRef.
- M. Bhateria, R. Ramakrishna, D. B. Pakala and R. S. Bhatta, Development of an LC-MS/MS method for simultaneous determination of memantine and donepezil in rat plasma and its application to pharmacokinetic study, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 1001, 131–139 CrossRef PubMed.
- M. Noetzli, N. Ansermot, M. Dobrinas and C. B. Eap, Simultaneous determination of antidementia drugs in human plasma: Procedure transfer from HPLC-MS to UPLC-MS/MS, J. Pharm. Biomed. Anal., 2012, 64, 16–25 CrossRef PubMed.
- A. H. Khuroo, S. J. Gurule, T. Monif, D. Goswami, A. Saha and S. K. Singh, ESI-MS/MS stability-indicating bioanalytical method development and validation for simultaneous estimation of donepezil, 5-desmethyl donepezil and 6-desmethyl donepezil in human plasma, Biomed. Chromatogr., 2012, 26, 636–649 CrossRef PubMed.
- M. W. Jann, K. L. Shirley and G. W. Small, Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors, Clin. Pharmacokinet., 2002, 41, 719–739 CrossRef PubMed.
- M. A. Radwan, H. H. Abdine, B. T. Al-Quadeb, H. Y. Aboul-Enein and K. Nakashima, Stereoselective HPLC assay of donepezil enantiomers with UV detection and its application to pharmacokinetics in rats, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2006, 830, 114–119 CrossRef PubMed.
- K. Nakashima, K. Itoh, M. Kono, M. N. Nakashima and M. Wada, Determination of donepezil hydrochloride in human and rat plasma, blood and brain microdialysates by HPLC with a short C 30 column, J. Pharm. Biomed. Anal., 2006, 41, 201–206 CrossRef PubMed.
- R. Koeber, H.-H. Kluenemann, R. Waimer, A. Koestlbacher, M. Wittmann, R. Brandl, A. Doerfelt, T. Jahner, D. Melchner and E. Haen, Implementation of a cost-effective HPLC/UV-approach for medical routine quantification of donepezil in human serum, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 881, 1–11 CrossRef PubMed.
- US Food and Drug Administration, Guidance for industry, Bioanalytical method validation, http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm, 2013.
- European Medicines Agency, Guideline on bioanalytical method validation, http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf, 2012.
- K. Yamaoka, T. Nakagawa and T. Uno, Application of Akaike's information criterion (AIC) in the evaluation of linear pharmacokinetic equations, J. Pharmacokinet. Pharmacodyn., 1978, 6, 165–175 CrossRef.
- L. Parnetti, S. Amici, A. Lanari, C. Romani, C. Antognelli, N. Andreasen, L. Minthon, P. Davidsson, H. Pottel and K. Blennow, Cerebrospinal fluid levels of biomarkers and activity of acetylcholinesterase (AChE) and butyrylcholinesterase in AD patients before and after treatment with different AChE inhibitors, J. Neurol. Sci., 2002, 23, s95–s96 CrossRef PubMed.
- G. L. Ellman, K. D. Courtney, V. Andres Jr and R. M. Featherstone, A new and rapid colorimetric determination of acetylcholinesterase activity, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef PubMed.
- O. H. Lowry, N. J. Rosebrough, A. L. Farr and R. J. Randall, Protein measurement with the Folin phenol reagent, J. Biol. Chem., 1951, 193, 265–275 Search PubMed.
- S. Sharma and R. Taliyan, Synergistic effects of GSK-3β and HDAC inhibitors in intracerebroventricular streptozotocin-induced cognitive deficits in rats, Naunyn-Schmiedeberg's Arch. Pharmacol., 2015, 388, 337–349 CrossRef PubMed.
- S. Zhan, B. Ding, Y. Ruan, X. Huang, G. Liu, X. Lv, X. Huang, M. Li, N. Jiang and Q. Shao, A simple blood microdialysis in freely-moving rats for pharmacokinetic–pharmacodynamic modeling study of Shengmai injection with simultaneous determination of drug concentrations and efficacy levels in dialysate, J. Pharm. Biomed. Anal., 2018, 154, 23–30 CrossRef PubMed.
- S. Zhan, W. Guo, Q. Shao, X. Fan, Z. Li and Y. Cheng, A pharmacokinetic and pharmacodynamic study of drug–drug interaction between ginsenoside Rg1, ginsenoside Rb1 and schizandrin after intravenous administration to rats, J. Ethnopharmacol., 2014, 152, 333–339 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03379j |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a
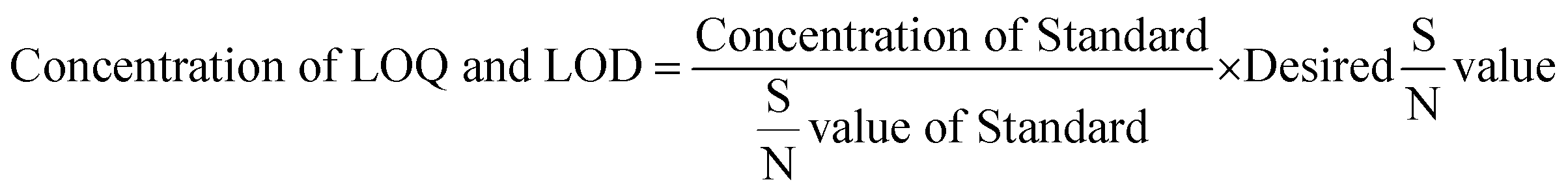
 , 1/y, 1/y2, 1/
, 1/y, 1/y2, 1/ ) wherein, best weighting factors were chosen as 1/x2 as these showed minimum percentage relative error (% RE) (ESI; Table S4†). The observed mean back calculated concentrations with accuracy (% bias) and precision (% CV) are presented in Table 2. At LLOQ (n = 6) accuracy (% bias) was in the range of 3.74% with a % CV of 6.19%. LOD and LLOQ were established as 20 ng mL−1 and 50 ng mL−1 respectively.
) wherein, best weighting factors were chosen as 1/x2 as these showed minimum percentage relative error (% RE) (ESI; Table S4†). The observed mean back calculated concentrations with accuracy (% bias) and precision (% CV) are presented in Table 2. At LLOQ (n = 6) accuracy (% bias) was in the range of 3.74% with a % CV of 6.19%. LOD and LLOQ were established as 20 ng mL−1 and 50 ng mL−1 respectively.